

Abstract
The neurotensin receptor 1, NTS1, is a G protein-coupled receptor with seven transmembrane domains (TM) that mediates most of the known effects of the neuropeptide. Our previous studies have pointed to extracellular loop 3 and adjacent TM7 as being potentially involved in agonist-induced activation of the NTS1.
Here we investigated residues in these domains that might be involved in transconformational activation of the rat NTS1. Single amino acid mutated receptors were expressed in COS cells and inositol phosphate (IP) and cyclic AMP productions were studied.
The F358A mutation in TM7 resulted in a time- and receptor concentration-dependent increase in spontaneous IP production. At expression levels of 12 pmol mg−1, agonist-independent IP production was increased 10 fold over basal for the F358A mutant receptor whereas the wild type NTS1 exhibited virtually no spontaneous activity at expression levels of 7.5 pmol mg−1.
Neurotensin remained agonist on the F358A mutant receptor with a maximal effect that amounted to greater than twice basal IP levels. SR 48692 was inverse agonist at the mutant receptor, reversing IP production almost back to the levels measured in wild type NTS1-transfected cells.
Cyclic AMP production was not constitutively activated with the F358A mutant receptor but was stimulated by neurotensin with the same concentration dependence as that observed with the wild type NTS1.
This is the first report, to our knowledge, of a constitutively active mutant of the NTS1. The data are consistent with TM7 being involved in the transconformational changes that lead to agonist-induced coupling of the NTS1 to Gq.
Keywords: Neurotensin, NTS1, G protein-coupled receptor, constitutive activity, mutation
Introduction
Constitutively active mutant receptors, whether naturally occurring or engineered by mutagenesis, have been described for a great number of G protein-coupled receptors (GPCRs) and have provided useful tools to understand the conformational changes associated with receptor activation (Gether & Kobilka, 1998; Scheer & Cotecchia, 1997). Our best knowledge of the intramolecular rearrangements that occur when GPCRs are activated comes from studies with rhodopsin and adrenergic receptors. In both cases, it has been reported that substitution of highly conserved residues, an acidic residue in transmembrane domain (TM) 3 and a basic residue in TM7, resulted in constitutive activity of the mutant receptors (Porter et al., 1996; Robinson et al., 1992). This and other experimental evidence led to the hypothesis that the residues form an inter-helical salt bridge that maintains the receptors in an inactive state and that disruption of the bridge by either photons for rhodopsin or agonist ligands for adrenoceptors leads to receptor activation (Porter & Perez, 1999). Relative movements of TM3 and TM6 were also reported to occur upon activation of rhodopsin or the beta2-adrenergic receptor (Farrens et al., 1996; Gether et al., 1997). From these data and from other studies with a number of GPCRs, it has been proposed that inter-helical constraints maintain GPCRs in an inactive state and that agonist binding relaxes the constraints, thereby allowing rearrangement of TMs and receptor activation by exposition of the intracellular domain(s) that interact with G protein(s) (Gether & Kobilka, 1998).
Neurotensin (NT) is a 13 aminoacid peptide that exerts neuromodulatory functions in the central nervous system and endocrine/paracrine actions in the periphery (Rostene & Alexander, 1997; Vincent, 1995). Three NT receptors, termed NTS1, NTS2 and NTS3 have been identified so far (Vincent et al., 1999). The NTS1 and NTS2 are GPCRs and share 60% homology while the NTS3 belongs to an entirely different family of proteins. The NTS1 has been the most extensively studied of the three NT receptor subtypes and is thought to mediate most of the known central and peripheral effects of the peptide (Vincent et al., 1999). SR 48692, a non peptide NT antagonist that preferentially binds to the NTS1 (Gully et al., 1993), has provided a useful tool to explore the biological role of this receptor (Rostene et al., 1997). Recently, we established tridimensional models of the SR 48692 and NT binding sites in the rat NTS1 (Barroso et al., 2000; Labbe-Jullie et al., 1998). We showed that NT and SR 48692 interact with common residues in TM6 and TM4, and that the agonist or antagonist behaviour of the ligands depends on their ability to make further strong interactions with either the C-terminal side of extracellular loop (E) 3 (agonist) or the connected N-terminal side of TM7 (antagonist). We also showed that the W339A and F344A mutations in E3 markedly decreased the potency of agonist-induced second messenger production (Barroso et al., 2000). This suggests that E3 might play an essential role not only for agonist binding but also for agonist-induced activation of the receptor, and that TM7 might be involved in the transconformational events that lead to G protein activation.
