

Introduction
Since the first description of bradykinin more than 50 years ago (Rocha e Silva et al., 1949), actions of the peptide in a variety of physiological and pathological responses have been extensively researched (reviewed in Bhoola et al., 1992; Wirth et al., 1997; Calixto et al., 2000). In the cardiovascular system, the classical action of bradykinin is vasodilatation, mediated in several vascular beds by the release of nitric oxide (NO) and prostacyclin (Hatta et al., 1997; Wirth et al., 1997). In the heart, exogenously-administered bradykinin is a potent coronary artery vasodilator substance, although the contribution of endogenous bradykinin to the regulation of coronary vascular tone is unclear. Several actions of bradykinin in the heart are of particular interest as they are independent of the vasodilator actions of the peptide. Such actions include the modulation of cell growth and division in the heart and the modulation of myocardial responses to ischaemia-reperfusion. The ability of bradykinin to act as an endogenous cytoprotective mediator in the ischaemic myocardium has received a great deal of attention in recent years. Much of this research on bradykinin has been fuelled by a growing appreciation of ischaemic preconditioning, an adaptive mechanism in which bradykinin plays an important role. This review focuses on the cytoprotective actions of bradykinin in the ischaemic and reperfused myocardium, discusses its role in the ischaemic preconditioning response, and examines the potential for manipulating endogenous bradykinin for therapeutic benefit in myocardial ischaemia.
Myocardial ischaemia-reperfusion injury
Acute thrombotic occlusion of a major coronary artery, leading to myocardial ischaemia, is a leading cause of death and morbidity in the industrialized and developing countries. Ischaemia rapidly produces profound metabolic, functional and morphological changes within myocardium, the severity of which are ultimately determined by the duration of impaired oxygenation and substrate delivery (Ganz & Braunwald, 1997). The principal metabolic changes centre around the failure of adequate adenosine triphosphate (ATP) generation by oxidative phosphorylation and the accumulation of byproducts of anaerobic glycolysis, particularly H+. The functional consequences of ATP depletion are rapidly manifested as a decrease in contractility and disturbances of a host of homoeostatic processes, including the activities of ion channels and exchangers, cell volume regulation and enzyme reactions. The electrical properties of ischaemic myocardium may be altered to the point where arrhythmogenic mechanisms can promote life theatening tachyarrhythmias. Ultrastructural changes may be detectable within several minutes of the onset of ischaemia. These alterations may be considered reversible if reperfusion of the tissue can be effected promptly. However, ischaemia lasting more than 20 – 30 min will result in irreversible cell injury (Schaper et al., 1992). Without reperfusion to salvage ischaemic myocardium, the most extreme manifestation of irreversible injury is tissue necrosis (myocardial infarction). Prompt reperfusion of the occluded vessel is required to save ischaemic myocardium from sustaining irreversible injury but, paradoxically, reperfusion may be associated with further cellular stress resulting in ‘reperfusion injury'. The development of therapeutic strategies that can attenuate ischaemia-reperfusion injury has been a keen area of research for more than 30 years.
Ischaemic preconditioning of myocardium
Brief antecedent episodes of ischaemia enhance tissue tolerance to a subsequent longer episode of ischaemia. This phenomenon was formally described by Murry et al. (1986) who coined the term ‘preconditioning with ischaemia'. They demonstrated in canine myocardium that four short non-injurious coronary artery occlusions, before a subsequent 40 min coronary occlusion, reduced the development of necrosis during the 40 min occlusion by almost 75% compared to the necrosis in non-preconditioned hearts. This powerful protective effect of antecedant ischaemia was not explained by changes in coronary collateral blood flow, suggesting a fundamental cellular alteration in the response to ischaemia. The protection conferred by preconditioning with ischaemia has been subsequently confirmed in many studies and has excited a huge effort to determine the underlying molecular mechanisms of protection (Cohen et al., 2000).
Preconditioning protocols vary somewhat from one laboratory to another and many endpoints of ischaemic injury have been adopted to assess the extent of protection conferred by preconditioning, including development of necrosis, severity of arrhythmias, post-ischaemic recovery of contractile function and cardiac enzyme release. Striking features of preconditioning are the temporal aspects of the protection. The protection conferred by preconditioning is not absolute in as much as preconditioning in canine myocardium limits infarction produced by a 40 min coronary occlusion but does not protect against a 180 min occlusion (Murry et al., 1986). Protection is lost if the reperfusion period between the brief preconditioning ischaemia and the long ischaemic period is extended beyond 2 or 3 h (van winkle et al., 1991; Kuzuya et al., 1993). However, a further period of protection may be detected many hours later suggesting a biphasic pattern of protection. The early phase of protection, of rapid onset and short duration is the classic preconditioning effect, originally described by Murry et al. (1986), while the delayed phase occurring many hours later and lasting much longer, has been termed ‘second window of protection', ‘delayed preconditioning' or ‘late preconditioning' (Bolli, 2000; Baxter & Ferdinandy, 2001).
Paracrine and autocrine triggers of classical preconditioning
Extensive research has revealed that several endogenous mediators of myocyte, endothelial and neural origin, are generated during the brief preconditioning period, and these act as co-activators (or ‘triggers') of a signal transduction cascade that rapidly results in the acquisition of tolerance to further ischaemia (see Figure 1). Detailed discussion of the involvement of multiple kinase families is beyond the scope of this review and has been comprehensively reviewed elsewhere (Cohen et al., 2000; Baines et al., 2001). The earliest mediator to be examined as an activator of preconditioning was adenosine. In several species, adenosine, released from myocytes as a consequence of ATP breakdown during preconditioning, acts on adenosine A1 receptors and possibly A3 receptors, initiating a signaling cascade. Early experimental evidence to support the involvement of adenosine came from Liu et al. (1991) who showed that adenosine receptor blockade with 8-p-(sulphophenyl)-theophylline (8-SPT) during preconditioning could abolish the infarct-limiting effect. Conversely, transient adenosine A1 receptor activation, but not A2 receptor activation, with selective agonists mimicked the preconditioning effect of brief coronary occlusion in the rabbit (Thornton et al., 1992; Tsuchida et al., 1993). A role for adenosine A3 receptor activation has been proposed by some workers (Armstrong & Ganote, 1994; Liu et al., 1994) but is not clearly resolved (Guo et al., 2001; Kilpatrick et al., 2001). It has since become clear that several other endogenously liberated autocrine/paracrine mediators contribute critically to initiating the process of cellular adaptation. All of these mediators are released or rapidly generated during relatively brief periods of myocardial ischaemia and they include catecholamines (Bankwala et al., 1994), opioid peptides (Schultz & Gross, 2001), reactive oxygen species (Baines et al., 1997) and bradykinin, which is the focus of this article.
Figure 1.
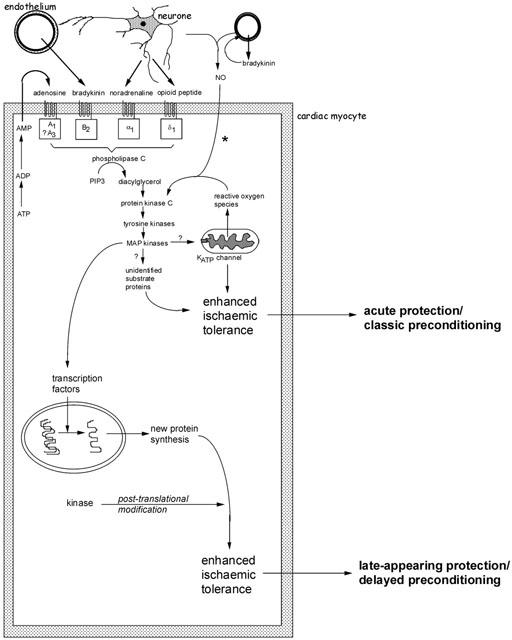
Schematic representation of the major identified pathways of early and delayed forms of preconditioning. Several autocrine/paracrine mediators released during the period of preconditioning ischaemia act on G-protein coupled receptors and are known to participate in the infarct-limiting effect of ischaemic preconditioning. These include adenosine released during ischaemia as a result of ATP breakdown, bradykinin released from vascular endothelium and mediators of neural origin (noradrenaline and opioid peptides). Reactive oxygen species, especially superoxide anion generated as a result of mitochondrial uncoupling, may also act as upstream mediators. A complex signal cascade is activated which involves activation of protein kinase C isoenzymes, tyrosine kinases and mitogen-activated protein kinases. The phosphorylation cascade is thought to result in activation of the ATP-sensitive potassium (KATP) channel on the mitochondrial inner membrane. At present it remains unknown how opening of this channel confers protection during ischaemia. The participation of other ‘cytoprotective' proteins has been proposed, including proteins that suppress or modulate apoptosis and proteins associated with cytoskeletal integrity (αB-crystallin and 27 kDa heat shock protein). *The participation of endogenous NO (of endothelial or neural origin) in initiating the classical preconditioning mechanism may be model specific. Early protection against cell death and infarction is not NO-dependent whereas preconditioning against arrhythmias does involve NO generation. For delayed preconditioning, evidence for the involvement of NO (possibly as a signalling intermediate downstream of bradykinin) is more persuasive and consistent. The distinguishing feature of delayed preconditioning is the co-ordinated regulation of a gene transcription programme as a result of upstream kinase signalling. The delayed phase of protection is dependent on de novo synthesis of inducible proteins. Those thought to be particularly important in the acquisition of delayed tolerance to ischaemia include iNOS, cyclo-oxygenase-2 and intracellular antioxidant enzymes such as manganese – superoxide dismutase. For more detailed discussion see Baxter & Ferdinandy (2001).
Formation and catalytic degradation of bradykinin
Bradykinin is one of several oligopeptides called kinins. The most important physiologically active kinins are kallidin (Lys-bradykinin), bradykinin, and des-Arg9-bradykinin. The interested reader is referred to a recent review by Blais et al. (2000) for a detailed account of kinin synthesis. Kallidin and bradykinin are synthesized by kallikreins acting on kininogen precursor molecules (summarized in Figure 2a). Precursors of kallikreins are found in plasma (pre-kallikreins) and in tissues (pro-kallikreins) and are activated by a variety of chemical and biological stimuli, including activated factor XII (Hageman factor). Circulating kininogens are synthesized primarily in liver and include a high molecular weight kininogen (88 – 115 kDa according to species) and a low molecular weight kininogen (50 – 68 kDa).
