

Abstract
This study was designed to compare the proarrhythmic activity of the antimalarial drug, halofantrine and the antihistamine, terfenadine, with that of clofilium a K+ channel blocking drug that can induce torsade de pointes.
Experiments were performed in pentobarbitone-anaesthetized, open-chest rabbits. Each rabbit received intermittent, rising dose i.v. infusions of the α-adrenoceptor agonist phenylephrine. During these infusions rabbits also received increasing i.v. doses of clofilium (20, 60 and 200 nmol kg−1 min−1), terfenadine (75, 250 and 750 nmol kg−1 min−1), halofantrine (6, 20 and 60 μmol kg−1) or vehicle.
Clofilium and halofantrine caused dose-dependent increases in the rate-corrected QT interval (QTc), whereas terfenadine prolonged PR and QRS intervals rather than prolonging cardiac repolarization. Progressive bradycardia occurred in all groups. After administration of the highest dose of each drug halofantrine caused a modest decrease in blood pressure, but terfenadine had profound hypotensive effects resulting in death of most rabbits.
The total number of ventricular premature beats was highest in the clofilium group. Torsade de pointes occurred in 6 out of 8 clofilium-treated rabbits and 4 out of 6 of those which received halofantrine, but was not seen in any of the seven terfenadine-treated rabbits.
These results show that, like clofilium, halofantrine can cause torsade de pointes in a modified anaesthetized rabbit model whereas the primary adverse effect of terfenadine was cardiac contractile failure.
Keywords: Torsade de pointes, QT prolongation, halofantrine, terfenadine, clofilium, α-agonist, ECG intervals, proarrhythmia
Introduction
Torsade de pointes is a form of polymorphic ventricular tachycardia (VT) that exhibits a characteristic twisting of QRS complexes around the isoelectric axis. Under normal circumstances this arrhythmia is rare, but it can occur when the QT interval of the ECG is prolonged, either as a consequence of a genetic alteration in ion channel behaviour or because of administration of certain drugs (Viskin, 1999). Drugs with class III antiarrhythmic actions prolong QT intervals, but a variety of other drugs used for non-cardiac indications have also been associated with this phenomenon, including the antihistamine terfenadine (Crumb et al., 1995) and the antimalarial halofantrine (Castot et al., 1993; Monlun et al., 1993; Nosten et al., 1993). Although there are a number of reports of the actions of terfenadine and halofantrine on QT intervals and information is available on their abilities to block ion channels, there is a lack of information about their actions in in vivo models of torsade de pointes. We have demonstrated previously that halofantrine prolongs rate-corrected QT (QTc) intervals in anaesthetized guinea-pigs (Batey et al., 1997) and rabbits (Lightbown et al., 2001) but torsade de pointes did not occur in either of these studies.
The in vivo models used most often to examine the potential of drugs to cause torsade de pointes involve acute administration of the drug of interest to anaesthetized dogs with chronic hypokalaemia and atrioventricular (AV) block and anaesthetized rabbits subject to α-adrenoceptor stimulation (Eckardt et al., 1998). The rabbit model was first described by Carlsson et al. (1990) and has been used by this group and others, e.g. (Buchanan et al., 1993; Farkas et al., 1998) to examine the proarrhythmic effects of various drugs. Carlsson's model involves anaesthesia with α-chloralose but the same protocol has been used in pentobarbitone-anaesthetized rabbits (Bril et al., 1996).
Clofilium has class III antiarrhythmic actions and has been used as a standard proarrhythmic drug in these anaesthetized rabbit models (Bril et al., 1996; Buchanan et al., 1993; Carlsson et al., 1990). The main aim of the present work was therefore to determine the effects of halofantrine and terfenadine and compare them with those of clofilium in pentobarbitone-anaesthetized, α-adrenoceptor stimulated rabbits. Before commencing the main experiments, pilot studies with clofilium were performed to establish that the model worked adequately in our laboratory. These studies led to modification of the model so that clofilium-induced torsade de pointes could be obtained in the majority of open-chest, pentobarbitone-anaesthetized rabbits. The modified protocol was then used to examine and compare the effects of terfenadine and halofantrine with those of clofilium.
Methods
Surgical preparation
Male New Zealand White rabbits (2.5 – 3.5 kg) were purchased from Charles River, Margate, Kent. The left marginal ear vein was anaesthetized locally by application of lignocaine ointment and 15 min later general anaesthesia was induced by i.v. administration of sodium pentobarbitone 30 to 50 mg kg−1 using a 30 mg ml−1 solution. The trachea was cannulated, and the animal ventilated (Bioscience pump, 38 strokes min−1, 6 to 7 ml kg−1) with room air if necessary. The right femoral vein and artery were cannulated for drug administration and measurement of blood pressure respectively. All cannulae in blood vessels were filled with 0.9% saline, containing 10 units ml−1 heparin to maintain patency. Arterial blood gases and pH were checked (Ciba-Corning 850 blood gas analyzer), and the pump volume adjusted to maintain PO2 > 80 mmHg. If necessary, the depth of anaesthesia was increased using 60 mg ml−1 sodium pentobarbitone. A sternal split and pericardotomy were then performed to expose the anterior surface of the heart. Any animals not already being ventilated were ventilated at this point, and a positive end expiratory pressure of 1 – 2 cm H2O was applied. Blood gases were checked again, the ventilation pump adjusted if necessary, and further samples taken for blood gas analysis to confirm adequacy of ventilation. Limb lead ECGs (leads I, II and III) were monitored using subcutaneous needle electrodes. The left carotid artery was isolated and a cannula inserted via this artery into the lumen of the left ventricle to monitor left ventricular pressure. An endocardial monophasic action potential recording was obtained by advancing an electrode via the right jugular vein into the apex of the right ventricle. An epicardial monophasic action potential was monitored using a suction electrode (10 – 15 mmHg), attached to the anterior surface of the left ventricle. Animals were allowed at least 10 min stabilization after completion of surgery.
