

Abstract
Human embryonic kidney (HEK)-293 cells expressing recombinant Gαi-coupled, human CXC chemokine receptor 2 (CXCR2) were used to study the elevation of the intracellular [Ca2+] ([Ca2+]i) in response to interleukin-8 (IL-8) following pre-stimulation of endogenously expressed P2Y1 or P2Y2 nucleotide receptors.
Pre-stimulation of cells with adenosine 5′-triphosphate (ATP) revealed a substantial Ca2+ signalling component mediated by IL-8 (Emax=83±8% of maximal ATP response, pEC50 of IL-8 response=9.7±0.1).
1 μM 2-methylthioadenosine 5′-diphosphate (2MeSADP; P2Y1 selective) and 100 μM uridine 5′-triphosphate (UTP; P2Y2 selective) stimulated equivalent maximal increases in [Ca2+]i elevation. However, UTP caused a sustained elevation, whilst following 2MeSADP [Ca2+]i rapidly returned to basal levels.
Both UTP and 2MeSADP increased the potency and magnitude of IL-8-mediated [Ca2+]i elevation but the effects of UTP (Emax of IL-8 response increased to 50±1% of the maximal response to ATP, pEC50 increased to 9.8±0.1) were greater than those of 2MeSADP (Emax increased to 36±2%, pEC50 increased to 8.7±0.2).
The potentiation of IL-8-mediated Ca2+ signalling by UTP was not dependent upon the time of IL-8 addition following UTP but was dependent on the continued presence of UTP. Potentiated IL-8 Ca2+ signalling was apparent in the absence of extracellular Ca2+, demonstrating the release of Ca2+ from intracellular stores.
Activation of P2Y1 and P2Y2 receptors also revealed Ca2+ signalling by an endogenously expressed, Gαs-coupled β-adrenoceptor.
In conclusion, pre-stimulation of P2Y nucleotide receptors, particularly P2Y2, facilitates Ca2+ signalling by either recombinant CXCR2 or endogenous β-adrenoceptors.
Keywords: Calcium, G proteins, CXCR2, P2Y receptors, FLIPR, potentiation
Introduction
The CXC chemokine receptor 2 (CXCR2; also known as the interleukin-8 receptor B) is a member of the super-family of heptahelical G-protein-coupled receptors (GPCRs). Of the more than 50 chemokines currently known, CXCR2 is activated by the CXC chemokines interleukin-8 (IL-8), epithelial-derived neutrophil-activating peptide-78, granulocyte chemotactic protein, neutrophil-activating peptide 2, lipopolysaccharide-induced CXC chemokine and growth-related oncogene α, β and γ. The expression and function of CXCR2 have been characterized predominantly in polymorphonuclear cells of the immune system including neutrophils, macrophages and eosinophils in which CXCR2 activation mediates chemotaxis and activation during the inflammatory process (for a full review of chemokine classification, expression and function see Murphy et al., 2000). The expression of CXCR2 is not restricted to cells of the immune system and it has also been implicated in the regulation of hematopoesis (Cacalano et al., 1994; Broxmeyer et al., 1996), foetal CNS development (Dame & Juul, 2000), communication between CNS neurons and glia (Horuk et al., 1997; Coughlan et al., 2000) and trophic/pro-survival signalling in adult neurons (Araujo & Cotman, 1993; Horuk et al., 1997). Additionally, aberrant expression or activity of either CXCR2 or its ligands has been implicated in pathological conditions including the progression of atherosclerotic plaque (Boisvert et al., 1998), psoriasis (Kulke et al., 1998), Alzheimer's disease (Xia et al., 1997; Xia & Hyman, 1999) and the angiogenesis associated with solid tumour formation and wound repair (Belperio et al., 2000; Addison et al., 2000).
CXCR2, whether expressed endogenously in leukocytes or as a recombinant protein in human embryonic kidney (HEK)-293 cells, couples preferentially to pertussis toxin (PTX)-sensitive G-proteins, particularly Gαi2 (Damaj et al., 1996). Indeed, the functional consequences of CXCR2 activation in neutrophils, including chemotaxis and degranulation, are abolished by PTX pre-treatment (Addison et al., 2000; Hall et al., 1999). Although the precise signal transduction events underlying the functional responses to CXCR2 activation remain to be defined, the elevation of intracellular [Ca2+] ([Ca2+]i) is likely to play a central role in many of these responses. Previous studies have shown that a number of other Gαi-coupled GPCRs have markedly potentiated Ca2+ signalling following prestimulation of co-expressed Gαq/11-coupled receptors (Megson et al., 1995; Connor et al., 1997; Dickenson & Hill, 1998; Yeo et al., 2001; Buckley et al., 2001). Such potentiation has not yet been reported for chemokine receptors, and may have important implications for the physiological and pathological functions of these receptors.
A number of cell types that express CXCR2, including neutrophils and monocytes, also express P2Y nucleotide receptors, most likely P2Y2, that couple via Gαq/11 to the activation of phospholipase C (PLC) (Boarder et al., 1995; Jin et al., 1998). Thus, there is clear scope for a cross-talk mechanism to exist in these cells. This has the potential for cellular responses to chemokines that are active at CXCR2 to be tailored according to the presence or absence of ATP. Thus, responses to CXCR2 activation could differ in inflammatory or non-traumatic lesions (where ATP release will be low) compared to traumatic or necrotic lesions (where ATP release from damaged cells will be high). In the present study we used a HEK-293 cell line co-expressing recombinant human CXCR2 receptors and endogenous P2Y nucleotide receptors to determine whether this chemokine receptor is subject to regulatory cross-talk, specifically whether activation of a PLC-coupled receptor potentiates CXCR2-mediated Ca2+ signalling. We also assessed the G-protein specificity of any cross talk by determining the interaction between P2Y nucleotide receptors and endogenously expressed Gαs-coupled (CTX-sensitive) adrenoceptors.
Methods
Materials
All cell culture reagents were obtained from Gibco BRL Life Technologies (Paisley, U.K.) except foetal calf serum (PAA Laboratories, Linz, Austria). Lipofectamine transfection reagent also came from Gibco BRL. The expression plasmid, pRc/CMV, was supplied by Invitrogen (Inchinnan, U.K.). Fura-2 acetoxymethyl ester (fura-2-AM), apyrase (Grade III), cholera toxin (CTX) and pertussis toxin (PTX) were obtained from Sigma Aldrich (Poole, U.K.). Fluo-3 acetoxymethyl ester (fluo-3-AM) was from TEF Labs (Austin, TX, U.S.A.) and pluronic F-127 was from Molecular Probes Ltd (Leiden, Holland). Cell culture plastics were obtained from Nalgene (Europe) Ltd (Hereford, U.K.) and black poly-D-lysine-coated 96-well plates for use in the fluorescent light imaging plate reader (FLIPR) were from Becton Dickinson (Bedford, MA, U.S.A.). Adenosine 5′-triphosphate (ATP), 2-methylthio-ATP (2MeSATP), adenosine 5′-diphosphate (ADP), 2-methylthio-ADP (2MeSADP), uridine 5′-triphosphate (UTP), uridine 5′-diphosphate (UDP) and (±)-arterenol were from Sigma Aldrich (Poole, U.K.). IL-8 was supplied by R&D Systems (Abingdon, U.K.). AR-C67085 (2-propylthio-beta, gamma-dichloromethylene-D-ATP) was provided by the Medicinal Chemistry Department, AstraZeneca R&D Charnwood. All other reagents were of analytical grade and were obtained from Sigma Aldrich (Poole, U.K.) or Fisher Scientific (Loughborough, U.K.).