Figure 2.
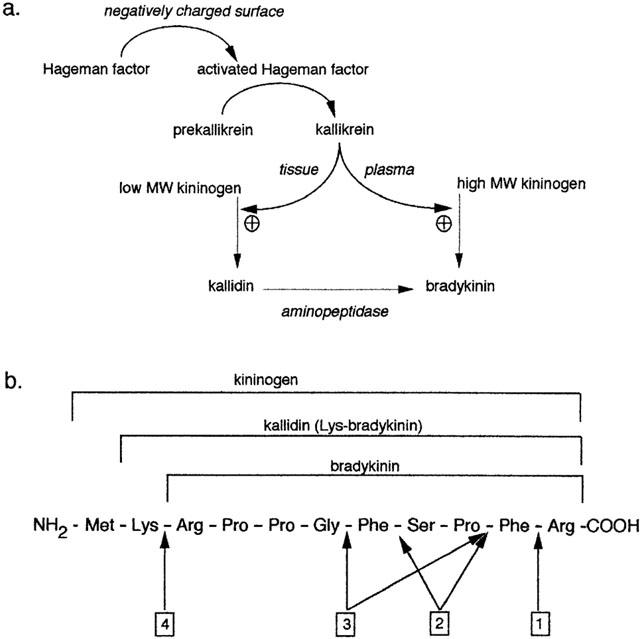
(a) Synthesis of bradykinin. The activated form of Hageman factor promotes the conversion of prekallikrein to kallikrein. In rat, both plasma and tissue kallikrein catalyse the formation of bradykinin. In humans, however, plasma kallikrein generates bradykinin using high molecular weight kininogens and tissue kallikrein generates kallidin using low molecular weight kininogens. (b) Basic amino acid sequence of bradykinin and precursors. Arrows designate the peptide bonds cleaved by: 1 kininase-I; 2 angiotensin converting enzyme (kininase-II); 3 neutral endopeptidase; 4 aminopeptidase.
Vascular endothelial cells are the primary source of bradykinin in the heart (Linz et al., 1996; Wirth et al., 1997). Enzymatic cleavage of pre-kallikrein generates kallikrein at the endothelial cell surface. Circulating kininogen is then cleaved by kallikrein to generate the kinin at the endothelial cell surface. The mechanisms by which pre-kallikrein is activated may be factor XII-dependent or -independent. In the absence of endothelial cell injury and hence contact binding of factor XII, the mechanism of kininogen attraction may involve a cell surface receptor complex. However, this mechanism is presently not well understood. It has been proposed that isolated cardiac myocytes can synthesize kinins (Matoba et al., 1999) but this possibility remains to be investigated more fully. A number of studies have provided evidence that even during brief preconditioning periods of ischaemia tissue and plasma bradykinin levels increase markedly (Linz et al., 1996; Schulz et al., 1998; Campbell, 2000; Pan et al., 2000). Bradykinin is generated in isolated tissues and endothelial cells in the absence of plasma (Baumgarten et al., 1993; Ahmad et al., 1996; Linz et al., 1996). Bradykinin released during ischaemia has been shown to primarily originate from endothelial cells (Linz et al., 1996; Wirth et al., 1997) but the precise molecular pathological mechanism leading to bradykinin generation during brief ischaemia is not understood.
Once released, bradykinin is rapidly degraded into inactive metabolites (Bhoola et al., 1992) (Figure 2b). Enzymes that degrade bradykinin are generically referred to as ‘kininases' or ‘kinin peptidases'. The most important of these metalloproteases are angiotensin converting enzyme (ACE; syn. kininase II; EC 3.4.15.1), neutral endopeptidase (NEP; syn. NEP 24.11; enkephalinase; EC 3.4.24.11), kininase I (syn. carboxypeptidase N; EC 3.4.17.3), carboxypeptidase M (syn. membrane-bound kininase I), and aminopeptidase P (syn. prolyl-aminopeptidase; EC 3.4.11.9). Other enzymes include endopeptidase (EC 3.4.24.15), endothelin converting enzyme (ECE) and prolyl endopeptidase but their contribution is small (Brown & Vaughan, 1998; Erdos & Skidgel, 1997; Piedimonte et al., 1994). ACE is regarded as the most important kininase in most species (Ahmad et al., 1996; Dumoulin et al., 1998; Ersahin & Simmons, 1997; Hornig et al., 1997; Kuoppala et al., 2000). ACE has a higher affinity for bradykinin than for angiotensin I, resulting in more favourable kinetics for bradykinin than for angiotensin I degradation (Zisman, 1998). Hence ACE may be regarded as being primarily a kininase rather than an angiotensinase (Blais et al., 2000).
The actions of kinins are mediated by two receptor subtypes, distinguishable on the basis of both pharmacological and molecular characterization (Hall, 1992; 1997). The bradykinin B2 receptor usually predominates, with the bradykinin B1 receptor only being expressed under pathological conditions (Bhoola et al., 1992). The B2 receptor belongs to the family of heptahelical G-protein coupled receptors, which initiate the generation of inositol triphosphate and diacylglycerol, with subsequent activation of PKC (Derian & Moskowitz, 1986; Minshall et al., 1995; Morgan-Boyd et al., 1987). Highly specific antagonists at the B2 receptor include the bradykinin-derivative icatibant (HOE140) and the non-peptide FR173657 (Aramori et al., 1997).
Endogenous bradykinin and its role in ischaemic preconditioning
Schoelkens et al. (1988) were the first to report the cardioprotective effects of exogenously administered bradykinin. In an isolated working rat heart preparation subjected to ischaemia followed by reperfusion, perfusion with bradykinin 10−10 mol l−1 resulted in better recovery of coronary flow and cardiac work during reperfusion, a reduction in the release of soluble markers of tissue injury, and improvement of metabolic efficiency. Following this report, intracoronary bradykinin administration, at a dose that did not induce coronary vasodilatation, was found to suppress both ischaemia- and reperfusion-induced arrhythmias in an anaesthetized canine model of epicardial coronary artery occlusion (Végh et al., 1991). In a porcine coronary occlusion model, infusion of bradykinin after the onset of coronary occlusion was found to attenuate plasma creatine kinase concentration. (Tio et al., 1991; Tobe et al., 1991). Subsequently, Végh et al. (1993) showed the anti-arrhythmic effect of bradykinin to be mediated by NO. These workers suggested that bradykinin might be a ‘primary mediator' of ischaemic preconditioning.
In 1994, two reports provided evidence for a primary role of endogenous bradykinin in mediating ischaemic preconditioning. Wall et al. (1994) reported that icatibant abolished the protective effects of preconditioning against infarction in an anaesthetized open-chest rabbit preparation with coronary artery occlusion. They also found that protection, comparable to that induced by preconditioning, could be produced by direct infusion of exogenous bradykinin (Wall et al., 1994). Almost simultaneously, Végh et al. (1994) documented the abrogation by icatibant of the anti-arrhythmic effects of preconditioning in the canine coronary occlusion model. Goto et al. (1995) subsequently confirmed the finding of Wall et al. (1994) that icatibant blocked the infarct-limiting effect of preconditioning in rabbit heart in vivo. However, they were unable to abolish the protective effect of preconditioning with icatibant in an isolated buffer-perfused rabbit heart preparation even when the preconditioning stimulus was increased from one to four 5 min cycles of ischaemia. This apparent non-participation of bradykinin in preconditioning of the buffer-perfused heart was attributed to the lack of blood-borne kininogens. Similarly, Bugge & Ytrehus (1996) found that icatibant did not block the protective effects of preconditioning in an isolated rat heart preparation. However, Bouchard et al. (1998) found that preconditioning of isolated rat heart attenuated post-ischaemic endothelial dysfunction. This effect was not abolished by icatibant but was reversed by Lys [Leu8] Des-Arg9-bradykinin, a bradykinin B1 receptor antagonist.
Despite these inconsistencies in the literature examining the participation of endogenous bradykinin in isolated heart preparations, the ability of exogenously administered bradykinin to mimic ischaemic preconditioning has been confirmed by numerous investigators in a variety of models. These include the isolated rabbit heart with infarct size as the endpoint (Goto et al., 1995); the isolated rat heart with infarct size (Goto et al., 1995; Bugge & Ytrehus, 1996; Starkopf et al., 1997; Feng & Rosenkranz, 1999; Feng et al., 2000; Ebrahim et al., 2000) and ischaemia-reperfusion arrhythmias (Hassanabad et al., 1998) as endpoints; in pigs subjected to infarction (Schulz et al., 1998); in isolated human cardiac tissue subjected to hypoxia and reoxygenation (Brew et al., 1995) and in humans undergoing coronary angioplasty with ST segment shift as the endpoint (Leesar et al., 1999).
Further evidence implying a central role for bradykinin in ischaemic preconditioning comes from mice with a targeted disruption of the bradykinin B2 receptor gene. Yang et al. (1997b), using B2 receptor knock-out mice, showed that ischaemic preconditioning did not confer protection against infarct size in these animals. These workers also demonstrated that rats deficient in high molecular weight kininogen did not display the preconditioning response (Yang et al., 1997b). A recent study provides compelling genetic evidence supporting a cytoprotective role of bradykinin in the ischaemic heart (Yoshida et al., 2000). The human tissue kallikrein gene was delivered into rats using adenoviral vector. One week following gene delivery, cardiac kinin levels were significantly increased. Hearts were subjected to coronary artery occlusion and reperfusion. It was observed that kallikrein gene delivery was associated with significant limitation of infarct size and attenuated severity of ventricular fibrillation. Finally, kallikrein gene delivery also attenuated apoptosis in the ischaemic zone, determined by terminal deoxynucleotidyl transferase-mediated nick end labelling. All the beneficial effects kallikrein overexpression were abolished by icatibant, implying a role for the bradykinin B2 receptor in the protection observed.
