

Abstract
Adenosine is a regulator of mesenteric vasodilation involved in auto-regulation and post-prandial hyperemia, but the adenosine receptor subtype involved in this relaxant effect is poorly characterized. We have now pharmacologically characterized this receptor in rabbit mesenteric arteries and investigated how this adenosine receptor response changes in portal hypertensive animals since the adenosine response is decreased.
The closest non-metabolisable adenosine analogue, 2-chloroadenosine (CADO), the mixed A1/A2 receptor agonist, 5′-ethylcarboxamidoadenosine (NECA), and the selective A2A receptor agonist, 2-[4-(2-p-carbonyethyl)phenylamino]-5′-N-ethylcarboxamidoadenosine (CGS 21680) (1 pM – 1 mM) relaxed noradrenaline pre-contracted arteries with a rank order of potency of CGS 21680 (EC50=20 nM)⩾NECA (60 nM)>>CADO (640 nM).
The selective A2A receptor antagonist, 4-(2-[7-amino-2-(2-furyl)-[1,2,4]-triazolo[2,3-a][1,3,5]-triazin-5-ylamino]ethyl)phenol (ZM 241385, 100 nM), shifted to the right the CADO concentration-response curve.
In portal hypertensive animals, there was mainly a decreased potency but also a decreased efficacy of all tested adenosine agonists compared to normal animals. Concomitantly, there was a decreased adenosine plasma level and a decreased binding density of [3H]-CGS 21680 and [3H]-ZM 241385 to mesenteric artery membranes from portal hypertensive compared to normal rabbits.
These results indicate that A2A receptor activation is required for the adenosine-induced mesenteric relaxation and that the decreased density of A2A receptors may contribute to the decreased relaxation induced by adenosine of mesenteric arteries in portal hypertensive animals.
Keywords: Portal hypertension, adenosine, rabbit, mesenteric artery, A2A receptors
Introduction
Adenosine is a potent vasodilator that mediates several physiological phenomena in the splanchnic circulation, including autoregulation of the cranial mesenteric artery (Lautt, 1986), the hepatic buffer response (Lautt, 1985; Lautt & Greenway, 1987), and post-prandial hyperemia (Sawmiller & Chou, 1988). There has been some controversy about the adenosine receptor involved in mesenteric artery vasodilatation, with claims of both A2A (Hiley et al., 1995) or A2B receptor involvement (Rubino et al., 1995; Prentice et al., 1997), although it is clear that adenosine-mediated mesenteric vasodilation is dependent on the presence of a functional endothelium (Hiley et al., 1995; Tabrizchi & Lupichuk, 1995; Villa de brito et al., 1999). In different vascular beds, adenosine vasodilation mostly involves A2A receptor activation, although in some instances A2B receptors may also play a role (reviewed by Olsson & Pearson, 1990; Belardinelli et al., 1998; Feoktistov & Biaggioni, 1997). The importance of this adenosine-induced vasodilation is best exemplified by the observation that chronic blockade of adenosine receptors leads to hypertension (Guimarães & Albino-Teixeira, 1996).
The rabbit model of partial portal vein ligation is an increasingly used means of study for portal hypertension characterized by the development of portal-systemic shunting (Chojkier & Groszmann, 1981; Atucha et al., 1998). The vascular responses to several vasoactive agents are modified in portal hypertension (Lee et al., 1992; Clària et al., 1994; Gadano et al., 1997). In a previous study, we showed that the vasodilation of the cranial mesenteric artery induced by adenosine is decreased in rabbits with portal hypertension both in vitro (Villa de brito et al., 1998) and in vivo (Marques et al., 1999).
To investigate the reasons of this decreased responsiveness to adenosine in hypertensive animals, we now decided to characterize the adenosine receptor involved in adenosine-induced mesenteric vasodilation and to investigate if this receptor activity was changed in portal hypertensive animals.
Methods
Animals and surgical procedures
Adult male New Zealand White rabbits (2.7 – 3.3 kg) were used for all studies. Rabbits were housed in a controlled environment and allowed free access to food and water. All experiments were performed according to the EU guidelines of care and use of laboratory animals.
Portal hypertension was produced by partial portal vein ligation (Chojkier & Groszmann, 1981). Briefly, a laparotomy was performed under ketamine (25 mg kg−1, i.m.) and medetomidine (0.5 mg kg−1, s.c.) anaesthesia. A ligature (3/0 silk) was placed, distal to the confluence of the right and left portal vein branches. An 18-gauge blunt needle was placed beside the portal vein, the ligature was tightened around both needle and vein, and the needle was removed, producing a standard, calibrated mechanical resistance. The abdomen was closed and the animal allowed recovery for 6 weeks. In sham-operated rabbits, the same procedure was carried out, with the exception that after the portal vein was isolated no ligature was placed.