In the present work, a number of rNTS1 receptors point mutated in E3 and TM7 were screened for their ability to constitutively activate G protein coupling. We report that mutating Phe358 in TM7, a residue that lies at the bottom of the SR 48692 binding pocket (Labbe-Jullie et al., 1998), conferred constitutive activity to the receptor with regard to inositol phosphate (IP) production. NT retained its ability to act as a potent agonist whereas, SR 48692 behaved as an inverse agonist on this receptor.
Methods
Materials
Neurotensin was from Neosystem and SR 48692 from Sanofi Recherche. Monoiodo-[125I-Tyr3]-neurotensin was prepared as described in (Bidard et al., 1993). Mutant receptor preparation was previously described (Labbe-Jullie et al., 1998).
Cell culture and transfection
COS M6 cells were grown in Dulbecco's modified Eagle's medium (Gibco) containing 8% foetal bovine serum (Dutcher) and 50 μg ml−1 gentamicine (Sigma) (culture medium). For transient transfection, 100 mm cell culture dishes seeded with 106 cells the day before were washed twice with Tris-buffered saline (mM: Tris 25, NaCl 140, CaCl2 2.3, MgCl2 0.5, Na2HPO4 0.4, pH 7.4) and incubated for 30 min with recombinant pcDNA3 plasmid, from 1 ng to 1 μg, in the presence of DEAE Dextran (0.5 mg ml−1) at room temperature. After 3 h in culture medium supplemented with 100 μM chloroquine, cells were washed twice with Tris-buffered saline, and cultured for 24 h in culture medium. Each batch of transfected cells were then trypsinized and seeded in culture medium for other 24 h in 12- or 6-well cell culture plates for IP or cyclic AMP assays respectively and in 100-mm cell culture dishes for binding assay.
Cell membranes preparation
Twenty-four hours after seeding in 100-mm cell culture dishes, transfected cells were washed twice with phosphate-buffered saline and collected in 5 mM ice cold Tris/HCl, pH 8. After homogenization by repeated passages through a syringe needle and centrifugation at 4°C for 30 min at 100,000×g, cell membranes were resuspended in 300 μl per dish of 5 mM Tris/HCl, pH 7.5, and stored at −20°C. Membrane protein concentration was determined by the Bio Rad Protein Assay.
Inositol phosphate determination
Six hours after seeding in 12-well cell culture plates, cells were labelled with 0.5 μCi 3H-myo-inositol (ICN) in culture medium for 18 h. After two washes with Earle buffer (mM: HEPES 25, Tris 25, NaCl 140, KCl 5, CaCl2 1.8, MgCl2 0.9, glucose 5 containing 0.1% bovine serum albumin), cells were incubated for 15 min at 37°C in 1 ml of Earle buffer. For IP production as a function of receptor expression, cells were then incubated for 30 min in Earle buffer supplemented by 20 mM LiCl (LiCl-Earle buffer). For IP production as a function of time, cells were then incubated for varying time ranging from 0 to 30 min in LiCl-Earle buffer. For NT or SR 48692 effect measurement, after 30 min in LiCl-Earle buffer, drugs were added in 10 μl of Earle buffer for additional 30 min incubation. The reaction was stopped by 750 μl of ice cold 10 mM HCOOH. After 1 h at 4°C, the supernatant was collected and neutralized by 3 ml of 5 mM NH4OH. Total [3H]-inositol phosphates (IP) were separated from free [3H]-inositol on Dowex AG1-X8 (Bio Rad) chromatography by eluting successively with 5 ml of water and 4 ml of 40 mM and 1 M ammonium formate buffer, pH 5.5. The radioactivity contained in the 1 M fraction was counted after addition of 5 ml Ecolume (ICN).