Although the majority of work implicates involvement of bradykinin B2 receptor activation in mediating the cardioprotective actions of bradykinin during ischaemia-reperfusion, there is limited evidence implying a role for bradkinin B1 receptor activation. The bradykinin B1 receptor is not constitutively expressed in most tissues but is inducible under certain pathological conditions such as inflammation and anoxia (Bhoola et al., 1992). Activation of this receptor sub-type has been proposed to mediate the vascular protection afforded by preconditioning. Bouchard et al. (1998) reported that the beneficial effects of ischaemic preconditioning on endothelial function was partly mediated by activation of the bradykinin B1 receptor. In addition to this, Chahine and colleagues found that bradykinin limited noradrenaline outflow and reduced the occurrence of arrhythmias in the isolated rat heart model (Chahine et al., 1993). This protective effect was not abrogated using Hoe 140 but with a specific bradykinin B1 receptor antagonist, Lys [Leu8] Des-Arg9-bradykinin, implying a role for the bradykinin B1 receptor as opposed to the bradykinin B2 receptor (Chahine et al., 1993).
Molecular mechanisms of bradykinin-induced acute cardioprotection
The mechanisms underlying the acute protective actions of bradykinin protection are not well understood. A number of agents have been proposed to participate in the protection including NO, prostaglandin I2, protein kinase C (PKC) and tyrosine kinases (Vegh et al., 1993; Goto et al., 1995; Zhu et al., 1995; Bugge & Ytrehus, 1996; Feng & Rosenkranz, 1999; Feng et al., 2000). There is almost universal consensus that B2 receptor activation is required for protection, since icatibant in most models abolishes the protection afforded by bradykinin. PKC activation is thought to be central in the preconditioning phenomenon and it has been proposed that once activated, it determines the phosphorylation of distal kinase and end effector proteins. Brew et al. (1995), Bugge & Ytrehus (1996) and Goto et al. (1995) have presented evidence that exogenously administered bradykinin protects against ischaemia-reperfusion injury through a PKC-dependent mechanism in isolated rat and isolated rabbit myocardium.
The role of NO in mediating the acute cardioprotective properties of both endogenous and exogenously administered bradykinin has been the subject of some interest. NO has been implicated in some studies as a mediator of the early cardioprotective actions of bradykinin (Schoelkens & Linz, 1992; Végh et al., 1993; Zhu et al., 1995; Feng et al., 2000). However, in contrast to these studies, Goto et al. (1995) found that bradykinin-induced infarct limitaion in rabbit heart was not abrogated by L-nitroarginine methyl ester (L-NAME). Similarly Bugge & Ytrehus (1996) found that bradykinin-induced acute cardioprotection in rat heart was not modified by L-nitro-ω-arginine, a NO synthase (NOS) inhibitor with a similar pharmacological profile to L-NAME. Thus, there may be important species and end-point variations in the role of NO in mediating the acute cardioprotective actions of bradykinin, with some models showing NO dependency. As we shall see subsequently, although the acute infarct-limiting effect of bradykinin does not appear to be NO-dependent, the delayed effect of bradykinin may be mediated by NO generation.
Opening of the mitochondrial KATP channel has been proposed as distal mediator of preconditioning (O'rourke, 2000) (see Figure 1). At present it is not known how opening of this channel might beneficially influence outcome from ischaemia. Some pharmacological evidence supports the notion that bradykinin may elicit protection through a mechanism involving mitochondrial KATP channel opening (Kita et al., 2000; Sanada et al., 2000). The mechanism by which this might occur is not clear. Several possibilities have been proposed including (i) the release of prostanoids, (ii) the generation of NO and (iii) the activation of a kinase cascade, downstream of PKC (as shown in Figure 1). By activating PLA2 bradykinin stimulates production of prostaglandins. Although various prostanoids have been shown to activate sarcolemmal KATP channels, which have also been suggested to participate in classical preconditioning (Bouchard et al., 1994; Sanada et al., 2000), there is little evidence at present to suggest that prostanoids influence mitochondrial KATP channel opening. Similarly, it is not clear to what extent NO generated as a result of bradykinin B2 receptor activation on endothelium participates in or contributes to mitochondrial KATP channel activation. There is some evidence that NO directly modulates mitochondrial KATP channel opening in cardiac myocytes (Sasaki et al., 2000)
Endogenous bradykinin in relation to other endogenous mediators: the ‘threshold' hypothesis of preconditioning
An important observation made by Goto et al. (1995) was the ‘dose-dependency' of bradykinin's involvement in preconditioning. Anaesthetized, open chest rabbits were subjected to coronary artery occlusion with infarct size as the endpoint of protection. If three 5 min cycles of ischaemia were used to elicit the preconditioning response then icatibant did not block protection. However, if one 5 min cycle of ischaemia was used to precondition the myocardium then protection was abrogated by icatibant. These workers, who had previously provided extensive evidence for the involvement of adenosine in ischaemic preconditioning, concluded that a ‘threshold' must be reached in order for the full protective response of preconditioning to occur. It was proposed that when only one brief cycle of ischaemia is used, then bradykinin plays a primary role in inducing protection such that B2 receptor blockade abolishes the effect. However, if three cycles are employed, other mediators are generated in sufficient quantity, such that the ‘threshold' can be attained even in the presence of the B2 receptor antagonist (see Figure 3). Experimental support for this important ‘threshold hypothesis' has been provided by several investigators in relation to the use of ACE inhibitor drugs which is discussed in more detail below.
Figure 3.
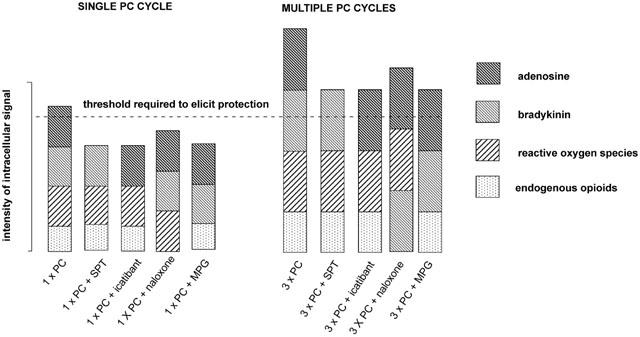
Schematic illustrating the multiple trigger hypothesis of classical preconditioning advanced by Goto et al. (1995). The hypothesis proposes that the preconditioning response is only elicited when a critical threshold of intracellular kinase activity is exceeded. All of the endogenous mediators known to act as triggers of the preconditioning response can independently activate the intracellular signal cascade but they act in concert to trigger the preconditioning response. When a single preconditioning cycle is used (1×PC), blockade of adenosine receptors with 8-sulphophenyltheophylin (SPT), blockade of bradykinin B2 receptors with icatibant, blockade of opioid receptors with (−)-naloxone or scavenging of free radicals with mercaptoprionylglycine (MPG) will be sufficient to reduce the intensity of the intracellular signal below the critical threshold. When multiple preconditioning cycles are used (e.g. 3×PC), relatively more of the endogenous triggers are released. Under these conditions, antagonism of any single trigger may still result in sufficient kinase activation to exceed the threshold which elicits protection.
ACE inhibitors and the ischaemic myocardium
The clearly-defined role of ACE as a kininase of primary importance is supported by studies in which ACE inhibitors have been shown to elevate circulating and tissue bradykinin concentrations (Baumgarten et al., 1993; Pellacani et al., 1994; Hornig & Drexler, 1997). Baumgarten et al. (1993) showed that ramiprilat increased bradykinin outflow from the isolated ischaemic rat heart. This study showed that a local kallikrein system exists in the rat heart but additionally suggests that ACE inhibitors are capable of increasing bradykinin levels by inhibiting its breakdown (see Figure 4). With this in mind, several studies indicate that ACE inhibitors can potentiate a sub-threshold preconditioning stimulus by increasing bradykinin levels (Miki et al., 1996; Morris & Yellon, 1997; Ebrahim et al., 2001a). A sub-threshold preconditioning stimulus is regarded as a short ischaemic period which liberates some or all of the triggers involved in classical preconditioning (including bradykinin) but their concentration is insufficient to trigger the protective response (see Figure 3). Miki et al. (1996) showed that captopril, combined with a subthreshold preconditioning protocol was sufficient to elicit the full preconditioning response in the open-chest rabbit coronary artery occlusion preparation, which was abrogated with icatibant, implying a role for the bradykinin B2 receptor. Similarly, Morris & Yellon (1997) found that both captopril and lisinopril were able to enhance subthreshold preconditioning by augmenting bradykinin levels, an effect also eliminated with icatibant. More recently Ebrahim et al. (2001a) have shown that captopril enhanced a subthreshold preconditioning to induce marked limitation of infarct size in the isolated rat heart.
Figure 4.
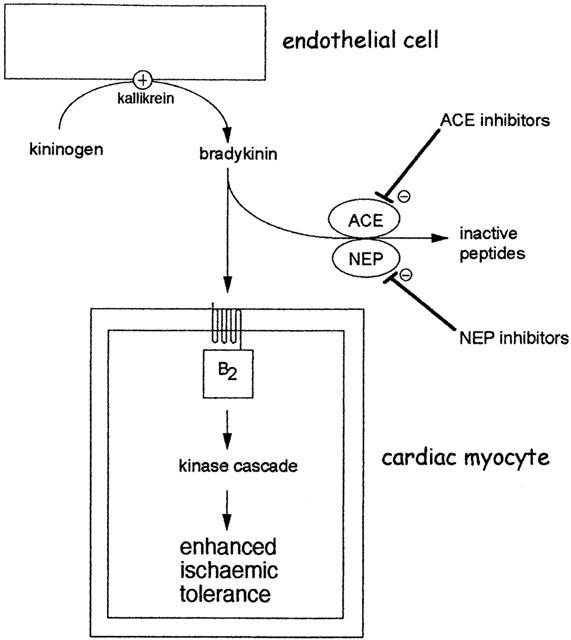
Augmentation of the preconditioning response by angiotensin converting enzyme (ACE) inhibitors or neutral endopeptidase (NEP) inhibitors. Bradykinin is efficiently and rapidly degraded by several enzymes especially ACE and NEP. Inhibition of these enzymes increases the availability of bradykinin at B2 receptors on cardiac myocytes. During very brief periods of ischaemia, interstitial bradykinin concentration may be insufficient to initiate the preconditioning mechanism. However, in the presence of an ACE or NEP inhibitor, augmentation of bradykinin concentration is sufficient to initiate preconditioning. The ability of ACE inhibitors to potentiate subthreshold ischaemic stimuli has been demonstrated for both early and delayed forms of preconditioning (see text).
