

Abstract
We examined the role of non-NMDA receptors in kainic acid (KA)-induced apoptosis in cultures of rat cerebellar granule cells (CGCs). KA (1 – 500 μM) induced cell death in a concentration-dependent manner, which was prevented by NBQX and GYKI 52466, non-NMDA receptor antagonists. Moreover, AMPA blocked KA-induced excitotoxicity, through desensitization of AMPA receptors.
Similarly, KA raised the intracellular calcium concentration of CGCs, which was inhibited by NBQX and GYKI 52466. Again, AMPA (100 μM) abolished the KA (100 μM)-induced increase in intracellular calcium concentration.
KA-induced cell death in CGCs had apoptotic features, which were determined morphologically, by DNA fragmentation, and by expression of the prostate apoptosis response-4 protein (Par-4).
KA (500 μM) slightly (18%) increased caspase-3 activity, which was strongly enhanced by colchicine (1 μM), an apoptotic stimulus. However, neither Z-VAD.fmk, a pan-caspase inhibitor, nor the more specific caspase-3 inhibitor, Ac-DEVD-CHO, prevented KA-induced cell death or apoptosis. In contrast, both drugs inhibited colchicine-induced apoptosis.
The calpain inhibitor ALLN had no effect on KA or colchicine-induced neurotoxicity.
Our findings indicate that colchicine-induced apoptosis in CGCs is mediated by caspase-3 activation, unlike KA-induced apoptosis.
Keywords: Apoptosis, kainic acid, cerebellar granule cells, Par-4, colchicine, caspases, calpain, Z-VAD.fmk, Ac-DEVD-CHO, glutamate
Introduction
Glutamate, the main excitatory neurotransmitter in the central nervous system (CNS), mediates a variety of neurological effects as a result of ionotropic glutamate receptor stimulation. Three classes of ionotropic glutamate receptors are involved in glutamate-induced neuronal cell death (excitotoxicity): the N-methyl-D-aspartate (NMDA) receptor, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, and the kainate receptor (Bettler & Mulle, 1995; Bleakman & Lodge, 1998; Meldrum, 2000). Although the role of NMDA receptor activation in neuronal cell death has been widely studied, that of AMPA and kainate receptors is less well known.
Cerebellar granule cells (CGCs), which are particularly vulnerable to excitotoxins, are used to study the mechanisms of neuronal cell death mediated by kainate receptors (Dykens et al., 1987; Kato et al., 1991). Concerning the nature of cell death, it has been shown that CGCs exposed to glutamate, AMPA or KA undergo apoptosis or necrosis depending on the intensity of exposure (Ankarcrona et al., 1995; Ankarcrona, 1998; Cebers et al., 1997; Simonian et al., 1996; Giardina et al., 1998). There is controversy as to which of the ionotropic glutamate receptors is involved in KA-induced neuronal cell death in CGCs. Some authors suggest a process mediated through AMPA receptors (Ambrosio et al., 2000; Leski et al., 2000) while others point to kainate receptors (Carroll et al., 1998; Giardina & Beart, 2001).
Whereas necrosis is a passive process of cell death in which the neuron ultimately lyses its contents into the immediate surroundings, apoptosis is an active mode of cell death. Apoptotic cell death is characterized by several morphological and biochemical features, including reduced cell volume, condensation of nuclear chromatin, and DNA fragmentation (Sastry & Rao, 2000). The most relevant enzymes involved in neuronal cell death are cysteine proteases. Among them, calpains participate in both necrotic and apoptotic cell death, whereas caspases are specifically activated only in the apoptotic process (Wang, 2000). Moreover, several lines of evidence suggest that caspase-3, a member of the CED-3 subfamily of caspases, controls apoptosis (Marks et al., 1998; Budd et al., 2000; Mattson, 2000).
Recently, it has been demonstrated that prostate apoptosis response-4 (Par-4) has a pro-apoptotic effect (Duan et al., 1999; 2000). This protein belongs to the family of immediate early gene products, such as c-Myc, c-Fos and c-Jun (Rangnekar, 1998). Unlike the other immediate early gene products, Par-4 induction appears to be induced exclusively by apoptotic stimuli (Camandola & Mattson, 2000). It has been proposed that Par-4 and caspases are critical mediators of neuronal apoptosis in neurodegenerative disorders (Liu & Zhu, 1999). Previous studies have implicated caspase-3 in apoptosis induced by glutamate (Du et al., 1997a), MPP+ (Du et al., 1997b) and 6-hydroxydopamine in CGCs (Dodel et al., 1999). In addition, caspase-3 inhibitors protect CGCs from the neurotoxic effect of these compounds. Moreover, caspase-3 activation contributes to neuronal apoptosis induced by potassium deprivation (D'mello et al., 1998; 2000; Marks et al., 1998; Moran et al., 1999). However, in mice lacking caspase-3, this cysteine protease is not necessary for neuronal death of CGCs induced by potassium deprivation, although it is involved in neuronal apoptosis (D'mello et al., 2000).
Thus, the apoptotic mechanism induced by glutamate is well known in CGCs, but the pathways involved in KA-induced apoptosis are less. Although KA may activate caspase-3 (Nath et al., 1998) and c-jun (Cheung et al., 1998), the routes involved in KA-induced apoptosis are unclear.
Here, we study both the ionotropic glutamate receptors involved in KA-induced apoptosis in CGCs and the role of caspase-3 and calcium-dependent proteases in KA-induced apoptosis. We demonstrate that KA-induced apoptosis is mediated mainly through activation of AMPA receptors and is not prevented by caspase inhibitors. We suggest that Par-4 plays a key role in KA-induced apoptosis in CGCs. Therefore, we propose a caspase-independent mechanism of apoptosis mediated by KA.
Methods
Drugs
Kainic acid (KA), colchicine, propidium iodide (PI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), HEPES and bovine serum albumin (BSA) were from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Benzyloxycarbonyl-val-ala-asp-(O-methyl)-fluormethylketone (Z-VAD.fmk) and Ac-Asp-Glu-Val-Asp-aldehyde (Ac-DEVD-CHO) were from Bachem AG (Bubendorf, Switzerland). N-acetyl-Leu-Leu-Nle-CHO (ALLN) was from Calbiochem. AMPA, (+)MK-801, GYKI 52466 and NBQX were purchased from TOCRIS (Ballwin, MO, U.S.A.). Fura-2 AM was from Molecular Probes (Eugene, OR, U.S.A.). Cell culture media and foetal calf serum (FCS) were obtained from GIBCO (Life Technologies, Paisley, U.K.). Cell culture salts, enzymes, Mowiol® 4 – 88 and Triton X-100 were from Sigma. Other chemical reagents were of analytical quality and purchased by Panreac Quimica (Barcelona, Spain).
Cerebellar granule cell cultures
Primary cultures of CGCs were prepared from 7-day-old Sprague Dawley rat pups following Nicoletti et al. (1986). Briefly, cerebella were quickly removed and cleaned of meninges, followed by manual slicing with a sterile blade, dissociation with trypsin and treatment with DNase. Cells were adjusted to 8×105 cells ml−1 and plated on both poly-L-lysine-coated 24-well plates and glass coverslips at a density of 320,000 cells cm−2. Cultures were grown in basal medium with Eagle's salts (BME) containing 10% FCS, 2 mM L-glutamine, 0.1 mg ml−1 gentamicin and 25 mM KCl, referred to as a complete medium. Cytosine arabinoside (10 μM) was added 16 – 18 h after plating to inhibit the growth of non-neuronal cells. Cultures prepared by this method were more than 95% enriched in granule neurons, as assessed by GFAP immunocytochemistry (data not shown).
Treatment of CGCs and survival assay
CGCs were used after 9 – 10 days in vitro. KA and colchicine were dissolved in complete culture medium, and neutralized with NaOH to pH 7.4 if necessary before addition to the cell culture (to a maxim volume of 10 μl). To investigate the effect of NBQX, GYKI 52466, (+)MK-801, cyclothiazide, concanavalin-A, Z-VAD.fmk and Ac-DEVD-CHO were added to the medium, at the precise concentrations, 30 min before addition of neurotoxins. Cell death was determined 24 h later by the MTT assay as follows.