Cyclic AMP determination
Six hours after seeding in 6-well cell culture plates, cells were labelled with 1 μCi [2,8-3H]-adenine (ICN) in culture medium for 18 h. After two washes with Earle buffer (mM: HEPES 25, Tris 25, NaCl 140, KCl 5, CaCl2 1.8, MgCl2 0.9, glucose 5 containing 0.1% bovine serum albumin), cells were incubated for 10 min at 37°C in 500 μl of Earle buffer supplemented by 1 mM 3-isobutyl-1-methylxanthine (IBMX-Earle buffer). Then, cells were incubated for 20 min with varying concentration of NT in IBMX-Earle buffer. The incubation medium was removed and the reaction was stopped by 500 μl of ice cold 5% TCA containing 2 mM of unlabelled cyclic AMP and 2 mM ATP as carriers. After 30 min at 4°C, the supernatant was collected and each well was washed with 500 μl of water and the wash was combined with the supernatant fraction (final sample volume: 1 ml). Samples were loaded on 1.2 ml Dowex 50W-X4 columns (200 – 400 mesh, hydrogen form, Fluka), pre-equilibrated in water. First, the columns were rinsed with 3 ml of H2O which eluted most of the [3H]-ATP. Then, the Dowex columns were eluted with 10 ml of H2O that were directly loaded on 0.8 ml alumina (Sigma) columns equilibrated in 100 mM imidazole-HCl buffer pH 7.5. Each alumina column was subsequently eluted with 6 ml of imidazole buffer. Eluates, containing [3H]-cAMP, were mixed with 7 ml of Ecolume (ICN) and counted for radioactivity.
Binding experiments
Saturation experiments were carried out for 20 min at room temperature with 0.01 to 2 nM monoiodo-[125I]-neurotensin and 1 to 10 μg of cell membrane proteins in a final volume of 250 μl of 50 mM Tris/HCl, pH 7.5, containing 0.1% bovine serum albumin and 0.8 mM 1,10-phenanthroline. The reaction was stopped by addition of 2 ml of ice cold buffer and filtration on cellulose acetate filter (0.2 μm, Sartorius) followed by two washes of the tube and filter with 2 ml of the same buffer. Non specific binding was determined in the presence of 1 μM unlabelled neurotensin. Data were analysed by the LIGAND software (Munson & Rodbard, 1980).
Results
In the course of mapping the binding sites of SR 48692 and NT in the rNTS1, we measured the binding affinity of both agonist and antagonist ligands on a number of single site mutated receptors in E3 and in TM7 (Barroso et al., 2000; Labbe-Jullie et al., 1998; Richard et al., 2002). Here, the mutant receptors (with mutations bearing on residues F331, Y333, W339, F344, F346, H348, Y349, F350 and F358) were transiently transfected in COS cells with 1 μg of plasmid and screened for their ability to constitutively activate IP production. At the level of expression obtained for each mutant receptor, only one mutation, F358A that lies in TM7 two helical turns below the cell surface, resulted in elevated basal intracellular IP levels and was characterized in more details with respect to constitutive activity. The data in Figure 1a show that basal IP levels rose linearly with time, increasing 5 fold over a 30-min interval. Such a time-dependent increase in IP concentrations was not seen with the wild type receptor (Figure 1a). In addition, basal IP production measured after a 30 min incubation was proportional to the amount of the F358A mutant expressed in COS cells whereas it barely varied as a function of wild type rNTS1 expression (Figure 1b). Altogether, these data are consistent with the F358A mutant rNTS1 being constitutively active.
Figure 1.

Constitutive inositol phosphate production in COS cells transfected with wild type rNTS1 or F358A mutant receptor. (a) Forty-eight hours after transfection with 1 μg recombinant pcDNA3 plasmid, basal IP production was measured as a function of time. (b) Twenty-four hours after transfection with increasing recombinant pcDNA3 plasmid, ranging from 1 ng to 1 μg, each batch of transfected cells was divided into two parts used 24 h later either to measure basal IP production after 30 min incubation or to measure receptor concentration by monoiodo-125I-neurotensin saturation experiments on cell membrane preparation. The values are the means±s.e.mean from three independent experiments.