Organ bath experiments
The rabbits were killed by an i.v. bolus of pentobarbitone (2 ml of a saturated solution), after which the cranial mesenteric arteries were removed, cleaned of fat and connective tissue, and cut into rings of approximately 3 mm length. The rings were suspended on tungsten wires under 2.0 g resting tension and allowed to equilibrate for 60 min in Krebs-Henseleit (mM): NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4 1.2; KH2PO4 1.2, NaHCO3 25.0 and glucose 11.1. Organ baths were maintained at 38°C and oxygenated with 95% O2 and 5% CO2. Tension was recorded with a Lectromed UF1 isometric transducer connected to a Lectromed polygraph 5041.
The mesenteric rings were sub-maximally contracted with noradrenaline (10 μM) and the integrity of the endothelium was confirmed by the response to acetylcholine (10 nM – 1 mM). The rings that did not relax to acetylcholine were discarded. Cumulative concentration-response curves to the tested adenosine receptor agonists (CGS 21680, NECA and CADO) were constructed by the addition of increasing concentrations of each drug (1 pM – 1 mM) to the organ bath. The responses to increasing concentrations of CADO were also evaluated, in normal rings, before and after incubation with ZM 241385 (100 nM). Since all these adenosine receptor ligands are hydrophobic, they were all dissolved in concentrated solutions of dimethylsulphoxide and we verified that dimethylsulphoxide, in the maximal concentration applied to the preparations (2%), was devoid of effects. The Emax and the EC50 values were calculated using a sigmoidal dose response curve with variable slope and no weighting with the logistic equation of Y=Emin+(Emax−Emin)/(1+10∧((LogEC50−X)*Hill slope)), where X is the log of the agonist concentration, Y is the response measured and Emin is fixed at 0, using the GraphPad Prism software (GraphPad Software, San Diego, U.S.A.).
Adenosine plasma levels quantification
Normal rabbits and rabbits with portal hypertension were anaesthetized with sodium pentobarbitone (40 mg kg−1, i.v.) and blood was obtained from the caudal vena cava near the liver, at the end of the suprahepatic vein, by venipuncture after laparotomy. Each blood sample (1.8 ml) was rapidly collected into a syringe containing 82 μl of a stopping solution consisting of 1 mM dilazep (to inhibit adenosine uptake into and release from red blood cells), 10 μM erythro-9-(2-hydroxy-3-nonyl)adenine (to block adenosine deaminase activity) and 525 μM α,β-methylene adenosine-5′-diphosphate (to inhibit ecto-5′-nucleotidase) (Slowiaczek & Tattersall, 1982; Mccann & Katholi, 1990; Miura et al., 1991; Zhang et al., 1991). Only the samples free of observable haemolysis were considered. The blood samples were immediately centrifuged at 14,000×g for 1 min. Aliquots (100 μl) of plasma were collected and deproteinated with 30 μl of 7% perchloric acid. Samples were centrifuged again for 5 min at 14,000 r.p.m., and 75 μl of clear supernatant were immediately neutralized with a 0.7 M KOH and 50 mM Tris solution. The samples were kept at −70°C until HPLC analysis.
Adenosine was separated by HPLC using a 5 μm reverse-phase C18 column and a 100 mM KH2PO4 with 15% (v v−1) methanol eluent (pH 6.5) at a flow rate of 1.75 ml min−1 and quantified by area integration of the peak detected spectrophotometrically at 254 nm (Cunha et al., 1989; Cunha & Sebastião, 1993).
Adenosine A2A receptor binding assays
Membranes were prepared basically as previously described (Cunha et al., 1999) from frozen arteries. Briefly, the tissue was mechanically ground and sonicated to achieve homogenization in 10 volumes of sucrose solution (0.32 M), containing (mM): Tris-HCl 50, EGTA 2 and dithiothreitol 1, pH 7.6. The homogenates were centrifuged at 1000×g for 10 min at 4°C. The supernatants were collected and centrifuged at 21,000×g for 20 min at 4°C. The pellets were then resuspended in a solution containing (mM): Tris-HCl 50, EGTA 2, EDTA 1 (pH 7.4 at 23°C) with 5 u ml−1 adenosine deaminase and incubated for 30 min at 37°C to remove endogenous adenosine. The mixture was then centrifuged at 21,000×g for 10 min at 4°C, and the pellets resuspended in the incubation solution containing 50 mM Tris and 10 mM MgCl2, pH 7.4, with 5 u ml−1 adenosine deaminase.
[3H]-ZM 241385 (0.1 – 6 nM) and [3H]-CGS 21680 (30 nM) binding studies were performed as previously described (Cunha et al., 1999). Briefly, binding reactions were carried out for 30 min (for [3H]-ZM 241385 binding) or 2 h (for [3H]-CGS 21680 binding) at room temperature (23 – 25°C) with 82 – 124 μg of membrane protein in a final volume of 300 μl of the incubation solution. The binding reactions were stopped by vacuum filtration through Whatman GF/C glass fibre filters, followed by washing of the filters and reaction tubes with 10 ml of the incubation solution, kept at 4°C. The filters were then placed in scintillation vials, and 5 ml of scintillation liquid (Scintran Cocktail T, Wallac) were added. Radioactivity bound to the filters was determined after 12 h with an efficiency of 50%. Results are expressed as specific binding, determined by subtraction of the non-specific binding, which was measured in the presence of 2 μM 8-{4-[(2-aminoethyl)amino]carbonylmethyl-oxyphenyl}xanthine, and normalysed per amount of protein, determined following the method of Peterson (1977). All binding assays were performed in duplicate. To calculate the binding parameters of [3H]-ZM 241385 binding, the saturation isotherm was adjusted by a rectangular hyperbola to estimate the KD and Bmax values using the GraphPad Prism software.