Assessment of neuronal injury
MTT was added to the cells to a final concentration of 250 μM and incubated for 1 h to reduce MTT to a dark blue formazan product (Hansen et al., 1989). The media were removed and cells were dissolved in dimethylsulphoxide. Formation of formazan was tested by measuring the amount of reaction product by absorbance change (595 nm) using a microplate reader (BioRad Laboratories, CA, U.S.A.). Viability results was expressed as a percentage of the absorbance measured in untreated cells.
Measurement of cytosolic calcium increases
The increase in intracellular free Ca2+ was determined in CGCs grown in glass cover slips (Corning Costar Corp., Acton, MA, U.S.A.) using a Mg2+-free, Locke-HEPES buffer (LH-BSA), which consisted of (in mM): NaCl 154, KCl 5.6, NaHCO3 3.6, CaCl2 1.3, D-glucose 5.6, HEPES 10 and 0.1% (w v−1) BSA (pH=7.35). After 9 – 10 days in culture, one cover slip was carefully transferred to a Petri dish containing 3 ml of LH-BSA buffer and 2 μM Fura-2 AM, and incubated at 37°C for 1 h on a cell incubator. For fluorescence recording, the cover slip was carefully rinsed in LH-BSA buffer, mounted on a specific holder (coverslip accessory L2250008, PerkinElmer, Inc., Wellesley, MA, U.S.A.) and placed in a quartz cuvette containing 1.3 ml LH-BSA buffer. Measurements were made at 37°C under continuous mild stirring in a LS50B PerkinElmer fluorescence spectrometer, equipped with a fast-filter accessory for Fura-2 fluorescence ratio measurements. Emission data (510 nm) were collected with alternate excitation at 340 and 380 nm, and the ratio F340/F380 was calculated in real time, using proprietary software (FL WinLab 2.0)
Analysis of apoptosis rate by flow cytometry
Apoptosis was measured 24 h after KA or colchicine addition. In brief, PI (10 μg ml−1) and 0.1% (v v−1) Triton X-100 were added to culture media 30 min before cytofluorometry analysis. Cells were collected from culture plates by pipetting. Flow cytometer experiments were carried out using an Epics XL flow cytometer (Coulter Corp. Hialeah, FL, U.S.A.). The instrument was set up with the standard configuration: excitation of the sample was performed using as a standard 488 nm air-cooled argon-ion laser at 15 mW power. Forward scatter (FSC), side scatter (SSC) and red (620 nm) fluorescence for PI were acquired. Optical alignment was based on optimized signal from 10 nm fluorescent beads (Immunocheck, Epics Division). Time was used as a control of the stability of the instrument. Red fluorescence was projected on a 1024 monoparametrical histogram.
Detection of apoptotic nuclei by propidium iodide staining
PI staining was used to detect morphological evidence of apoptosis (Atabay et al., 1996). CGCs were grown on glass coverslips and after treatment with KA (500 μM) or colchicine (1 μM), they were fixed in 4% (w v−1) paraformaldehyde/phosphate buffered saline solution (PBS), pH 7.4 for 1 h at room temperature. After washing with PBS, they were incubated for 3 min with a solution of PI in PBS (10 μg ml−1). Coverslips were mounted in Mowiol® 4 – 88. Stained cells were visualized under u.v. illumination using the 20×objective (Leica DMRB fluo microscope, Leica Microsystems AG, Germany) and their digitized images were captured.
Apoptotic cells showed shrunken, brightly fluorescent, apoptotic nuclei showing with high fluorescence and condensed chromatin, compared with nonapoptotic cells. Apoptotic cells were scored by counting at least 500 cells of six fields for each sample in three experiments.
Assay of caspase-3 enzymatic activity
We used the colorimetric substrate Ac-DEVD-p-nitroaniline (Oncogen) for the determination of caspase-3 activity described below. Twelve, 18 or 24 h after treatment with 500 μM KA and 24 h after treatment with 1 μM colchicine, CGCs were collected in lysis buffer (HEPES 50 mM, NaCl 100 mM, 0.1% (w v−1) CHAPS and EDTA 0.1 mM, pH 7.4). 0.5 μg μl−1 of protein was incubated with 200 μM Ac-DEVD-p-nitroaniline in assay buffer (HEPES 50 mM, NaCl 100 mM, 0.1% (w v−1) CHAPS, 10 mM dithiothreitol and, 0.1 mM EDTA, pH 7.4) in 96-well plates at 37°C for 24 h. Absorbance of the cleaved product was measured at 405 nm in a microplate reader (BioRad). Results were expressed as percentages of the absorbance measured in vehicle-treated cells.
Western blot analysis
Aliquots of cells homogenate, containing 30 μg of protein per sample, were placed in sample buffer (0.5 M Tris-HCl pH 6.8, 10% (w v−1) glycerol, 2% (w v−1) SDS, 5% (v v−1) 2-β-mercaptoethanol and, 0.05% bromophenol blue) and denatured by boiling at 95 – 100°C for 5 min.
Samples were separated by electrophoresis on 10% (w v−1) acrylamide gels. Proteins were then transferred to polyvinylidene fluoride (PVDF) sheets (Immobilon™-P, Millipore Corp., Bedford, MA, U.S.A.) using a transblot apparatus (BioRad). Membranes were blocked overnight with 5% (w v−1) nonfat milk dissolved in TBS-T buffer (Tris 50 mM; NaCl 1.5% (w v−1); Tween 20 0.05% (v v−1), pH 7.5). Membranes were then incubated with a primary rabbit polyclonal antibody against Par-4 (1 : 1000, Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). After 90 min, blots were washed thoroughly in TBS-T buffer and incubated for 1 h with a peroxidase-conjugated anti rabbit IgG antibody (Amersham Corp., Arlington Heights, IL, U.S.A.). Immunoreactive protein was visualized using a chemiluminiscence-based detection kit following the manufacturer guidelines (ECL kit; Amersham Corp.).
Immunocytochemistry against Par-4
CGCs were grown on sterile glass slides. After stimuli, cells were washed twice in PBS and fixed in 4% (w v−1) paraformaldehyde/PBS, pH 7.4 for 1 h at room temperature and then permeabilized in 0.3% (v v−1) Triton X-100 for 10 min. After blocking with goat serum (1 h at room temperature), cells were incubated with rabbit anti-Par-4 (1 : 400, Santa Cruz Biotechnology) overnight at 4°C. They were then washed extensively and incubated with secondary antibody (anti-rabbit-FITC conjugated (1 : 100, Sigma) for 1 h at room temperature. Coverslips were thoroughly washed and mounted in Mowiol® 4 – 88 and immunosignal analysis was performed using a confocal microscope at 60× magnification (Leica, TCS, 4D).
Statistics
The data are presented as the mean±s.e.mean. For statistical comparisons, one-way analysis of variance followed by Tukey's test was used for multiple comparison and Student's test for pairs of data. Probability values lower than 0.05 was accepted as statistically significant.
Results
Characterization of kainic acid-induced toxicity in cultured rat CGCs
Exposure of cultured CGCs to various concentrations of KA (1 – 500 μM) for 24 h was neurotoxic in a concentration-dependent manner, as revealed by the MTT assay. Maximal toxicity was obtained at a concentration of 500 μM, which decreased viability to 27±5.7% (n=6) (Figure 1). Nonlinear analysis of viability values yielded an EC50 of 68±8.9 μM (n=6). The AMPA/kainate receptors antagonists NBQX and GYKI 52466 significantly prevented KA-induced (100 μM) cell death (Figure 2A,B). However, (+)MK-801 (10 μM), a selective antagonist of NMDA receptors, did not prevent KA-induced cell death (Figure 2A).
Figure 1.