We then tested the effects of NT and SR 48692 on IP production after a 30 min incubation. As shown in Figure 2a, despite the constitutive activity of the F358A receptor, NT was still able to stimulate IP production with an EC50 value of 2.1±0.9 nM (n=3) and a maximal effect that amounted to greater than twice basal IP levels. For comparison, the EC50 value for NT-stimulated IP production with the wild type rNTS1 was 0.8±0.2 nM (n=3), as previously reported (Barroso et al., 2000). Interestingly, SR 48692 concentration-dependently inhibited the constitutive activity of the mutant receptor (Figure 2b) with an IC50 value of 14±7 nM (n=3), in good agreement with the previously published Kd value (9±2 nM, n=3) of the antagonist for the mutant receptor (Labbe-Jullie et al., 1998). The non peptide compound behaved as an efficient inverse agonist as IP production was reversed almost back to the levels measured in wild type NTS1-transfected cells.
Figure 2.

Concentration-response curves for the effect of neurotensin and SR 48692 on inositol phosphate production in COS cells transfected with the F358A mutant rNTS1. (a) Concentration-response curve for NT-stimulated IP production. (b) Concentration response curve for SR 48692-induced inhibition of IP production. The values are the means±s.e.mean from three independent experiments.
Previous studies have shown that high expression of the NTS1 in transfected cell systems led to its coupling to Gs, though the potency of NT for stimulating cyclic AMP production was lower than that for inducing IP production (Yamada et al., 1994). In agreement with these data, Figure 3 shows that NT stimulated cyclic AMP production in wild type rNTS1-expressing COS cells with an EC50 value of 10±6 nM (n=3), that was 13 fold higher than that obtained for IP production. The F358A receptor was also tested for its ability to increase basal cyclic AMP production in COS cells as a function of incubation time and receptor expression levels. No constitutive stimulation of cyclic AMP production was observed with the mutant at the longest incubation time and highest expression level (Table 1). However, NT concentration-dependently increased cyclic AMP levels with an EC50 value of 12±5 nM (n=3) that was similar to that obtained with the wild type receptor (Figure 3).
Figure 3.
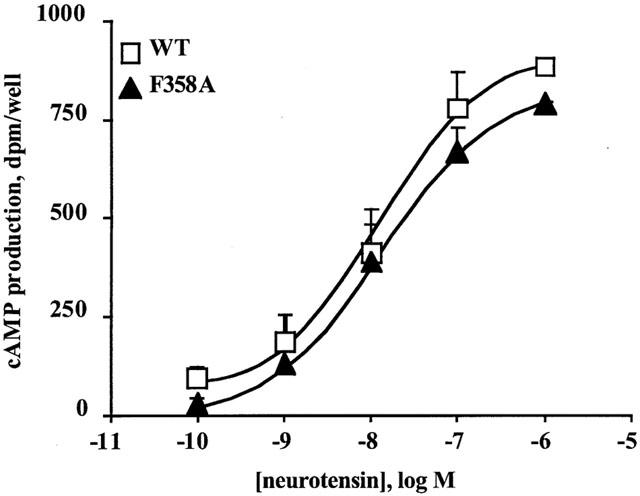
Effect of neurotensin on cyclic AMP production in COS cells transfected with wild type rNTS1 or F358A mutant receptor. Concentration-response curves for NT-stimulated cyclic AMP production were performed with the wild type receptor or F358A mutant receptor. Cyclic AMP values are expressed as the difference between NT-stimulated cyclic AMP levels minus basal cyclic AMP levels, in the presence of 1 mM IBMX. The values are the means±s.e.mean from three independent experiments.
Table 1.