The ability of ACE inhibitors to confer protection in the absence of a preconditioning stimulus is more controversial (Heusch et al., 1997). In the studies previously described, administration of either captopril (Miki et al., 1996; Morris & Yellon, 1997; Ebrahim et al., 2001a) or lisinopril (Morris & Yellon, 1997) alone prior to the index ischaemic event did not result in protection. Those findings are in contrast to several other studies in which ACE inhibitor treatment alone was shown to protect against ischaemia-reperfusion injury (Ertl et al., 1982; Massoudy et al., 1994; Anderson et al., 1996; Dogan et al., 1998; Matoba et al., 1999; Jin & Chen, 2000; Weidenbach et al., 2000). The reasons for the lack of effect of ACE inhibitors in some studies are not clear. Anderson et al. (1996) showed that captopril but not enalapril was protective in the isolated rat heart and attenuated lipid peroxidation. Indeed, it has been proposed that ACE inhibitors, such as captopril, that possess sulfhydryl moieties are able to act as scavengers or reactive oxygen species and as a consequence are protective when administered alone (Anderson et al., 1996). However, the picture is complicated by the fact that some ACE inhibitors that do not possess a sulphydryl group have been reported to be cardioprotective in some models (Birincioglu et al., 1997; Matoba et al., 1999). Matoba et al. (1999) showed that cilazaprilat, a non-sulfhydryl containing ACE inhibitor, protected directly against hypoxia-reoxygenation injury in cultured rat myocytes and enhanced bradykinin production in the culture media of the myocytes.
The question of whether ACE inhibitors are independently cytoprotective in experimental acute myocardial ischemia without preconditioning remains unanswered. However, the recent Heart Outcomes Prevention Evaluation (HOPE) trial demonstrated that ramipril reduced risk of death in patients with coronary artery disease (HOPE Investigators, 2000), an effect that appears to be unrelated to blood pressure reduction alone.
Neutral endopeptidase inhibition
Of the several enzymes other than ACE that contribute to the inactivation of bradykinin, NEP is probably the most important (Ura et al., 1987; Piedimonte et al., 1994; Kokkonen et al., 1999). NEP, like ACE, is a cell surface zinc metalloprotease, but unlike ACE its concentration in endothelium is low. NEP is highly concentrated in the epithelial cells of the kidney, it is also found in lung, liver and myocardium (Bhoola et al., 1992; Piedimonte et al., 1994). Studies with NEP inhibitors have found that these agents can evoke cardioprotection (Schriefer et al., 1996; Yang et al., 1997a). Yang et al. (1997a) in an in vivo rat model of coronary artery occlusion found that the NEP inhibitor CGS24592 was able to induce cardioprotection comparable to that induced by an ACE inhibitor using infarct size as an end point.
Dual inhibitors of ACE and NEP are novel compounds often referred to as ‘vasopeptidase inhibitors', a colloquial marketing term with no scientific provenance. They have been developed recently for the treatment of hypertension and heart failure (Fink et al., 1996; Robl et al., 1997; Weber, 1999; Asher & Naftilan, 2000; van Veldhuisen & van Gilst, 2000). Omapatrilat (BMS 186716) is the first in this new class of agents. In isolated rat hearts, using infarct size as an experimental end point, Ebrahim et al. (2001a) found that omapatrilat could potentiate a sub-threshold preconditioning stimulus sufficiently to evoke cardioprotection, which was also abrogated with icatibant. Interestingly, omapatrilat administered alone, provided moderate but significant cardioprotection, an effect also abrogated with icatibant. Ebrahim et al. (2001a) compared the effects of omapatrilat with a conventional ACE inhibitor, captopril. Although they found that captopril was able to enhance the sub-threshold preconditioning stimulus, similar to omapatrilat, when administered alone it was not cardioprotective. This implies that dual inhibition of ACE and NEP may have additional cardiovascular benefits when compared with ACE inhibition alone.
Rastegar et al. (2000a) reported that a dual ACE and NEP inhibitor, Z13752A, produced a protective effect in an in vivo dog model of coronary artery occlusion, using arrhthymia prevalence as an end point. They also found icatibant abolished the cardioprotective effect of Z13752A. Additionally, Schriefer et al. (1996) in an anaesthestized, open-chest rabbit model of coronary artery occlusion found that dual inhibition of ACE and NEP produced cardioprotective effects over and above treatment with just an ACE or NEP inhibitor alone. As these beneficial effects were blocked with icatibant, bradykinin-mediated cardiprotection is most likely. Since NEP is also responsible for the catalytic degradation of various other vasodilator peptides including atrial natriuretic peptide (ANP), type-B natriuretic peptide (BNP), type-C natriuretic peptide (CNP) and substance P (Piedimonte et al., 1994; Ozaki et al., 1999), it is feasible that any of these other vasodilator peptides may be involved in the cardioprotective effect observed with NEP inhibitors. ANP has been shown to exert cardioprotective effects during ischaemia-reperfusion although these studies are equivocal. In one study (Rastegar et al., 2000b) ANP treatment was found to reduce ischaemia-reperfusion arrhythmias in canine heart. In another canine study (Takagi et al., 2000), no reduction in arrhythmias was observed but infarction was substantially limited by ANP treatment. BNP has been reported recently to limit infarct size in a concentration dependent manner in rat heart (D'souza et al., 2002). However, the contribution of natriuretic peptides to the cardioprotective effects of ACE and NEP inhibitors appears to be overwhelmed by the contribution of bradykinin. Yang et al. (1997a) also reported that all protection afforded by a NEP inhibitor was abrogated using icatibant, implying a role only for bradykinin and not any of the other peptides augmented as a consequence of NEP inhibition. In addition, Yang et al. (1997a) used a selective natriuretic peptide receptor antagonist and were not able to abrogate the protection afforded by dual ACE and NEP inhibition, although it was slightly attenuated. Rastegar et al. (2000a) and Ebrahim et al. (2001a) found that the protection afforded by dual ACE/NEP inhibitors was lost in the presence of icatibant, strongly implicating a role for bradykinin rather than the other peptides.
Aminopeptidase P inhibition
Although ACE and NEP appear to play primary roles in bradykinin catabolism, recent reports imply that aminopeptidase P may be an important contributor to endogenous bradykinin turnover. The aminopeptidase inhibitor, apstatin, which has little affinity for ACE or NEP, was shown to be cardioprotective against ischaemia-reperfusion injury in isolated rat heart (Ersahin et al., 1999) and to limit infarct size in rat heart in vivo (Wolfrum et al., 2001). In both models, the protection afforded by apstatin was comparable to that seen with a selective ACE inhibitor and appeared to be bradykinin-mediated since icatibant abolished the protective properties of apstatin.
Bradykinin and delayed myocardial protection
Preconditioning the myocardium with ischaemia induces protection in two distinct phases; an early phase, which occurs immediately following the preconditioning stimulus and lasts for up to 2 h (classical preconditioning) and a late phase which occurs 24 h following a preconditioning stimulus and lasts for up to 72 h (Bolli, 2000; Baxter & Ferdinandy, 2001). Molecular triggers of classical preconditioning including adenosine, opioid peptides, and catecholamines have all been shown to also induce a delayed preconditioning-like effect (Baxter et al., 1994; Fryer et al., 1999; Meng et al., 1999). Preliminary evidence that bradykinin might act as a trigger of pacing-induced delayed protection against ischaemia-reperfusion arrhythmias was reviewed by Parratt et al. (1997). A role for a bradykinin in eliciting delayed protection against infarction has been established in two recent studies. Ebrahim et al. (2001b) administered 40 μg kg−1 bradykinin intravenously to rats and 24 h later studied responses to ischaemia-reperfusion. Using infarct size as the experimental end point, it was shown that hearts from bradykinin pretreated animals exhibited smaller infarct size and tendency towards better coronary flow (Ebrahim et al., 2001b). Kositprapa et al. (2001) showed that delayed ischaemic preconditioning against infarction in rabbit heart was abolished when icatibant was administered during the preconditioning stimulus. Conversely intra-atrial infusion of bradykinin (50 μg kg−1 min−1 for 15 min) resulted in significant limitation of infarction during coronary occlusion 24 h later. Interestingly, Ebrahim et al. (2001b) found that this late protective effect of bradykinin treatment was completely abrogated when bradykinin was administered following a NOS inhibitor, L-NAME (see Figure 5). This finding is compliant with the hypothesis proposed by Bolli and colleagues who have provided persuasive evidence that NO acts as a trigger of delayed preconditioning (see Bolli, 2000 for review). Indeed, it seems plausible that bradykinin acts as a primary trigger of delayed preconditioning, and that this effect is mediated by generation of NO as a signalling intermediate (Figure 5).
Figure 5.
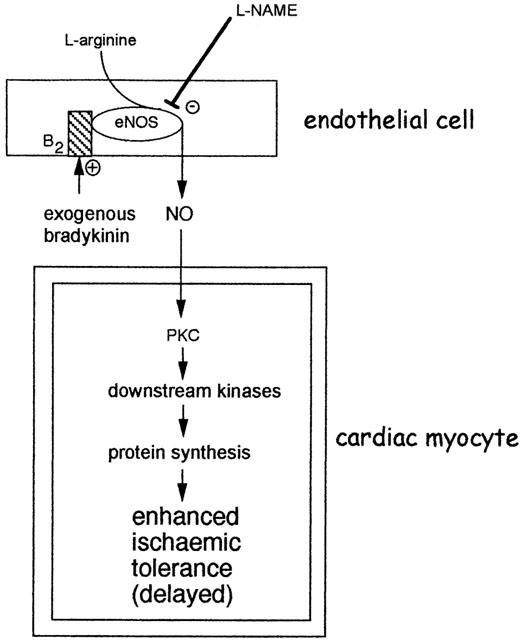
Proposed mechanism for the induction of delayed cardioprotection by bradykinin. The immediate activation of NOS as a result of bradykinin B2 receptor activation leads to the generation of NO. The most likely NOS isoform is eNOS. According to prevailing the delayed preconditioning hypothesis, NO could subsequently trigger an adaptive response in cardiac myocytes, involving the activation of protein kinase C (PKC) isoforms and other kinases. The subsequent induction of unknown mediators of protection results in enhanced tolerance to ischaemia 24 h following liberation or application of bradykinin. Adapted from Ebrahim et al. (2001b).