Arterial and left ventricular pressures were recorded with Bell & Howell type 4-422 transducers connected to a Grass 7P122 or 7P1/7DA amplifiers. ECG and monophasic action potential signals were amplified by Grass 7P1, 7P4 or 7P6 preamplifiers with 7DA driver amplifiers. All signals from the Grass amplifiers were continuously recorded at 1000 Hz, on a Po-Ne-Mah computerized data acquisition and analysis system (Linton, Diss, Norfolk, U.K.). Files were subsequently archived on compact disk.
Experimental protocol
Preliminary experiments were carried out to establish a suitable protocol for the induction of torsade de pointes in pentobarbitone-anaesthetized, open-chest rabbits given clofilium and an α-adrenoceptor agonist. Combinations of various infusion rates of α-adrenoceptor agonist and doses of clofilium were investigated and the protocol detailed below (see Figure 1) was chosen. The effects of clofilium, terfenadine and halofantrine were then compared with those of a vehicle. The doses of drugs tested in this study were clofilium 20, 60 and 200 nmol kg−1 min−1, terfenadine 75, 250 and 750 nmol kg−1 min−1 and halofantrine 6, 20 and 60 μmol kg−1. The terfenadine vehicle served as the control. A cannula was primed with phenylephrine (0.9 mg ml−1), and introduced into the right marginal ear vein. At time zero, phenylephrine infusion was begun at 75 nmol kg−1 min−1 (Syringe infusion pump 22, Harvard Apparatus, Edenbridge, Kent, U.K.). After 5 min, infusion of the lowest dose of the drug of interest was begun. At +15 min, the phenylephrine infusion rate was increased to 150 nmol kg−1 min−1. At +18 and +21 min the rate increased to 225 and then 300 nmol kg−1 min−1. At +24 min, both infusion pumps were turned off. This protocol was repeated with two higher doses of the drug of interest, with 10 min intervals between doses.
Figure 1.
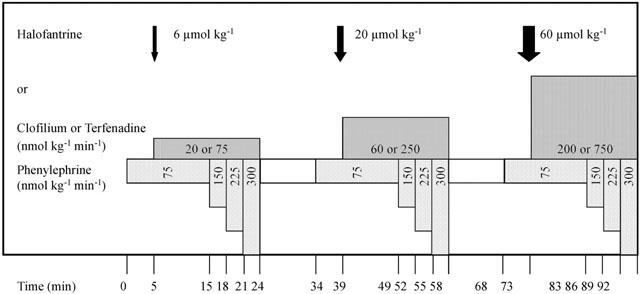
The experimental protocol. After preparation and stabilization, infusion of phenylephrine at 75 nmol kg−1 min−1 was started at time 0 min. At 5 min the first dose of the drug of interest was given, either as a bolus dose (e.g. halofantrine) or as a continuous infusion (e.g. clofilium or terfenadine). At 15 min the infusion rate for phenylephrine was increased to 150 nmol kg−1 min−1 with further increases to 225 and 300 nmol kg−1 min−1 at 18 and 21 min. Both infusions were switched off at 24 min and a 10 min drug-free interval allowed. This cycle of drug administration was repeated twice more using the same infusion rates for phenylephrine but increasing the dose of the drug of interest by approximately 3 fold each time.
Post experiment analysis
Data was replayed on a personal computer, for retrieval of haemodynamic parameters and arrhythmia identification and counting. Any areas of unusual rhythm were printed out for independent identification by a second investigator. Ventricular tachycardia (VT) was defined according to the Lambeth Conventions (Walker et al., 1988) as four or more consecutive ventricular premature beats (VPBs). Torsade de pointes was defined as a polymorphic VT where clear twisting of QRS complexes around the isoelectric line could be seen in at least one ECG lead (see Figure 2 for examples). In almost all cases of torsade de pointes the arterial blood pressure declined towards zero with very little pulsatile activity whereas, when polymorphic VT that did not twist around the isoelectric line occurred, the blood pressure was usually pulsatile.
Figure 2.
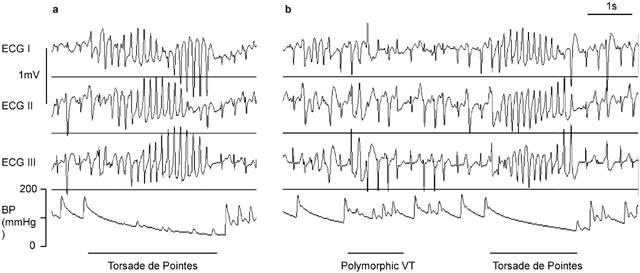
Examples of arrhythmias in an anaesthetized rabbit which was receiving phenylephrine and clofilium. The traces shown are Leads I, II and III of the limb lead ECG and the arterial blood pressure (BP). Panel (a) is from the 2nd cycle of drug administration and shows a short episode of torsade de pointes, where the twisting of the QRS complexes around the isoelectric line can be seen clearly. In panel (b) which is from the 3rd dosing cycle, there is a short episode of polymorphic VT that is not torsade de pointes and shortly after there is another episode of torsade de pointes. Note that during the polymorphic VT the blood pressure signal remains pulsatile whereas during the episodes of torsade de pointes there is little pulsatile activity and the blood pressure is declining towards zero.
ECG intervals were measured manually using on screen cursors, taking the mean of at least four ECG complexes at each time point measured. ECG intervals were only measured in beats that originated from the sino-atrial node. The PR interval was measured from the onset of the P wave to the onset of the R wave (i.e. Q), the QRS interval from the beginning of the Q wave to the point at which the ST segment crossed the isoelectric line, and the QT interval from Q to the end of the T wave (including any U wave, if present). At some time points the T (or U) wave overlapped the P wave of the subsequent complex, because of either a relatively high heart rate or extensive QT prolongation. In these cases the end of the TU wave was extrapolated from the curve of the TU wave to the isoelectric line under the P wave. At some time points ECG intervals could not be obtained from all animals because marked arrhythmic activity precluded accurate measurement of ECG intervals.