Values of basal cyclic AMP levels for the wild-type rNTS1 and F359A mutant receptors as a function of time and receptor expression levels

Discussion
The major finding of the present study is that one single amino acid mutation (F358A) in TM7 of the rNTS1 conferred constitutive activity to the receptor with regard to IP production. This is to our knowledge the first report on a constitutively active mutant of the NTS1. At expression levels of 12 pmol mg−1, agonist-independent IP production was increased 10 fold over basal for the F358A mutant receptor. As the wild type NTS1 exhibited virtually no spontaneous activity at the highest expression level tested (7.5 pmol mg−1), the F358A mutation appears quite efficient in activating the NTS1. The F358A mutant receptor shared a number of pharmacological properties with other constitutively active GPCRs. First, the natural agonist NT retained the ability to efficiently activate the mutant receptor more than 2 fold over its spontaneous activity. It is a most common observation that agonists are still able to stimulate constitutively active GPCRs (Burstein et al., 1997; Gaudin et al., 1998; Rosenkilde & Schwartz, 2000). Second, the non peptide antagonist SR 48692 behaved as an efficient inverse agonist, decreasing IP production almost back to the levels observed in wild type receptor-transfected cells. Again, this is a widely observed property of constitutively active GPCRs, and, concerning neuropeptide GPCRs in particular, it has often been observed that non peptide molecules defined as antagonists at wild type receptors (that are often devoid of measurable spontaneous actitvity) turned out to be inverse agonists at constitutively active mutant receptors (Chidiac et al., 1994; Shryock et al., 1998). Third, the F358A mutant constitutively activated the IP-generating pathway but did not influence basal cyclic AMP production. Such a preference of the constitutively active state of a GPCR for a transduction pathway has been documented for other receptors that can signal through different G proteins (Cohen et al., 1997; Parma et al., 1995).
Mutations that produce constitutively active receptors have been described for a number of GPCRs (Pauwels & Wurch, 1998) and fall in three categories: (i) those located in extracellular domains, as reported for the thrombin and thyroid-stimulating hormone receptors (Nanevicz et al., 1996; Parma et al., 1995), that presumably induce a change in receptor conformation similar to that caused by agonist binding; (ii) those located in the intracellular domains that are thought to interact with G proteins, as examplified by studies with adrenergic receptors (Scheer & Cotecchia, 1997); and (iii) mutations within the TMs that presumably disrupt helix-helix interactions and relax the receptor in an agonist-independent active state (Porter & Perez, 1999). The best documented example of the latter class of mutations concerns rhodopsin in which the chromophore retinal is covalently linked to the amine group of Lys296 in TM7 through a protonated Schiff base that makes an ionic link with Glu113 in TM3, thereby maintaining the photoreceptor in an inactive state (Cohen et al., 1992). Light triggers the isomerization of retinal, thus disrupting the salt bridge and activating rhodopsin. Disruption of the salt bridge by mutation of either Lys296 or Glu113 results in constitutive activity (Cohen et al., 1992). It was further shown that activation of rhodopsin involves movements of TM7 and TM6 relative to TM3. Similar molecular mechanisms have been reported to operate for adrenergic receptors (Gether et al., 1997). In the case of the NTS1, the activating mutation concerns a residue, Phe358, that lies in TM7 at the bottom of the SR 48692 binding pocket and whose side chain points toward TM6 (Labbe-Jullie et al., 1998). Interestingly, sequence alignment of TM7 in the rNTS1 and rhodopsin shows that Phe358 occupies a position that is one residue away from that of Lys296 in rhodopsin. Although Lys296 in rhododpsin points toward TM3 whereas Phe358 of the NTS1 points toward TM6, it may be suggested by analogy with the photoreceptor that Phe358 may be involved in maintaining the NTS1 in an inactive state through TM-TM interactions. Disruption by mutagenesis of a presumed interaction between two residues in TM3 and TM7 of the AT1A angiotensin II receptor was also reported to elicit constitutive activity (Groblewski et al., 1997). We have previously noticed that the binding mode of NT on the NTR1 resembled that of peptide agonists at the delta opioid receptor (Barroso et al., 2000; Valiquette et al., 1996). It was recently reported that mutating Tyr308 in TM7 of the delta opioid receptor, a position equivalent to that of Phe358 in the rNTS1, constitutively activated the former (Befort et al., 1999), thus further underlying the similarities between the two receptors. Altogether, these observations suggest that TM7 may play a key role in the activation process of a number of GPCRs.