Generation of cell line and cell culture
The coding sequence for CXCR2 was cloned into the expression plasmid pRc/CMV and transfected into HEK-293 cells using Lipofectamine in accordance with the manufacturer's instructions. Clones were selected using geneticin and positive clones expressing CXCR2 identified using [125I]-IL-8 binding. Expression of CXCR2 in the clone used for these studies was approximately 50,000 sites/cell (data not shown). HEK-CXCR2 cells were maintained routinely in Dulbecco's Modified Eagle's Medium (containing 25 mM D-glucose, 4 mM L-alanyl-L-glutamine and 1 mM sodium pyruvate) and supplemented with 10% foetal calf serum, 1% non-essential amino acids and geneticin (0.4 mg ml−1). Cells were grown in 175 cm2 tissue culture flasks at 37°C in a 5% CO2 humidified atmosphere.
[Ca2+]i measurement – Fluorescence Imaging Plate Reader (FLIPR)
Cells were seeded at 50,000 cells per well in poly-D-lysine coated 96-well plates and grown overnight to approximately 80% confluence. Preliminary experiments indicated that this density was optimal for detection of agonist-induced changes in fluorescence in subsequent FLIPR assays (data not shown). Cells were incubated for 1 h at room temperature in a balanced salts solution (BSS, composition (mM): NaCl, 130; KCl, 5.4; NaHCO3, 16; NaH2PO4, 1.3; MgCl2, 0.8; CaCl2, 1.8; HEPES, 10; D-glucose, 5.5; and bovine serum albumin, 1%; pH 7.4) containing 5 μM fluo-3-AM and 0.044% pluronic F-127. Cells were washed once with BSS at 37°C, 100 μl of BSS added to each well and the plate transferred to a FLIPR (Molecular Devices Ltd, U.S.A.) for assay at 37°C. Following the determination of basal fluorescence for 10 s, an addition of a nucleotide receptor agonist or its vehicle control (BSS) was made, followed by addition of either IL-8 or the stable noradrenaline analogue, (±)-arterenol. All additions were made in a 50 μl volume at a rate of 40 μl s−1. Fluo-3-loaded cells were excited at 505 nm, with emission recorded at 530 nm every 2 s.
The Kd of fluo-3 is 450 nM at 37°C and the region of the Ca2+ binding curve that approximates to linearity (20 – 80%) will, therefore, be within the magnitude of the [Ca2+]i responses in these cells. Furthermore we have shown that in fluo-3-loaded HEK cells, ionomycin (by elevation of [Ca2+]i) causes an increase in fluorescence that is twice that seen with a maximal concentration of UTP (data not shown). Thus, agonists produce elevations of [Ca2+]i that are considerably below dye saturation. Additionally, over the range of fluorescence values within our experiments, the relationship between [fluo-3] and fluorescence in nominally Ca2+-free BSS is linear as measured by FLIPR (data not shown) indicating linearity of the fluorescence detection system. Thus, we believe that fluo-3 fluorescence values will be directly proportional to [Ca2+]i over the range encountered in these experiments and allow direct determination of both pEC50 and Emax values.
[Ca2+]i measurement – fluorimetry
HEK-CXCR2 cells were grown to confluence, harvested with trypsin and washed once with BSS. Viable cells (>95%) were counted using trypan blue exclusion, re-suspended in BSS at 5×106 viable cells per ml and loaded with 5 μM of fura-2-AM in the presence of 0.044% pluronic F-127. Aliquots of 3.5×106 cells were taken, centrifuged at 7000 r.p.m. for 5 s and re-suspended in 2 ml BSS at 37°C in a cuvette. Following a 5 – 10 min incubation at 37°C to enhance intracellular de-esterification of fura-2-AM, cuvettes were transferred to a Fluoromax I fluorimeter (Jobin-Yvon Ltd, Middlesex, U.K.) in which the cell suspension was maintained at 37°C and mixed continually using a magnetic follower. Cells were alternately excited at wavelengths of 340 nm and 380 nm with emission determined at 510 nm. Data are presented as a ratio of the 340/380 values that were collected every 1.5 s.
Ca2+ measurement - Confocal microscopy
Cells were seeded onto 22 mm diameter poly-D-lysine coated glass coverslips and cultured for 48 h. Cells were then loaded with fluo-3-AM (using the conditions described above for FLIPR-based [Ca2+]i measurement) and the coverslips mounted in a chamber on the stage of an Olympus IX70-S1F inverted microscope. The chamber was perfused at a rate of 5 ml min−1 with BSS or drug solutions and the temperature maintained at 37°C using a peltier unit. Using an UltraVIEW confocal imaging system (PerkinElmer Life Sciences, Cambridge, U.K.), cells were excited with a krypton/argon laser at 488 nm and emitted light collected above 510 nm. Confocal images were captured by cooled CCD camera at a rate of approximately one frame per second.
Data analysis
The response to all agonists was expressed as the initial peak increase in fluorescence (fluo-3) or fluorescence ratio (fura-2) that occurred following addition of agonist (see Figure 1). These measurements provide an index of the increase in [Ca2+]i calculated as the difference between the lowest point on the baseline in the 10 s preceding agonist addition and the highest point of elevation in the 30 s following the addition. Unless stated otherwise, data were normalized against the response achieved by a maximal concentration of the pre-stimulating P2Y nucleotide receptor agonist (⩾100 μM for ATP or UTP; ⩾1 μM for 2MeSADP). All data are expressed as the mean of three or more experiments±s.e.mean. Where representative data are shown these are also from experiments performed to n⩾3. Concentration-response curves were fitted using a four-parameter logistic equation with equal weighting to all points using GraphPad Prism (GraphPad Software, San Diego, CA, U.S.A.). The pEC50 value (negative logarithm of the molar concentration of agonist giving 50% of the maximal response to that agonist) and Emax values (the maximal response relative to the maximal response achieved by the pre-stimulating agonist unless otherwise stated) were determined from these curves and the individual values from each experimental set used to calculate mean values. Unless otherwise stated, statistical analysis was by Student's two-tailed, unpaired, t-test. For all tests, significance was accepted at P<0.05.
Figure 1.
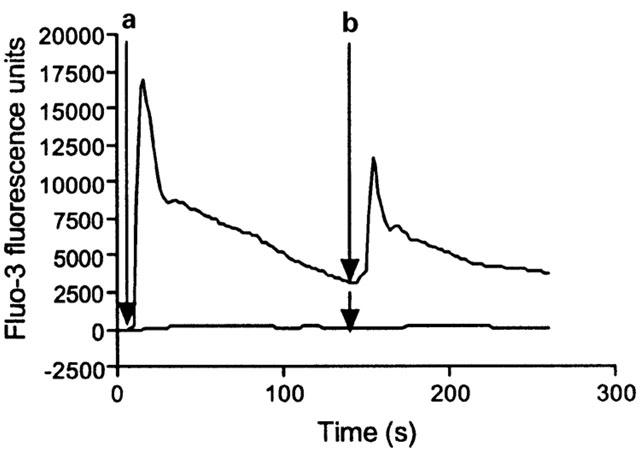
Potentiation of IL-8-mediated elevation of [Ca2+]i in HEK-CXCR2 cells following ATP pre-stimulation. Representative traces from a FLIPR experiment showing fluo-3 fluorescence as an index of [Ca2+]i. Arrow A (10 s) indicates addition of 100 μM ATP (upper trace only) and arrow B (150 s) indicates addition of 30 nM IL-8 (upper and lower traces). Data are raw FLIPR data expressed in fluo-3 fluorescence units.
Results
Potentiation of IL-8- and arterenol-mediated Ca2+ signalling by ATP
Initial studies using the FLIPR demonstrated that challenge of HEK-CXCR2 cells with 30 nM IL-8 alone was unable to elevate [Ca2+]i (Figure 1). In contrast, challenge with the same concentration of IL-8, 150 s following (and in the continued presence of) 100 μM ATP resulted in a substantial elevation of [Ca2+]i (Figure 1). The initial challenge with ATP produced an increase in [Ca2+]i consisting of a rapid, transient peak followed by a slower declining phase such that [Ca2+]i was still elevated at the point of IL-8 addition. Similarly, challenge of endogenously expressed adrenoceptors with (±)-arterenol did not elevate [Ca2+]i unless ATP was present (data not shown).