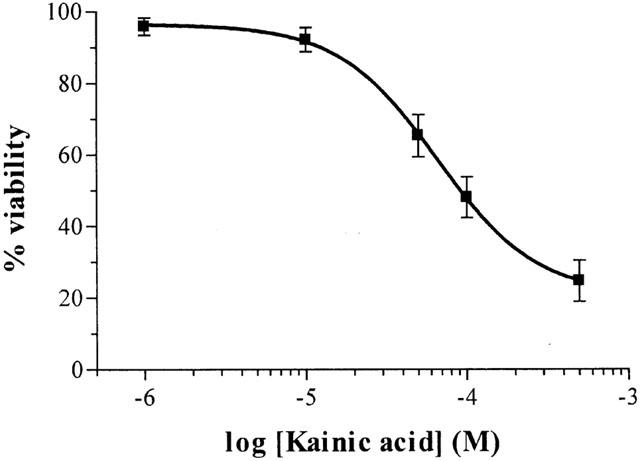
Effect of exposure to KA (1 – 500 μM) for 24 h on CGCs viability, based on MTT assay. Each point is the mean±s.e.mean of 5 – 6 cultures, carried out in quadruplicate.
Figure 2.
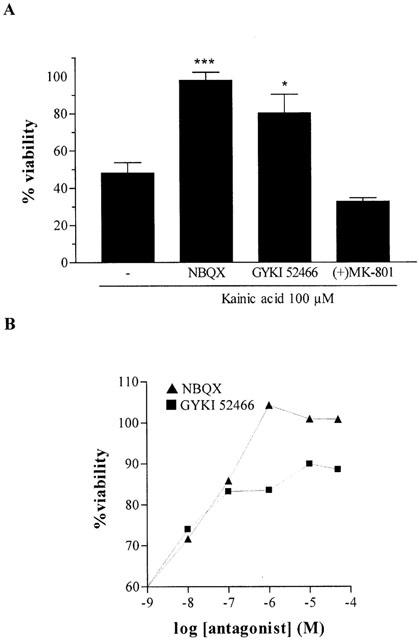
(A) Protective effect of AMPA/kainate receptor antagonists (10 μM) on KA-induced toxicity. CGCs were pre-treated with drugs 30 min before KA addition (100 μM). The data represent the mean±s.e.mean of four independent experiments carried out in quadruplicate, and are expressed as the percentage change of control cells, arbitrarily set at 100%. When necessary, the statistical analysis was carried out with the one-way ANOVA followed by Tukey's test *P<0.05, ***P<0.001 vs KA. (B) Concentration-response curves of NBQX and GYKI 52466 vs KA-induced toxicity (100 μM) in CGCs.
We studied the implication of kainate receptors in the neurotoxic effects of KA using concanavalin A (Con A, ranging from 1 μg ml−1 to 250 μg ml−1), a lectin that inhibits the desensitization of kainate receptors. Con A neither decreased nor enhanced KA toxicity in CGCs (Figure 3A).
Figure 3.
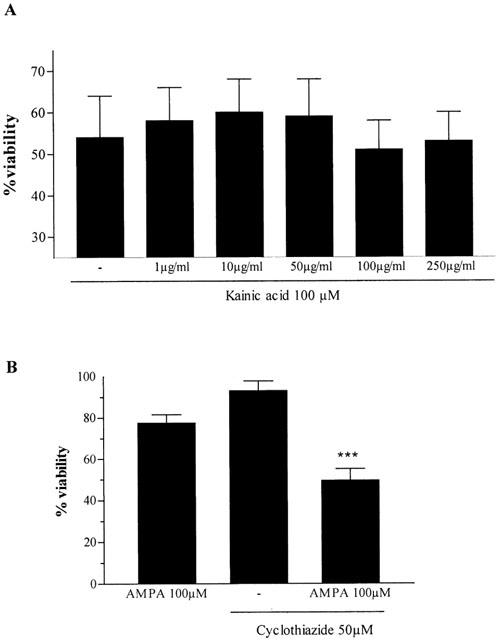
(A) Effect of various concentrations of concanavalin A on KA-induced toxicity in CGCs. (B) Cyclothiazide potentiated the effect of AMPA on CGCs viability. Data was obtained from 3 – 4 experiments and are the mean±s.e.mean of the percentage change of control cells. The statistical analysis was carried out using the one-way ANOVA followed by Tukey's test *P<0.05, ***P<0.001 vs KA.
Exposure of CGCs to AMPA (100 μM) slightly decreased cell viability. Cyclothiazide (CYZ, 50 μM), a specific inhibitor of AMPA receptor desensitization, potentiated AMPA-induced cell death (Figure 3B). However, CYZ alone had no effect on cell survival. To further demonstrate that KA neurotoxicity is mediated by interaction with AMPA receptors, KA (100 μM) was incubated in the presence of increasing concentrations of AMPA (10 – 100 μM). Viability assays showed that KA toxicity was significantly inhibit by 100 μM AMPA (P<0.05) (Figure 4).
Figure 4.
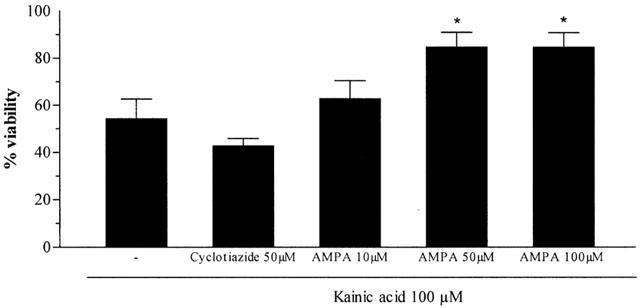
Effect of AMPA receptor modulators on KA-induced neurotoxicity. Each point is the mean±s.e.mean of four wells of three cultures, expressed as the percentage change of control cells, arbitrarily set at 100%. The statistical analysis was carried out using the one-way ANOVA followed by Tukey's test *P<0.05 vs KA.
We studied effect of CYZ on KA toxicity. Again, CYZ (50 μM) slightly promoted the neurotoxic effects of KA. Although the difference was not significant, viability decreased from 54±8.4 (n=3) to 43±3.1 (n=6) in the presence of CYZ (Figure 4).
Kainic acid-induced effects on intracellular Ca2+ concentration in CGCs
We also evaluated the effect of KA on cytoplasmic calcium in CGCs using the specific probe Fura-2. KA (10 – 100 μM) strongly increased intracellular calcium concentration. The nonlinear regression analysis yielded an EC50 of 128.8±3.3 μM (n=3) (Figure 5A). The effect of various concentrations of GYKI 52466 and NBQX (0.01 – 10 μM) were tested on the KA (100 μM)-induced increase in calcium concentration, yielding IC50 values of 9.1±0.87 μM and 2.8±0.9 μM, respectively (n=3). To assess the implication of AMPA receptors in KA effects in CGCs, the effect of KA on intracellular calcium increases was assessed in the presence or absence of AMPA (100 μM). Added 5 min before KA (100 μM), AMPA completely blocked the increase in cytosolic calcium concentration. (Figure 5B,C). These results further support the hypothesis that KA-induced responses are mainly due to the activation of AMPA receptors (Leski et al., 2000).
Figure 5.