Previous mutagenesis studies have shown that the C-terminal side of the third intracellular loop (I3) of the NTS1 is involved in the activation of Gq but not Gs (Yamada et al., 1994). The C-terminal part of I3 is connected to TM6. Given the locations of the agonist and antagonist binding sites and of the Gq-activating F358A mutation in the NTS1, it is tempting to speculate that agonist-induced receptor activation is initiated, at least in part, through relative movements of TM6 and TM7. NT, by anchoring strongly to the TM6/E3 junction and to the C-terminal part of E3 would favour these movements, whereas SR 48692, by binding tightly to TM6 and TM7 would prevent them and lock the receptor in an inactive state. The F358A mutation in TM7 would disrupt some of the interactions that maintain the receptor in the inactive state and partially relax it in an active state. SR 48692 binding to the F358A mutant receptor would bring it back to the inactive state, thus revealing the inverse agonist properties of the non peptide compound. Although the model proposed here for the transconformation of the rNTS1 remains speculative, it is consistent with a number of mutagenesis studies proposing that TM/TM interactions are important for maintaining GPCRs in an inactive state (Gether & Kobilka, 1998). Further work will be required for validating our hypothesis. In particular, it should be interesting to search for other TM mutations that activate the receptor and to identify residue(s) that could possibly interact with Phe358 to constrain the receptor in its inactive state.
Acknowledgments
We are grateful to Paul Vigne for fruitful discussions and to Gisèle Jarretou for expert technical assistance.
Abbreviations
- E
extracellular loop
- GPCR
G protein-coupled receptor
- I
intracellular loop
- IP
inositol phosphate
- NT
neurotensin
- NTS1
neurotensin receptor 1
- NTS2
neurotensin receptor 2
- NTS3
neurotensin receptor 3
- TM
transmembrane domain
References
- BARROSO S., RICHARD F., NICOLAS-ETHEVE D., REVERSAT J.L., BERNASSAU J.M., KITABGI P., LABBE-JULLIE C. Identification of residues involved in neurotensin binding and modeling of the agonist binding site in neurotensin receptor 1. J. Biol. Chem. 2000;275:328–336. doi: 10.1074/jbc.275.1.328. [DOI] [PubMed] [Google Scholar]
- BEFORT K., ZILLIOX C., FILLIOL D., YUE S., KIEFFER B.L. Constitutive activation of the delta opioid receptor by mutations in transmembrane domains III and VII. J. Biol. Chem. 1999;274:18574–18581. doi: 10.1074/jbc.274.26.18574. [DOI] [PubMed] [Google Scholar]
- BIDARD J.N., DE NADAI F., ROVERE C., MOINIER D., LAUR J., MARTINEZ J., CUBER J.C., KITABGI P. Immunological and biochemical characterization of processing products from the neurotensin/neuromedin N precursor in the rat medullary thyroid carcinoma 6-23 cell line. Biochem. J. 1993;291:225–233. doi: 10.1042/bj2910225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURSTEIN E.S., SPALDING T.A., BRANN M.R. Pharmacology of muscarinic receptor subtypes constitutively activated by G proteins. Mol. Pharmacol. 1997;51:312–319. doi: 10.1124/mol.51.2.312. [DOI] [PubMed] [Google Scholar]
- CHIDIAC P., HEBERT T.E., VALIQUETTE M., DENNIS M., BOUVIER M. Inverse agonist activity of beta-adrenergic antagonists. Mol. Pharmacol. 1994;45:490–499. [PubMed] [Google Scholar]