As QT intervals are influenced by heart rate, baseline data for heart rate and QT intervals were used to determine the relationship between RR interval and QT interval. These data were obtained from 133 pentobarbitone-anaesthetized rabbits prepared as described above, including those used here in the main study and others used in similar studies in our laboratory (Batey & Coker, 2000; Farkas & Coker, 2000; 2001). Simple linear regression revealed a positive correlation between QT and RR intervals, with QT=0.704RR+16.364 (Figure 3a). In a manner similar to that described by Carlsson et al. (1993), this equation was rearranged to allow the calculation of the rate-corrected QT interval (QTc-Liverpool (QTcL)) at an RR interval of 250 ms (i.e. a heart rate of 240 beats min−1) using the formula QTcL=QT − 0.704(RR-250). With this equation, plotting QTcL against RR interval produces a regression line with a slope of zero (Figure 3b) indicating that this correction removes the influence of heart rate. Thus, all QTc intervals in this study were corrected according to this ‘Liverpool' formula.
Figure 3.
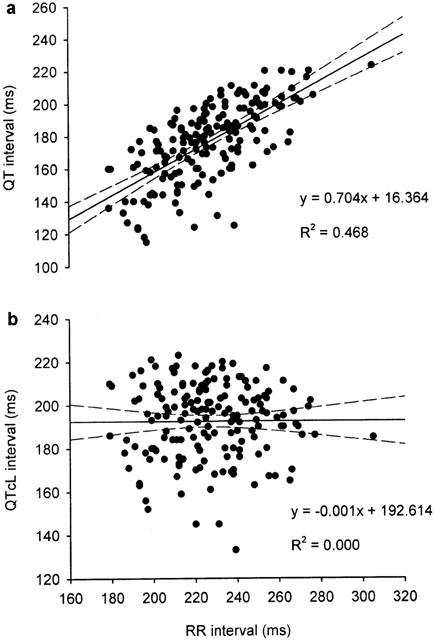
The relationships between RR interval and (a) QT interval and (b) QTc interval corrected using the Liverpool formula (QTcL) where QTcL=QT-0.704(RR-250). Each panel contains 169 baseline data points obtained from 133 male, pentobarbitone-anaesthetized, open-chest rabbits. Dashed lines indicate the 95% confidence intervals of the regression line.
Statistics
Where appropriate data are expressed as mean±s.e.mean of n experiments. Shapiro-Wilk tests revealed that some data were not distributed normally. Within group comparisons were made with Friedman's tests whereas Kruskal-Wallis tests were used for comparisons between groups at the beginning, mid-point and end of each cycle (i.e. time 0 min, 15, 24, 34, 49, 58, 68, 83 and 92 min). Fisher's exact tests were used to compare the incidence of events. A probability value of P<0.05 was considered to be significant.
Drugs
Anaesthetics, chemicals and drugs were purchased from the following suppliers: clofilium tosylate, L-phenylephrine, (±)-terfenadine, polyethylene glycol 200 - Sigma, Poole, Dorset, U.K.; lignocaine ointment (Lignocaine ointment BP 5%, Biorex), heparin (Multiparin®, CP Pharmaceuticals) - Royal Liverpool University Hospital Pharmacy; sodium pentobarbitone (Sagatal®) - Rhône Mérieux, Dagenham, Essex, U.K.; D-glucose - FSA Laboratory Supplies, Loughborough, U.K.. (±)-Halofantrine HCl (formula 9, batch no. 3D01HP; a 50 mg ml−1 solution) was a gift from SmithKline Beecham Pharmaceuticals, Brentford Middlesex, U.K.
Clofilium tosylate was dissolved in 0.9% w v−1 NaCl solution, at a stock concentration of 1.02 mg ml−1. Terfenadine was dissolved in 20% of final volume polyethylene glycol 200, acidified with 1 M HCl, and sonicated at room temperature for 30 min. The volume was then made up with 5% w v−1 D-glucose solution, to make a stock solution of 3.54 mg ml−1. Further dilutions were made in 5% D-glucose solution as required. The terfenadine vehicle was given in appropriate concentrations to the control group. The halofantrine formulation consisted of halofantrine HCl dissolved in dimethylacetamide (40%)/propylene glycol (60%) v v−1, at 50 mg ml−1. Immediately prior to administration of each dose, the appropriate volume of stock drug solution was diluted with 5% w v−1 D-glucose solution containing 10 units ml−1 heparin to give a total volume for administration of 1 ml kg−1. Each dose was given by slow intravenous injection (∼30 s) and flushed through the cannula with heparinized D-glucose solution. It has been shown previously that the vehicle for halofantrine does not alter heart rate, blood pressure or ECG intervals in anaesthetized rabbits (Lightbown et al., 2001).
Results
Model development
In the first six preliminary experiments the α-agonist methoxamine was infused at 70 nmol kg−1 min−1 continuously and clofilium was infused at 63 nmol kg−1 min−1 for 30 min, then at 630 nmol kg−1 min−1 for a further 30 min. Although VPBs were observed in all six rabbits, none had torsade de pointes. In further experiments the rate of infusion of methoxamine was increased to 140 nmol kg−1 min−1 and sometimes to higher rates. Phenylephrine was also used in some experiments instead of methoxamine. Torsade de pointes occurred in six out of 15 rabbits. Sudden changes in the rate of α-agonist infusion appeared to act as a trigger for torsade de pointes. Bursts of torsade de pointes often coincided with the switching on or off of the α-agonist infusion, or with changing the rate of infusion, either up or down. Stable rhythms such as normal sinus rhythm, bigeminy, or AV block could be interrupted by changing the rate of infusion of the α-agonist, often inducing torsade de pointes. In phenylephrine stimulated animals it was noted that AV block was much less common than with methoxamine. For these reasons it was decided to make use of a protocol which included changing phenylephrine infusion rates.