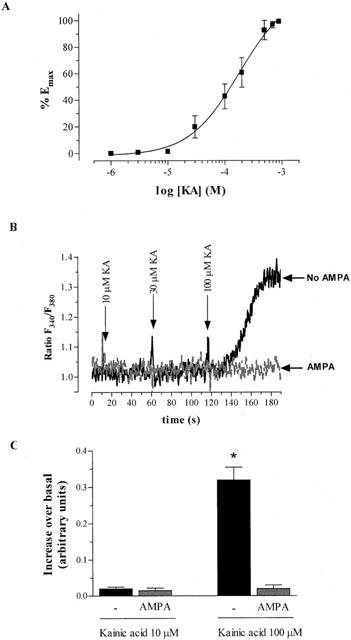
Effect of KA on intracellular calcium concentration in cultured CGCs. Recordings of the ratio of the fluorescent emission of Fura-2, under the excitation at 340 and 380 nm were measured as described in the Methods section. The ratio F340/F380 is indicative of changes in cytosolic free calcium. (A) Concentration-response curve of the increase in intracellular calcium concentration induced by KA. (B) Effect of KA on intracellular calcium concentration in the absence or presence of AMPA 100 μM. Tracings are a representative experiment using a sample first preincubated with AMPA and exposed to increasing concentrations of KA (grey tracing). After recording, cells were washed twice and the experiment was repeated in the absence of AMPA (black tracing). (C) Bar graph showing the data from three experiments using the same procedure as in (B). Each value is the mean±s.e.mean. The statistical analysis was carried out using Student's test *P<0.05 vs KA.
Kainic acid activates caspase-3 in CGCs
Exposure of CGCs to KA (500 μM) for 24 h induced a slight, but significant increase (18±8.7%; n=6) in caspase-3 activity as soon as 12 h after KA addition to cultures (Figure 6). KA (100 μM) did not induce caspase-3 activity. Colchicine (1 μM), used as a positive control of caspase-3 activation, strongly enhanced (150±40.5%, n=6) caspase-3 activation.
Figure 6.
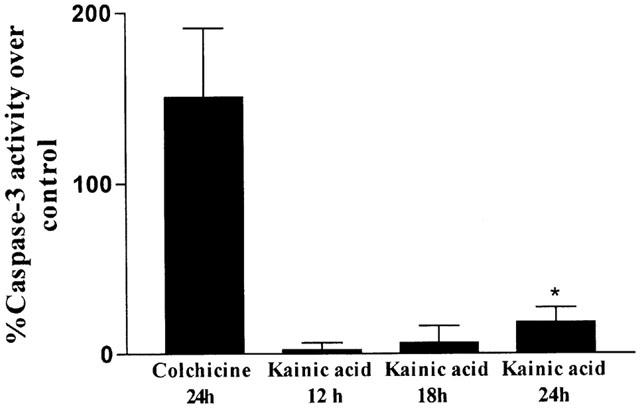
Bar chart showing the percentage of caspase-3-like activity in CGCs exposed to KA (500 μM) and colchicine (1 μM). Results are the mean±s.e.mean of four cultures. The statistical analysis was carried out using Student's test *P<0.05.
Caspase inhibitors do not prevent kainic acid-induced toxicity
To investigate the involvement of the apoptotic process in excitotoxic neuronal death mediated by KA, we used two compounds, the pan-specific caspase family inhibitor Z-VAD.fmk and the specific caspase-3 inhibitor Ac-DEVD-CHO. Neither Z-VAD.fmk (0.1 μM) nor Ac-DEVD-CHO (100 μM) prevented the excitotoxic effects induced by KA (500 μM, 24 h). However, Z-VAD.fmk (but not Ac-DEVD-CHO) inhibited the decrease in viability induced by colchicine (1 μM). We also studied the involvement of calpain-1 in KA neurotoxicity. Treatment of CGCs with the calpain-I inhibitor ALLN (1 μM) did not protect the cells from KA-induced neuronal damage (Figure 7).
Figure 7.
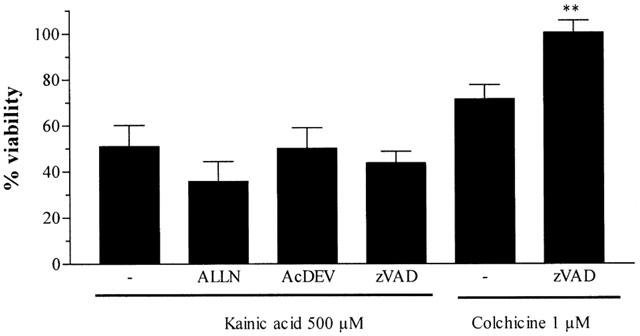
Effect of caspase (Z-VAD.fmk, 0.1 μM, and Ac-DEVD-CHO, 100 μM) and calpain (ALLN, 10 μM) inhibitors on KA or colchicine-induced toxicity. CGCs were pre-treated with drugs 30 min before KA (500 μM) or colchicine (1 μM) addition. The data represent the mean±s.e.mean of three/four independent experiments carried out in quadruplicate, and are expressed as the percentage change of control cells, arbitrarily set at 100%. The statistical analysis was carried out using the one-way ANOVA followed by Tukey's test **P<0.05 vs colchicine.
Kainic acid-induced apoptosis in CGCs is not prevented by caspase inhibitors
KA-induced apoptosis in CGCs was evaluated by two methods: DNA fragmentation by flow cytometry and counting the fraction of cells with nuclear condensation. When NBQX (10 μM) or GYKI 52466 (10 μM) were co-incubated with KA (500 μM, 24 h), they markedly reduced the percentage of apoptotic cells (Figure 8). On the other hand, Z-VAD.fmk (0.1 μM) and Ac-DEVD-CHO (100 μM) did not modify the percentage of the hypodiploid population. However, co-incubation of colchicine (1 μM) with Z-VAD.fmk or Ac-DEV-CHO decreased the percentage of apoptotic cells from 41±2.1 to 15±3.3 and 18±2.8, respectively (data are the mean±s.e.mean of 4 – 8 experiments performed in duplicate).
Figure 8.
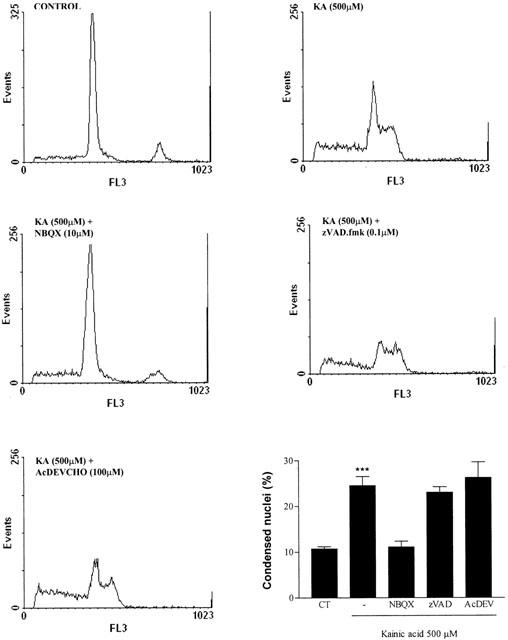
Flow cytometry analysis of KA-induced apoptosis in permeabilized CGCs shown by propidium iodide fluorescence histograms. Bar chart shows the percentage of apoptotic cells in the conditions tested. The statistical analysis was carried out using the one-way ANOVA followed by Tukey's test ***P<0.001 vs control.
Apoptotic features were also characterized by changes in the morphology of the nuclei, after staining with PI observed under fluorescence. The number of cells with chromatin condensation increased after treatment with KA (500 μM, 24 h). NBQX prevented KA effects on nuclear morphology. However, neither Z-VAD.fmk (0.1 μM) nor Ac-DEVD-CHO (100 μM) blocked KA-induced nuclear condensation (Figure 9).
Figure 9.
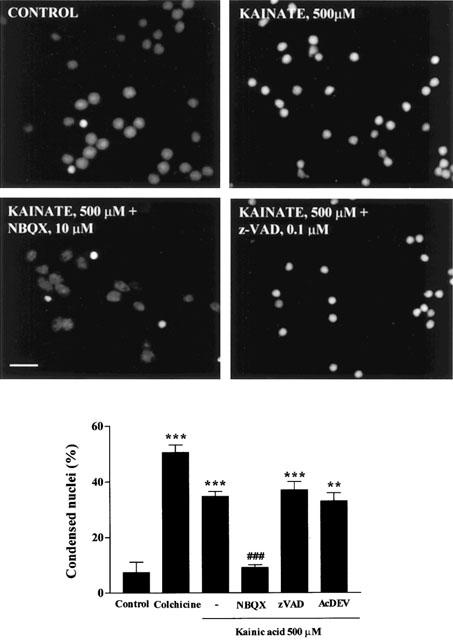
Chromatin condensation in permeabilized CGCs exposed to KA (500 μM) for 24 h. After exposure to KA, the CGCs were fixed, stained with propidium iodide and photographed under the fluorescence microscope, calibration bar, 10 μM. The nuclei were counted under the fluorescence microscope, distinguishing the normal from the condensed nuclei with the criteria stated in Methods. The statistical analysis was carried out using one-way ANOVA followed by Tukey's test **P<0.01, ***P<0.001 vs Control; ###P<0.001 vs KA 500 μM. Colchicine, 1 μM.
