

Abstract
Desferrithiocin (DFT) is an orally effective Fe chelator, with a similar high affinity and selectivity for Fe to desferrioxamine (DFO), which has been shown clinically to possess antineoplastic activity. In this study, DFT was assessed for antineoplastic potential in hepatocellular carcinoma cell lines (HCC). This was done as there are few treatments for this aggressive neoplasm. The effects of DFT on cell proliferation, cell cycle progression, Fe uptake and toxicity were examined. To establish whether DFT was selective for cancer cells a comparison was made with normal (non-proliferating) hepatocytes and non-tumorigenic (proliferating) fibroblasts (SWISS-3T3). DFT was a potent inhibitor of HCC proliferation (IC50∼40 μM). DFO also inhibited HCC proliferation under the same conditions, but was much less active (IC50=110 – 210 μM). When saturated with Fe, the activity of DFT, like DFO, was greatly diminished, suggesting it may act by depriving the cells of Fe or inactivating essential Fe pool(s). Indeed DFT rapidly decreased Fe uptake from Tf-59Fe by hepatoma cells and also by normal hepatocytes. However, DFT (and DFO) had much less effect on cell survival in hepatocytes and fibroblasts than in hepatoma cells. DFT may, like DFO, inhibit the cell cycle in the S phase of DNA synthesis. Both chelators showed low toxicity. These results indicate that DFT has potent antineoplastic activity in HCC. Further investigation into the DFT class of Fe chelators seems warranted, particularly in view of its high activity in relation to DFO, a chelator which is already in clinical trial for neuroblastoma.
Keywords: Desferrithiocin, desferrioxamine, Fe chelators, Fe uptake, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer morbidity and mortality in the world, accounting for well over one million deaths per year (Lau et al., 1997). Prognosis of patients diagnosed with HCC is poor, with long-term survival quite rare (Rustgi, 1998). Liver resection is the only curative treatment at present but remains applicable in only 10 – 15% of cases (Schafer & Sorrell, 1999). Hence investigation into alternative treatment methods and strategies such as chelation therapy for treatment of HCC are necessary.
Traditionally, Fe chelators have been investigated as potential therapeutic agents for diseases of Fe overload e.g. thalassemia. Desferrioxamine (DFO), a siderophore isolated from Streptomyces pilosus, is the only chelator in regular clinical use for the treatment of Fe overload. In several previous studies DFO has been shown to inhibit cellular proliferation in a number of neoplastic cell types in vitro by preventing Fe uptake (Becton & Bryles, 1988; Bierer & Nathan, 1990; Blatt & Stitely, 1987; Blatt et al., 1988; Kaplinsky et al., 1987; Nastruzzi et al., 1989; Richardson et al., 1995). For example Blatt & Stitely (1987) reported the anti-neoplastic activity of DFO against two neuroblastoma cell lines and found the activity of DFO to be dose-dependent (Blatt & Stitely, 1987).
In addition DFO has been shown to be an effective anti-cancer agent in vivo, halting tumour advancement in a 6 week old infant with acute lymphoblastic leukaemia (Donfrancesco et al., 1993). Subsequent clinical studies on neuroblastoma patients have confirmed this potent anti-neoplastic activity (Donfrancesco et al., 1990; 1992; 1993; 1995). For example, in one study involving 65 patients with advanced neuroblastoma (Donfrancesco et al., 1995), DFO (80 mg kg−1 per day), administered by intravenous infusion, caused a complete or partial response in 88% of patients. Despite its demonstrated activity as an antineoplastic agent, the effectiveness of DFO is limited by its relatively slow membrane permeability and its susceptibility to hydrolysis, which precludes oral administration (Hoffbrand, 1994).
Another more lipophilic chelator, desferrithiocin (DFT), is also a siderophore, isolated from Streptomyces antibioticus (Anderegg & Raber, 1990). Despite being structurally unrelated to DFO, DFT has a similar selectivity and very high binding affinity for Fe (Hann et al., 1990; Peter, 1985) and hence may also prevent tumour cell proliferation. Originally, DFT was investigated for the long-term treatment of Fe overload and was shown to be a highly effective and orally active Fe chelator (Baker et al., 1992b; Bergeron et al., 1990; Bergeron et al., 1990; Longueville & Crichton, 1986; Wolfe et al., 1989). Unfortunately, when administered for long time intervals at concentrations sufficient to decrease Fe overload, there was evidence of toxicity (probably caused by the Fe chelate, ferrithiocin (FT) (Baker et al., 1992b; Bergeron et al., 1993), resulting in DFT being withdrawn from development for long-term therapy. However, DFT or one of its analogues may hold potential as a short-term chemotherapeutic agent, alone or in conjunction with other agents e.g. hydroxyurea.