Further evidence supporting the involvement of bradykinin as a trigger of delayed preconditioning comes from recent work with the ACE inhibitor perindoprilat. Jaberansari et al. (2001) have reported that pretreatment of pigs with perindoprilat, potentiated a sub-threshold preconditioning stimulus (two 2 min coronary artery occlusions) sufficiently to induce delayed preconditioning comparable to that induced by four 5 min coronary occlusions. Although this study does not provide direct evidence for the involvement of bradykinin, the result was compatible with the hypothesis that bradykinin (or other peptides catalytically inactivated by ACE) might be implicated in triggering the delayed phase of preconditioning. As with other ACE-inhibitor studies, this demonstration of an association with delayed preconditioning may have important implications for our perceptions of ACE inhibitors as cardioprotective agents.
Bradykinin, apoptosis and attenuation of reperfusion injury
The majority of cardioprotective agents including bradykinin have to be administered prior to the ischaemic insult in order to limit injury. However, as it is difficult to predict when most patients are likely to encounter an ischaemic event, it is rarely feasible to administer these agents to patients. Intermittent administration to high-risk patients such as those with unstable angina is feasible. Greatest benefit in the clinic would be observed if agents could be given at reperfusion and thus limit reperfusion injury. Ischaemia / reperfusion injury results in necrosis and apoptosis. While, reperfusion of the jeopardized myocardium is imperative, reperfusion itself can result in irreversible cell injury, either through necrosis and/or apoptosis (Yellon & Baxter, 1999). Most agents that limit ischaemia-reperfusion injury must be administered prior to the onset of ischaemia insult to be effective. However, some agents may influence the the apoptotic programme which is activated or enhanced during reperfusion. Inhibitors of the apoptotic cascade can be administered at the onset of reperfusion or just prior to the onset of reperfusion (Mocanu et al., 2000). Peptide growth factors such as transforming growth factor-β1, insulin-like growth factor, insulin, cardiotrophin and fibroblast growth factors limit reperfusion injury at least partially through activation of anti-apoptotic ‘survival' signal pathways. These include phosphatidyl inositide 3′-OH kinase (PI3 kinase), Akt/protein kinase B (PKB) and p42/p44 mitogen activated protein kinases (Yellon & Baxter, 1999). Recent evidence has demonstrated that bradykinin can activate PI3 kinase in myocytes (Ritchie et al., 1999). Hence, it appears feasible that bradykinin may limit reperfusion injury by activating at least one ‘survival' kinase pathway. Massoudy et al. (1994) demonstrated that bradykinin and or ramiprilat administered at reperfusion improved recovery of mechanical function in a guinea pig isolated heart preparation. In studies by Yang et al. (1997a; 1999) and by Shrieffer et al. (1996), ACE inhibitors, NEP inhibitors or dual ACE/NEP inhibitors were cardioprotective when given at reperfusion. However, in eNOS knockout mice, the cardioprotective of an ACE inhibitor at reperfusion was absent, suggesting that the protection afforded by enhanced bradykinin at reperfusion was NO-dependent (Yang et al., 1999). The bradykinin B2 receptor is coupled to eNOS and in the presence of bradykinin, eNOS is uncoupled leading to its activation (Marrero et al., 1999). We have observed that bradykinin administered at reperfusion in an isolated rat heart model limited infarct size, an effect abrogated by an inhibitor of PI3 kinase, wortmannin (unpublished data). Thus, bradykinin may be effective in attenuating reperfusion injury, which in turn implies that agents that increase bradykinin levels, such as ACE inhibitors, could theoretically be applied to limit reperfusion injury.
Conclusion and future perspectives
Bradykinin exerts a unique and robust pattern of injury limiting actions in the ischaemic and reperfused heart. A wealth of evidence suggests that bradykinin administered prior to the onset of myocardial ischaemia exerts a cardioprotective effect in animal and human experimental models. Endogenous bradykinin participates in classical preconditioning in many experimental models. Recently, a potential role for bradykinin in eliciting a delayed phase of preconditioning has emerged. Bradykinin has also been shown to limit reperfusion injury, an action which may be beneficial in patients receiving reperfusion therapy for acute myocardial infarction. Of current therapeutic relevance, agents that inhibit the breakdown of bradykinin, notably ACE inhibitors and the newly introduced combined ACE/NEP inhibitors, may display valuable protective effects both experimentally and clinically. Thus, administration of exogenous bradykinin in some clinical settings and augmentation of endogenous bradykinin levels may soon become feasible and valuable approaches to the treatment of ischaemic heart disease.
Acknowledgments
The authors gratefully acknowledge the financial support of the British Heart Foundation which has funded key aspects of their work on ischaemic preconditioning and bradykinin through studentship FS/98075 and programme grant RG/98002.
Abbreviations
- ACE
angiotensin converting enzyme
- ATP
adenosine triphosphate
- MPG
mercaptopropionylglycine
- NEP
neutral endopeptidase
- NO
nitric oxide
- NOS
nitric oxide synthase
- PKC
protein kinase C
- 8-SPT
8-(p-sulphophenyl)theophylline
References
- AHMAD M., ZEITLIN I.J., PARRATT J.R. The release of kininase from rat isolated hearts during myocardial ischaemia. Immunopharmacology. 1996;33:299–300. doi: 10.1016/0162-3109(96)00047-1. [DOI] [PubMed] [Google Scholar]
- ANDERSON B., KHAPER N., DHALLA A.K., SINGAL P.K. Anti-free radical mechanisms in captopril protection against reperfusion injury in isolated rat hearts. Can. J. Cardiol. 1996;12:1099–1104. [PubMed] [Google Scholar]
- ARAMORI I., ZENKOH J., MORIKAWA N., O'DONNELL N., ASANO M., NAKAMURA K., IWAMI M., KOJO H., NOTSU Y. Novel subtype-selective nonpeptide bradykinin receptor antagonists FR167344 and FR173657. Mol. Pharmacol. 1997;51:171–176. doi: 10.1124/mol.51.2.171. [DOI] [PubMed] [Google Scholar]
- ARMSTRONG S., GANOTE C.E. Adenosine receptor specificity in preconditioning of isolated rabbit cardiomyocytes: evidence of A3 receptor involvement. Cardiovasc. Res. 1994;28:1049–1056. doi: 10.1093/cvr/28.7.1049. [DOI] [PubMed] [Google Scholar]
- ASHER J.R., NAFTILAN A.J. Vasopeptidase inhibition: a new direction in cardiovascular treatment. Curr. Hypertens. Rep. 2000;2:384–391. doi: 10.1007/s11906-000-0042-y. [DOI] [PubMed] [Google Scholar]
- BAINES C.P., GOTO M., DOWNEY J.M. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J. Mol. Cell. Cardiol. 1997;29:207–216. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- BAINES C.P., PASS J.M., PING P. Protein kinases and kinase-modulated effectors in the late phase of ischemic preconditioning. Basic Res. Cardiol. 2001;96:207–218. doi: 10.1007/s003950170051. [DOI] [PubMed] [Google Scholar]
- BANKWALA Z., HALE S.L., KLONER R.A. Alpha-adrenoceptor stimulation with exogenous norepinephrine or release of endogenous catecholamines mimics ischemic preconditioning. Circulation. 1994;90:1023–1028. doi: 10.1161/01.cir.90.2.1023. [DOI] [PubMed] [Google Scholar]
- BAUMGARTEN C.R., LINZ W., KUNKEL G., SCHÖLKENS B.A., WEIMER G. Ramiprilat increases bradykinin outflow from isolated hearts of rat. Br. J. Pharmacol. 1993;108:293–295. doi: 10.1111/j.1476-5381.1993.tb12797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAXTER G.F., FERDINANDY P. Delayed preconditioning of myocardium: current perspectives. Basic Res. Cardiol. 2001;96:329–344. doi: 10.1007/s003950170041. [DOI] [PubMed] [Google Scholar]
- BAXTER G.F., MARBER M.S., PATEL V.C., YELLON D.M. Adenosine receptor involvement in a delayed phase of myocardial protection 24 h after ischaemic preconditioning. Circulation. 1994;90:2993–3000. doi: 10.1161/01.cir.90.6.2993. [DOI] [PubMed] [Google Scholar]