Drugs
Noradrenaline-L-hydrogen tartrate, acetylcholine chloride, NECA (5′-ethylcarboxamido adenosine), CADO (2-chloroadenosine), adenosine deaminase (type VI, 1803 u ml−1, EC 3.5.4.4.), dilazep, α,β-methylene adenosine-5′-diphosphate and erythro-9-(2-hydroxy-3-nonyl)adenine were from Sigma, CGS 21680 (2-p-(2-carbonylethyl)phenylethylamino-5′-N-ethyl carboxamidoadenosine) and 8-{4-[(2-aminoethyl)amino] carbonylmethyloxyphenil}xanthine were from Research Biochemicals Inc., 4-(2-[7-amino-2-(2-furyl)-[1,2,4]-triazolol[2,3-a][1,3,5]-triazin-5-ylamino]ethyl)phenol (ZM 241385) and [3H]-ZM 241385 (specific activity of 17 Ci mmol−1) were from Tocris and [3H]-CGS 21680 (specific activity 37.5 Ci mmol−1) was from DuPont-New England Nuclear.
The solutions of all adenosine receptor ligands were prepared in dimethylsulphoxide and diluted in Krebs buffer on the day of use.
Statistical analysis
Results are expressed as mean±s.e.mean, except the KD values, which are presented as mean (95% confidence interval). Statistical analysis was performed using ANOVA (parameters derived from the concentration response curves and adenosine plasma levels) and unpaired Student's t-test (binding data). A P value of less than 0.05 was considered statistically significant.
Results
Pharmacological characterization of the adenosine-induced mesenteric relaxation
All adenosine receptor agonists tested produced arterial vasodilation in a concentration dependent manner (Figure 1a) with similar maximal response (Table 1). However, the potency of the tested adenosine receptor agonists differed. The selective A2A receptor agonist, CGS 21680, was the most potent, closely followed by the mixed A1/A2 receptor agonist, NECA, both of them markedly (P<0.05) more potent than the closest chemical analogue of adenosine, CADO (Figure 1a and Table 1). This observed rank order of potency is indicative of the involvement of an A2A receptor in the adenosine-induced mesenteric relaxation.
Figure 1.
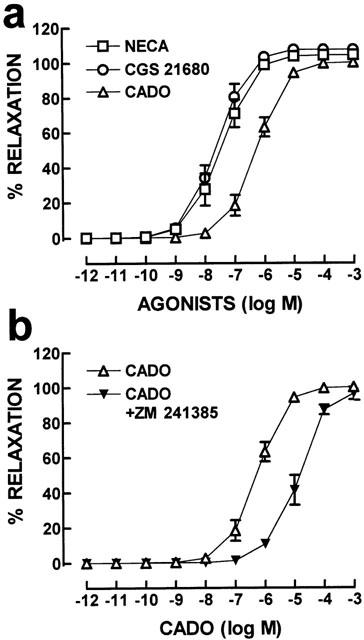
Relative potency of adenosine receptor agonists to relax rabbit cranial mesenteric arteries. The mesenteric artery rings were pre-contracted with noradrenaline (10 μM) and the functionality of the endothelium was verified by testing the response to acetylcholine. Then, cumulative concentration-response curves with increasing concentrations of the selective A2A receptor agonist, CGS 21680, of the mixed A1/A2 receptor agonist, NECA, or with the closest non-metabolizable adenosine analogue, CADO, were carried out in (a). In (b), cumulative concentration-response curves with increasing concentrations of CADO were performed either in the absence or in the presence of the selective A2A receptor antagonist, ZM 241385 (100 nM). The data are mean±s.e.mean of 5 – 6 experiments.
Table 1.
Different potency and efficacy of the adenosine receptor agonist (NECA, CGS 21680 and CADO) to relax noradrenaline-precontracted cranial mesenteric artery rings from control and portal hypertensive rabbits

To further confirm the main involvement of the A2A receptor subtype in mesenteric artery response to adenosine, concentration-dependent vasodilation caused by CADO, the closest chemical analogue of adenosine that is non-metabolizable, was examined in the absence and presence of a selective A2A adenosine receptor antagonist, ZM 241385. As illustrated in Figure 1b, ZM 241385 (100 nM) shifted to right (P<0.05) the concentration-response curve to CADO (EC50 increased to 8.2±2.1 μM, n=6) with no change (P>0.05) in efficacy (Emax=98.4±3.9%, n=6). By itself, ZM 241385 (100 nM) caused a slight non-significant (P>0.05) relaxation of noradrenaline pre-contracted arteries (n=6).