Besides DFO, investigations into the use of iron chelators in chemotherapy have so far been limited to a few compounds including pyridoxal isonicotinoyl hydrazone (PIH) and a number of its analogues (Darnell & Richardson, 1999; Richardson et al., 1995; Richardson & Milnes, 1997), Deferiprone (L1) (Chenoufi et al., 1998; Cragg et al., 1998; Hileti et al., 1995) and most recently, Tachypyr (Torti et al., 1998). The present study is an investigation of the potential antineoplastic activity of DFT, assessing both rat and human hepatoma cell lines in comparison with a non-tumorigenic proliferating cell line (fibroblasts) and normal non-proliferating rat hepatocytes. Chelators assessed included DFT and its ferric complex FT, and other less lipophilic Fe chelator maltol (MAL) and its ferric complex (MALF). Maltol, a product of carbohydrate degradation, has been shown to induce dose-dependent toxicity in neuroblastoma cell lines (Hironishi et al., 1996) and was thus included in this study for comparison with DFT. DFO was used as reference chelator in all experiments because of its clinically demonstrated antineoplastic activity (Becton & Bryles, 1988; Bierer & Nathan, 1990; Blatt & Stitely, 1987; Blatt et al., 1988; Kaplinsky et al., 1987; Nastruzzi et al., 1989; Richardson et al., 1994). Chelators were assessed in vitro in four ways: first, by observing their effect on cell survival and proliferation using the MTT assay (Mosmann, 1983) and second, by directly examining their effect on Fe uptake from 59Fe-Tf (Baker et al., 1992a). In addition, their site of action was investigated by comparing cell cycle progression in cells exposed to the different chelators, and their toxicity assessed from the release of cytoplasmic aspartate amino transferase (AST).
Methods
Materials
The radioisotope 59Fe (FeCl3) was purchased from Dupont (Sydney, NSW, Australia). Human apo-transferrin, dexamethasone and bovine serum albumin (BSA) were purchased from Sigma Chemical Company (St Louis, MO, U.S.A.). Minimum Essential Medium (MEM), foetal calf serum (FCS), penicillin G, streptomycin sulphate and trypsin-EDTA were obtained from Gibco – BRL (Melbourne, Victoria, Australia). Insulin was supplied by Commonwealth Serum Laboratories (Melbourne, Victoria, Australia). Pronase was purchased from Boehringer Mannheim Biochemica (Mannheim, Germany). Non-essential amino acid concentrate (100×) for MEM Eagle was obtained from ICN Biochemicals (CA, U.S.A.), fungizone (amphotericin B) from Trace Bioscience Pty Ltd (Australia) and Triton X-100 from Ajax Chemicals (NSW, Australia). Balanced salt solution (BSS) was prepared using the method of Hanks & Wallace (1949). The synthetic medium, Ultroser G was obtained from Sepracor (la Garenne, France). All chemicals used in this investigation were of analytical grade.
Chelators
The iron chelator desferrioxamine mesylate (DFO) was purchased from Sigma Chemical Company, (St Louis, MO, U.S.A.). DFT (2-(3-hydroxypyrid-2yl)-4-methyl-Δ2-thiazoline-4(S)-carboxylic acid) and FT were synthesized and provided by Novartis Pharma, (Switzerland) and prepared according to the method described in Baker et al. (1992b). Novartis was also the supplier of Maltol (MAL (3-hydroxy-2-methyl-4-pyrone)) and MALF. Structures of chelators are shown in Figure 1.
Figure 1.

Structures of Fe chelators. Chelators assessed included the hexadentate DFO, the tridentate DFT, and the bidentate maltol.
Cell culture
Experiments were performed on human and rat hepatocellular carcinoma cell lines which had different levels of differentiation and doubling times (Table 1) (Aden et al., 1979; Nakabayshi et al., 1982). These included the rat hepatoma cell lines HTC and McArdle 7777 (McArdle Labs, WI, U.S.A.) and the human hepatoma cell lines wild type HepG2 (ATCC) and HuH7 (courtesy of Dr D. Trinder, Department of Physiology, UWA, Australia). The mouse fibroblast cell line (SWISS-3T3) was kindly donated by the Skin Culture Unit (Princess Margaret Hospital, Perth, Australia). All hepatoma cells were grown in MEM solution containing FCS (10% v v−1), penicillin (100 u ml−1), streptomycin (100 μg ml−1), and fungizone (0.28 μg ml−1). The fibroblast cell line was grown in DMEM/Hams solution containing FCS (10% v v−1), penicillin (100 u ml−1), and streptomycin (100 μg ml−1). All cells were grown in a Forma Scientific incubator at 37°C in an atmosphere of 5% CO2/95% air.
Table 1.