Concentration-response curves were generated to IL-8 and (±)-arterenol following either no addition, addition of 1 mM ATP or vehicle addition. In the absence of any pre-addition, neither IL-8 nor (±)-arterenol elevated [Ca2+]i (Figure 2a,b). Following addition of 1 mM ATP, IL-8 evoked a concentration-dependent increase in [Ca2+]i with a pEC50 of 9.7±0.1 and Emax 83±8% (Figure 2a; n=3), whilst (±)-arterenol elevated [Ca2+]i with a pEC50 of 6.8±0.1 and Emax of 67±3% (Figure 2b; n=3). Addition of vehicle (BSS) alone also revealed [Ca2+]i responses to IL-8 and (±)-arterenol although these were significantly less than the responses observed in the presence of 1 mM ATP (P<0.05 for both pEC50 and Emax values) (Figure 2a,b; n=3). Vehicle pre-treatment caused IL-8 to elevate [Ca2+]i with a pEC50 of 8.4±0.4 and Emax 34±3% whilst arterenol elevated [Ca2+]i with a pEC50 of 6.2±0.1 and Emax of 11±1%.
Figure 2.
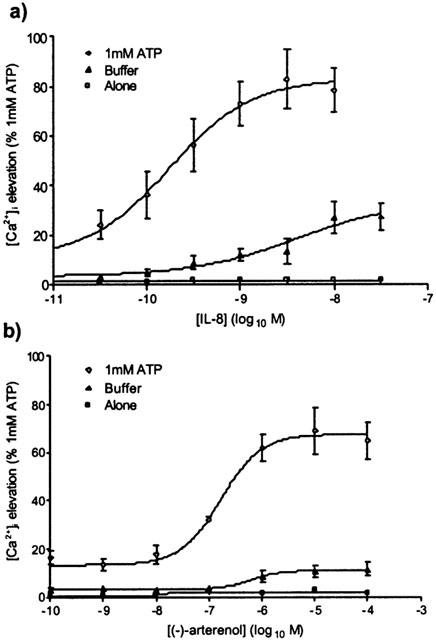
Potentiation by ATP of IL-8-mediated (a) and (±)-arterenol-mediated (b) [Ca2+]i responses in HEK-CXCR2 cells. Using the FLIPR, cells were pre-stimulated at t=10 s with either 1 mM ATP, buffer control or were not pre-stimulated, and at t=150 s, either IL-8 (a) or (±)-arterenol (b) was added. Changes in fluo-3 fluorescence were measured as an index of [Ca2+]i. Data are expressed as the percentage of the response to 1 mM ATP and are the mean±s.e.mean of three experiments, each performed in duplicate.
It has been shown previously that Ca2+-signalling by δ-opioid receptors in stirred suspensions of SH-SY5Y cells is sensitive to apyrase (ATP diphosphohydrolase, which converts nucleotide polyphosphates to nucleoside monophosphates (Curdova et al., 1982)), suggesting that endogenous ATP release may act as a potentiating factor (Yeo et al., 2001). We therefore investigated the possibility that release of endogenous nucleotides from the cell monolayer following addition of buffer was responsible for the effects of vehicle pre-treatment. In the absence of apyrase, the addition of vehicle revealed responses to IL-8 or (±)-arterenol that were 28.4±1.4% and 26.9±2.0% (n=3), respectively, of the response to 1 mM ATP (Figure 3). These responses were reduced to 10.4±0.2% and 5.5±0.6% (n=3), respectively, in the presence of 10 u ml−1 apyrase (Figure 3). Pre-addition of buffer did not reveal a response to a subsequent addition of buffer in either the presence or absence of apyrase (Figure 3). Because the addition of buffer per se caused a small potentiation, responses revealed by nucleotide pre-addition were always compared with cells to which a pre-addition of buffer had been made.
Figure 3.
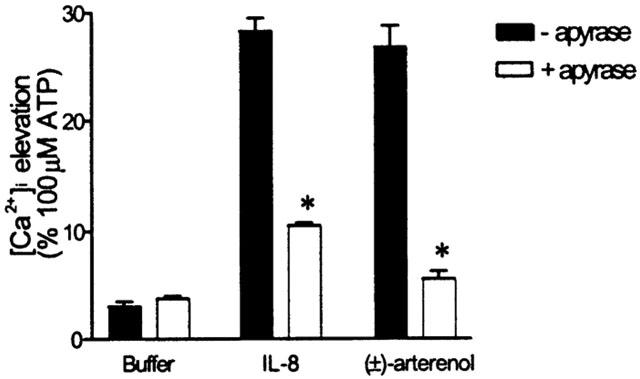
Effects of apyrase on buffer-induced potentiation of IL-8- and (±)-arterenol-mediated Ca2+ signalling. Using the FLIPR, cells were exposed to an addition of buffer (t=10 s) followed (t=150 s) by either an addition of buffer, 10 nM IL-8 or 10 μM (±)-arterenol. Apyrase (10 u ml−1, grade III) was either absent or present for the duration of the experiment. Fluo-3 fluorescence was recorded as an index of [Ca2+]i. The elevation of [Ca2+]i in response to the second addition (t=150 s) is expressed as a percentage of the response to 100 μM ATP. Data are mean±s.e.mean, n=3 and * denotes P<0.001 versus controls not treated with apyrase.
Characterization of P2Y nucleotide receptor expression in HEK-CXCR2 cells
In order to further characterize the receptor type that revealed IL-8- and arterenol-mediated [Ca2+]i responses, we constructed concentration-response curves for a number of P2Y receptor agonists. The rank order of potency was determined as: 2MeSADP>2MeSATP>ADP>ATP=UTP (Figure 4 and Table 1). UDP (10 nM – 1 mM) did not elevate [Ca2+]i (data not shown). These data are consistent with previous studies (Schachter et al., 1997) and demonstrate the expression of P2Y1 and P2Y2 receptors in HEK cells. AR-C67085, a potent P2Y11 agonist (Communi et al., 1999), did not elevate [Ca2+]i in our HEK-CXCR2 cell line confirming the absence of P2Y11 receptor expression.
Figure 4.
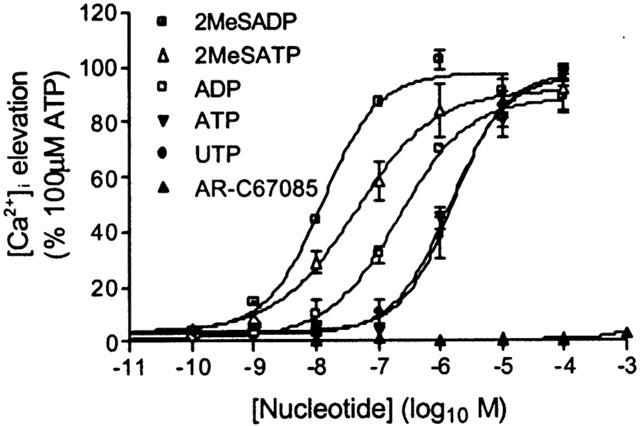
[Ca2+]i elevation in HEK-CXCR2 cells challenged with P2Y nucleotide receptor agonists. Using the FLIPR, changes in fluo-3 fluorescence were recorded as an index of [Ca2+]i following the addition of different P2Y nucleotide receptor agonists. The maximal change was determined in the 30s following agonist addition and expressed as a percentage of [Ca2+]i response to 100 μM ATP. Data are mean±s.e.mean, n=4.
Table 1.