- COHEN D.P., THAW C.N., VARMA A., GERSHENGORN M.C., NUSSENZVEIG D.R. Human calcitonin receptors exhibit agonist-independent (constitutive) signaling activity. Endocrinology. 1997;138:1400–1405. doi: 10.1210/endo.138.4.5046. [DOI] [PubMed] [Google Scholar]
- COHEN G.B., OPRIAN D.D., ROBINSON P.R. Mechanism of activation and inactivation of opsin: role of Glu113 and Lys296. Biochemistry. 1992;31:12592–12601. doi: 10.1021/bi00165a008. [DOI] [PubMed] [Google Scholar]
- FARRENS D.L., ALTENBACH C., YANG K., HUBBELL W.L., KHORANA H.G. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- GAUDIN P., MAORET J.J., COUVINEAU A., ROUYER-FESSARD C., LABURTHE M. Constitutive activation of the human vasoactive intestinal peptide 1 receptor, a member of the new class II family of G protein-coupled receptors. J. Biol. Chem. 1998;273:4990–4996. doi: 10.1074/jbc.273.9.4990. [DOI] [PubMed] [Google Scholar]
- GETHER U., KOBILKA B.K. G protein-coupled receptors. II. Mechanism of agonist activation. J. Biol. Chem. 1998;273:17979–17982. doi: 10.1074/jbc.273.29.17979. [DOI] [PubMed] [Google Scholar]
- GETHER U., LIN S., GHANOUNI P., BALLESTEROS J.A., WEINSTEIN H., KOBILKA B.K. Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. EMBO J. 1997;16:6737–6747. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROBLEWSKI T., MAIGRET B., LARGUIER R., LOMBARD C., BONNAFOUS J.C., MARIE J. Mutation of Asn111 in third transmembrane domain of the AT1A angiotensin II receptor induces its constitutive activation. J. Biol. Chem. 1997;272:1822–1826. doi: 10.1074/jbc.272.3.1822. [DOI] [PubMed] [Google Scholar]
- GULLY D., CANTON M., BOIGEGRAIN R., JEANJEAN F., MOLIMARD J.C., PONCELET M., GUEUDET C., HEAULME M., LEYRIS R., BROUARD A., PELAPRAT D., LABBÉ-JULLIÉ C., MAZELLA J., SOUBRIÉ P., MAFFRAND J.P., ROSTÈNE W., KITABGI P., LE FUR G. Biochemical and pharmacological profile of a potent and selective nonpeptide antagonist of the neurotensin receptor. Proc. Natl. Acad. Sci. U.S.A. 1993;90:65–69. doi: 10.1073/pnas.90.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LABBE-JULLIE C., BARROSO S., NICOLAS-ETEVE D., REVERSAT J.L., BOTTO J.M., MAZELLA J., BERNASSAU J.M., KITABGI P. Mutagenesis and modeling of the neurotensin receptor NTR1. Identification of residues that are critical for binding SR 48692, a nonpeptide neurotensin antagonist. J. Biol. Chem. 1998;273:16351–16357. doi: 10.1074/jbc.273.26.16351. [DOI] [PubMed] [Google Scholar]
- MUNSON P.J., RODBARD D. Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- NANEVICZ T., WANG L., CHEN M., ISHII M., COUGHLIN S.R. Thrombin receptor activating mutations. Alteration of an extracellular agonist recognition domain causes constitutive signaling. J. Biol. Chem. 1996;271:702–706. doi: 10.1074/jbc.271.2.702. [DOI] [PubMed] [Google Scholar]
- PARMA J., VAN SANDE J., SWILLENS S., TONACCHERA M., DUMONT J., VASSART G. Somatic mutations causing constitutive activity of the thyrotropin receptor are the major cause of hyperfunctioning thyroid adenomas: identification of additional mutations activating both the cyclic adenosine 3′,5′-monophosphate and inositol phosphate-Ca2+cascades. Mol. Endocrinol. 1995;9:725–733. doi: 10.1210/mend.9.6.8592518. [DOI] [PubMed] [Google Scholar]