Biochemical profiles of the hepatoma cell lines used in the present study

Isolation and culture of rat hepatocytes
The hepatomas' normal (non-proliferating) cell counterpart, the hepatocyte was studied in parallel with the cancer cells. The primary culture of rat hepatocytes (from 7-week-old male Wistar rats) was performed using standard techniques previously described (Baker et al., 1992a) and was done in accordance with the guidelines of the Animal Experimentation Ethics Committee (AEEC) of the University of Western Australia. Isolated hepatocytes were grown in MEM containing fungizone (2.5 μg ml−1), penicillin G, and streptomycin at 100 u ml−1 and 100 μg ml−1 respectively, dexamethasone (0.4 μg ml−1), and insulin (0.2 μg ml−1), supplemented with 10% (v v−1) FCS. Cells were plated out at a density of 106 cells ml−1 and maintained in an atmosphere of 5% CO2/95% air at 37°C for 4 h. After that, cells were washed four times with ice-cold BSS to remove non-viable cells and a synthetic medium, Ultroser G (2% v v−1 in MEM containing all the above except the substituted FCS) was added to the plates. The hepatocytes (70 – 90% confluent) were used for uptake studies the next day.
Protein labelling
Human apo-transferrin was radiolabelled with 59Fe according to the methods of Hemmaplardh & Morgan (1977).
Cell survival and proliferation
Cell survival and proliferation were evaluated using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium) assay (Mosmann, 1983). Cells were seeded aseptically into a 96-well microtitre plate at a density of 104 cells in 110 μl in MEM containing FCS (10%), fungizone (0.28 μg ml−1), penicillin G (100 u ml−1), streptomycin (100 μg ml−1), non-essential amino acids (100×) (1% v v−1) and human diferric transferrin (1.25 μM). This density was shown previously to yield an exponential growth of cells in the absence of chelators over a 48 h time period (data not shown). The chelators were first dissolved in the MEM solution mentioned above and added to the cell suspension over a range of concentrations (0 – 500 μM). Both a positive control (cells without the chelator) and a negative control (media only) were also plated out and the plates incubated over a 48 h period at 37°C. MTT (5 mg ml−1) was then added to all wells and the cells incubated for 2 h after which 110 μl of a solution containing SDS (10%) and isobutanol (50%) in 0.1 M HCl was added and the plates reincubated for at least another 2 h at 37°C. The level of dissolved formazan product was then read at 595 nm on a BIORAD 3550 microplate reader and the concentration causing 50% inhibition of cell growth (IC50) determined.
Chelator selectivity
Specificity of chelator activity for hepatoma cells was assessed by incubating the different cell types with varying concentrations of Fe chelators over 48 h at 37°C. An MTT assay (see above) was then performed to assess viability.
Iron uptake
Cells in the exponential phase of growth were incubated in a MEM solution (pH 7.4) containing HEPES-Tris buffer (20 mM), BSA (10 mg ml−1), the Fe chelator (0 – 500 μM) previously dissolved in MEM, and 59Fe-Tf (2.5 μM Fe) for up to 24 h. After the incubation period had elapsed, the cells were washed five times with ice cold BSS. Pronase (1 mg ml) was then added and the cells incubated for a further 30 min at 4°C. The suspension was centrifuged for approximately 1 min at 10,000×g to separate the membrane bound (Pronase sensitive) and internalized (Pronase resistant) 59Fe-Tf fractions which were then counted on a LKB Wallac 1282 Compugamma universal γ counter (Turku, Finland). Parallel studies were conducted on the primary cultures of adult rat hepatocytes to assess the effects of Fe chelators on Fe uptake by the normal non-proliferating cell counterpart of hepatoma cells.
Toxicity
Toxicity was assessed by measuring the release of cytosolic aspartate aminotransferase (AST) from cells incubated with or without chelators, using the AST assay kit from Sigma. Cells in exponential phase of growth rate were incubated in MEM (pH 7.4) containing FCS (10% v v−1), penicillin (100 u ml−1), streptomycin (100 μg ml−1), fungizone (0.28 μg ml−1) and the chelator ([ ]=IC50 or 500 μM) for either 2 or 24 h. The incubation medium (efflux fraction) was collected, the cells were washed five times in cold BSS, and Pronase added (1 mg ml−1) to separate the membrane-bound and internalized fractions. The amount of AST in each fraction was measured according to the manufacturer's instructions (Sigma Chemical Co, U.S.A.).