The pEC50, slope and Emax values for stimulation of [Ca2+]i elevation by various P2Y receptor agonists
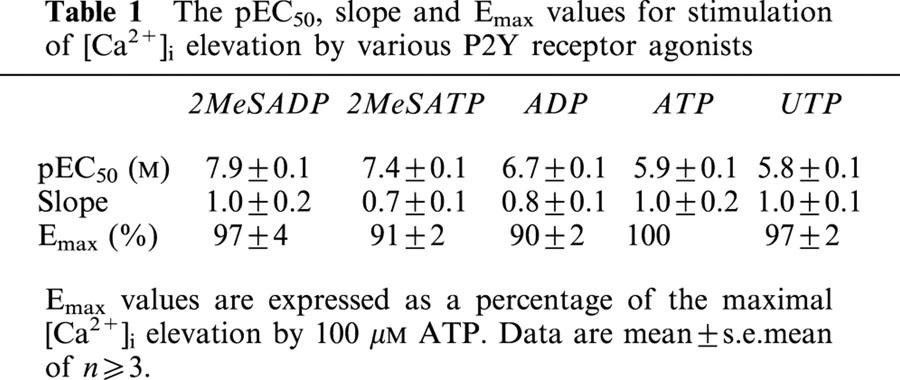
P2Y1 and P2Y2 receptor activation evoke different temporal patterns of [Ca2+]i elevation and have differential effects on Ca2+ signalling by IL-8 and (±)-arterenol
The profiles of [Ca2+]i elevation following addition of the P2Y receptor-selective agonists 2MeSADP (P2Y1 selective) or UTP (P2Y2 selective) (Nicholas et al., 1996) were markedly different. 2MeSADP caused a rapid, transient elevation of [Ca2+]i followed by a rapid fall back to basal levels (Figure 5; t½ value, for decay of Ca2+ elevation to 50% of maximum: 17±1 s after addition, n>10). In contrast, UTP caused a rapid, transient elevation of [Ca2+]i followed by a much slower declining phase (t½ value for decay of Ca2+ elevation to 50% of maximum: 72±4 s after addition, n>10) such that the [Ca2+]i was still elevated above basal (26±6% of maximum, n>10) at the time at which the addition of either IL-8 or (±)-arterenol was made (Figure 5). A second addition of a maximal concentration of either 2MeSADP or UTP following an initial maximal addition of the same agonist did not cause a further elevation of [Ca2+]i (Figure 5).
Figure 5.
Comparison of [Ca2+]i elevation profile for UTP and 2MeSADP. Cells were stimulated in the FLIPR at t=10 s with either 100 μM UTP or 1 μM 2MeSADP and the change in fluo-3 fluorescence recorded as an index of [Ca2+]i. The cells were then re-stimulated with an identical concentration of the same agonist at t=150 s. Each trace is the average of at least 10 traces from three separate experiments.
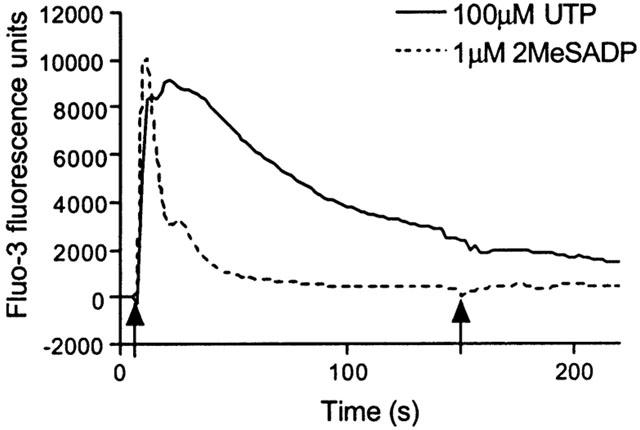
Cells were exposed to a range of concentrations of either 2MeSADP (0.1 nM – 10 μM) or UTP (1 nM – 1 mM), before being stimulated with a sub-maximal concentration of IL-8 (1 nM). The response to IL-8 was measured and expressed as a percentage of the response to 100 μM UTP. This concentration of UTP is maximal for [Ca2+]i elevation and gives a response equivalent to that of 1 mM ATP (Figure 4). Compared to addition of buffer, UTP significantly (P<0.001 by one-way analysis of variance (ANOVA)) increased the [Ca2+]i response to 1 nM IL-8 with a pEC50 for the potentiation of 5.0±0.1 (Figure 6a). The Emax achieved by 1 nM IL-8 following stimulation with a maximal concentration of UTP (100 μM) was 73±3%. 2MeSADP also significantly (P<0.002, one-way ANOVA) increased the response to 1 nM IL-8 (pEC50 for the potentiation=7.6±0.1, Emax=22±1%, n=3) (Figure 6a). Similarly, UTP and 2MeSADP both significantly (P<0.001, one way ANOVA) potentiated the [Ca2+]i response to 10 μM arterenol (pEC50 values for the potentiation=5.5±0.1 and 7.8±0.1 (n=3), respectively; Emax 82±1% and 30±1% (n=3), respectively) (Figure 6b).
Figure 6.
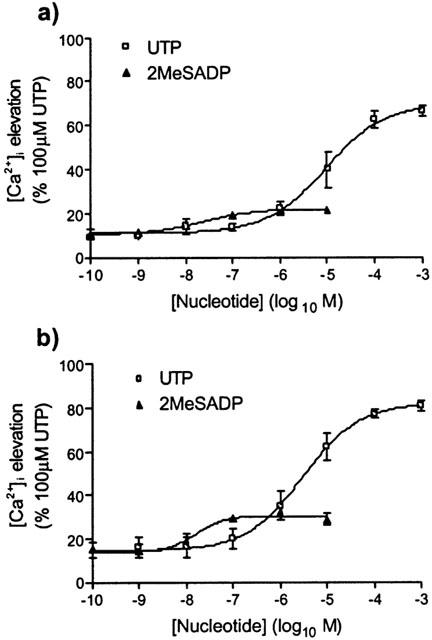
Ca2+ signalling following CXCR2 or adrenoceptor activation is potentiated by both P2Y2 and P2Y1 receptor activation. Using the FLIPR, cells were exposed to 1 nM IL-8 (a) or 10 μM (±)-arterenol (b) at t=150 s following pre-stimulation at t=10 s with increasing concentrations of a subtype-selective P2Y receptor agonist (P2Y2-selective: UTP; P2Y1-selective: 2MeSADP). Changes in fluo-3 fluorescence were recorded as an index of [Ca2+]i in response to the IL-8 or (±)-arterenol addition and expressed as a percentage of the maximal Ca2+ response to 100 μM ATP. All data are mean±s.e.mean, n=3.
To further investigate the relative abilities of P2Y1 and P2Y2 receptors to potentiate [Ca2+]i signalling by IL-8 and (±)-arterenol, we used concentrations of either 2MeSADP (1 μM) or UTP (100 μM) for the pre-stimulation that were maximal and equi-effective in terms of the spike [Ca2+]i response (Figure 4). Both the potency and Emax of IL-8- and (±)-arterenol-mediated [Ca2+]i elevations were significantly greater following prestimulation with 100 μM UTP compared to prestimulation with 1 μM 2MeSADP (Figure 7a,b. See tables accompanying Figure 7 for values).
Figure 7.
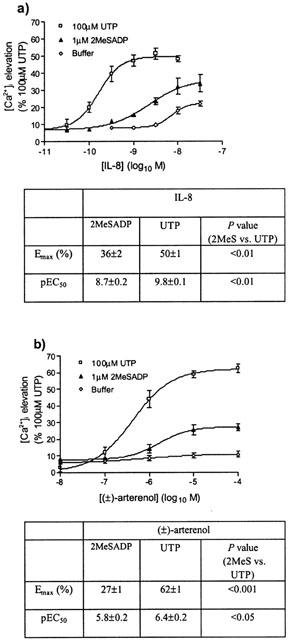
(a,b) Potentiation of IL-8- and (±)-arterenol-mediated responses by concentrations of P2Y receptor subtype-selective ligands that elicit equivalent maximal increases in [Ca2+]i. Cells were pre-stimulated with concentrations of UTP or 2MeSADP that gave equivalent elevation of [Ca2+]i (100 μM and 1 μM, respectively), or with vehicle. Cells were subsequently stimulated with increasing concentrations of IL-8 (a) or (±)-arterenol (b). Changes in [Ca2+]i in response to IL-8 and (±)-arterenol were measured by increases in fluo-3 fluorescence and are presented as a percentage of the maximal Ca2+ response to 100 μM UTP. Data are mean±s.e.mean, n=3.