- BHOOLA K.D., FIGUEROA C.D., WORTHY K. Bioregulation of kinins: kallikreins, kininogens and kininases. Pharmacol. Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- BIRINCIOGLU M., AKSOY T., OLMEZ E., ACET A. Protective effect of ACE inhibitors on ischaemia-reperfusion-induced arrythmias in rats: is this effect related to the free radical scavenging action of these drugs. Free Radic. Res. 1997;27:389–396. doi: 10.3109/10715769709065778. [DOI] [PubMed] [Google Scholar]
- BLAIS C., MARCEAU F., ROULEAU J-L., ADAM A. The kallikrein-kininogen-kinin system: lessons from quantification of endogenous kinins. Peptides. 2000;21:1903–1940. doi: 10.1016/s0196-9781(00)00348-x. [DOI] [PubMed] [Google Scholar]
- BOLLI R. The late phase of preconditioning. Circ. Res. 2000;87:972–983. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- BOUCHARD J.F., CHOUINARD J., LAMONTAGE D. Role of kinins in the endothelial protective effects of ischaemic preconditioning. Br. J. Pharmacol. 1998;123:413–420. doi: 10.1038/sj.bjp.0701619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOUCHARD J.F., DUMONT E., LAMONTAGNE D. Evidence that prostaglandins I2, E2 and D2 may activate ATP sensitive potassium channels in the isolated rat heart. Cardiovasc. Res. 1994;28:901–905. doi: 10.1093/cvr/28.6.901. [DOI] [PubMed] [Google Scholar]
- BREW E., MITCHELL M.B., REHRING T.F., GAMBONI-ROBERTSON F., MC INTYRE R.C., HARKEN A.H., BANERJEE A. Role of bradykinin in cardiac functional protection after global ischaemia-reperfusion in rat heart. Am. J. Physiol. 1995;269:H1370–H1378. doi: 10.1152/ajpheart.1995.269.4.H1370. [DOI] [PubMed] [Google Scholar]
- BROWN N.J., VAUGHAN D.E. Angiotensin-converting enzyme inhibitors. Circulation. 1998;97:1411–1420. doi: 10.1161/01.cir.97.14.1411. [DOI] [PubMed] [Google Scholar]
- BUGGE E., YTREHUS K. Bradykinin protects against infarction but does not mediate ischaemic preconditioning in the isolated rat heart. J. Mol. Cell. Cardiol. 1996;28:2333–2341. doi: 10.1006/jmcc.1996.0226. [DOI] [PubMed] [Google Scholar]
- CALIXTO J.B., CABRINI D.A., FERREIRA J., CAMPOS M.M. Kinins in pain and inflammation. Pain. 2000;87:1–5. doi: 10.1016/S0304-3959(00)00335-3. [DOI] [PubMed] [Google Scholar]
- CAMPBELL D.J. Towards understanding the kallikrein-kinin system: insights from measurement of kinin peptides. Braz. J. Med. Biol. Res. 2000;33:665–677. doi: 10.1590/s0100-879x2000000600008. [DOI] [PubMed] [Google Scholar]
- CHAHINE R., ADAM A., YAMAGUCHI N., GASPO R., NADEAU R. Protective effects of bradykinin on the ischaemic heart: implication of the B1 receptor. Br. J. Pharmacol. 1993;108:318–322. doi: 10.1111/j.1476-5381.1993.tb12802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN M.V., BAINES C.P., DOWNEY J.M. Ischaemic preconditioning: from adenosine receptor to KATP channel. Annu. Rev. Physiol. 2000;62:79–109. doi: 10.1146/annurev.physiol.62.1.79. [DOI] [PubMed] [Google Scholar]
- DERIAN C.K., MOSKOWITZ M.A. Polyphosphoinositide hydrolysis in endothelial cells and carotid artery segments: bradykinin B2 receptor stimulation is calcium-independent. J. Biol. Chem. 1986;261:3831–3837. [PubMed] [Google Scholar]
- DOGAN R., SARIGUL A., ISBIR S., FARSAK B., TUNCER M., KILINE K., DEMIRCIN M. Beneficial effect of captopril against ischaemia-reperfusion injury in isolated guinea pig hearts. Scand. J. Clin. Lab. Invest. 1998;58:119–126. doi: 10.1080/00365519850186698. [DOI] [PubMed] [Google Scholar]
- D'SOUZA S.P., YELLON D.M., BAXTER G.F.Type-B natriuretic peptide limits infarct size in rat isolated heart Br. J. Pharmacol. 2002. in press (abstract)
- DUMOULIN M.J., ADAM A., BLAIS C., JR, LAMONTAGNE D. Metabolism of bradykinin by the rat coronary vascular bed. Cardiovasc. Res. 1998;38:229–236. doi: 10.1016/s0008-6363(98)00006-6. [DOI] [PubMed] [Google Scholar]
- EBRAHIM Z., YELLON D.M., BAXTER G.F.Bradykinin-induced cardioprotection is attenuated in the hypertrophied rat heart Br. J. Pharmacol. 2000129suppl191P(abstract) [Google Scholar]
- EBRAHIM Z., YELLON D.M., BAXTER G.F.Omapatrilat lowers the threshold for induction of ischaemic preconditioning via activation of the bradykinin B2 receptor Br. J. Pharmacol. 2001a133suppl10P(abstract) [Google Scholar]
- EBRAHIM Z., YELLON D.M., BAXTER G.F. Bradykinin elicits ‘second window' myocardial protection in rat heart through an NO-dependent mechanism. Am. J. Physiol. 2001b;281:H1458–H1464. doi: 10.1152/ajpheart.2001.281.3.H1458. [DOI] [PubMed] [Google Scholar]
- ERDOS E.G., SKIDGEL R.A.Metabolism of bradykinin by peptidases in health and disease Handbook of immunopharmacology: the kinin system 1997London: Academic Press; 111–141.Farmer, S.G. (ed) pp [Google Scholar]
- ERTL G., KLONER R.A., ALEXANDER R.W., BRAUNWALD E. Limitation of experimental infarct size by an angiotensin-converting enzyme inhibitor. Circulation. 1982;65:40–48. doi: 10.1161/01.cir.65.1.40. [DOI] [PubMed] [Google Scholar]
- ERSAHIN C., EULER D.E., SIMMONS W.H. Cardioprotective effects of the aminopeptidase P inhibitor apstatin: studies on ischaemia/reperfusion injury in the isolated rat heart. J. Cardiovasc. Pharmacol. 1999;34:604–611. doi: 10.1097/00005344-199910000-00019. [DOI] [PubMed] [Google Scholar]
- ERSAHIN C., SIMMONS W.H. Inhibition of both aminopeptidase P and angiotensin-converting enzyme prevents bradykinin degradation in the rat coronary circulation. J. Cardiovasc. Pharmacol. 1997;30:96–101. doi: 10.1097/00005344-199707000-00014. [DOI] [PubMed] [Google Scholar]
- FENG J., ROSENKRANZ E.R. Bradykinin pretreatment improves ischaemia tolerance of the rabbit heart by tyrosine mediated pathways. Ann. Thorac. Surg. 1999;68:1567–1572. doi: 10.1016/s0003-4975(99)01041-3. [DOI] [PubMed] [Google Scholar]
- FENG J.F., LI H., ROSENKRANZ E.R. Bradykinin protects the rabbit heart after cardioplegic ischaemia via NO-dependent pathways. Ann. Thorac. Surg. 2000;70:2119–2124. doi: 10.1016/s0003-4975(00)02148-2. [DOI] [PubMed] [Google Scholar]
- FINK C.A., CARLSON J.E., MCTAGGART P.A., QIAO Y., WEBB R., CHATELAIN A.Y., TRAPANI A.J. Mercaptoacyl dipeptides as orally active dual inhibitors of angiotensin-converting enzyme and neutral endopeptidase. J. Med. Chem. 1996;39:3158–3168. doi: 10.1021/jm960323z. [DOI] [PubMed] [Google Scholar]
- FRYER R.M., HSU A.K., EELLS J.T., NAGASE H., GROSS G.J. Opioid - induced second window of cardioprotection. Potential role of KATP channnels. Circ. Res. 1999;84:846–851. doi: 10.1161/01.res.84.7.846. [DOI] [PubMed] [Google Scholar]
- GANZ P., BRAUNWALD E.Coronary blood flow and myocardial ischemia Heart Disease: A Textbook of Cardiovascular Medicine 1997Philadelphia: WB Saunders; 1161–1183.Braunwald, E. (ed) pp [Google Scholar]
- GOTO M., LIU Y., YANG X.M., ARDELL J.L., COHEN M.V., DOWNEY J.M. Role of bradykinin in protection of ischaemic preconditioning in rabbit hearts. Circ. Res. 1995;77:611–621. doi: 10.1161/01.res.77.3.611. [DOI] [PubMed] [Google Scholar]
- GUO Y., BOLLI R., BAO W., WU W.J., BLACK R.G., MURPHREE S., SALVATORE C.A., JACOBSON M.A., AUCHAMPACH J.A. Targeted deletion of the A3 adenosine receptor confers resistance to myocardial ischaemic injury and does not prevent early preconditioning. J. Mol. Cell. Cardiol. 2001;33:825–830. doi: 10.1006/jmcc.2001.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALL J.M. Bradykinin receptors: pharmacological properties and biological roles. Pharmacol. Ther. 1992;56:131–190. doi: 10.1016/0163-7258(92)90016-s. [DOI] [PubMed] [Google Scholar]
- HALL J.M. Bradykinin receptors. Gen. Pharmacol. 1997;28:1–6. doi: 10.1016/s0306-3623(96)00174-7. [DOI] [PubMed] [Google Scholar]
- HASSANABAD Z.F., FURMAN B.L., PARRATT J.R., AUGHEY E. Coronary endothelial dysfunction increases the severity of ischaemia-induced ventricular arrhythmias in rat isolated perfused hearts. Basic Res. Cardiol. 1998;93:241–249. doi: 10.1007/s003950050091. [DOI] [PubMed] [Google Scholar]
- HATTA E., RUBIN L.E., SEYEDI N., LEVI R. Bradykinin and cardioprotection: don't set your heart on it. Pharmacol. Res. 1997;35:531–536. doi: 10.1006/phrs.1997.0182. [DOI] [PubMed] [Google Scholar]
- HEUSCH G., ROSE J., EHRING T. Cardioprotection by ACE inhibitors in myocardial ischaemia/reperfusion. The importance of bradykinin. Drugs. 1997;54:31–34. doi: 10.2165/00003495-199700545-00006. [DOI] [PubMed] [Google Scholar]
- HOPE INVESTIGATORS, THE Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N. Engl. J. Med. 2000;342:145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- HORNIG B., DREXLER H. Endothelial function and bradykinin in humans. Drugs. 1997;54:42–47. doi: 10.2165/00003495-199700545-00007. [DOI] [PubMed] [Google Scholar]