Arrhythmias
In the control group, infusion of rising doses of phenylephrine in combination with the vehicle caused VPBs, but these were restricted primarily to the first dosing cycle (Figure 4) and occurred mainly as single VPBs with very occasional pairs. No VT or torsade de pointes was observed (Figure 5). When clofilium was infused with phenylephrine there was a marked increase in arrhythmic activity with significantly greater numbers of VPBs occurring in each dosing cycle (Figure 4). The arrhythmias observed with clofilium were also more severe and included monomorphic VT and torsade de pointes (Figure 5). Although the greatest number of VPBs occurred in the first dosing cycle, torsade de pointes was most likely to occur during the second dosing cycle. The numbers of VPBs occurring in each cycle in the terfenadine and halofantrine groups were similar (Figure 4) but terfenadine failed to cause torsade de pointes, whereas this arrhythmia was observed in four of the six rabbits which received halofantrine (Figure 5).
Figure 4.
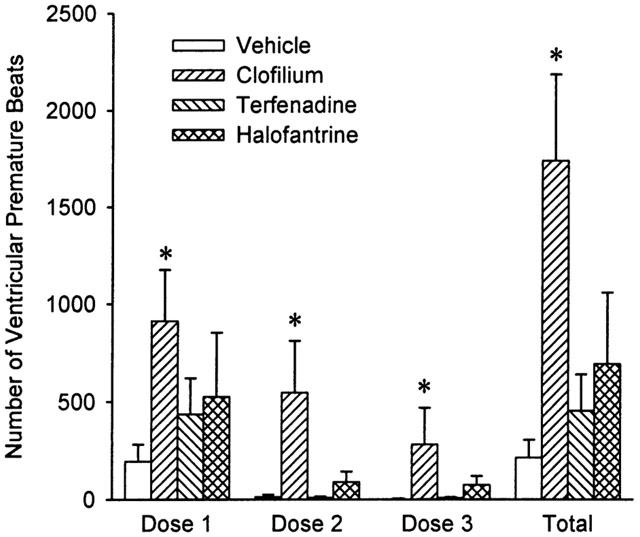
The number of ventricular premature beats (VPBs) counted during each of the three dosing cycles and the total number of VPBs that occurred in each group. Values are means with vertical bars indicating s.e.mean. Group n values are vehicle=5, clofilium=8, terfenadine=7 and halofantrine=6. *P<0.05 compared to vehicle control group, Kruskal-Wallis test.
Figure 5.
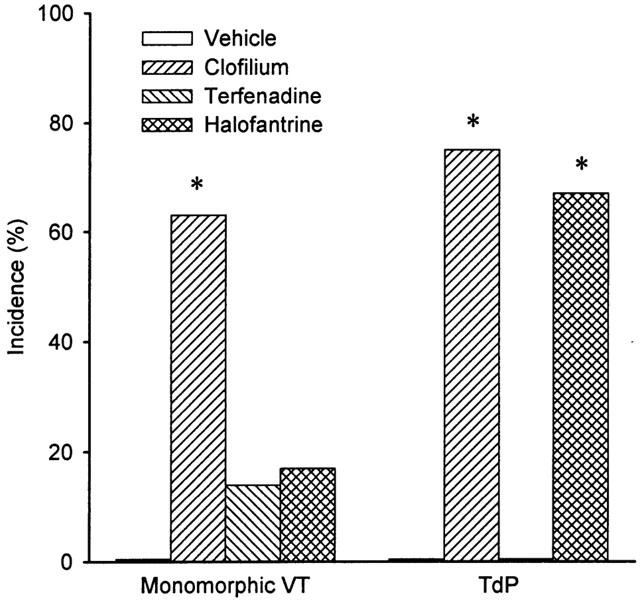
The incidences of monomorphic ventricular tachycardia (VT) and torsade de pointes (TdP). Group n values are vehicle=5, clofilium=8, terfenadine=7 and halofantrine=6. *P<0.05 compared to vehicle control group, Fisher's exact test.
Haemodynamics
Phenylephrine dose-dependently increased arterial blood pressure. During the first dosing cycle there were no differences in mean blood pressure among the groups (Figure 6). In the second and third cycles commencing infusion of clofilium and halofantrine tended to delay the phenylephrine-induced pressor response and this effect was more marked with terfenadine (Figure 6). At the end of the second cycle the mean blood pressure was significantly lower in the halofantrine and terfenadine groups. There appeared to be further blunting of the pressor response to phenylephrine in the third cycle in the halofantrine group and the depressor effect of terfenadine was so marked that it led to death of the majority of the rabbits before the end of the experiment.
Figure 6.
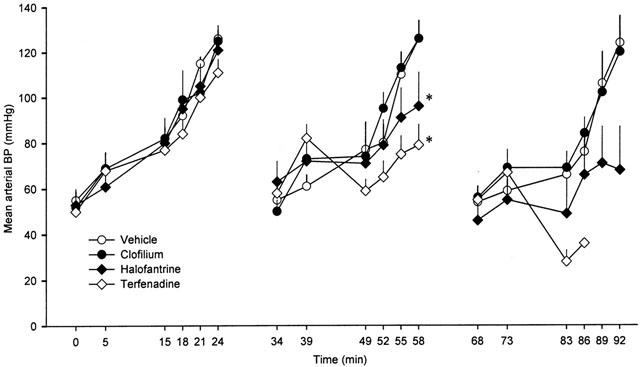
Mean arterial blood pressure (BP) in anaesthetized rabbits during each of the three consecutive dosing cycles. Values are means with vertical bars indicating s.e.mean. Group n values are vehicle=5, clofilium=8, terfenadine=7 and halofantrine=6. *P<0.05 compared to vehicle control group, Kruskal-Wallis test.
As the experiments progressed bradycardia developed in all groups (Table 1). There was some variation in baseline values for heart rate among the groups. The data have therefore been plotted as per cent changes to allow comparison between groups (Figure 7). During the first and second dosing cycles there were no significant differences among the groups but in the third cycle the reduction in heart rate was greater with terfenadine. It is also interesting to note that the pattern of changes in heart rate does not mirror the changes in blood pressure. At the end of each cycle the blood pressures returned to baseline values, whereas there was no reversal of the changes in heart rate, suggesting that these were unlikely to be solely a consequence of reflex activity.
Table 1.
Heart rate, QT and QTcL intervals measured during normal sinus rhythm at various time during the experiments

Figure 7.