- PAUWELS P.J., WURCH T. Review: amino acid domains involved in constitutive activation of G- protein-coupled receptors. Mol. Neurobiol. 1998;17:109–135. doi: 10.1007/BF02802027. [DOI] [PubMed] [Google Scholar]
- PORTER J.E., HWA J., PEREZ D.M. Activation of the alpha1b-adrenergic receptor is initiated by disruption of an interhelical salt bridge constraint. J. Biol. Chem. 1996;271:28318–28323. doi: 10.1074/jbc.271.45.28318. [DOI] [PubMed] [Google Scholar]
- PORTER J.E., PEREZ D.M. Characteristics for a salt-bridge switch mutation of the alpha(1b) adrenergic receptor. Altered pharmacology and rescue of constitutive activity. J. Biol. Chem. 1999;274:34535–34538. doi: 10.1074/jbc.274.49.34535. [DOI] [PubMed] [Google Scholar]
- RICHARD F., BARROSO S., NICOLAS-ETHEVE D., KITABGI P., LABBE-JULLIE C.Impaired G protein coupling of the neurotensin receptor by mutations in extracellular loop 3 Eur. J. Pharmacol. 2002. in press [DOI] [PubMed]
- ROBINSON P.R., COHEN G.B., ZHUKOVSKY E.A., OPRIAN D.D. Constitutively active mutants of rhodopsin. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-b. [DOI] [PubMed] [Google Scholar]
- ROSENKILDE M.M., SCHWARTZ T.W. Potency of ligands correlates with affinity measured against agonist and inverse agonists but not against neutral ligand in constitutively active chemokine receptor. Mol. Pharmacol. 2000;57:602–609. doi: 10.1124/mol.57.3.602. [DOI] [PubMed] [Google Scholar]
- ROSTENE W., ALEXANDER M. Neurotensin and neuroendocrine regulation. Front. Neuroendocrinol. 1997;18:115–173. doi: 10.1006/frne.1996.0146. [DOI] [PubMed] [Google Scholar]
- ROSTENE W., AZZI M., BOUDIN H., LEPEE I., SOUAZE F., MENDEZUBACH M., BETANCUR C., GULLY D. Use of nonpeptide antagonists to explore the physiological roles of neurotensin. Focus on brain neurotensin/dopamine interactions. Ann. N.Y. Acad. Sci. 1997;814:125–141. doi: 10.1111/j.1749-6632.1997.tb46151.x. [DOI] [PubMed] [Google Scholar]
- SCHEER A., COTECCHIA S. Constitutively active G protein-coupled receptors: potential mechanisms of receptor activation. J. Recept. Signal. Transduct. Res. 1997;17:57–73. doi: 10.3109/10799899709036594. [DOI] [PubMed] [Google Scholar]
- SHRYOCK J.C., OZECK M.J., BELARDINELLI L. Inverse agonists and neutral antagonists of recombinant human A1 adenosine receptors stably expressed in chinese hamster ovary cells. Mol. Pharmacol. 1998;53:886–893. [PubMed] [Google Scholar]
- VALIQUETTE M., VU H.K., YUE S.Y., WAHLESTEDT C., WALKER P. Involvement of Trp-284, Val-296, and Val-297 of the human delta-opioid receptor in binding of delta-selective ligands. J. Biol. Chem. 1996;271:18789–18796. doi: 10.1074/jbc.271.31.18789. [DOI] [PubMed] [Google Scholar]
- VINCENT J., MAZELLA J., KITABGI P. Neurotensin and neurotensin receptors. Trends Pharmacol. Sci. 1999;20:302–309. doi: 10.1016/s0165-6147(99)01357-7. [DOI] [PubMed] [Google Scholar]
- VINCENT J.P. Neurotensin receptors: binding properties, transduction pathways, and structure. Cell. Mol. Neurobiol. 1995;15:501–512. doi: 10.1007/BF02071313. [DOI] [PubMed] [Google Scholar]
- YAMADA M., YAMADA M., WATSON M.A., RICHELSON E. Deletion mutation in the putative third intracellular loop of the rat neurotensin receptor abolishes polyphosphoinositide hydrolysis but not cyclic AMP formation in CHO-K1 cells. Mol. Pharmacol. 1994;46:470–476. [PubMed] [Google Scholar]