- HORNIG B., KOHLER C., DREXLER H. Role of bradykinin in mediating vascular effects of angiotensin-converting enzyme inhibitors in humans. Circulation. 1997;95:115–1118. doi: 10.1161/01.cir.95.5.1115. [DOI] [PubMed] [Google Scholar]
- JABERANSARI M.T., BAXTER G.F., MULLER C.M., LATOUF S.E., ROTH E., OPIE L.H., YELLON D.M. ACE inhibition enhances a subthreshold stimulus to elicit delayed preconditioning in pig myocardium. J. Am. Coll. Cardiol. 2001;37:1996–2001. doi: 10.1016/s0735-1097(01)01232-3. [DOI] [PubMed] [Google Scholar]
- JIN Z.Q., CHEN X. Pretreatment with ramiprilat induces cardioprotection against free radical injury in guinea-pig isolated heart: Involvement of bradykinin, protein kinase C and prostaglandins. Clin. Expt. Pharm. Physiol. 2000;27:257–262. doi: 10.1046/j.1440-1681.2000.03233.x. [DOI] [PubMed] [Google Scholar]
- KILPATRICK E.L., NARAYAN P., MENTZER R.M., LASLEY R.D. Adenosine A3 agonist cardioportection in isolated rat and rabbit hearts is blocked by the A1 antagonist DPCPX. Am. J. Physiol. 2001;281:H847–H853. doi: 10.1152/ajpheart.2001.281.2.H847. [DOI] [PubMed] [Google Scholar]
- KITA H., MIURA T., MIKI T., GENDA S., TANNO M., FUKUMA T., SHIMAMOTO K. Infarct size limitation by bradykinin receptor activation is mediated by mitochondrial but not the sarcolemmal K (ATP) channel. Cardiovasc. Drugs Ther. 2000;14:497–502. doi: 10.1023/a:1007837022300. [DOI] [PubMed] [Google Scholar]
- KOKKONEN J.O., KUOPPALA A., SAARINEN J., LINDSTEDT K.A., KOVANEN P.T. Kallidin- and bradykinin-degrading pathways in human heart. Circulation. 1999;99:1984–1990. doi: 10.1161/01.cir.99.15.1984. [DOI] [PubMed] [Google Scholar]
- KOSITPRAPA C., OCKAILI R.A., KUKREJA R.C. Bradykinin B2 receptor is involved in the late phase of preconditioning in rabbit heart. J. Mol. Cell. Cardiol. 2001;33:1355–1362. doi: 10.1006/jmcc.2000.1396. [DOI] [PubMed] [Google Scholar]
- KUOPPALA A., LINDSTEDT K.A., SAARINEN J., KOVANEN P.T., KOKKONEN J.O. Inactivation of bradykinin by angiotensin-converting enzyme and by carboxyepeptidase N in human plasma. Am. J. Physiol. 2000;278:H1069–H1074. doi: 10.1152/ajpheart.2000.278.4.H1069. [DOI] [PubMed] [Google Scholar]
- KUZUYA T., HOSHIDA S., YAMASHITA N., FUJI H., OE H., HORI M., KAMADA T., TADA M. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ. Res. 1993;72:1293–1299. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- LEESAR M.A., STODDARD M.F., MANCHIKALAPUDI S., BOLLI R. Bradykinin-induced preconditioning in patients undergoing coronary angioplasty. J. Am. Coll. Cardiol. 1999;34:639–650. doi: 10.1016/s0735-1097(99)00297-1. [DOI] [PubMed] [Google Scholar]
- LINZ W., WEIMER G., SCHÖLKENS B.A. Role of kinins in the pathophysiology of myocardial ischaemia. In vitro and in vivo studies. Diabetes. 1996;45:51–58. doi: 10.2337/diab.45.1.s51. [DOI] [PubMed] [Google Scholar]
- LIU G.S., RICHARDS S.C., OLSSON R.A., MULLANE K., WALSH R.S., DOWNEY J.M. Evidence that adenosine A3 receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovasc. Res. 1994;28:1057–1061. doi: 10.1093/cvr/28.7.1057. [DOI] [PubMed] [Google Scholar]
- LIU G.S., THORNTON J., VAN WINKLE D.M., STANLEY A.W., OLSSON R.A., DOWNEY J.M. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- MARRERO M.B., VENEMA V.J., JU H., HE H., LIANG H., CALDWELL R.B., VENEMA R.C. Endothelial nitric oxide synthase interactions with G-protein coupled receptors. Biochem. J. 1999;343:335–340. [PMC free article] [PubMed] [Google Scholar]
- MASSOUDY P., BECKER B.F., GERLACH E. Bradykinin accounts for improved postischaemic function and decreased glutathione release of guinea pig heart treated with the angiotensin-converting enzyme inhibitor ramiprilat. J. Cardiovasc. Pharmacol. 1994;23:632–639. doi: 10.1097/00005344-199404000-00017. [DOI] [PubMed] [Google Scholar]
- MATOBA S., TASUMI T., KEIRA N., KAWAHARA A. Cardioprotective effect of angiotensin converting enzyme inhibition against hypoxia/reoxygenation injury in cultured rat myocytes. Circulation. 1999;99:817–822. doi: 10.1161/01.cir.99.6.817. [DOI] [PubMed] [Google Scholar]
- MENG X., SHAMES B.D., PULIDO E.J., MELDRUM D.R., AO L., JOO K.S., HARKEN A.H., BANERJEE A. Adrenergic induction of bimodal myocardial protection: signal transduction and cardiac gene reprogramming. Am. J. Physiol. 1999;276:R1525–R1533. doi: 10.1152/ajpregu.1999.276.5.R1525. [DOI] [PubMed] [Google Scholar]
- MIKI T., MIURA T., URA N., OGAWA T., SUZUKI K., SHIMAMOTO K., IIMURA O. Captopril potentiates the myocardial infarct size-limiting effect of ischaemic preconditioning through bradykinin B2 receptor activation. J. Am. Coll. Cardiol. 1996;28:1616–1622. doi: 10.1016/s0735-1097(96)00371-3. [DOI] [PubMed] [Google Scholar]
- MINSHALL R.D., NAKAMURA F., BECKER R.P., RABITO S.F. Characterisation of bradykinin B2 receptors in adult myocardium and neonatal rat cardiomyocytes. Circ. Res. 1995;76:773–780. doi: 10.1161/01.res.76.5.773. [DOI] [PubMed] [Google Scholar]
- MOCANU M.M., BAXTER G.F., YELLON D.M. Capase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br. J. Pharmacol. 2000;130:197–200. doi: 10.1038/sj.bjp.0703336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORGAN-BOYD R., STEWART J.M., VAVAREK R.J., HASSID A. Effects of bradykinin and angiotensin II on intracellular Ca2+ dynamics in endothelial cells. Am. J. Physiol. 1987;253:C588–C598. doi: 10.1152/ajpcell.1987.253.4.C588. [DOI] [PubMed] [Google Scholar]
- MORRIS S.D., YELLON D.M. Angiotensin-converting enzyme inhibitors potentiate preconditioning through bradykinin B2 receptor activation in human heart. J. Am. Coll. Cardiol. 1997;29:1599–1606. doi: 10.1016/s0735-1097(97)00087-9. [DOI] [PubMed] [Google Scholar]
- MURRY C.E., JENNINGS R.B., REIMER K.A. Preconditioning with ischaemia: a delay of lethal cell injury in ischaemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- O'ROURKE B. Myocardial KATP channels in preconditioning. Circ. Res. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- OZAKI J., SHIMIZU H., HASHIMOTO Y., ITOH H., NAKAO K., INUI K. Enzymatic inactivation of major circulating forms of arterial and brain natriuretic peptide. Eur. J. Pharmacol. 1999;370:307–312. doi: 10.1016/s0014-2999(99)00115-6. [DOI] [PubMed] [Google Scholar]
- PAN H.-L., CHEN S.R., SCICLIM G.M., CARRETERO O.A. Cardiac interstitial bradykinin release during ischaemia is enhanced by ischaemic preconditioning. Am. J. Physiol. 2000;279:H116–H121. doi: 10.1152/ajpheart.2000.279.1.H116. [DOI] [PubMed] [Google Scholar]
- PARRATT J.R., VEGH A., ZEITLIN I.J., AHMAD M., OLDROYD K., KASZALA K., PAPP J.G. Bradykinin and endothelial-cardiac myocyte interactions in ischemic preconditioning. Am. J. Cardiol. 1997;80 suppl 3A:124A–131A. doi: 10.1016/s0002-9149(97)00467-0. [DOI] [PubMed] [Google Scholar]
- PELLACANI A., BRUNNER H.R., NUSSBERGER J. Plasma kinins increase after angiotensin-converting enzyme inhibition in human subjects. Clin. Sci. (Colch). 1994;87:567–574. doi: 10.1042/cs0870567. [DOI] [PubMed] [Google Scholar]
- PIEDIMONTE G., NADEL J.A., LONG C.S., HOFFMAN J.I.E. Neutral endopeptidase in the heart. Neutral endopeptidase inhibition prevents isoproterenol-induced myocardial hypoperfusion in rats by reducing bradykinin degradation. Circ. Res. 1994;75:770–779. doi: 10.1161/01.res.75.4.770. [DOI] [PubMed] [Google Scholar]
- RASTEGAR M.A., MARCHINI F., MORAZZONIA G., VÉGH A., PAPP J.G., PARRATT J.R. The effects of Z13752A, a combined ACE/NEP inhibitor, on responses to coronary artery occlusion; a primary protective role for bradykinin. Br. J. Pharmacol. 2000a;129:671–680. doi: 10.1038/sj.bjp.0703109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RASTEGAR M.A., VEGH A., PAPP J.G., PARRATT J.R. Atrial natriuretic peptide reduces the severe consequences of coronary artery occlusion in anaesthetized dogs. Cardiovasc. Drugs Ther. 2000b;14:471–479. doi: 10.1023/a:1007828804553. [DOI] [PubMed] [Google Scholar]
- RITCHIE R.H., MARSH J.D., SCHIEBINGER R.J. Bradykinin-stimulated protein synthesis by myocytes is dependent on the MAP kinase pathway and P70S6K. Am. J. Physiol. 1999;276:H1393–H1398. doi: 10.1152/ajpheart.1999.276.4.H1393. [DOI] [PubMed] [Google Scholar]