Cell cycle analysis
Cells were analysed using flow cytometry (FACSCalibur® Becton-Dickinson, CA, U.S.A.) to measure the chelators' effects on the cell cycle. Cells were seeded and maintained in 25 cm2 flasks until they reached the exponential phase of growth. Once in this phase, cells were incubated with the chelators at their IC50 concentration or 500 μM for 24 h, under standard conditions. Cells were then harvested, washed once with PBS, and then fixed with 70% ethanol for 1 h on ice. Cells were washed again before Ribonuclease A (0.5 mg ml−1) in the presence of 0.2% Triton X-100 in H2O, was added and left to incubate at 37°C for 30 min. This was done in order to digest all RNA present and prevent any non-specific binding. Cells were finally stained with propidium iodide (50 μg/ml−1) for 10 min on ice, then filtered through a 100 μm cell strainer prior to analysis.
Statistical analysis
Unless stated otherwise, all results were compared using the Student's paired t-test at the 95% confidence level. Experiments were conducted at least twice and all values from the uptake experiments are the mean of triplicates within each experiment. IC50 concentrations were determined from two experiments, each containing eight replicates.
Results
Cell proliferation
DFT was the most effective chelator, consistently inhibiting cell proliferation in all hepatoma types, with an IC50 value of approximately 40 μM under these conditions (Table 2). In contrast its ferric form, FT, was only 10% as effective and inhibited cellular proliferation in only two of the cell lines (Table 2). It is worth noting that DFO was far less effective than DFT under the same conditions, and varied widely in activity between the different hepatomas, being most active in the fastest growing hepatoma, Q7. The other Fe chelator, MAL, was ineffective at inhibiting cell proliferation in any cell type (IC50>500 μM). However its Fe complex (MALF) showed slight activity (IC50=210 – 420 μM), presumably inhibiting cellular proliferation by a different mechanism than Fe chelation.
Table 2.
Effect of Fe chelators on hepatoma proliferation

The level of differentiation in the cell lines had little impact on the overall antiproliferative activity of DFT. Firstly, the effect of DFT on cellular proliferation was not species dependent, with similar antiproliferative effects in rat and human hepatoma cells (Table 2). Nor was the effect of the different carcinogens used to induce tumour growth in the rat (Table 1) an influential factor on antiproliferative activity (Table 2). Cell protein expression patterns and doubling times also differed markedly between the two rat HCC lines, but without effect on the chelators' activity.
Interestingly, DFO activity in the hepatoma cell lines was more varied than DFT. Like DFT, the antiproliferative activity of DFO was not species specific, however the proliferation rate was important with higher activity (Table 2) in the more rapidly proliferating Q7 cells (doubling time 14 h, Table 1) than in HTC, HepG2 or HuH7 cells (doubling times ≈20 h, Table 1). Since DFT was clearly the only chelator to have a marked effect on cell proliferation in all cell types, all subsequent experiments centred on this ligand to further investigate its mechanism of action.
Effect of DFT on cell survival: comparison with normal rat hepatocytes and fibroblasts
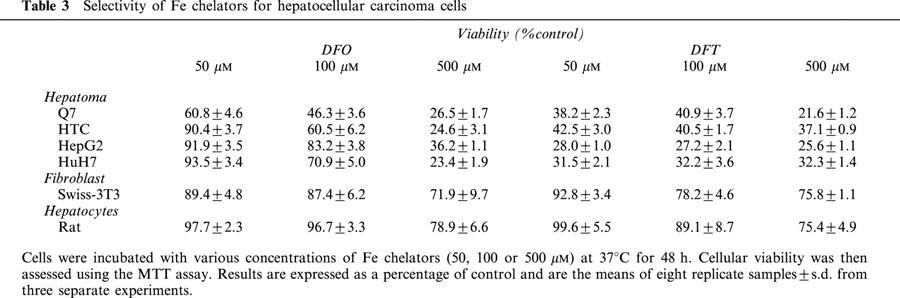
DFT caused a marked concentration-dependent decrease in hepatoma cell viability after 48 h exposure (Table 3) decreasing to 28 – 42% of control values at 50 μM. A much greater effect was observed when hepatoma cells were exposed to DFT than DFO, paralleling differences in their IC50 concentrations determined previously (Table 2). In contrast, when SWISS-3T3 fibroblasts were incubated with low concentrations (50 – 100 μM) of DFT and DFO over a similar period, cellular viability was only reduced slightly (78 – 93% of control). Similarly, viability of primary cultures of hepatocytes was also only decreased slightly to 89 – 99% of control at the same concentrations. When exposed to a very high concentration of either DFT or DFO (500 μM), the viability of the fibroblasts was still high (between 72 – 76% of control), being approximately 3 fold greater than the hepatoma viability (21 – 37%) (Table 3). Similarly, when hepatocytes were exposed to either DFT or DFO at the high concentration, viability was reduced to 75 – 78% of control, but still was markedly greater than that observed in hepatoma cells (Table 3).
Table 3.