We used a cuvette-based spectrofluorimeter to examine the temporal characteristics of the potentiation and found that the elevation of [Ca2+]i by IL-8 was potentiated irrespective of the time of addition of IL-8 between 50 s and 270 s (the longest time point used) after the addition of 100 μM UTP (Figure 8a).
Figure 8.
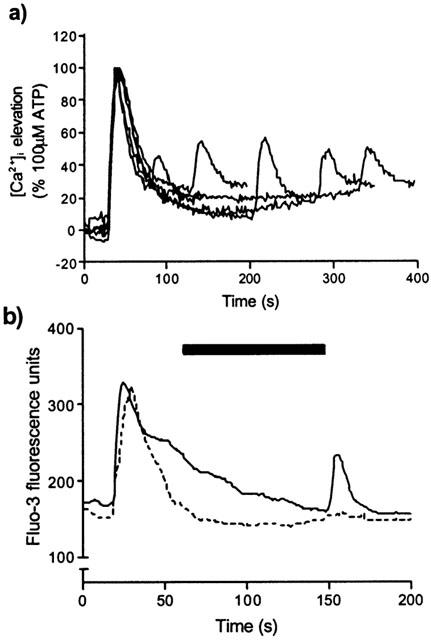
(a) The [Ca2+]i response to stimulation of CXCR2 is potentiated by P2Y2 receptor activation independently of the time of addition of IL-8 following UTP. Fura-2-loaded HEK-CXCR2 cells were pre-stimulated at 25 s with 100 μM UTP followed, after varying time intervals, by 10 nM IL-8. Using a cuvette-based fluorimeter, cells were excited alternately at 340 and 380 nm, with emission collected at 510 nm as an index of [Ca2+]i. Graph is a representative experiment (n=3) showing changes in [Ca2+]i expressed as the 340/380 ratio. In each case the [Ca2+]i response to IL-8 occurred within 10 s of its addition. (b) The potentiation of CXCR2-mediated Ca2+ signalling requires the continued activation of nucleotide receptors. Changes in [Ca2+]i were determined in fluo-3-loaded HEK-CXCR2 cells on 22 mm coverslips by confocal microscopy. The solid line represents an initial stimulation with 100 μM UTP (10 s) followed by a second stimulation (in the continued presence of UTP) with 10 nM IL-8 (150 s). The dashed line represents the same protocol except that UTP was washed out between additions by perfusing with buffer in the 90 s prior to the second addition (represented by solid bar). Data shown are representative of at least three experiments.
Potentiation requires continued presence of nucleotide
In order to assess the requirement for continued presence of the pre-stimulating agonist, we used a perfusion system and confocal microscopy to analyse changes in [Ca2+]i. In the absence of a P2Y nucleotide receptor agonist, neither IL-8 (10 nM) nor (±)-arterenol (10 μM) elevated [Ca2+]i (data not shown). In the presence of UTP (100 μM), IL-8 (10 nM) stimulated an [Ca2+]i elevation (Figure 8b). However, if UTP was washed out for 60 s before the addition of IL-8, no [Ca2+]i elevation was seen in response to IL-8 (Figure 8b). Similar results were seen when (±)-arterenol rather than IL-8 was used (data not shown).
Sensitivity of Ca2+ signalling to pertussis and cholera toxins
Treatment of cells for 20 h with 100 ng ml−1 pertussis toxin (PTX) (which ADP-ribosylates and inactivates Gαi) had no effect on [Ca2+]i responses to 100 μM UTP (data not shown), but abolished [Ca2+]i responses to 10 nM IL-8 following UTP pre-stimulation (47±4% in the absence of PTX, 1±0.5% in the presence of PTX; Figure 9a). In contrast, PTX had no effect on the response to 10 μM (±)-arterenol in the presence of 100 μM UTP (Figure 9a). Treatment of cells for 20 h with 2 μg ml−1 cholera toxin (CTX) (which ADP-ribosylates and, with extended exposure, down-regulates Gαs-mediated responses (Seidel et al., 1999)) had no effect on [Ca2+]i responses to 100 μM UTP (data not shown), but reduced the [Ca2+]i response to 10 μM (±)-arterenol in the presence of 100 μM UTP from 44±2% to 19±2% (Figure 9b). CTX pre-treatment caused a small but significant increase in the response to 10 nM IL-8 in the presence of 100 μM UTP (15±1.0%; P<0.05).
Figure 9.
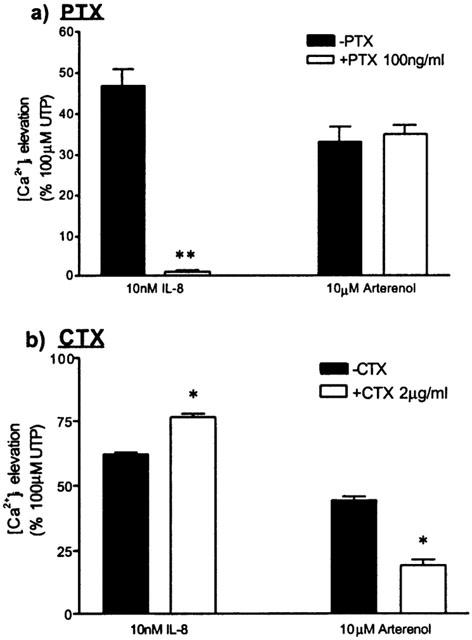
Effects of PTX and CTX on the potentiation of CXCR2- and adrenoceptor-mediated Ca2+ signalling by UTP. (a) HEK-CXCR2 cells were cultured with or without 100 ng ml−1 PTX for 20 h prior to FLIPR assay. The data represent [Ca2+]i responses to either 10 nM IL-8 or 10 μM (±)-arterenol following pre-stimulation with 100 μM UTP. (b) Identical experiment to that shown in (a) with the exception that the cells were cultured with or without 2 μg ml−1 CTX for 20 h prior to FLIPR assay. Results are mean±s.e.mean n=3. For *P<0.01 and **P<0.001 compared to the appropriate controls not treated with toxin.
Dependence of the potentiation effect and IL-8-mediated Ca2+ signalling on extracellular calcium
To determine if Ca2+ flux across the plasma membrane was required for the ability of P2Y receptor activation to potentiate IL-8- or (±)-arterenol-mediated [Ca2+]i signalling, experiments were performed in BSS without added Ca2+. Under these conditions, the magnitude of the spike [Ca2+]i responses to either UTP or 2MeSADP were unaffected (Figure 10a,b). However, the [Ca2+]i returned quickly to basal levels following stimulation with either nucleotide (Figure 10a,b). In the absence of extracellular Ca2+, the addition of IL-8 following either UTP or 2MeSADP still provoked an elevation of [Ca2+]i that was not significantly decreased compared to that in the presence of added extracellular Ca2+ (Figure 10c). Similarly the absence of extracellular Ca2+ had no effect on the ability of UTP to potentiate the [Ca2+]i response to (±)-arterenol (Figure 10d).
Figure 10.