Kainic acid induces the expression of the prostate apoptosis response-4 (Par-4) protein
In previous studies, a correlation has been shown between the induction of Par-4 expression and neuronal apoptosis (Duan et al., 1999; 2000). To assess whether Par-4 is a common signal expressed after various apoptotic stimuli in CGCs, immunohistochemical and Western blot analyses were performed. Immunoblot analysis revealed a marked increase in the level of Par-4 protein expression after KA (500 μM) or colchicine (1 μM) treatment (Figure 10). This was confirmed by immunocytochemistry, since KA-treated CGCs showed a marked increase in Par-4 expression. NBQX (10 μM) prevented the increase in KA-induced Par-4 expression.
Figure 10.
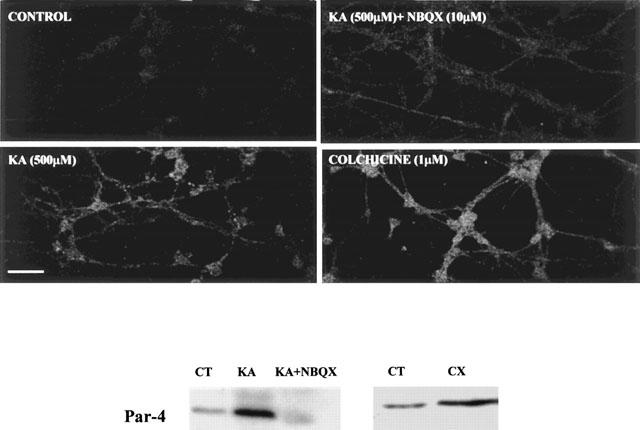
Evidence that Par-4 mediates KA-induced apoptosis in CGCs. Upper panels show confocal images of Par-4 immunoreactivity in CGCs, calibration bar, 30 μM. Lower panel shows levels of Par-4 assessed by Western blot. Control (CT), kainate (KA, 500 μM), NBQX (10 μM) colchicine (CX, 1 μM).
Discussion
Our results show that KA-induced apoptosis in cerebellar granule neurons is mediated through AMPA receptors and is not prevented by caspase inhibitors. The type of receptor responsible for KA effects on neuronal damage and/or apoptosis in CGCs is a matter controversy. Some authors suggest that kainate receptors are involved in KA-induced neuronal damage in several cell types (Carroll et al., 1998), while others implicate AMPA receptors (Ohno et al., 1997; 1998; Leski et al., 2000). Recently, Gasull et al. (2001) have demonstrated that AMPA receptors, but not kainate receptors, are responsible for KA-induced neuronal damage in rat cortical neurons. Likewise, other investigators have found similar results on hippocampal neurons (Ohno et al., 1997; Ambrosio et al., 2000) or retina (Ferreira et al., 1998). Previous studies have detected specific kainate receptors in CGCs by electrophysiological methods (Pemberton et al., 1998; Savidge et al., 1999) or by measuring changes in intracellular calcium concentration (Savidge et al., 1997; Savidge & Bristow, 1998). Moreover, KA, through kainate receptors, enhances the activation of the AP-1 transcription factor, and this effect is increased by concanavalin A, whereas cyclothiazide has no effect (Kovács et al., 2000).
However, few reports examine which of the receptors that are activated by KA are responsible for neuronal death in CGCs. Our study demonstrates that KA-induced toxicity is mediated by AMPA receptors, as supported by several findings. First, addition of AMPA to the culture medium protects cells from KA neurotoxicity and prevents the increase in cytosolic calcium concentration induced by KA. Since this agonist quickly evokes AMPA receptor desensitization, the inhibition of KA-induced neuronal damage in CGCs demonstrates that the induction of desensitization at this receptor impedes KA effects. Moreover, the selective inhibitor of the desensitization of the AMPA receptor cyclothiazide promoted KA-induced neurotoxicity, although the effect was not significant. Probably, KA does not totally desensitize the AMPA receptor, and when desensitization is blocked, its effects are boosted. However, when desensitization is by AMPA incubation, KA-induced neurodegeneration is prevented. Conversely, the specific inhibitor of kainate receptor desensitization concanavalin A did not affect KA neurotoxicity. Finally, GYKI 52466, the selective AMPA antagonist, prevented KA-induced neuronal death. These results strongly suggest that KA or AMPA-induced neurotoxicity in CGCs is due to the activation of AMPA receptors.
In vivo studies support the hypothesis that kainate receptors are involved in the excitotoxic process because they enhance the release of glutamate (Malva et al., 1998). To rule out that the effect of KA depended on NMDA receptor activation, we used the selective NMDA antagonist (+)MK-801. In agreement with other authors, this compound did not prevent KA-induced cell death in cultures of CGCs (Pemberton et al., 1998; Savidge et al., 1999).
Thus, our data reveal the key role of AMPA receptors in excitotoxicity and suggest that whereas NMDA and AMPA receptors are essential to glutamate-mediated excitotoxicity, the function of kainate receptors in excitotoxicity remains to be determined.
Caspase-3 has been identified as a central executioner of programmed cell death in neurons from Alzheimer's disease patients (Stadelmann et al., 1999; Shimohama et al., 1999), Parkinson's disease patients (Tatton, 2000) and after traumatic brain injury (Yakovlev et al., 1997). Moreover, caspase-3 is activated in CGCs by glutamate (Du et al., 1997a), dopamine (Dodel et al., 1999), MPP+ (Du et al., 1997b), colchicine (Bonfoco et al., 1995) and serum/potassium deprivation (Marks et al., 1998). There is evidence that NMDA increases caspase-3 activity in CGCs but this increase is much lower than that observed in low potassium-treated cells (Nath et al., 1998). The same authors showed that KA-induced caspase activation by detecting an α-spectrin breakdown product and NMDA clearly increases caspase activity, whereas KA-treated cells show only a slight increase. On the other hand, in cultures of cortical neurons, NMDA receptor activation dramatically increases caspase-3 activity and also induces apoptosis. Stimulation of AMPA/kainate receptors slightly modifies these parameters (Hirashima et al., 1999).
Here, we demonstrate that KA induces a modest increase in caspase-3 activity compared with colchicine. These results suggest that caspase activation does not play a pivotal role in KA-induced neuronal cell death in CGCs. This hypothesis is supported by the inability of Z-VAD.fmk (broad acting inhibitor of caspases) and Ac-DEVD-CHO (caspase-3 specific inhibitor) to prevent KA-induced apoptosis. KA may exert its neurotoxic effects in CGCs via a caspase-independent pathway. KA excitotoxicity may be associated with damage of the plasma membrane due to cell swelling (Kiedrowski, 1998; Rago et al., 2001), whereas glutamate excitotoxicity is associated with a prolonged alteration of the mitochondrial membrane potential. The authors also suggest that the intracellular sodium increase through the stimulation of AMPA/kainate receptors is essential to KA-induced excitotoxicity in CGCs. Conversely, NMDA receptor-induced excitotoxicity, involves a rise in cytoplasmic calcium concentration, and the intracellular calcium concentration is altered in CGCs exposed to glutamate. The differences in caspase-3 activation between glutamate and KA may be due to the distinct ion permeability of NMDA and AMPA receptors (Savidge et al., 1997; 1999; Savidge & Bristow, 1997; 1998; Rago et al., 2001).