Selectivity of Fe chelators for hepatocellular carcinoma cells
Effects of Fe chelators on Fe uptake
Kinetic studies were performed to determine whether a correlation existed between the inhibition of cellular survival and proliferation and inhibition of Fe uptake. Results obtained showed that DFT inhibited Fe uptake in all cells (Figure 2). Comparing the four cell lines tested, after 20 min exposure to DFT (500 μM), Fe uptake varied from 12 – 42% of control (P<0.05). In contrast, DFO (500 μM) had very little effect on Fe uptake over the same period. Over a 24 h period, DFT inhibited Fe uptake in a concentration-dependent manner (Figure 3), to around 20% of the control even at 50 μM. In contrast to its lack of activity over the short-term incubations, DFO over 24 h incubation also inhibited Fe uptake in a concentration-dependent manner, although much less than DFT.
Figure 2.

Effect of short-term incubation (up to 20 min) with DFO and DFT on Fe uptake. Cells were incubated with 59Fe-Tf ([Fe]=2.5 μM), at 37°C with either DFO or DFT at 500 μM and Fe internalization measured. Results are the means of triplicate samples±s.d. from two experiments and are expressed as a percentage of the control.
Figure 3.

Effect of long-term incubation (24 h) with DFO and DFT on Fe uptake. Cells were incubated with 59Fe-Tf ([Fe}=2.5 μM), over 24 h with 50, 100, or 500 μM of DFO or DFT. 59Fe internalization was measured and expressed as a percentage of the control. Each value is the mean of triplicate samples±s.d. from at least two experiments. #, † and * denote a significant difference at the 0.05, 0.01 and 0.001 confidence levels, respectively.
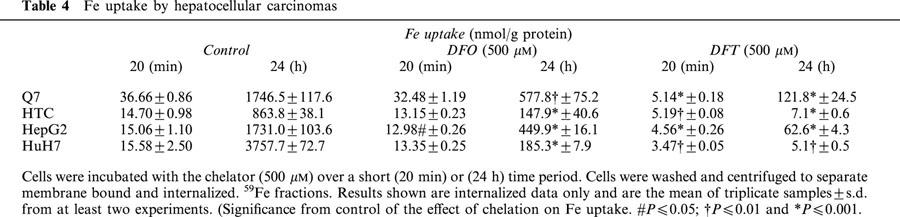
Fe uptake by the hepatoma cells over short-term incubations appeared to be inversely related to their proliferation rates, being greatest in Q7 hepatoma cells with the shortest doubling time (Table 4). However this was not the only factor since Fe uptake into all cell lines over a longer period of incubation (24 h) did not correspond to their doubling periods. For example despite having the fastest proliferation rate, Fe uptake into Q7 cells was lower at 24 h than that of HuH7 cells, which have a slower doubling period (Tables 1 and 4). In contrast, DFT was far more active than DFO, reducing Fe uptake between 70 – 85% over the short-term incubation (Table 4) and 83 – 100% when incubated over 24 h.
Table 4.
Fe uptake by hepatocellular carcinomas
When levels of inhibition estimated from the MTT assay and Fe uptake studies were compared (Figure 4), it was observed that generally the inhibition in proliferation with DFO correlated with a similar decrease in Fe uptake. This suggested that the antiproliferative effects of DFO were entirely associated with its ability to inhibit Fe uptake by cells (Figure 4). In contrast, the levels of inhibition of Fe uptake observed with DFT were consistently greater than the corresponding decrease in cell proliferation.
Figure 4.

Comparison of the effects of Fe chelators on cell proliferation and Fe uptake. Cells were incubated with the chelator (500 μM) and a MTT assay conducted after 48 h incubation and 59Fe internalization measured after 24 h. Results are the mean (of triplicate samples for Fe uptake or mean of eight samples from two separate experiments for MTT)±s.d. and are expressed as a percentage of controls. #, † and * denote a significant difference at the 0.05, 0.01 and 0.001 confidence levels, respectively.
Comparison of the effects of Fe chelators on Fe uptake by normal rat hepatocytes and hepatoma cells
To determine whether there was any selectivity for neoplastic cells in the activity of the chelators in relation to Fe uptake, hepatoma cells and hepatocytes were compared over 24 h. Fe uptake into hepatocytes as well as hepatomas was significantly affected by both DFT (P<0.001) and DFO (P<0.01) (Figure 5) with the inhibitory effect of DFT being much greater than that of DFO at all concentrations. At a low concentration (50 μM) the effect of DFT on Fe uptake in hepatocytes differed significantly from the rat hepatoma (P<0.01) but was comparable to the effects seen on the human hepatoma cell line. Effects of DFT at higher concentrations were more comparable. Interestingly, the effects of DFT on the rat and human HCC cell lines also differed significantly (P<0.01). DFO also decreased Fe uptake in a concentration dependent manner in both hepatocytes and hepatomas, with a greater effect being observed in the hepatocyte cultures at low concentrations (P<0.01) (Figure 5).