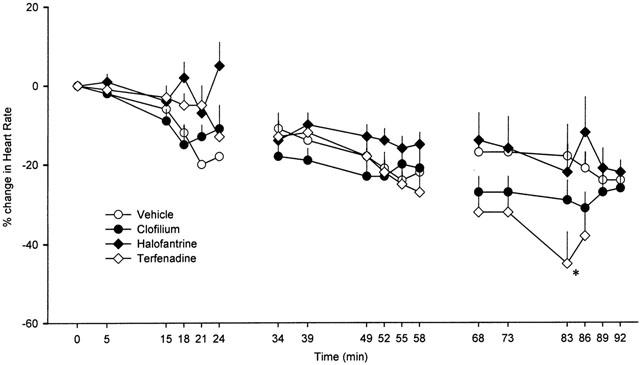
Per cent change in heart rate in anaesthetized rabbits during each of the three consecutive dosing cycles. Values are means with vertical bars indicating s.e.mean. Group n values are vehicle=5, clofilium=8, terfenadine=7 and halofantrine=6. At some time points n is less than the total for the group because marked arrhythmic activity prevented accurate measurement of heart rate. Where data are missing n<3 due to arrhythmias or in the case of terfenadine, contractile failure. *P<0.05 compared to vehicle control group, Kruskal-Wallis test.
ECG intervals
The QT intervals increased with time in all groups (Table 1), however, as noted above, heart rate also declined in all groups as the experiments progressed. After correction for variations in heart rate, only clofilium and halofantrine produced significant increases in QTc intervals (Table 1). Although terfenadine appeared to increase QTc intervals there was more variability among these data and the apparent differences failed to reach statistical significance. As there was also a difference in QTc intervals between groups at baseline, the values have been plotted as per cent changes to allow comparison among groups (Figure 8). During the second cycle of drug administration the per cent change in QTc interval was greater in the clofilium group. In the control group PR and QRS intervals remained steady throughout the duration of the experiments. Clofilium caused a small increase in PR interval whereas a numerically similar increase with the highest dose of halofantrine did not reach statistical significance (Table 2). Terfenadine, however, caused extensive elongation of both the PR and QRS intervals (Table 2).
Figure 8.
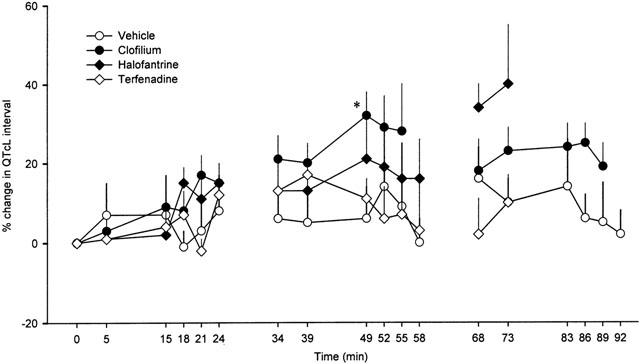
Per cent change in the QT interval corrected for heart rate using the Liverpool formula (QTcL) in anaesthetized rabbits during each of the three consecutive dosing cycles. Values are means with vertical bars indicating s.e.mean. Group n values are vehicle=5, clofilium=8, terfenadine=7 and halofantrine=6. At some time points n is less than the total for the group because marked arrhythmic activity prevented accurate measurement of ECG intervals. Where data are missing n<3 due to arrhythmias or in the case of terfenadine, contractile failure. *P<0.05 compared to vehicle control group, Kruskal-Wallis test.
Table 2.
PR and QRS intervals measured during normal sinus rhythm at various points during the experiments

Discussion
The results of these experiments demonstrate clearly that halofantrine and clofilium can cause torsade de pointes in an appropriate animal model, whereas terfenadine failed to cause torsade de pointes but did cause a fatal decline in cardiac contractility. In order to obtain this information the model originally described by Carlsson et al. (1990) had to be modified so that a reasonable incidence of torsade de pointes could be obtained using clofilium as a positive control in pentobarbitone-anaesthetized, open-chest rabbits.
Model modification
The results of the first preliminary experiments were disappointing as no torsade de pointes occurred using the standard protocol of infusion of methoxamine and clofilium. The use of a different anaesthetic is unlikely to account for the failure to induce torsade de pointes in these preliminary experiments as clofilium caused torsade de pointes in ketamine/xylazine anaesthetized rabbits (Mazur et al., 1999) and in pentobarbitone-anaesthetized rabbits (Bril et al., 1996). It was noted, however, that despite using the same doses, the changes in blood pressure and heart rate induced by methoxamine seemed to be lower than those reported by others for methoxamine (Bril et al., 1996; Carlsson et al., 1990; Mazur et al., 1999) or phenylephrine (Farkas et al., 1998), so the dose of α-agonist was increased. Most authors who have published on the α-adrenoceptor stimulated anaesthetized rabbit model have used closed-chest animals with only a few experiments from open-chest animals having been reported (Carlsson et al., 1990). It is likely that in order to achieve a depth of anaesthesia sufficient to perform a thoracotomy there was greater suppression of α-adrenoceptor responsiveness in the present study. This could explain why higher doses of α-agonist had to be used to produce sufficient increases in blood pressure and reductions in heart rate.
We used open-chest rabbits to allow recording of epicardial monophasic action potentials. Although delays in repolarization were observed in animals with QT prolongation, signal quality deteriorated when complex arrhythmias occurred making it difficult to measure action potential duration accurately at these times. Subsequently, in other studies we have used a different type of electrode and been able to quantify action potential duration satisfactorily (Farkas & Coker, 2000).
Halofantrine
The present study is the first demonstrating that halofantrine does cause torsade de pointes in an appropriate in vivo animal model. Since 1993 there have been a number of clinical reports of adverse cardiac events including QTc prolongation and sudden death, (Castot et al., 1993; Monlun et al., 1993; Nosten et al., 1993). More recently, halofantrine has been shown to block the rapidly activating delayed rectifier potassium current (IKr) in feline ventricular myocytes (Wesche et al., 2000) and a corresponding cloned channel (HERG) expressed in Chinese hamster ovary cells (Tie et al., 2000). In previous in vivo studies we found that halofantrine prolonged QTc intervals in anaesthetized guinea-pigs and rabbits (Batey et al., 1997; Lightbown et al., 2001). Although AV block and delays in cardiac repolarization were observed in these studies, torsade de pointes did not occur. The present study therefore proves, that in an appropriate model, halofantrine can cause torsade de pointes and provides evidence linking the effects of halofantrine on IKr in vitro to the adverse cardiac events observed in patients.