Nevertheless, a caspase-independent apoptotic mechanism has also been reported in other apoptotic processes, since caspase inhibitors do not protect from neuronal cell death induced by β-amyloid peptide (Sáez-Valero et al., 2000) in mice lacking caspase-3 (apoptosis is prevented but not neuronal cell death) (D'mello et al., 2000). Moreover, the mechanism of neuronal cell death induced by methylmercury in CGCs (Castoldi et al., 2000; Daré et al., 2000), serum deprivation in cortical neurons (Hamabe et al., 2000) and lack of Bax in mice (Miller et al., 1997) seems to be caspase-independent. Calpain I is activated during apoptosis (Chan & Mattson, 1999). ALLN, a calpain I inhibitor, did not prevent KA-induced neurotoxity. Thus, we conclude that this protein is not involved in this apoptotic/necrotic process.
The mechanism by which KA induces apoptosis is not well understood, but it is known that KA induces the expression of some apoptotic signals such as c-Jun, the cell-cycle regulatory proteins p21 and p53 (Uberti et al., 1998; Chan & Mattson, 1999; Grilli & Memo, 1999) and cyclin D activation (Giardina et al., 1998). To further characterize the apoptotic process induced by KA in CGCs, we studied the expression of Par-4, an immediate early gene involved in the neuronal apoptotic biochemical cascade. Our results indicate that both KA and colchicine-incubation evoke the expression of the pro-apoptotic protein Par-4. Moreover, this increase in Par-4 expression depends on AMPA/kainate receptor activation, since it is prevented by NBQX. Par-4 is a protein identified in prostate tumour cells undergoing apoptosis (Sells et al., 1997). It plays a key role in the apoptotic process induced by several toxins and excitatory amino acids (Duan et al., 1999; Camandola & Mattson, 2000). Moreover, both in Alzheimer's disease and in amyotrophic lateral sclerosis, Par-4 levels increase (Guo et al., 1998; Pedersen et al., 2000). However, the mechanism by which Par-4 evokes neuronal apoptosis in unclear, but it is characterized by a mitochondrial alteration (Duan et al., 1999). We would like to highlight that mitochondrial dysfunction releases cyt-c and AIF (apoptotic induction factor), which activate the apoptosis executioner machinery.
In conclusion, caspase-3 activation is not a crucial event in KA-induced apoptosis in CGCs, since caspase inhibitors do not prevent apoptotic features like nuclear condensation. Moreover, the expression of Par-4 may play a key role in CGCs apoptosis.
Acknowledgments
We thank the Language Advice Service of the University of Barcelona for revising the manuscript. We also thank Dr Jaume Comas and Ms Rosario González, from the Scientific-Technical Services of the University of Barcelona, for their technical assistance in flow cytometry. This study was supported by the CICYT Grant PM98-0195 and Fundació La Marató de TV3. E. Verdaguer is the recipient of a fellowship from the University of Barcelona.
Abbreviations
- Ac-DEVD-CHO
Ac- Asp- Glu- Val- Asp- aldehyde
- ALLN
N-acetyl-Leu-Leu-Nle-CHO
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- BME
Basal medium with Eagle's salts
- BSA
Bovine serum albumin
- CGCs
Cerebellar granule cells
- CNS
Central nervous system
- Con A
Concanavalin A
- CYZ
Cyclothiazide
- FSC
Foetal calf serum
- FSC
Forward scatter
- KA
Kainic acid
- LH-BSA
Locke-HEPES buffer
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NMDA
N-methyl-D-aspartate
- Par-4
Prostate apoptosis response-4
- PBS
Phosphate buffered saline solution
- PI
Propidium iodide
- SSC
Side scatter
- Z-VAD.fmk
Benzyloxycarbonyl-val-ala-asp-(O-methyl)-fluormethylketone
References
- AMBROSIO A.F., SILVA A.P., MALVA J.O., MESQUITA J.F., CARVALHO A.P., CARVALHO C.M. Role of desensitisation of AMPA receptors on the neuronal viability and the [Ca2+]i changes in cultured rat hippocampal neurons. Eur. J. Neurosci. 2000;12:2021–2031. doi: 10.1046/j.1460-9568.2000.00091.x. [DOI] [PubMed] [Google Scholar]
- ANKARCRONA M. Glutamate induced cell death: Apoptosis or necrosis. Prog. Brain Res. 1998;116:265–272. doi: 10.1016/s0079-6123(08)60442-2. [DOI] [PubMed] [Google Scholar]
- ANKARCRONA M., DYPBUKT J.M., BONFOCO E., ZHIVOTOVSKY B., ORRENIUS S., LIPTON S.A., NICOTERA P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- ATABAY C., CAGNOLY C.M., KHARLAMOV E., IKONOMOVIC M.D., MENEV H. Removal of serum from primary cultures of cerebellar granule neurons induces oxidative stress and DNA fragmentation: protection with antioxidants and glutatamate receptor antagonists. J. Neurosci. Res. 1996;43:465–475. doi: 10.1002/(SICI)1097-4547(19960215)43:4<465::AID-JNR7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- BETTLER B., MULLE C. AMPA and Kainate receptors. Neuropharmacology. 1995;34:123–139. doi: 10.1016/0028-3908(94)00141-e. [DOI] [PubMed] [Google Scholar]
- BLEAKMAN D., LODGE D. Neuropharmacology of AMPA and kainate receptors. Neuropharmacology. 1998;37:1187–1204. doi: 10.1016/s0028-3908(98)00139-7. [DOI] [PubMed] [Google Scholar]
- BONFOCO E., CECCATELLI S., MANZO L., NICOTERA P. Colchicine induces apoptosis in cerebellar granule cells. Exp. Cell Res. 1995;218:189–200. doi: 10.1006/excr.1995.1147. [DOI] [PubMed] [Google Scholar]
- BUDD S.L., TENNETI L., LISHNAK T., LIPTON S.A. Mitochondrial and extramitochondrial apoptotic signalling pathways in cerebrocortical neurons. Proc. Nat. Acad. Sci. U.S.A. 2000;97:6161–6166. doi: 10.1073/pnas.100121097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMANDOLA S., MATTSON M.P. Pro-apoptotic action of PAR-4 involves inhibition of NF-kappaB activity and suppression of BCL-2 expression. J. Neurosci. Res. 2000;61:134–139. doi: 10.1002/1097-4547(20000715)61:2<134::AID-JNR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- CARROLL F.Y., CHEUNG N.S., BEART P.M. Investigations of non-NMDA receptor-induced toxicity in serum-free antioxidant-rich primary cultures of murine cerebellar granule cells. Neurochem. Int. 1998;33:23–28. doi: 10.1016/s0197-0186(05)80004-x. [DOI] [PubMed] [Google Scholar]
- CASTOLDI A.F., BARNI S., TURIN I., GANDINI C., MANZO L. Early acute necrosis, delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granule neurons exposed to methylmercury. J. Neurosci. Res. 2000;59:775–787. doi: 10.1002/(SICI)1097-4547(20000315)59:6<775::AID-JNR10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- CEBERS G., ZHIVOTOVSKY B., ANKARCRONA M., LILJEQUIST S. AMPA neurotoxicity in cultured cerebellar granule neurons: mode of cell death. Brain Res. Bull. 1997;43:393–403. doi: 10.1016/s0361-9230(97)00025-7. [DOI] [PubMed] [Google Scholar]