Figure 5.

The effect of DFO and DFT on Fe uptake by hepatocytes compared to hepatomas. Cells were incubated with the chelators at 50, 100 or 500 μM for 24 h. Internalized 59Fe was then measured and is expressed as a percentage of the control. Results are means±s.d. of triplicates from at least two experiments. #, † and * denote a significant difference at the 0.05, 0.01 and 0.001 confidence levels, respectively. (a) denotes the difference between the effect of chelators in hepatocytes and hepatomas.
Toxicity studies
The toxicity of DFT and DFO in HCC cells was determined by measuring AST released when cells were incubated with the chelators for 2 or 24 h at their IC50. AST release was also measured at 500 μM for comparison with other studies. Results showed that toxicity was dependent on both length of exposure and chelator concentration (data not shown). Toxicity was negligible when cells were incubated with DFO or DFT for 2 h or 24 h at the IC50 (data not shown). Toxicity was observed when cells were incubated with a high concentration of each chelator (500 μM), although more evident over 24 h than at 2 h incubation (data not shown). Overall the toxicity of DFO (in clinical use) and DFT were similar.
Cell cycle analysis
The effects of Fe chelators upon the cell cycle was assessed after incubating cells with the chelators at their IC50 μM (as shown in Table 2) over short (2 h) and long (24 h) periods. The effects of DFT as well as DFO on the cell cycle were time dependent. Over the 2 h incubation, both DFO and DFT had very little effect on cell cycle distribution (data not shown) despite being very effective at inhibiting Fe uptake at this time point. After 24 h incubation with DFT, the proportion of cells in G0 - G1 phase had decreased with a corresponding increase in the proportion of cells in the S phase (Table 5). This was less evident in HepG2 and HuH7 cells. Similarly when cells were exposed to DFO over 24 h, the proportion of cells in G0-G1 phase decreased, which corresponded to the increase in the proportion of cells in the S phase.
Table 5.
Effect of Fe chelator on the cell cycle

Discussion
Often characterized by a rapid growth rate, neoplastic cells require a number of different growth factors including Fe in order to proliferate. Hence restricting the supply of these factors e.g. Fe with the use of Fe chelators, may hold potential as a novel form of cancer therapy. The aim of this investigation was to examine the effects of one particular Fe chelator, DFT, for its potential anti-proliferative activity using hepatoma cells as an example of an aggressive cancer type. This compound is of interest as it has a similar affinity and specificity for Fe to DFO (which is in clinical use for the treatment of Fe overload disease and clinical trial as an antineoplastic agent), with the advantage of being more lipophilic and active when given orally (Baker et al., 1992b; Bergeron et al., 1993; Wolfe et al., 1989). From the results obtained in this study, DFT was found to have high antineoplastic activity and was significantly more active than DFO.
Results from this investigation suggest DFT to be a broad acting antineoplastic agent, inhibiting cell proliferation similarly in all hepatomas at a low concentration (IC50≈40 μM). The other chelators tested were either ineffective (MAL) at inhibiting cellular proliferation, or less effective (e.g. DFO) and showed more selective action to particular cell lines. The mechanism of action of DFT was in part related to its ability to deplete cells of Fe essential for proliferation, as FT is much less active. It is also important to note though that inhibition of Fe uptake did vary between the cell lines despite having similar effects on proliferation, indicating that the activity of DFT is only partly explained by its ability to bind to Fe and inhibit uptake. Interestingly, Fe complexes of both DFT and MAL (FT and MALF respectively) appeared to show slight activity, whether by general toxicity or some other mechanism (Baker et al., 1992b; Bergeron et al., 1994). In addition, a wide difference in activity of DFT and FT compared to that of MAL and MALF was observed. Since these latter chelators were only effective at the upper end of the concentration range examined, they were eliminated from further investigation. However, it is of interest that MAL has been shown to be much more active in neuroblastomas (Hironishi et al., 1996) than in HCC (Table 2).