Terfenadine
Despite being given in doses sufficient to cause profound hypotension, terfenadine did not cause torsade de pointes. These effects of terfenadine are in agreement with those of Lu et al. (2000), published since we commenced our studies, which indicate that terfenadine also failed to cause torsade de pointes in chloralose-anaesthetized, methoxamine-stimulated, closed-chest rabbits. These authors also reported that high doses of terfenadine caused cardiac arrest. The cardiac contractile failure induced by high doses of terfenadine is likely to be a consequence of blockade of L-type Ca2+ channels or Na+ channels. The structure of terfenadine is similar to that of phenylalkylamine Ca2+ channel blockers and in vitro studies have provided evidence that terfenadine blocks L-type Ca2+ channels in cardiac myocytes (Liu et al., 1997; Ming & Nordin, 1995). Blockade of Na+ channels by terfenadine (Lu & Wang, 1999; Ming & Nordin, 1995) could also contribute to negative inotropic actions.
Blockade of L-type Ca2+ channels is also a plausible explanation for the failure to demonstrate prolongation of QTc intervals with terfenadine in the present study. Although terfenadine may have reduced IKr which would be expected to increase action potential duration, this could have been offset by a reduced action potential duration resulting from blockade of L-type Ca2+ channels. A number of reports indicate that terfenadine can block Na+, Ca2+ and K+ channels in cardiac myocytes (Berul & Morad, 1995; Crumb et al., 1995; Liu et al., 1997; Lu & Wang, 1999; Ming & Nordin, 1995; Salata et al., 1995). The marked increases in PR and QRS intervals seen with terfenadine suggest that the doses administered here were sufficient to reduce currents flowing through Na+ (INa) and Ca2+ (ICaL) channels. It has been shown previously that BRL-32872, a compound which blocks both ICaL and IKr, had limited ability to induce torsade de pointes in pentobarbitone-anaesthetized, α-adrenoceptor stimulated rabbits (Bril et al., 1996).
In the present study terfenadine increased QRS duration without significantly increasing QTc intervals. This is in agreement with the findings of Lu et al. (2000) who reported that terfenadine did not alter JT intervals, and that the observed increases in QT intervals were due to increases in QRS duration. The failure of terfenadine to induce torsade de pointes in both of these rabbit models does not necessarily suggest that either of the models is inappropriate. Although terfenadine has been associated with sudden cardiac death, documented cases of torsade de pointes are very rare and have usually occurred when terfenadine has been given in combination with an inhibitor of CYP3A4 (Burkhart & Freiman, 1995; Pratt et al., 1994). It has also been suggested that most sudden deaths with terfenadine were due to cardiac arrest (Pratt et al., 1995), but it is not clear whether this was a consequence of arrhythmia or contractile failure. Thus, given the very low prevalence of torsade de pointes relative to the exposure of many millions of individuals to terfenadine, and the lack of selective prolongation of cardiac repolarization in the present study, it is perhaps not surprising that terfenadine did not cause torsade de pointes.
Clofilium
Clofilium has been used as a standard proarrhythmic drug by several groups (Bril et al., 1996; Carlsson et al., 1990; Lu et al., 2000; Mazur et al., 1999). The present study confirms that clofilium also has proarrhythmic activity in open-chest anaesthetized rabbits when phenylephrine is used as the α-adrenoceptor agonist, provided that the doses of phenylephrine that are given are sufficient to cause adequate changes in blood pressure and heart rate.
In their recent publication Lu et al. (2000) suggest that clofilium causes torsade de pointes because it is a selective blocker of IKr whereas terfenadine does not cause torsade de pointes because it is a ‘non-selective IKr blocker'. Whilst terfenadine certainly blocks a wide range of ion channels it may be inappropriate to assume that clofilium causes torsade de pointes by selective blockade of IKr. As well as blocking IKr clofilium has been reported to block other K+ currents such as the slowly activating delayed rectifier potassium current (IKs) (Li et al., 2001) and the transient outward potassium current (Ito) (Castle, 1991). In addition, it also blocks Na+ and Ca2+ channels (Li et al., 1996). Small but significant changes in PR interval did occur with clofilium although QRS intervals were not altered, which may suggest a small degree of blockade of Ca2+ channels. The assumption that only selective IKr blockers can cause torsade de pointes in the ‘Carlsson' model is also challenged by the reports of torsade de pointes occurring with ibutilide, which enhances an inward Na+ current (Buchanan et al., 1993), and with RP-58866 which blocks the inward rectifier potassium current (IK1) and IKr (Bril et al., 1996). Using the present model, terikalant (RP-62719, the active enantiomer of RP58866) also caused torsade de pointes (Farkas & Coker, 2000).
Correcting QT intervals for changes in heart rate
A novel correction factor was applied to the present data because neither Carlsson's nor Bazett's correction factor removed the relationship between RR interval and heart rate in the baseline data. When the correction factor used by Carlsson et al. (1993) was applied to our baseline data it revealed that Carlsson's correction formula (QTc=QT-0.175(RR-300) did not correct our data adequately as there was still a positive correlation between QTcC (Carlsson) and RR interval (R2=0.332). Similarly, Bazett's correction factor (QTc=QT/√HRR) also failed to correct adequately for the influence of heart rate. Once again there was still some positive correlation between QTcB (Bazett) and RR interval (R2=0.149). Interestingly, in our previous studies on halofantrine in rabbits anaesthetized with Hypnorm and pentobarbitone (Lightbown et al., 2001), Bazett's correction factor did correct baseline QT intervals adequately for variations in heart rate, as there was no correlation between QTcB and RR interval (R2=0.001). This emphasises that it is best to determine an appropriate correction factor for each laboratory and particular set of conditions. In the present study all QTc intervals are the QT intervals corrected according to the Liverpool formula.