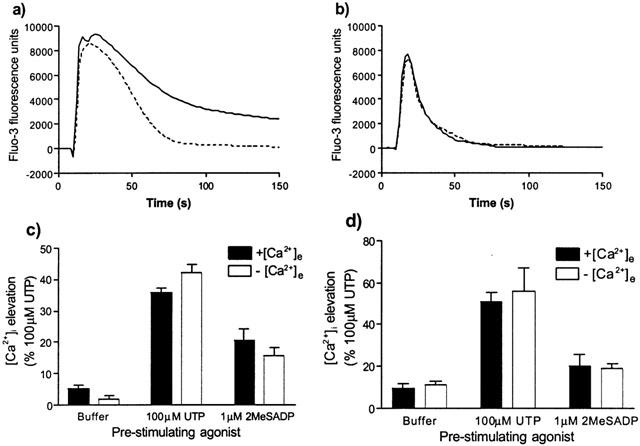
Effects of removal of extracellular Ca2+ on Ca2+ signalling by P2Y2 and P2Y1 receptors (a and b, respectively) and the nucleotide receptor-mediated potentiation of signalling by CXCR2 (c) and β-adrenoceptors (d). Following loading with fluo-3, cells were washed with BSS containing no added Ca2+ and then transferred to the FLIPR with each well of the plate containing 100 μl of BSS or BSS with no added Ca2+. Following collection of basal fluorescence values for 10 s, cells were stimulated by the addition of either UTP (a) or 2MeSADP (b) in either BSS or in BSS with no added Ca2+. Profiles are the average of 20 or more similar traces over three different experiments. To determine the effect of extracellular Ca2+ on the potentiation response cells were pre-treated with either 100 μM UTP, 1 μM 2MeSADP or buffer at t=10 s, followed at t=150 s by 10 nM IL-8 (c) or 10 μM (±)-arterenol (d) in the presence or absence of extracellular Ca2+. The data show the maximal response to the second addition (IL-8 or (±)-arterenol) as a percentage of the maximal response to 100 μM UTP. Data are mean±s.e.mean n=3.
Discussion
This study demonstrates that activation of endogenously expressed, PLC-coupled nucleotide receptors in HEK-293 cells markedly potentiates Ca2+ signalling by recombinant, co-expressed human CXCR2 that signal through PTX-sensitive G-proteins. P2Y receptor activation enhances both the potency and magnitude of CXCR2-mediated signalling consistent with an increased functional receptor reserve. Similarly, P2Y receptor activation potentiates Ca2+ signalling by endogenously expressed receptors for (±)-arterenol that signal through CTX-sensitive G-proteins. We also demonstrate that the potentiation effect is greater following activation of P2Y2 than P2Y1 nucleotide receptors, that sustained P2Y receptor-mediated signalling is required, and that influx of extracellular Ca2+ is not a pre-requisite for IL-8- or (±)-arterenol-mediated signalling.
Although our HEK cell line expresses both endogenous P2Y1 and P2Y2 nucleotide receptors, we show that potentiation of CXCR2 and β-adrenoceptor Ca2+ signalling is mediated predominantly by P2Y2 receptors. The differential effect of P2Y1 and P2Y2 receptors is unrelated to the magnitude of the initial [Ca2+]i elevation as a concentration of UTP that gives an equivalent spike response to 2MeSADP causes greater potentiation of both the potency and Emax of the subsequent response to IL-8 or (±)-arterenol. A possible explanation of this differential effect is that P2Y1 and P2Y2 receptors desensitize to different extents over the time-frame of these experiments. Thus, the relatively rapid return of [Ca2+]i to basal levels following activation of P2Y1 receptors with 2MeSADP indicates that, at least at this level of signalling, this receptor fully desensitizes. In contrast, P2Y2 receptor activation results in a sustained [Ca2+]i elevation. Although differences in the cellular handling of Ca2+ during P2Y1 and P2Y2 receptor stimulation could account for the different profiles of [Ca2+]i elevation (for example, differential coupling to the plasma membrane Ca2+-ATPase), these data suggest that P2Y1 receptors may desensitize more fully than P2Y2 receptors over the time-frame of our experiments. The consequence of these two different profiles of receptor desensitization would be that at the point of IL-8 or (±)-arterenol addition, the P2Y1 receptor would be fully desensitized, whilst signalling via the P2Y2 receptor would be maintained. This need for sustained receptor signalling is in agreement with the finding that washout of UTP results in a loss of potentiation of IL-8-mediated Ca2+ signalling. Furthermore, removal of extracellular Ca2+ does not affect the ability of P2Y receptor activation to potentiate Ca2+ signalling by CXCR2 or β-adrenoceptors. This indicates both that influx of extracellular Ca2+ is not required to mediate the potentiation effect, and that activation of CXCR2 or β-adrenoceptors, in the presence of P2Y receptor stimulation, can mobilize Ca2+ from an intracellular store.
The potentiating effect of P2Y receptor activation on CXCR2-mediated Ca2+ signalling is not a consequence of the over-expression of a recombinant receptor as the level of CXCR2 expression (approximately 50,000 sites per cell) is similar to that in neutrophils (approximately 50 – 90,000 sites per cell; Lee et al., 1992). Furthermore, signalling by endogenously expressed β-adrenoceptors is also potentiated. The Ca2+ signalling evoked by (±)-arterenol in the presence of P2Y receptor activation is inhibited by CTX but not PTX whereas CXCR2 signalling is inhibited by PTX and not CTX. Thus, (±)-arterenol signalling appears to be through a Gαs-coupled β-adrenoceptor. This is consistent with evidence demonstrating the expression of these but not other adrenoceptor sub-types in HEK cells (Premont et al., 1992). Further, our data are consistent with other reports (Hall et al., 1999; Damaj et al., 1996) that CXCR2 is coupled via Gαi. Activation of Gαq/11-coupled receptors is, therefore, able to potentiate Ca2+ signalling through both Gαi- and Gαs-coupled receptors.
Although UTP and 2MeSADP produce similar increases in the initial peak Ca2+ elevation, and their relative potencies for this response are similar to those for their ability to facilitate CXCR2- and β-adrenoceptor-mediated Ca2+ signalling, the mechanism of potentiation appears to be independent of the size of the initial [Ca2+]i ‘spike'. Concentrations of the two nucleotide agonists that elevate [Ca2+]i to equivalent levels are markedly different in terms of their ability to potentiate CXCR2 signalling. In addition, removal of Ca2+ from the extracellular milieu, which prevents sustained P2Y2 receptor-mediated Ca2+ signalling, has no effect on the ability of P2Y2 receptors to mediate potentiation of Ca2+ signalling by CXCR2. This suggests that neither the initial peak Ca2+ elevation nor the sustained plateau of Ca2+ elevation are required (or at least are not sufficient) for this potentiation of CXCR2 signalling. In support of this, Yeo et al. (2001) show that a similar type of crosstalk occurring between δ-opioid receptors and muscarinic m3 receptors in SH-SY5Y cells cannot be mimicked by a lysophosphatidic acid-induced elevation of [Ca2+]i via sphingosine kinase, indicating that potentiation cannot be brought about simply by evoking an intracellular Ca2+ response.
The mechanism by which nucleotide receptors allow the activation of CXCR2 or β-adrenoceptors to elevate [Ca2+]i is unclear. Gαi-coupled receptors have been demonstrated to cause the release of intracellular Ca2+ through the release of Gβγ-subunits from PTX-sensitive G-proteins and the subsequent activation of βγ-sensitive PLC isoforms (Lee et al., 1993; Dickenson & Hill, 1998). This mechanism does not explain the requirement for sustained Gαq/11-mediated signalling shown in the present study. However, Gαq/11 can cause a reversible conformational change in PLCβ that reveals an activation site for Gβγ (Okajima et al., 1993; Smrcka & Sternweis, 1993; Chan et al., 2000) and this could account for the requirement of ongoing Gαq/11-coupled receptor activation for the potentiating effect. Such mechanisms presume that it is the Gαi- or Gαs-coupled receptor that is directly responsible for the Ca2+ signalling. Alternatively, this Ca2+ signalling could be directly dependent on the Gαq/11 signalling pathway with CXCR2 or β-adrenoceptor stimulation enhancing signalling by the Gαq/11 pathway. Currently we have no evidence of the level at which such interaction may be occurring within the signalling pathway. A number of potential sites have been suggested previously including the increased supply of substrate (PtdIns(4,5)P2) for PLC by PTX-sensitive G-proteins (Schmidt et al., 1996) and a Gαs-mediated shunting of Ca2+ between intracellular stores that allows Gαq-coupled receptors to access an apparently larger Ca2+ store (Short & Taylor, 2000). Another possibility is that heterodimerization of receptors results in an alteration in the functional properties of the receptors involved. Of particular interest, recent work has shown heterodimerization between Gαi-coupled CCR2 and CCR5 chemokine receptors and demonstrated that this results in a gain of PTX-resistant Ca2+ signalling (Mellado et al., 2001).