Results obtained in this study show that DFT is more effective than DFO, consistently inhibiting growth of all tumour cells at a much lower concentration. In contrast, DFO inhibited cellular proliferation to a varying extent in the different cell lines with higher IC50 values of 110 – 210 μM determined. These correspond with estimates obtained (by extrapolation) by Hann et al. (1990) but are higher than those obtained by Voest et al. (1993) (IC50≈100 μM). Reports using hepatoma cell lines suggest a lower IC50 varying between 20 – 60 μM (Kim et al., 1993; 1994). DFO has also been reported to be active in vitro in neuroblastomas (IC50=2.5 – 50 μM) (Becton & Bryles, 1988; Brodie et al., 1993; Selig et al., 1993; Timeus et al., 1994), glioblastomas (IC50=10 – 20 μM) (Brodie et al., 1993; Renton & Jeitner, 1996), neuroepitheliomas (IC50=20 μM) (Timeus et al., 1994) and leukaemic cells (IC50=5 – 50 μM) (Bomford et al., 1986; Kaplinsky et al., 1987). In this study, the activity of DFT is significantly greater than DFO which has already been shown to be clinically effective, yet the level of activity of DFO (110 – 210 μM) is much higher than its activity reported in other investigations. This variation in absolute activity suggests that the MTT assay is fairly insensitive. It may also explain why DFT appears only to inhibit growth to 75%. It may also be possible that DFT is not a high potency growth inhibitor over a 48 h incubation but may require longer for a complete inhibition of growth. Further studies are required using a more sensitive clonogenic assay as well as measuring the effects of DFT on apoptosis and DNA synthesis in the different cell types.
To be an ideal chemotherapeutic agent, an Fe chelator should have little or no effect on normal cells. In this investigation selectivity of action of DFT for cancer cells was assessed using two criteria: cell survival and Fe uptake. Both DFT and DFO exhibited greater activity against hepatoma proliferation than the proliferation of a normal non-tumorigenic cell, normal fibroblasts. Other investigations have found DFO to be carcinoma-specific in that the antiproliferative activity of DFO was seen only in liver cancer cells and not in normal proliferating human diploid cell lines derived from embryonic lungs or newborn foreskin (WI-38 and MRHF respectively) (Blatt & Stitely, 1987; Hann et al., 1990). The antiproliferative activity observed in the hepatoma cells could not be directly compared to that of their normal cell counterpart due to the very slowly proliferating nature of the normal adult hepatocyte. However, the viability of primary cultures of rat hepatocytes was only slightly reduced even when incubated over long periods with either DFT or DFO. Whilst the exact cause of DFT's apparent selectivity for tumorigenic cells over normal cells remains uncertain, the fact the ribonucleotide reductase (an Fe-containing enzyme required for the conversion of ribonucleotides into deoxynucleotides for DNA synthesis) shows the greatest increase in activity in tumour cells compared to normal cells, may partly explain the greater sensitivity of neoplastic cells to Fe chelators in general (Chitambar & Seligman, 1986; Cory et al., 1985; Weber, 1977; Witt et al., 1979).
From this study it was observed that DFT lacked any selectivity for hepatomas over hepatocytes with respect to Fe uptake. Although not identical, the extremely significant inhibitory effects of DFT observed on hepatoma Fe uptake was paralleled in normal rat hepatocytes, under similar conditions. DFO also inhibited Fe uptake in both hepatocytes and hepatomas, although a greater effect was observed in hepatocytes. Despite this hepatocyte viability was only slightly reduced. Although not an ideal Fe chelator, DFO is used clinically long-term to treat iron overload, despite its limitations. Short-term chemotherapeutic regimens have been associated with transient increases in serum F3 levels (Bierer & Nathan, 1990). Administration of DFT, DFO or any other Fe chelator over similar periods should not significantly reduce body Fe stores resulting in clinically important manifestations. Whilst to our knowledge attainable plasma levels of DFT have not been measured, numerous studies have measured marked increases in Fe clearance in its presence in vivo. Although its level of chronic toxicity is unacceptable, DFT has been shown to be a highly effective ligand at clearing Fe from rodents and primates when administered orally (Bergeron et al., 1990; 1994).
Signs of toxicity from any compound with potential clinical application are of major concern. This aspect was addressed by measuring the effect of the chelators on the proportion of the intracellular enzyme AST released into the culture media. Results showed that any observed toxicity was determined by the cell type, length of exposure and chelator concentration. Previous in vitro studies found DFT to be cytotoxic in, for example, peripheral blood mononuclear cells, above concentrations of 350 μM (Stahel et al., 1988). Baker et al. (1992b) observed that DFT was only toxic to rat hepatocytes at very high concentrations (>500 μM). At these concentrations, DFT caused membrane disruption of rat hepatocytes resulting in the release of intracellular AST. A similar study by Stahel et al. (1988) found no signs of DFT toxicity in cultured rat hepatocytes using a concentration range between 50 – 200 μM. It is important to note that toxicity in this study was only observed at concentrations far above current clinically acceptable and attainable levels. However in a previous long-term in vivo study (Bergeron et al., 1993), DFT given orally at a dose of 150 μmol kg−1 per day over a 10 day period to rats was found to be nephrotoxic. This and a subsequent study (Bergeron et al., 1996) suggested that both concentration and length of exposure to DFT were critical factors influencing toxicity. Results from this study confirm this finding and need to be addressed when DFT is assessed in vivo for antineoplastic activity.