- CHAN S.L., MATTSON M.P. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J. Neurosci. Res. 1999;58:167–190. [PubMed] [Google Scholar]
- CHEUNG N.S., CARROLL J.A., LARM J.A., BEART P.M., GIARDINA S.F. Kainate-induced apoptosis correlates with c-Jun activation in cultured cerebellar granule cells. J. Neurosci. Res. 1998;52:69–82. doi: 10.1002/(SICI)1097-4547(19980401)52:1<69::AID-JNR7>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- D'MELLO S.R., AGLIECO F., ROBERTS M.R., BORODEZT K., HAYCOCK J.W. A DEVD-inhibited caspase other than CPP32 is involved in the commitment of cerebellar granule neurons to apoptosis induced by K+ deprivation. J. Neurochem. 1998;70:1809–1818. doi: 10.1046/j.1471-4159.1998.70051809.x. [DOI] [PubMed] [Google Scholar]
- D'MELLO S.R., KUAN C.Y., FLAVELL R.A., RAKIC P. Caspase-3 is required for apoptosis-associated DNA fragmentation but not for cell death in neurons deprived of potassium. J. Neurosci. Res. 2000;59:24–31. [PubMed] [Google Scholar]
- DARÉ E., GÖTZ M.E., ZHIVOTOVSKY B., MANZO L., CECCATELLI S. Antioxidants J811 and 17β-Estradiol protect cerebellar granule cells from methylmercury-induced apoptotic cell death. J. Neurosci. Res. 2000;62:557–565. doi: 10.1002/1097-4547(20001115)62:4<557::AID-JNR10>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- DODEL R.C., DU Y., BALES K.R., LING Z., CARVEY P.M., PAUL S.M. Caspase-3-like proteases and 6-hydroxydopamine induced neuronal cell death. Brain Res. Mol. Brain Res. 1999;64:141–148. doi: 10.1016/s0169-328x(98)00318-0. [DOI] [PubMed] [Google Scholar]
- DU Y., BALES K.R., DODEL R.C., HAMILTON-BYRD E., HORN J.W., CZILLI D.L., SIMMONS L.K., NI B., PAUL S.M. Activation of caspase 3-related cysteine protease is required for glutamate-mediated apoptosis in cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 1997a;94:11,657–11,662. doi: 10.1073/pnas.94.21.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DU Y., DODEL R.C., BALES K.R., JEMMERSON R., HAMILTON-BYRD E., PAUL S.M. Involvement of a caspase-3-like cysteine protease in 1-Methyl-4-phenylpyridinium-mediated apoptosis of cultured cerebellar granule neurons. J. Neurochem. 1997b;69:1382–1388. doi: 10.1046/j.1471-4159.1997.69041382.x. [DOI] [PubMed] [Google Scholar]
- DUAN W., GUO, ZHIHONG G., MATTSON M.P. Participation of Par-4 in the degeneration of striatal neurons induced by metabolic compromise with 3-Nitropropionic acid. Exp. Neurol. 2000;165:1–11. doi: 10.1006/exnr.2000.7434. [DOI] [PubMed] [Google Scholar]
- DUAN W., RANGNEKAR V.M., MATTSON M.P. Prostate apoptosis response-4 production in synaptic compartments following apoptotic and excitotoxic insults: evidence for a pivotal role in mitochondrial dysfunction and neuronal degeneration. J. Neurochem. 1999;72:2312–2322. doi: 10.1046/j.1471-4159.1999.0722312.x. [DOI] [PubMed] [Google Scholar]
- DYKENS J.A., STERN A., TRENKNER E. Mechanisms of kainate toxicity to cerebellar neurons in vitro is analogous to reperfusion tissue injury. J. Neurochem. 1987;49:1222–1228. doi: 10.1111/j.1471-4159.1987.tb10014.x. [DOI] [PubMed] [Google Scholar]
- FERREIRA I.L., DUARTE C.B., CARVALHO A.P. Kainate-induced retina amacrine cell damage is mediated by AMPA receptors. Neuroreport. 1998;9:3471–3475. doi: 10.1097/00001756-199810260-00025. [DOI] [PubMed] [Google Scholar]
- GASULL T., DEGREGORIO-ROCASOLANO N., TRULLAS R. Overactivation of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate and N-methyl-D-aspartate but not kainate receptors inhibits phosphatidylcholine synthesis before excitotoxic neuronal death. J. Neurochem. 2001;77:13–22. doi: 10.1046/j.1471-4159.2001.t01-2-00187.x. [DOI] [PubMed] [Google Scholar]
- GIARDINA S.F., BEART P.M. Excitotoxic profiles of novel, low affinity kainate receptors agonist in primary cultures of murine cerebellar granule cells. Neuropharmacology. 2001;41:421–432. doi: 10.1016/s0028-3908(01)00086-7. [DOI] [PubMed] [Google Scholar]
- GIARDINA S.F., CHEUNG N.S., REID M.T., BEART P.M. Kainate-induced apoptosis in cultured murine cerebellar granule cells elevates expression of the cell cycle gene cyclin D1. J. Neurochem. 1998;71:1325–1328. doi: 10.1046/j.1471-4159.1998.71031325.x. [DOI] [PubMed] [Google Scholar]
- GRILLI M., MEMO M. Possible role of NF-kappaB and p53 in the glutamate-induced pro-apoptotic neuronal pathway. Cell Death Differ. 1999;6:22–27. doi: 10.1038/sj.cdd.4400463. [DOI] [PubMed] [Google Scholar]
- GUO Q., FU W., XIE J., LUO H., SELLS S.F., GEDDES J.W., BONDADA V., RANGNEKAR V.M., MATTSON M.P. Par-4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer's disease. Nat. Med. 1998;4:957–962. doi: 10.1038/nm0898-957. [DOI] [PubMed] [Google Scholar]
- HAMABE W., FUKUSHIMA N., YOSHIDA A., UEDA H. Serum-free induced neuronal apoptosis-like cell death is independent of caspase activity. Mol. Brain Res. 2000;78:186–191. doi: 10.1016/s0169-328x(00)00074-7. [DOI] [PubMed] [Google Scholar]
- HANSEN M.B., NIELSEN S.E., BERG K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Meth. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- HIRASHIMA Y., KURIMOTO M., NOGAMI K., ENDO S., SAITOH M., OHTANI O., NAGATA T., MURAGUCHI A., TAKAKU A. Correlation of glutamate-induce apoptosis with caspase activities in cultured rat cerebral cortical neurons. Brain Res. 1999;849:109–118. doi: 10.1016/s0006-8993(99)02009-0. [DOI] [PubMed] [Google Scholar]
- KIEDROWSKI L. The difference between mechanisms of kainate and glutamate excitotoxicity in vitro: osmotic lesion versus mitochondrial depolarization. Res. Neurol. Neurosci. 1998;12:71–79. [PubMed] [Google Scholar]
- KATO K., PUTTFARCKEN P.S., LYONS W.E., COYLE J.T. Developmental time course and ionic dependence of kainate mediated toxicity in rat cerebellar granule cell cultures. J. Pharmacol. Exp. Ther. 1991;256:402–411. [PubMed] [Google Scholar]
- KOVÁCS A.D., CEBERS G., LILJEQUIST S. Kainate receptor-mediated activation of the AP-1 transcription factor complex in cultured rat cerebellar granule cells. Brain Res. Bull. 2000;52:127–133. doi: 10.1016/s0361-9230(00)00247-1. [DOI] [PubMed] [Google Scholar]
- LESKI M.L., VALENTINE S.L., BAER J.D., COYLE J.T. Insulin-like growth factor I prevents the development of sensitivity to kainate neurotoxicity in cerebellar granule cells. J. Neurochem. 2000;75:1548–1556. doi: 10.1046/j.1471-4159.2000.0751548.x. [DOI] [PubMed] [Google Scholar]
- LIU X., ZHU X.Z. Roles of p53, c-Myc, Bcl-2, Bax and caspases in glutamate-induced neuronal apoptosis and the possible neuroprotective mechanism of basic fibroblast growth factor. Brain Res. Mol. Brain Res. 1999;71:210–216. doi: 10.1016/s0169-328x(99)00186-2. [DOI] [PubMed] [Google Scholar]