Many chemokine receptors are activated by more than one chemokine, whilst many ligands activate more than one type of chemokine receptor. This suggests either redundancy or a fine-tuning that would allow tailored responses to different inflammatory stimuli. This fine-tuning may include not only the selective expression of chemokines and their receptors (Murphy et al., 2000) but also receptor ‘cross-talk'. For example, inhibitory desensitization has been demonstrated between CXCR1 and CXCR2 (Richardson et al., 1998). In the current study we demonstrate cross-talk between a non-chemokine receptor and CXCR2 that results in the potentiation of at least the Ca2+ signalling element of the CXCR2 transduction pathway. Although we show that this effect is not restricted to chemokine receptors, the interaction between P2Y receptors and CXCR2 could have a significant impact in the inflammatory process. Thus, in cells such as neutrophils and monocytes that co-express P2Y2 receptors and CXCR2, the release of ATP from damaged cells and/or activated platelets could potentiate CXCR2-mediated Ca2+ signalling. In human neutrophils CXCR2 mediates a substantial Ca2+ response (Schorr et al., 1999). However, it should be noted that the protocols used to isolate neutrophils in that study, and indeed other studies, most likely releases endogenous ATP. Neutrophils also express P2Y2 receptors and it is currently unclear if released ATP acts to potentiate CXCR2-mediated Ca2+ signalling in these cells. An alternative potential function of such crosstalk is that the coincidence of ligands for different receptor types may allow the activation of receptors that might otherwise be silent. For example, dendritic cells express both CXCR2 and CXCR1 but neither receptor mediates Ca2+ signalling or chemotaxis (Sozzani et al., 1997). Priming of these cells by activation of nucleotide or other receptor types may result in the coupling of these receptors to a functional response. Whether such regulation is indeed relevant to customizing the immune response is unclear but the current data suggest an additional level of complexity and potentially a novel therapeutic opportunity.
We conclude that [Ca2+]i signalling by recombinant CXCR2 in HEK cells is potentiated by prior activation of endogenous P2Y nucleotide receptors. Such interaction has implications for signalling in native cells although the mechanism and physiological significance have yet to be elucidated.
Acknowledgments
This work was jointly funded by AstraZeneca R&D Charnwood and the British Biological Sciences Research Council (BBSRC). The confocal microscope was purchased through an award from The Wellcome Trust (grant number 061050).
Abbreviations
- ADP
adenosine 5′-diphosphate
- ANOVA
analysis of variance
- ATP
adenosine 5′-triphosphate
- BSA
bovine serum albumin
- BSS
balanced salts solution
- Ca2+
calcium ion
- [Ca2+]i
intracellular calcium concentration
- CTX
cholera toxin
- CXCR2
C-X-C chemokine receptor 2 or IL-8 receptor B
- FLIPR
fluorescent light imaging plate reader
- fluo-3-AM
fluo-3 acetoxymethylester
- fura-2-AM
fura-2 acetoxymethylester
- GPCR
G-protein-coupled receptor
- IL-8
interleukin-8
- Ins(1,4,5)P3
inositol 1,4,5-trisphosphate
- 2MeSADP
2′-methylthioadenosine 5′-diphosphate
- 2MeSATP
2′-methylthioadenosine 5′triphosphate
- PTX
pertussis toxin
- UDP
uridine 5′-diphosphate
- UTP
uridine 5′-triphosphate.
References
- ADDISON C.L., DANIEL T.O., BURDICK M.D., LIU H., EHLERT J.E., XUE Y.Y., BUECHI L., WALZ A., RICHMOND A., STRIETER R.M. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR(+) CXC chemokine-induced angiogenic activity. J. Immunol. 2000;165:5269–5277. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- ARAUJO D.M., COTMAN C.W. Trophic effects of interleukin-4, -7 and -8 on hippocampal neuronal cultures: potential involvement of glial-derived factors. Brain Res. 1993;600:49–55. doi: 10.1016/0006-8993(93)90400-h. [DOI] [PubMed] [Google Scholar]
- BELPERIO J.A., KEANE M.P., ARENBERG D.A., ADDISON C.L., EHLERT J.E., BURDICK M.D., STRIETER R.M. CXC chemokines in angiogenesis. J. Leukoc. Biol. 2000;68:1–8. [PubMed] [Google Scholar]
- BOARDER M.R., WEISMAN G.A., TURNER J.T., WILKINSON G.F. G protein-coupled P2 purinoceptors: from molecular biology to functional responses. Trends Pharmacol. Sci. 1995;16:133–139. doi: 10.1016/s0165-6147(00)89001-x. [DOI] [PubMed] [Google Scholar]
- BOISVERT W.A., SANTIAGO R., CURTISS L.K., TERKELTAUB R.A. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J. Clin. Invest. 1998;101:353–363. doi: 10.1172/JCI1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROXMEYER H.E., COOPER S., CACALANO G., HAGUE N.L., BAILISH E., MOORE M.W. Involvement of interleukin-8 (IL-8) receptor in negative regulation of myeloid progenitor cells in vivo: evidence from mice lacking the murine IL-8 receptor homologue. J. Exp. Med. 1996;184:1825–1832. doi: 10.1084/jem.184.5.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUCKLEY K.A., WAGSTAFF S.C., MCKAY G., GAW A., HIPSKIND R.A., BILBE G., GALLAGHER J.A., BOWLER W.B. Parathyroid hormone potentiates nucleotide-induced [Ca2+]i release in rat osteoblasts independently of Gq activation or cyclic monophosphate accumulation. A mechanism for localizing systemic responses in bone. J. Biol. Chem. 2001;276:9565–9571. doi: 10.1074/jbc.M005672200. [DOI] [PubMed] [Google Scholar]
- CACALANO G., LEE J., KIKLY K., RYAN A.M., PITTS-MEEK S., HULTGREN B., WOOD W.I., MOORE M.W. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- CHAN J.S., LEE J.W., HO M.K., WONG Y.H. Preactivation permits subsequent stimulation of phospholipase C by Gi-coupled receptors. Mol. Pharmacol. 2000;57:700–708. doi: 10.1124/mol.57.4.700. [DOI] [PubMed] [Google Scholar]
- COMMUNI D., ROBAYE B., BOEYNAEMS J.M. Pharmacological characterization of the human P2Y11 receptor. Br. J. Pharmacol. 1999;128:1199–1206. doi: 10.1038/sj.bjp.0702909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONNOR M., YEO A., HENDERSON G. Neuropeptide Y Y2 receptor and somatostatin sst2 receptor coupling to mobilization of intracellular calcium in SH-SY5Y human neuroblastoma cells. Br. J. Pharmacol. 1997;120:455–463. doi: 10.1038/sj.bjp.0700920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUGHLAN C.M., MCMANUS C.M., SHARRON M., GAO Z., MURPHY D., JAFFER S., CHOE W., CHEN W., HESSELGESSER J., GAYLORD H., KALYUZHNY A., LEE V.M., WOLF B., DOMS R.W., KOLSON D.L. Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience. 2000;97:591–600. doi: 10.1016/s0306-4522(00)00024-5. [DOI] [PubMed] [Google Scholar]
- CURDOVA E., JECHOVA V., HOSTALEK Z. Properties of apyrase and inorganic pyrophosphatase in Streptomyces aureofaciens. Folia Microbiol. 1982;27:159–166. doi: 10.1007/BF02877394. [DOI] [PubMed] [Google Scholar]