Changes in adenosine responsiveness in portal hypertension
As illustrated for CGS 21680 in Figure 2, we observed that the relaxant response to all tested adenosine receptor agonists was decreased in cranial mesenteric arteries of rabbits subjected to partial portal vein ligation. There was a significant (P<0.05) reduction mainly of the potency of all tested agonists, as well as a decrease in their maximal response (see Table 1). It should be noted that the artery rings from the two groups of animals displayed a similar (P>0.05) contractible response to 10 μM noradrenaline (0.96±0.06 g in control and 1.00±0.04 g in hypertensive animals, n=6).
Figure 2.
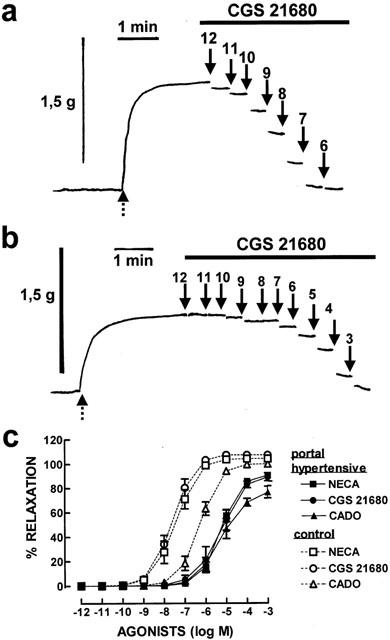
Modification of the potency of adenosine receptor ligands in cranial mesenteric arteries from portal hypertensive compared to control rabbits. In (a) and (b) are shown contractile recordings illustrating a cumulative concentration-response curve of the effect of CGS 21680 in cranial mesenteric arteries from control (a) and portal hypertensive rabbits (b). The mesenteric artery rings were pre-contracted with noradrenaline (10 μM, as shown by the dashed arrows in a and b). Then, a cumulative concentration-relaxant response to increasing concentration of CGS 21680 was carried out, as indicated by the upper bar in (a) and (b), and the recordings correspond to the last 30 s of recording out of each of the 5 min of recording after starting the superfusion of each concentration of CGS 21680, whose concentration is presented (as −log concentration) above each arrow. In (c) are shown the average results in preparations of control (open symbols, dashed lines) and portal hypertensive (filled symbols, filled lines) rabbits of the cumulative concentration-response curves with increasing concentrations of the selective A2A receptor agonist, CGS 21680, with the mixed A1/A2 receptor agonist, NECA, and with the closest non-metabolizable adenosine analogue, CADO. The data are mean±s.e.mean of 5 – 6 experiments.
The decreased relaxant responsiveness to adenosine analogues of mesenteric arteries from portal hypertensive animals is not due to a general decrease of its relaxation properties. Thus, the acetylcholine-induced relaxation was observed not to be statistically (P>0.05) different in mesenteric arteries from control and portal hypertensive rabbits. Thus, acetylcholine displayed an EC50 of 0.57±0.26 μM and of 0.80±0.18 μM and a Emax of 107.7±2.7% and of 100.8±2.1% in control and portal hypertensive animals (n=12). This is in accordance with the previous observation of an increased nitric oxide synthase-mediated responsiveness in mesenteric arteries of portal hypertensive animals (Heinemann & Stauber, 1996).
To further investigate the reasons underlying this diminished relaxant response to adenosine in mesenteric arteries from portal hypertensive animals, we investigated if this could either be due to an increase in the levels of endogenous extracellular adenosine or to a decrease in the density of the receptors involved in this effect of adenosine, i.e. A2A receptors.
Adenosine plasma levels, quantified in the vena cava near the liver, were significantly reduced (P<0.001) in portal hypertensive rabbits, when compared to those obtained in normal rabbits. Thus, the measured concentrations of adenosine were 244±13 nM in control and 78±5 nM in portal hypertensive rabbits (n=8). This makes it unlikely that the lower potency of adenosine receptor agonists might be due to an increased tonic activation of adenosine receptors by higher levels of endogenous extracellular adenosine.
In relation to the density of adenosine A2A receptors, there was a significant (P<0.05) decrease in the binding density of the selective A2A receptor antagonist, [3H]-ZM 241385, to membranes of mesenteric arteries of portal hypertensive (Bmax=384±29 fmol mg−1 protein, n=4) when compared to control rabbits (Bmax=522±28 fmol mg−1 protein, n=5). The saturation curves of [3H]-ZM 241385 binding (Figure 3) show that the tested A2A receptor antagonist bound to mesenteric artery membranes from control rabbits to a single binding site with a KD of 0.29 nM (95% confidence interval: 0.22 – 0.37 nM, n=5) that was unchanged (P>0.05) in portal hypertensive rabbits (KD=0.26 nM; 95% confidence interval: 0.18 – 0.33 nM, n=4). We also found that the single concentration of the selective A2A receptor agonist used, [3H]-CGS 21680 (30 nM), showed a reduced (P<0.05) amount of binding in mesenteric artery membranes prepared from portal hypertensive (233±29 fmol mg−1 protein, n=6) when compared to control rabbits (344±12 fmol mg−1 protein, n=6).