- MALVA J.O., CARVALHO A.P., CARVALHO C.M. Kainate receptors in hippocampal CA3 subregion: evidence for a role in regulating neurotransmitter release. Neurochem. Int. 1998;32:1–6. doi: 10.1016/s0197-0186(97)00046-6. [DOI] [PubMed] [Google Scholar]
- MARKS N., BERG M.J., GUIDOTTI A., SAITO M. Activation of caspase-3 and apoptosis in cerebellar granule cells. J. Neurosci. Res. 1998;52:334–341. doi: 10.1002/(SICI)1097-4547(19980501)52:3<334::AID-JNR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- MATTSON M.P. Apoptotic and anti-apoptotic synaptic signalling mechanisms. Brain Pathology. 2000;10:300–212. doi: 10.1111/j.1750-3639.2000.tb00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELDRUM B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- MILLER T.M., MOULDER K.L., KNUDSON C.M., CREEDON D.J., DESHMUKH M., KORSMEYER S.J., JOHNSON E.M. Bax deletion further orders the cell death pathway in cerebellar granule cells suggests a caspase-independent pathway to cell death. J. Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORAN J., ITOH T., REDDY U.R., CHEN M., ALNEMRI E.S., PLEASURE D. Caspase-3 expression by cerebellar granule neurons is regulated by calcium and cyclic AMP. J. Neurochem. 1999;73:568–577. doi: 10.1046/j.1471-4159.1999.0730568.x. [DOI] [PubMed] [Google Scholar]
- NATH R., PROBERT A., Jr, MCGINNIS K.M., WANG K.K. Evidence for activation of caspase-3-like protease in excitotoxin- and hypoxia/hypoglycemia-injured neurons. J. Neurochem. 1998;71:186–195. doi: 10.1046/j.1471-4159.1998.71010186.x. [DOI] [PubMed] [Google Scholar]
- NICOLETTI F., WROBLEWSKI J.T., NOVELLI A., ALHO H., GUIDOTTI A., COSTA E. The activation of inositol phospholipid metabolism as a signal-transducing system for excitatory amino acids in primary cultures of cerebellar granule cells. J. Neurosci. 1986;6:1905–1911. doi: 10.1523/JNEUROSCI.06-07-01905.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHNO K., OKADA M., TSUTSUMI R., KOHARA A., YAMAGUCHI T. Kainate excitotoxicity is mediated by AMPA but not kainate-preferring receptors in embryonic rat hippocampal cultures. Neurochem. Int. 1997;31:715–722. doi: 10.1016/s0197-0186(97)00011-9. [DOI] [PubMed] [Google Scholar]
- OHNO K., OKADA M., TSUTSUMI R., MATSUMOTO N., YAMAGUCHI T. Characterization of cyclotyhiazide-enhanced kainate excitotoxicity in rat hippocampal cultures. Neurochem. Int. 1998;32:265–271. doi: 10.1016/s0197-0186(97)00098-3. [DOI] [PubMed] [Google Scholar]
- PEDERSEN W.A., LUO H., KRUMAN I., KASARSKIS E., MATTSON M.P. The prostate apoptosis response-4 protein participates in motor neuron degeneration in amyotrophic lateral sclerosis. FASEB J. 2000;14:913–924. doi: 10.1096/fasebj.14.7.913. [DOI] [PubMed] [Google Scholar]
- PEMBERTON K.E., BELCHER S.M., RIPELLINO J.A., HOWE J.R. High-affinity kainate-type ion channels in rat cerebellar granule cells. J. Physiol. 1998;510:401–420. doi: 10.1111/j.1469-7793.1998.401bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAGO A.C., WARD M.W., NICHOLLS D.G. Mitochondria control AMPA/kainate receptor-induce cytoplasmatic calcium deregulation in rat cerebellar granule cells. J. Neurosci. 2001;21:1893–1901. doi: 10.1523/JNEUROSCI.21-06-01893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANGNEKAR V.M. Apoptosis mediated by a novel leucine zipper of the leucine zipper protein Par-4. Apoptosis. 1998;3:61–66. doi: 10.1023/a:1009666705875. [DOI] [PubMed] [Google Scholar]
- SÁEZ-VALERO J., ANGERETTI N., FORLONI G. Caspase-3 activation by β-amyloid and prion protein peptides is independent from their neurotoxic effect. Neurosci. Lett. 2000;293:207–210. doi: 10.1016/s0304-3940(00)01532-9. [DOI] [PubMed] [Google Scholar]
- SASTRY P.S., RAO K.S. Apoptosis and the nervous system. J. Neurochem. 2000;74:1–20. doi: 10.1046/j.1471-4159.2000.0740001.x. [DOI] [PubMed] [Google Scholar]
- SAVIDGE J.R., BRISTOW R. Calcium permeability and joro spider toxin sensitivity of AMPA and kainate-receptors on cerebellar granule cells. Eur. J. Pharmacol. 1998;351:131–138. doi: 10.1016/s0014-2999(98)00280-5. [DOI] [PubMed] [Google Scholar]
- SAVIDGE J.R., BLEAKMAN D., BRISTOW R. Identification of kainate receptor mediated intracellular calcium increases in cultured rat cerebellar granule cells. J. Neurochem. 1997;69:1763–1766. doi: 10.1046/j.1471-4159.1997.69041763.x. [DOI] [PubMed] [Google Scholar]
- SAVIDGE J.R., BRISTOW R. Distribution of Ca2+ permeable AMPA receptors among cultured rat cerebellar granule cells. Neuroreport. 1997;8:1877–1882. doi: 10.1097/00001756-199705260-00017. [DOI] [PubMed] [Google Scholar]
- SAVIDGE J.R., STURGESS N.C., BRISTOW D.R., LOCK E.A. Characterisation of kainate receptor mediated whole-cell currents in rat cultured cerebellar granule cells. Neuropharmacology. 1999;38:375–382. doi: 10.1016/s0028-3908(98)00202-0. [DOI] [PubMed] [Google Scholar]
- SELLS S.F., HAN S.S., MUTHUKKUMAR S., MADDIWAR N., JOHNSTONE R., BOGHAERT E., GILLIS D., LIU G., NAIR P., MONNIG S., COLLINI P., MATTSON M.P., SUKHATME V.P., ZIMMER S.G., WOOD D.P., MCROBERTS J.W., SHI Y., RANGNEKAR V.M. Expression and function of the leucine zipper protein Par-4 in apoptosis. Mol. Cell. Biol. 1997;17:3823–3832. doi: 10.1128/mcb.17.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMOHAMA S., TANINO H., FUJIMOTO S. Changes in caspase expression in Alzheimer's disease: comparison with development and aging. Biochem. Biophys. Res. Commun. 1999;256:381–384. doi: 10.1006/bbrc.1999.0344. [DOI] [PubMed] [Google Scholar]
- SIMONIAN N.A., GETZ R.L., LEVEQUE J.C., KONRAD C., COYLE J.T. Kainate induces apoptosis in neurons. Neuroscience. 1996;74:675–683. doi: 10.1016/0306-4522(96)00141-8. [DOI] [PubMed] [Google Scholar]
- STADELMANN C., DECKWERTH T.L., SRINIVASAN A., BANCHER C., BRUCK W., JELLINGER K., LASSMANN H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am. J. Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TATTON N.A. Increased caspase-3 and bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson's disease. Exp. Neurol. 2000;166:29–43. doi: 10.1006/exnr.2000.7489. [DOI] [PubMed] [Google Scholar]
- UBERTI D., BELLONI M., GRILLI M., SPANO P.F., MEMO M. Induction of tumor-supressor phosphoprotein p53 in the apoptosis of cultured rat cerebellar neurons triggered by excitatory amino acids. Eur. J. Neurosci. 1998;10:246–254. doi: 10.1046/j.1460-9568.1998.00042.x. [DOI] [PubMed] [Google Scholar]
- WANG K.K.W. Calpain and caspase a: can you tell the difference. Trends Neurosci. 2000;23:20–26. doi: 10.1016/s0166-2236(99)01536-2. [DOI] [PubMed] [Google Scholar]
- YAKOVLEV A.G., KNOBLACH S.M., FAN L., FOX G.B., GOODNIGHT R., FADEN A.I. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J. Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]