- DAMAJ B.B., MCCOLL S.R., MAHANA W., CROUCH M.F., NACCACHE P.H. Physical association of Gi2α with interleukin-8 receptors. J. Biol. Chem. 1996;271:12783–12789. doi: 10.1074/jbc.271.22.12783. [DOI] [PubMed] [Google Scholar]
- DAME J.B., JUUL S.E. The distribution of receptors for the pro-inflammatory cytokines interleukin (IL)-6 and IL-8 in the developing human fetus. Early Hum. Dev. 2000;58:25–39. doi: 10.1016/s0378-3782(00)00064-5. [DOI] [PubMed] [Google Scholar]
- DICKENSON J.M., HILL S.J. Involvement of G-protein βγ subunits in coupling the adenosine A1 receptor to phospholipase C in transfected CHO cells. Eur. J. Pharmacol. 1998;355:85–93. doi: 10.1016/s0014-2999(98)00468-3. [DOI] [PubMed] [Google Scholar]
- HALL D.A., BERESFORD I.J., BROWNING C., GILES H. Signalling by CXC-chemokine receptors 1 and 2 expressed in CHO cells: a comparison of calcium mobilization, inhibition of adenylyl cyclase and stimulation of GTPγS binding induced by IL-8 and GROα. Br. J. Pharmacol. 1999;126:810–818. doi: 10.1038/sj.bjp.0702329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HORUK R., MARTIN A.W., WANG Z., SCHWEITZER L., GERASSIMIDES A., GUO H., LU Z., HESSELGESSER J., PEREZ H.D., KIM J., PARKER J., HADLEY T.J., PEIPER S.C. Expression of chemokine receptors by subsets of neurons in the central nervous system. J. Immunol. 1997;158:2882–2890. [PubMed] [Google Scholar]
- JIN J., DASARI V.R., SISTARE F.D., KUNAPULI S.P. Distribution of P2Y receptor subtypes on haematopoietic cells. Br. J. Pharmacol. 1998;123:789–794. doi: 10.1038/sj.bjp.0701665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KULKE R., BORNSCHEUER E., SCHLUTER C., BARTELS J., ROWERT J., STICHERLING M., CHRISTOPHERS E. The CXC receptor 2 is over-expressed in psoriatic epidermis. J. Invest. Dermatol. 1998;110:90–94. doi: 10.1046/j.1523-1747.1998.00074.x. [DOI] [PubMed] [Google Scholar]
- LEE J., HORUK R., RICE G.C., BENNETT G.L., CAMERATO T., WOOD W.I. Characterization of two high affinity human interleukin-8 receptors. J. Biol. Chem. 1992;267:16283–16287. [PubMed] [Google Scholar]
- LEE S.B., SHIN S.H., HEPLER J.R., GILMAN A.G., RHEE S.G. Activation of phospholipase C β2 mutants by G protein αq and βγ subunits. J. Biol. Chem. 1993;268:25952–25957. [PubMed] [Google Scholar]
- MEGSON A.C., DICKENSON J.M., TOWNSEND-NICHOLSON A., HILL S.J. Synergy between the inositol phosphate responses to transfected human adenosine A1-receptors and constitutive P2-purinoceptors in CHO-K1 cells. Br. J. Pharmacol. 1995;115:1415–1424. doi: 10.1111/j.1476-5381.1995.tb16632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELLADO M., RODRIGUEZ-FRADE J.M., VILA-CORO A.J., FERNANDEZ S., MARTIN DE ANA A., JONES D.R., TORAN J.L., MARTINEZ-A C. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001;20:2497–2507. doi: 10.1093/emboj/20.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY P.M., BAGGIOLINI M., CHARO I.F., HEBERT C.A., HORUK R., MATSUSHIMA K., MILLER L.H., OPPENHEIM J.J., POWER C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- NICHOLAS R.A., LAZAROWSKIE R., WATT W.C., LI Q., BOYER J., HARDEN T.K. Pharmacological and second messenger signalling selectivities of cloned P2Y receptors. J. Autonom. Pharmacol. 1996;16:319–323. doi: 10.1111/j.1474-8673.1996.tb00044.x. [DOI] [PubMed] [Google Scholar]
- OKAJIMA F., TOMURA H., KONDO Y. Enkephalin activates the phospholipase C/Ca2+ system through cross-talk between opioid receptors and P2-purinergic or bradykinin receptors in NG 108-15 cells. A permissive role for pertussis toxin-sensitive G-proteins. Biochem. J. 1993;290:241–247. doi: 10.1042/bj2900241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PREMONT R.T., CHEN J., MA H.W., PONNAPALLI M., IYENGAR R. Two members of a widely expressed subfamily of hormone-stimulated adenylyl cyclases. Proc. Natl. Acad. Sci. 1992;89:9809–9813. doi: 10.1073/pnas.89.20.9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RICHARDSON R.M., PRIDGEN B.C., HARIBABU B., ALI H., SNYDERMAN R. Differential cross-regulation of the human chemokine receptors CXCR1 and CXCR2. Evidence for time-dependent signal generation. J. Biol. Chem. 1998;273:23830–23836. doi: 10.1074/jbc.273.37.23830. [DOI] [PubMed] [Google Scholar]
- SCHACHTER J.B., SROMEK S.M., NICHOLAS R.A., HARDEN T.K. HEK293 human embryonic kidney cells endogenously express the P2Y1 and P2Y2 receptors. Neuropharmacology. 1997;36:1181–1187. doi: 10.1016/s0028-3908(97)00138-x. [DOI] [PubMed] [Google Scholar]
- SCHMIDT M., NEHLS C., RUMENAPP U., JAKOBS K.H. m3 Muscarinic receptor-induced and Gi-mediated heterologous potentiation of phospholipase C stimulation: role of phosphoinositide synthesis. Mol. Pharmacol. 1996;50:1038–1046. [PubMed] [Google Scholar]
- SCHORR W., SWANDULLA D., ZEILHOFER H.U. Mechanisms of IL-8-induced Ca2+ signaling in human neutrophil granulocytes. Eur. J. Immunol. 1999;29:897–904. doi: 10.1002/(SICI)1521-4141(199903)29:03<897::AID-IMMU897>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- SEIDEL M.G., KLINGER M., FREISSMUTH M., HÖLLER C. Activation of mitogen-activated protein kinase by the A2A-adenosine receptor via a rap1-dependent and via a p21ras-dependent pathway. J. Biol. Chem. 1999;274:25833–25941. doi: 10.1074/jbc.274.36.25833. [DOI] [PubMed] [Google Scholar]
- SHORT A.D., TAYLOR C.W. Parathyroid hormone controls the size of the intracellular Ca2+ stores available to receptors linked to inositol trisphosphate formation. J. Biol. Chem. 2000;275:1807–1813. doi: 10.1074/jbc.275.3.1807. [DOI] [PubMed] [Google Scholar]
- SMRCKA A.V., STERNWEIS P.C. Regulation of purified subtypes of phosphatidylinositol-specific phospholipase Cβ by G protein α and βγ subunits. J. Biol. Chem. 1993;268:9667–9674. [PubMed] [Google Scholar]
- SOZZANI S., LUINI W., BORSATTI A., POLENTARUTTI N., ZHOU D., PIEMONTI L., D'AMICO G., POWER C.A., WELLS T.N., GOBBI M., ALLAVENA P., MANTOVANI A. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J. Immunol. 1997;159:1993–2000. [PubMed] [Google Scholar]
- XIA M.Q., HYMAN B.T. Chemokines/chemokine receptors in the central nervous system and Alzheimer's disease. J. Neurovirol. 1999;5:32–41. doi: 10.3109/13550289909029743. [DOI] [PubMed] [Google Scholar]
- XIA M., QIN S., MCNAMARA M., MACKAY C., HYMAN B.T. Interleukin-8 receptor B immunoreactivity in brain and neuritic plaques of Alzheimer's disease. Am. J. Pathol. 1997;150:1267–1274. [PMC free article] [PubMed] [Google Scholar]
- YEO A., SAMWAYS D.S., FOWLER C.E., GUNN-MOORE F., HENDERSON G. Coincident signalling between the Gi/Go-coupled delta-opioid receptor and the Gq-coupled m3 muscarinic receptor at the level of intracellular free calcium in SH-SY5Y cells. J. Neurochem. 2001;76:1688–1700. doi: 10.1046/j.1471-4159.2001.00185.x. [DOI] [PubMed] [Google Scholar]