Figure 3.
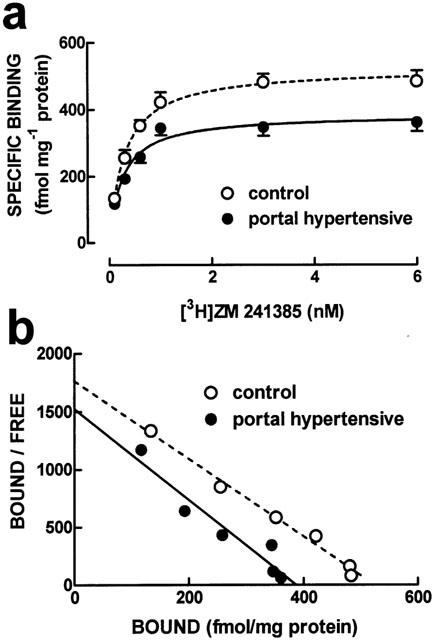
Average saturation curves (a) and corresponding Scatchard plots (b) of the binding of the A2A receptor antagonist, ZM241385, to whole membranes of mesenteric arteries from control (open symbols and dashed lines) and portal hypertensive rabbits (filled symbols and filled lines). The ordinates in (a) represent the specific binding of [3H]-ZM 241385 on subtraction of non-specific binding, determined in the presence of 2 μM 8-{4-[(2-aminoethyl)amino]carbonylmetyloxyphenil} xanthine, from total binding. The curves in (a) were generated from the average binding parameters obtained on fitting by non-linear regression assuming a single binding site, whereas the curves in (b) correspond to linearizations of the data points. Results are mean±s.e.mean of 4 – 5 experiments. The s.e.mean values are not presented in the Scatchard plots for the sake of simplicity.
Discussion
The present results indicate the involvement of adenosine A2A receptors in the adenosine-induced vasodilation in rabbit mesenteric arteries. Furthermore, the previously observed decreased ability of adenosine to cause vasodilation in mesenteric arteries of portal hypertensive rabbits (Villa de brito et al., 1998) was now confirmed to also occur for all tested adenosine receptor agonists and likely involves a decreased density of adenosine A2A receptors in rabbit mesenteric artery membranes.
The conclusion of a main involvement of adenosine A2A receptors in adenosine-mediated vasodilation of mesenteric arteries stems from the observation that the prototypical A2A receptor agonist, CGS 21680, has a potency in the low nanomolar range and is equipotent to NECA. Furthermore, the selective A2A receptor antagonist, ZM 241385, antagonized the relaxant effect caused by CADO, the closest non-metabolizable adenosine analogue. Finally, the parallel between the decrease in the binding density of A2A receptors and the decreased potency of the tested adenosine receptor agonists in portal hypertensive animals is another argument favouring our conclusion that A2A receptors mediate adenosine-induced vasodilation in cranial mesenteric arteries. This conclusion is in good agreement with previous observations in other studies performed in mesenteric arteries but using different experimental conditions and different animal specie (Balwierczak et al., 1991; Hiley et al., 1995). However, some other studies in mesenteric artery preparations could not obtain evidence for the involvement of A2A receptors, and concluded that adenosine-induced mesenteric vasodilation was mediated mostly via A2B receptor (Rubino et al., 1995; Prentice et al., 1997) with the contribution of an yet undefined intracellular site (Prentice et al., 1997). The studies that concluded on the involvement of A2B rather than A2A receptors in the vasodilation induced by adenosine in the mesenteric artery were carried out in a different specie, i.e. the rat (Rubino et al., 1995; Prentice et al., 1997). However, another study, also carried out in the rat, clearly shows relaxant responses caused by the selective A2A receptor agonist, CGS 21680 (Hiley et al., 1995). There are experimental differences between these three studies that may explain the opposite conclusion reached in regard to the adenosine receptor responsible for the relaxant response in mesenteric arteries of the rat. Two of the studies were carried out with bolus injections of different doses of CGS 21680 in perfused mesenteric beds (Hiley et al., 1995; Rubino et al., 1995). The study that observed relaxant effects of CGS 21680 used a lower perfusion rate (Hiley et al., 1995) and it is known from other systems that CGS 21680 equilibrates slowly with rat preparations (e.g. Cunha et al., 1997). Also, the concentration range where CGS 21680 is selective for A2A receptors is in the low nanomolar range and one of the studies, which failed to detect CGS 21680-induced relaxation, started to superfuse CGS 21680 at a concentration of 10 μM (Prentice et al., 1997). In different rat preparations, it has been observed that CGS 21680-induced responses, pharmacologically identified as being mediated by A2A receptors, cannot be revealed by the use of high (micromolar) concentrations of CGS 21680, possibly due to A2A receptor desensitization (reviewed in Cunha, 2001). Therefore, it can be concluded that, when the necessary precautions are taken to safely use CGS 21680 as a selective A2A receptor agonist, an A2A receptor-mediated relaxation of mesenteric arteries can be observed in the rat, as it is now described to occur in the rabbit. The possible involvement of ‘atypical' A2A binding sites, which have been described in the CNS (Johansson & Fredholm, 1995; Cunha et al., 1996), cannot be ruled out from the present set of data. However, it is important to note that these ‘atypical' A2A binding sites, which do not yet have a molecular correlate, appear to function mostly as ‘typical' A2A receptors although coupling to different transducing systems (e.g. Cunha & Ribeiro, 2000). Thus, irrespective of whether they are ‘typical' or ‘atypical', the present result extend to the mesenteric bed the major role played by A2A receptors in the adenosine-induced vasodilation that has been observed in different vascular beds (reviewed by Olsson & Pearson, 1990; Belardinelli et al., 1998; Feoktistov & Biaggioni, 1997).