- ROBL J.A., SUN C-C., STEVENSON J. Dual metalloprotease inhibitors: mercaptoacetyl-based fused heterocyclic dipeptide mimetics as inhibitors of angiotensin converting enzyme and neutral endopeptidase J. Med. Chem. 1997401570–1577.et al [DOI] [PubMed] [Google Scholar]
- ROCHA E SILVA M., BERALDO W.T., ROSENFELD G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am. J. Physiol. 1949;156:261–273. doi: 10.1152/ajplegacy.1949.156.2.261. [DOI] [PubMed] [Google Scholar]
- SASAKI N., SATO T., OHLER A., O'ROURKE B., MARBAN E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation. 2000;101:439–445. doi: 10.1161/01.cir.101.4.439. [DOI] [PubMed] [Google Scholar]
- SANADA S., KITAKAZE M., ASANUMA H., HARADA K., OGITA H., NODE K., TAKASHIMA S., SAKATA Y., ASAKURA M., SHINOZAKI Y., MORI H., KUZUYA T., HORI M. Role of mitochondrial and sarcolemmal K(ATP) channels in ischaemic preconditioning of the canine heart. Am. J. Physiol. 2000;280:H256–H2263. doi: 10.1152/ajpheart.2001.280.1.H256. [DOI] [PubMed] [Google Scholar]
- SCHAPER J., HEIN S., HEINRICHS C.M., WEIHRAUCH D. Myocardial Response to Acute Injury, Parratt, J.R. (ed)pp. London: MacMillan Press; 1992. Myocardial injury and repair; pp. 1–16. [Google Scholar]
- SCHOELKENS B.A., LINZ W. Bradykinin-mediated metabolic effects in isolated perfused rat hearts. Agents Actions Suppl. 1992;38:36–42. [PubMed] [Google Scholar]
- SCHOELKENS B.A., LINZ W., KONIG W. Effects of the angiotensin converting enzyme inhibitor, ramipril, in isolated ischaemic rat heart are abolished by a bradykinin antagonist. J. Hypertens. Suppl. 1988;6:25–28. doi: 10.1097/00004872-198812040-00004. [DOI] [PubMed] [Google Scholar]
- SCHULTZ J.E., GROSS G.J. Opioids and cardioprotection. Pharmacol. Ther. 2001;89:123–137. doi: 10.1016/s0163-7258(00)00106-6. [DOI] [PubMed] [Google Scholar]
- SCHULZ R., POST H., VAHLHAUS C., HEUSCH G. Ischaemic preconditioning in pigs: a graded phenomenon. Its relation to adenosine and bradykinin. Circulation. 1998;98:1022–1029. doi: 10.1161/01.cir.98.10.1022. [DOI] [PubMed] [Google Scholar]
- SHRIEFER J.A., BROUDY E.P., HASSEN A.H. Endopeptidase inhibitors decrease myocardial ischaemia/reperfusion injury in an in vivo rabbit model. J. Pharm. Exp. Ther. 1996;278:1034–1039. [PubMed] [Google Scholar]
- STARKOPF J., BUGGE E., YTREHUS K. Preischaemic bradykinin and ischaemic preconditioning in functional recovery of globally ischaemic rat heart. Cardiovasc. Res. 1997;33:63–70. doi: 10.1016/s0008-6363(96)00195-2. [DOI] [PubMed] [Google Scholar]
- TAKAGI G., LIUCHI K., ENDO T., YAMAMOTO T., SATO N., NEJIMA J., TAKANO T. α-Human atrial natriuretic peptide, carperitide, reduces infarct size but not arrhythmias after coronary occlusion/reperfusion in dogs. J. Cardiovasc. Pharmacol. 2000;36:22–30. doi: 10.1097/00005344-200007000-00003. [DOI] [PubMed] [Google Scholar]
- THORNTON J.D., LIU G.S., OLSSON R.A., DOWNEY J.M. Intravenous pretreatment with A1-selective adenosine analogues protects the heart against infarction. Circulation. 1992;85:659–665. doi: 10.1161/01.cir.85.2.659. [DOI] [PubMed] [Google Scholar]
- TIO R.A., TOBE T.J., BEL K.J., DE LANGEN C.D., VAN GILST W.H., WESSELING H. Beneficial effects of bradykinin on porcine ischaemic myocardium. Basic Res. Cardiol. 1991;86:107–116. doi: 10.1007/BF02190543. [DOI] [PubMed] [Google Scholar]
- TOBE T.J., DE LANGEN C.D., TIO R.A., BEL K.J., MOOK P.H., WESSELING H. In vivo effect of bradykinin during ischaemia and reperfusion: improved electrical stability two weeks after myocardial infarction in the pig. J. Cardiovasc. Pharmacol. 1991;17:600–607. doi: 10.1097/00005344-199104000-00012. [DOI] [PubMed] [Google Scholar]
- TSUCHIDA A., LIU G.S., WILBORN W.H., DOWNEY J.M. Pretreatment with the adenosine A1 selective agonist, 2-chloro-N6-cyclopentyladenosine (CCPA), causes a sustained limitation of infarct size in rabbits. Cardiovasc. Res. 1993;27:652–656. doi: 10.1093/cvr/27.4.652. [DOI] [PubMed] [Google Scholar]
- URA N., CARRETERO O.A., ERDOS E.G. Role of renal endopeptidase 24.11 in kinin metabolism in vitro and in vivo. Kidney Int. 1987;32:507–513. doi: 10.1038/ki.1987.239. [DOI] [PubMed] [Google Scholar]
- VAN VELDHUISEN D.J., VAN GILST W.H. Vasopeptidase inhibition in heart failure. Lancet. 2000;28:1526. doi: 10.1016/S0140-6736(05)73277-3. [DOI] [PubMed] [Google Scholar]
- VAN WINKLE D.M., THORNTON J., DOWNEY J.M. Cardioprotection from ischaemic preconditioning is lost following prolonged reperfusion in the rabbit. Coron Art Dis. 1991;2:613–619. [Google Scholar]
- VÉGH A., PAPP J.G.Y., PARRATT J. Attenuation of the antiarrhythmic effects of ischaemic preconditioning by blockade of bradykinin B2 receptors. Br. J. Pharmacol. 1994;113:1167–1172. doi: 10.1111/j.1476-5381.1994.tb17120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VEGH A., PAPP J.G., SZEKERES L., PARRATT J.R. Prevention by an inhibitor of the L-arginine-nitric oxide pathway of the antiarrhythmic effects of bradykinin in anaesthetized dogs. Br. J. Pharmacol. 1993;110:18–19. doi: 10.1111/j.1476-5381.1993.tb13764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VÉGH A., SZEKERES L., PARRATT J.R. Local intracoronary infusions of bradykinin profoundly reduce the severity of ischaemia-induced arrhythmias in anaesthetised dogs. Br. J. Pharmacol. 1991;104:294–295. doi: 10.1111/j.1476-5381.1991.tb12424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALL T.M., SHEEHY R., HARTMAN J.C. Role of bradykinin in myocardial preconditioning. J. Pharmacol. Exp. Ther. 1994;270:681–689. [PubMed] [Google Scholar]
- WEBER M. Emerging treatments for hypertension: potential role for vasopeptidase inhibition. Am. J. Hypertens. 1999;12:139–147. doi: 10.1016/s0895-7061(99)00205-8. [DOI] [PubMed] [Google Scholar]
- WEIDENBACH R., SCHULZ R., GRES P., BEHRENDS M., POST H., HEUSCH G. Enhanced reduction of myocardial infarct size by combined ACE inhibition and AT1-receptor antagonism. Br. J. Pharmacol. 2000;131:138–144. doi: 10.1038/sj.bjp.0703544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIRTH K.J., LINZ W., WEIMER G., SCHOELKENS B.A. Kinins and cardioprotection. Pharmacol. Res. 1997;35:527–530. doi: 10.1006/phrs.1997.0181. [DOI] [PubMed] [Google Scholar]
- WOLFRUM S., RICHARDT G., DOMINIAK P., KATUS H.A., DENDORFER A. Apstatin, a selective inhibitor of aminopeptidase P, reduces myocardial infarct size by a kinin-dependent pathway. Br. J. Pharmacol. 2001;134:370–374. doi: 10.1038/sj.bjp.0704236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG X.P., LIU Y.H., PETERSON E., CARRETERO O.A. Effect of neutral endopeptidase 24.11 inhibition on myocardial ischemia/reperfusion injury: the role of kinins. J. Cardiovasc. Pharmacol. 1997a;29:250–256. doi: 10.1097/00005344-199702000-00014. [DOI] [PubMed] [Google Scholar]
- YANG X.P., LIU Y.H., SCICLI G.M., WEBB C.R., CARRETERO O.A. Role of kinins in the cardioprotective effect of preconditioning: study of myocardial ischaemia/reperfusion injury in B2 receptor knockout mice and kininogen-deficient rats. Hypertension. 1997b;30:735–740. doi: 10.1161/01.hyp.30.3.735. [DOI] [PubMed] [Google Scholar]
- YANG X.P., LIU Y.H., SHESELY E.G., BULAGANNAWAR M., LIU F., CARRETERO O.A. Endothelial nitric oxide gene knockout mice. Cardiac phenotypes and effect of angiotensin-converting enzyme inhibitor on myocardial ischaemia/reperfusion injury. Hypertension. 1999;34:24–30. doi: 10.1161/01.hyp.34.1.24. [DOI] [PubMed] [Google Scholar]
- YELLON D.M., BAXTER G.F. Reperfusion injury revisited: is there a role for growth factor signaling in limiting lethal reperfusion injury. Trends Cardiovasc. Med. 1999;9:245–259. doi: 10.1016/s1050-1738(00)00029-3. [DOI] [PubMed] [Google Scholar]
- YOSHIDA H., ZHANG J.J., CHAO L., CHAO J. Kallikrein gene delivery attenuates myocardial infarction and apoptosis after myocardial ischaemia and reperfusion. Hypertension. 2000;35:25–31. doi: 10.1161/01.hyp.35.1.25. [DOI] [PubMed] [Google Scholar]
- ZHU P., ZAUGG C.E., SIMPER D., HORNSTEIN P., ALLEGRINI P.R., BUSER P.T. Bradykinin improves postischaemic recovery in the rat heart: role of high energy phosphates, nitric oxide and prostacyclin. Card. Res. 1995;29:658–663. [PubMed] [Google Scholar]
- ZISMAN L. Inhibiting tissue angiotensin-converting enzyme, a pound of flesh without the blood. Circulation. 1998;98:2788–2790. doi: 10.1161/01.cir.98.25.2788. [DOI] [PubMed] [Google Scholar]