Comparison of the data on QT and QTc intervals in Table 1 reveals the importance of applying an appropriate correction factor to QT values to account for changes in heart rate. QT intervals increased significantly in all groups, whereas QTc values were not significantly increased in the vehicle group. This indicates that phenylephrine did not prolong the QT interval per se and that phenylephrine-induced changes in QT intervals were just a consequence of reduced heart rate. With clofilium and halofantrine, however, the prolongation of QTc intervals reveals a genuine increase in QT intervals independent of heart rate.
Conclusions
The results of these experiments demonstrate, for the first time, that halofantrine can cause torsade de pointes in an in vivo model, and that its proarrhythmic activity is comparable to that of clofilium. In contrast, high doses of terfenadine did not elicit torsade de pointes, but did cause cardiac contractile failure. The data presented above also indicate that pentobarbitone-anaesthetized, open-chest, α-adrenoceptor stimulated rabbits are suitable for studying the effects of potentially proarrhythmic drugs, provided that an appropriate positive control group is included for comparison.
Acknowledgments
This work was supported by the British Heart Foundation (PG/96100). We thank Dr R. J. Horton, International Medical Division, SmithKline Beecham Pharmaceuticals Ltd., Brentford for arranging the supply of halofantrine and Dr Derek T. Connelly, Cardiothoracic Centre, Liverpool for advice on ECG and arrhythmia interpretation.
Abbreviations
- AV
atrioventricular
- ECG
electrocardiogram
- ICaL
L-type calcium current
- IK1
inward rectifier potassium current
- IKr
rapidly activating delayed rectifier potassium current
- IKs
slowly activating delayed rectifier potassium current
- INa
sodium current
- Ito
transient outward potassium current
- QTc
rate-corrected QT interval
- VPBs
ventricular premature beats
- VT
ventricular tachycardia
References
- BATEY A.J., COKER S.J. Lack of torsade de pointes with terfenadine compared to clofilium in an in vivo model. Br. J. Pharmacol. 2000;131:14P. doi: 10.1038/sj.bjp.0704550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BATEY A.J., LIGHTBOWN I.D., LAMBERT J.P., EDWARDS G., COKER S.J. Comparison of the acute cardiotoxicity of the antimalarial drug halofantrine in vitro and in vivo in anaesthetized guinea-pigs. Br. J. Pharmacol. 1997;122:563–569. doi: 10.1038/sj.bjp.0701402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERUL C.I., MORAD M. Regulation of potassium channels by nonsedating antihistamines. Circulation. 1995;91:2220–2225. doi: 10.1161/01.cir.91.8.2220. [DOI] [PubMed] [Google Scholar]
- BRIL A., GOUT B., BONHOMME M., LANDAIS L., FAIVRE J.F., LINEE P., POYSER R.H., RUFFOLO R.R., JR Combined potassium and calcium channel blocking activities as a basis for antiarrhythmic efficacy with low proarrhythmic risk: experimental profile of BRL-32872. J. Pharmacol. Exp. Ther. 1996;276:637–646. [PubMed] [Google Scholar]
- BUCHANAN L.V., KABELL G., BRUNDEN M.N., GIBSON J.K. Comparative assessment of ibutilide, D-sotalol, clofilium, E-4031, and UK-68,798 in a rabbit model of proarrhythmia. J. Cardiovasc. Pharmacol. 1993;22:540–549. [PubMed] [Google Scholar]
- BURKHART G.A., FREIMAN J.F. The risk of life-threatening cardiovascular events with terfenadine. Am. J. Cardiol. 1995;75:213–214. doi: 10.1016/s0002-9149(00)80087-9. [DOI] [PubMed] [Google Scholar]
- CARLSSON L., ABRAHAMSSON C., ANDERSSON B., DUKER G., SCHILLER-LINHARDT G. Proarrhythmic effects of the class III agent almokalant: importance of infusion rate, QT dispersion, and early afterdepolarisations. Cardiovasc. Res. 1993;27:2186–2193. doi: 10.1093/cvr/27.12.2186. [DOI] [PubMed] [Google Scholar]
- CARLSSON L., ALMGREN O., DUKER G. QTU-prolongation and torsades de pointes induced by putative class III antiarrhythmic agents in the rabbit: etiology and interventions. J. Cardiovasc. Pharmacol. 1990;16:276–285. doi: 10.1097/00005344-199008000-00014. [DOI] [PubMed] [Google Scholar]
- CASTLE N.A. Selective inhibition of potassium currents in rat ventricle by clofilium and its tertiary homolog. J. Pharmacol. Exp. Ther. 1991;257:342–350. [PubMed] [Google Scholar]
- CASTOT A., RAPOPORT P., LECOZ P. Prolonged QT interval with halofantrine. Lancet. 1993;341:1541–1541. [PubMed] [Google Scholar]
- CRUMB W.J., WIBLE B., ARNOLD D.J., PAYNE J.P., BROWN A.M. Blockade of multiple human cardiac potassium currents by the antihistamine terfenadine - possible mechanism for terfenadine-associated cardiotoxicity. Mol. Pharmacol. 1995;47:181–190. [PubMed] [Google Scholar]
- ECKARDT L., HAVERKAMP W., BORGGREFE M., BREITHARDT G. Experimental models of torsade de pointes. Cardiovasc. Res. 1998;39:178–193. doi: 10.1016/s0008-6363(98)00043-1. [DOI] [PubMed] [Google Scholar]
- FARKAS A., COKER S.J. Comparison of erythromycin, terikalant and clofilium in an in vivo model of torsade de pointes. Br. J. Pharmacol. 2000;131:93P. [Google Scholar]