The present results also confirm our previous observation that adenosine-induced vasodilation of cranial mesenteric artery is decreased in rabbits with portal hypertension both in vitro (Villa de brito et al., 1998) and in vivo (Marques et al., 1999). In fact, we now have found that the relaxant response to all adenosine receptor agonists tested was also decreased in mesenteric arteries from portal hypertensive rabbits. Portal hypertension is characterized by pronounced arterial vasodilation and increased systemic and mesenteric artery blood flow. This vasodilation leads to systemic hypotension that occurs despite an increased blood volume and cardiac output (Bosch et al., 1989; Groszmann, 1994). According to the liver artery buffer response (Lautt, 1985), when portal blood flow decreases, adenosine accumulates in the space of Mall and induces hepatic artery vasodilation. Therefore, it would be expected that the concentration of adenosine in the caudal vena cava would be increased or at least maintained in our study. Thus, we hypothesize that hypertension-induced changes in the uptake and/or metabolism of plasmatic adenosine may have contributed for the significant decrease in adenosine plasma levels observed in our study. The most likely explanation for the observed decrease in the potency and efficacy of adenosine receptor agonists to induce mesenteric relaxation in portal hypertensive rabbits is based on the observed decreased binding density of adenosine A2A receptors in mesenteric artery membranes of portal hypertensive rabbits. Indeed, theoretical studies (Kenakin, 1993) predict that in systems where the receptor is significantly distributed between two different affinity states for the agonists, as is the case for the adenosine A2A receptor (e.g. Cunha et al., 1999), a decrease in receptor number may produce a dextral displacement of the concentration-response curve together with a decrease of the maximal response. It is important to note that the overall response to A2A receptor agonists results from the number of A2A receptors as well as from the efficiency of the transdution systems operated by the receptor and of the effector pathways targeted. Thus, one would not expect that the percentage change in receptor number would match the percentage change in the efficiency of the tested adenosine agonists and, although the decreased number of A2A receptors is the prime explanation for the decreased efficiency of A2A receptor agonists in portal hypertensive animals, it cannot be excluded that other factors may help contributing for the decreased relaxant effect of adenosine receptor agonists in the mesenteric artery of portal hypertensive animals.
In conclusion, the present results indicate that the adenosine receptor responsible for the adenosine-induced relaxation of mesenteric arteries is of the A2A subtype and suggest that the reduced density of these A2A receptors may contribute for the reduced relaxant response to adenosine in the mesenteric arteries of portal hypertensive animals.
Acknowledgments
This study was supported by grants from CIISA, Faculty of Veterinary Medicine and by FCT.