- FARKAS A., COKER S.J. Prostaglandin E2 prevents clofilium-induced torsade de pointes. Br. J. Pharmacol. 2001;133:116P. doi: 10.1016/s0014-2999(03)01910-1. [DOI] [PubMed] [Google Scholar]
- FARKAS A., LEPRAN I., PAPP J.G. Comparison of the antiarrhythmic and the proarrhythmic effect of almokalant in anaesthetised rabbits. Eur J Pharmacol. 1998;346:245–253. doi: 10.1016/s0014-2999(98)00067-3. [DOI] [PubMed] [Google Scholar]
- LI Q., HIMMEL H.M., RAVENS U. Selectivity of class-III antiarrhythmic action of clofilium in guinea pig ventricular myocytes. J. Cardiovasc. Pharmacol. 1996;27:401–410. doi: 10.1097/00005344-199603000-00013. [DOI] [PubMed] [Google Scholar]
- LI R.A., MIAKE J., HOPPE U.C., JOHNS D.C., MARBAN E., NUSS H.B. Functional consequences of the arrhythmogenic G306R KvLQT1 K+ channel mutant probed by viral gene transfer in cardiomyocytes. J. Physiol.-London. 2001;533:127–133. doi: 10.1111/j.1469-7793.2001.0127b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIGHTBOWN I.D., LAMBERT J.P., EDWARDS G., COKER S.J. Potentiation of halofantrine-induced QTc prolongation by mefloquine: correlation with blood concentrations of halofantrine. Br. J. Pharmacol. 2001;132:197–204. doi: 10.1038/sj.bjp.0703823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU S., MELCHERT R.B., KENNEDY R.H. Inhibition of L-type Ca2+ channel current in rat ventricular myocytes by terfenadine. Circ. Res. 1997;81:202–210. doi: 10.1161/01.res.81.2.202. [DOI] [PubMed] [Google Scholar]
- LU H.R., REMEYSEN P., DE CLERCK F. Nonselective IKr-blockers do not induce torsades de pointes in the anesthetized rabbit during alpha1-adrenoceptor stimulation. J. Cardiovasc. Pharmacol. 2000;36:728–736. doi: 10.1097/00005344-200012000-00007. [DOI] [PubMed] [Google Scholar]
- LU Y., WANG Z. Terfenadine block of sodium current in canine atrial myocytes. J. Cardiovasc. Pharmacol. 1999;33:507–513. doi: 10.1097/00005344-199903000-00023. [DOI] [PubMed] [Google Scholar]
- MAZUR A., RODEN D.M., ANDERSON M.E. Systemic administration of calmodulin antagonist W-7 or protein kinase A inhibitor H-8 prevents torsade de pointes in rabbits. Circulation. 1999;100:2437–2442. doi: 10.1161/01.cir.100.24.2437. [DOI] [PubMed] [Google Scholar]
- MING Z., NORDIN C. Terfenadine blocks time-dependent Ca2+, Na+, and K+ channels in guinea-pig ventricular myocytes. J. Cardiovasc. Pharmacol. 1995;26:761–769. doi: 10.1097/00005344-199511000-00013. [DOI] [PubMed] [Google Scholar]
- MONLUN E., PILLET O., COCHARD J.F., GARRIGUES J.C.F., LEBRAS M. Prolonged QT interval with halofantrine. Lancet. 1993;341:1541–1542. [PubMed] [Google Scholar]
- NOSTEN F., TERKUILE F.O., LUXEMBURGER C., WOODROW C., KYLE D.E., CHONGSUPHAJAISIDDHI T., WHITE N.J. Cardiac effects of antimalarial treatment with halofantrine. Lancet. 1993;341:1054–1056. doi: 10.1016/0140-6736(93)92412-m. [DOI] [PubMed] [Google Scholar]
- PRATT C.M., HERTZ R.P., ELLIS B.E., CROWELL S.P., LOUV W., MOYE L. Risk of developing life-threatening ventricular arrhythmia associated with terfenadine in comparison with over-the-counter antihistamine, ibuprofen and clemastine. Am. J. Cardiol. 1994;73:346–352. doi: 10.1016/0002-9149(94)90006-x. [DOI] [PubMed] [Google Scholar]
- PRATT C.M., MOYE L., HERTZ R.P. The risk of life-threatening cardiovascular events with terfenadine - reply. Am. J. Cardiol. 1995;75:214. doi: 10.1016/s0002-9149(00)80087-9. [DOI] [PubMed] [Google Scholar]
- SALATA J.J., JURKIEWICZ N.K., WALLACE A.A., STUPIENSKI R.F., GUINOSSO P.J., LYNCH J.J. Cardiac electrophysiological actions of the histamine H1-receptor antagonists astemizole and terfenadine compared with chlorpheniramine and pyrilamine. Circ. Res. 1995;76:110–119. doi: 10.1161/01.res.76.1.110. [DOI] [PubMed] [Google Scholar]
- TIE H., WALKER B.D., SINGLETON C.B., VALENZUELA S.M., BURSILL J.A., WYSE K.R., BREIT S.N., CAMPBELL T.J. Inhibition of HERG potassium channels by the antimalarial agent halofantrine. Br. J. Pharmacol. 2000;130:1967–1975. doi: 10.1038/sj.bjp.0703470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VISKIN S. Long QT syndromes and torsade de pointes. Lancet. 1999;354:1625–1633. doi: 10.1016/S0140-6736(99)02107-8. [DOI] [PubMed] [Google Scholar]
- WALKER M.J.A., CURTIS M.J., HEARSE D.J., CAMPBELL R.W.F., JANSE M.J., YELLON D.M., COBBE S.M., COKER S.J., HARNESS J.B., HARRON D.W.G., HIGGINS A.J., JULIAN D.G., LAB M.J., MANNING A.S., NORTHOVER B.J., PARRATT J.R., RIEMERSMA R.A., RIVA E., RUSSELL D.C., SHERIDAN D.J., WINSLOW E., WOODWARD B. The Lambeth Conventions–Guidelines for the study of arrhythmias in ischemia, infarction, and reperfusion. Cardiovasc. Res. 1988;22:447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- WESCHE D.L., SCHUSTER B.G., WANG W.X., WOOSLEY R.L. Mechanism of cardiotoxicity of halofantrine. Clin. Pharmacol. Ther. 2000;67:521–529. doi: 10.1067/mcp.2000.106127. [DOI] [PubMed] [Google Scholar]