Abbreviations
- CADO
2-chloroadenosine
- CGS 21680
2-p-(2-carbonyl-ethyl)phenylethylamino-5′-N-ethylcarboxamidoadenosine
- NECA
5′-ethylcarboxamido adenosine
- ZM 241385
4-(2-[7-amino-2-(2-furyl)-[1,2,4]-triazolo[2,3-a][1,3,5]-triazin-5-ylamino]ethyl)phenol
References
- ATUCHA N.M., CLARA ORTIZ M., FORTEPIANI L.A., RUIZ F.M., MARTINEZ C., GARCIA-ESTAN A. Role of cyclic guanosine monophosphate and K+ channels as mediators of the mesenteric vascular hyporesponsiveness in portal hypertensive rats. Hepatology. 1998;27:900–905. doi: 10.1002/hep.510270402. [DOI] [PubMed] [Google Scholar]
- BALWIERCZAK J.L., SHARIF R., KRULAN C.M., FIELD F.P., WEISS G.B., MILLER M.J.S. Comparative effects of a selective adenosine A2 receptor agonist, CGS 21680, and nitroprusside in vascular smooth muscle. Eur. J. Pharmacol. 1991;196:117–123. doi: 10.1016/0014-2999(91)90416-n. [DOI] [PubMed] [Google Scholar]
- BELARDINELLI L., SHRYOCK J.C., SNOWDY S., ZHANG Y., MONOPOLI A., LOZZA G., ONGINI E., OLSSON R.A., DENNIS D.M. The A2A adenosine receptor mediates coronary vasodilation. J. Pharmacol. Exp. Ther. 1998;284:1066–1073. [PubMed] [Google Scholar]
- BOSCH J., NAVASA M., GARCIA PAGÁN J.C., DELACY A.M., RODÉS J. Portal hypertension. Med. Clin. North Am. 1989;73:931–953. doi: 10.1016/s0025-7125(16)30646-0. [DOI] [PubMed] [Google Scholar]
- CHOJKIER M., GROSZMANN R.J. Measurement of portal-systemic shunting in the rat by using gamma-labeled microspheres. Am. J. Physiol. 1981;240:G371–G375. doi: 10.1152/ajpgi.1981.240.5.G371. [DOI] [PubMed] [Google Scholar]
- CLÀRIA J., JIMÉNEZ W., ROS J., RIGOL M., ANGELI P., ARROYO V., RIVERA F., RODÉS J. Increased nitric oxide-dependent vasorelaxation in aortic rings of cirrhotic rats with ascites. Hepatology. 1994;20:1615–1621. doi: 10.1002/hep.1840200635. [DOI] [PubMed] [Google Scholar]
- CUNHA R.A. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem. Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- CUNHA R.A., CONSTANTINO M.D., RIBEIRO J.A. ZM241385 is an antagonist of the facilitatory responses produced by the A2A adenosine receptor agonists CGS21680 and HENECA in the rat hippocampus. Br. J. Pharmacol. 1997;122:1279–1284. doi: 10.1038/sj.bjp.0701507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNHA R.A., CONSTANTINO M.D., RIBEIRO J.A. G protein coupling of CGS 21680 binding sites in the rat hippocampus and cortex is different from that of A1 and striatal A2A receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;359:295–302. doi: 10.1007/pl00005355. [DOI] [PubMed] [Google Scholar]
- CUNHA R.A., JOHANSSON B., CONSTANTINO M.D., SEBASTIÃO A.M., FREDHOLM B.B. Evidence for high-affinity binding sites for the A2A receptor agonist [3H]CGS 21680 in the rat hippocampus and cerebral cortex that are different from striatal A2A receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1996;353:261–271. doi: 10.1007/BF00168627. [DOI] [PubMed] [Google Scholar]
- CUNHA R.A., RIBEIRO J.A. Purinergic modulation of [3H]GABA release from rat hippocampal nerve terminals. Neuropharmacology. 2000;39:1156–1167. doi: 10.1016/s0028-3908(99)00237-3. [DOI] [PubMed] [Google Scholar]
- CUNHA R.A., SEBASTIÃO A.M. Adenosine and adenine nucleotides are independently released from both the nerve terminals and muscle fibres upon electrical stimulation of innervated skeletal muscle of the frog. Pflügers Arch. Eur. J. Physiol. 1993;424:503–510. doi: 10.1007/BF00374914. [DOI] [PubMed] [Google Scholar]
- CUNHA R.A., SEBASTIÃO A.M., RIBEIRO J.A. Separation of adenosine tri-phosphate and its degradation products in innervated muscle of the frog by reverse phase high-performance liquid chromatography. Chromatographia. 1989;28:610–612. [Google Scholar]
- FEOKTISTOV I., BIAGGIONI I. Adenosine A2B receptors. Pharmacol. Rev. 1997;49:381–402. [PubMed] [Google Scholar]
- GADANO A.C., SOGNI P., YANG S., CAILMAIL S., MOREAU R., NEPVEUX P., COUTURIER D., LEBREC D. Endothelial calcium-calmodulin dependent nitric oxide synthase in the in vitro vascular hyporeactivity of portal hypertensive rats. J. Hepatology. 1997;26:678–686. doi: 10.1016/s0168-8278(97)80435-7. [DOI] [PubMed] [Google Scholar]
- GROSZMANN R.J. Hyperdynamic circulation of liver disease 40 years later: pathophysiology and clinical consequences. Hepatology. 1994;20:1359–1363. [PubMed] [Google Scholar]
- GUIMARÃES S., ALBINO-TEIXEIRA A. Hypertension due to chronic blockade of P1-purinoceptors. J. Auton. Pharmacol. 1996;16:367–370. doi: 10.1111/j.1474-8673.1996.tb00055.x. [DOI] [PubMed] [Google Scholar]
- HEINEMANN A., STAUBER R.E. Vasodilator responses to nitric oxide are enhanced in mesenteric arteries of portal hypertensive rats. Eur. J. Clin. Invest. 1996;26:824–826. doi: 10.1046/j.1365-2362.1996.2340557.x. [DOI] [PubMed] [Google Scholar]
- HILEY C.R., BOTTRILL F.E., WARNOCK J., RICHARDSON P.J. Effects of pH on responses to adenosine, carbachol and nitroprusside in the isolated perfused superior mesenteric arterial bed of the rat. Br. J. Pharmacol. 1995;116:2641–2646. doi: 10.1111/j.1476-5381.1995.tb17220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHANSSON B., FREDHOLM B.B. Further characterization of the binding of the adenosine receptor agonist [3H]CGS 21680 to rat brain using autoradiography. Neuropharmacology. 1995;34:393–403. doi: 10.1016/0028-3908(95)00009-u. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Pharmacologic Analysis of Drug-Receptor Interaction. Raven Press, New York; 1993. pp. 229–238. [Google Scholar]
- LAUTT W.W. Mechanism and role of intrinsic regulation of hepatic arterial blood flow: the hepatic buffer response. Am. J. Physiol. 1985;249:G449–G456. doi: 10.1152/ajpgi.1985.249.5.G549. [DOI] [PubMed] [Google Scholar]
- LAUTT W.W. Autoregulation of superior mesenteric artery is blocked by adenosine antagonisms. Can. J. Physiol. Pharmacol. 1986;64:1291–1295. doi: 10.1139/y86-218. [DOI] [PubMed] [Google Scholar]
- LAUTT W.W., GREENWAY C.V. Conceptual review of hepatic vascular bed. Hepatology. 1987;7:952–963. doi: 10.1002/hep.1840070527. [DOI] [PubMed] [Google Scholar]
- LEE F.Y., ALBILLOS A., COLOMBATO L., GROSZMANN R.J. The role of nitric oxide in the vascular hyporesposiveness to methoxamine in portal hypertensive rats. Hepatology. 1992;16:1043–1048. doi: 10.1002/hep.1840160430. [DOI] [PubMed] [Google Scholar]
- MARQUES M.C., VILLA DE BRITO M.T., COSTA P., SILVA-CARVALHO L. Role of adenosine and nitric oxide on splanchic haemodynamics in portal hypertension. J. Physiol. 1999;523:P39. [Google Scholar]
- MCCANN P.W., KATHOLI R.E. Control of artifacts in plasma adenosine determinations. Proc. Soc. Exp. Biol. & Med. 1990;194:314–319. doi: 10.3181/00379727-194-43097. [DOI] [PubMed] [Google Scholar]
- MIURA K., OKUMURA M., YUKIMURA T., YAMAMOTO K. Fluorimetric determination of plasma adenosine concentrations using high-performance liquid chromatography. Anal. Biochem. 1991;196:84–88. doi: 10.1016/0003-2697(91)90121-9. [DOI] [PubMed] [Google Scholar]
- OLSSON R.A., PEARSON J.D. Cardiovascular purinoceptors. Physiol. Rev. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- PETERSON G.L. A simplification of the protein assay method of Lowry et al., which is more generally applicable. Anal. Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- PRENTICE D.J., PAYN S.E., HOURANI S.M. Activation of two sites by adenosine receptor agonists to cause relaxation in rat isolated mesenteric artery. Br. J. Pharmacol. 1997;122:1509–1515. doi: 10.1038/sj.bjp.0701524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUBINO A., RALEVIC V., BURNSTOCK G. Contribuition of P1- (A2B subtype) and P2-purinoceptors to the control of vascular tone in the isolated mesenteric arterial bed. Br. J. Pharmacol. 1995;111:648–652. doi: 10.1111/j.1476-5381.1995.tb14981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAWMILLER D.R., CHOU C.C. Adenosine plays a role in food induced jejunal hyperemia. Am. J. Physiol. 1988;255:G168–G174. doi: 10.1152/ajpgi.1988.255.2.G168. [DOI] [PubMed] [Google Scholar]
- SLOWIACZEK P., TATTERSALL M.H.N. The determination of purine levels in human and mouse plasma. Anal. Biochem. 1982;125:6–12. doi: 10.1016/0003-2697(82)90376-1. [DOI] [PubMed] [Google Scholar]
- TABRIZCHI R., LUPICHUK S.M. Vasodilation produced by adenosine in isolated rat perfused mesenteric artery: role for endothelium. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;352:412–418. doi: 10.1007/BF00172778. [DOI] [PubMed] [Google Scholar]
- VILLA DE BRITO M.T., DUARTE CORREIA J.H., MARQUES M.C. Effect of nitric oxide on the vascular response to adenosine. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358 Suppl. 1:R258. [Google Scholar]
- VILLA DE BRITO M.T., DUARTE CORREIA J.H., MARQUES M.C. Role of endothelium on abnormal vascular responses to adenosine in portal hypertension. Br. J. Pharmacol. 1999;127 Suppl. P:P106. [Google Scholar]
- ZHANG Y., GEIGER J.D., LAUTT W.W. Improved high-pressure liquid chromatographic-fluorimetric assay for measurement of adenosine in plasma. Am. J. Physiol. 1991;260:G658–G664. doi: 10.1152/ajpgi.1991.260.4.G658. [DOI] [PubMed] [Google Scholar]