Preliminary findings made in this investigation examining the effects of chelators on the cell cycle revealed that DFO inhibited DNA synthesis in the S phase. DFT blocked cell cycle progression in a similar fashion. Other investigations have found that DFO prevents progression from the early S phase of the cell proliferation cycle in several cell types (Brodie et al., 1993; Dezza et al., 1989; Kim et al., 1994; Lederman et al., 1984; Nocka & Pelus, 1988; Renton & Jeitner, 1996). Both chelators may act by inactivating intracellular Fe pool(s) essential for the function of ribonucleotide reductase, which is rate limiting in DNA synthesis. The changes in S phase distribution do not fully parallel results obtained in the cell proliferation studies and may be due to timing differences in experimental protocol, since growth inhibition (and Fe uptake) studies were estimated at 48 h whereas effects on cell cycle was conducted at 24 h.
In summary, this investigation has shown DFT to be a highly effective antiproliferative agent, which functions similarly to DFO but is much more potent. Although less active than some other chelators currently under examination (Darnell & Richardson, 1999; Richardson et al., 1995; Richardson & Milnes, 1997), the large gap between the concentration of DFT showing effective antineoplastic activity and the concentration causing a decrease in viability in normal cells suggests further investigation into DFT's antineoplastic activity in vivo is warranted. In addition, analogues of DFT are being assessed. Bergeron et al. (1999a, 1999b, 1999c) in a series of studies have shown that specific deletions and substitutions on the DFT molecule can alter its efficacy and toxicity in relation to its use in Fe overload diseases. Hence DFT's activity as an antineoplastic agent may be enhanced further with manipulation of its chemical structure, as currently observed with PIH and its analogues (Darnell & Richardson, 1999; Richardson et al., 1995; Richardson & Milnes, 1997).
Acknowledgments
Dr Matthew Wikstrom is thanked for skilful technical assistance using the flow cytometer. We would also like to thank Ms Sharyn Baker for technical assistance and Dr Elizabeth Kicic-Starcevich for constructive criticism. This work was supported by a grant from the National Health and Medical Research Council of Australia (961344).
Abbreviations
- AST
aspartate amino transferase
- BSA
bovine serum albumin
- BSS
balanced salt solution
- DFO
desferrioxamine
- DFT
desferrithiocin
- FCS
foetal calf serum
- FT
ferrithiocin
- g
gram
- h
hours
- HCC
hepatocellular carcinoma
- IC50
inhibitory concentration at 50%
- MAL
maltol
- MALF
ferric complex of maltol
- MEM
minimum essential medium
- min
minutes
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium
- μM
micromolar
- Tf
transferrin
References
- ADEN D., FOGEL A., PLOTKIN S., DAMJANOV I., KNOWLES B. Controlled synthesis of HbsAg in a differentiated human liver carcinoma-derived cell line. Nature. 1979;282:615–616. doi: 10.1038/282615a0. [DOI] [PubMed] [Google Scholar]
- ANDEREGG G., RABER M. Metal complex formation of a new siderophore desferrithiocin and of three related ligands. J. Chem. Soc. Chem. Commun. 1990;17:1194–1196. [Google Scholar]
- BAKER E., RICHARDSON D.R., GROSS S., PONKA P. Evaluation of the iron chelation potential of hydrazones of pyridoxal, salicylaldehyde and 2-hydroxy-1-napththylaldehyde using the hepatocyte in culture. Hepatology. 1992a;15:492–501. doi: 10.1002/hep.1840150323. [DOI] [PubMed] [Google Scholar]
- BAKER E., WONG A., PETER H., JACOBS A. Desferrithiocin is an effective iron chelator in vivo and in vitro but ferrithiocin is toxic. Br. J. Haematol. 1992b;81:424–431. doi: 10.1111/j.1365-2141.1992.tb08251.x. [DOI] [PubMed] [Google Scholar]
- BECTON D.L., BRYLES P. Desferrioxamine inhibition of human neuroblastoma viability and proliferation. Cancer Res. 1988;48:7189–7192. [PubMed] [Google Scholar]
- BERGERON R.J., STREIFF R.R., WIEGAND J., VINSON T., LUCHETTA G., EVANS K.M., PETER H., HANS-BEAT J. A comparative evaluation of iron clearance models. Ann. N.Y. Acad. Sci. 1990;612:378–393. doi: 10.1111/j.1749-6632.1990.tb24325.x. [DOI] [PubMed] [Google Scholar]
- BERGERON R.J., STREIFF R.R., CREARY E.A., DANIELS R.D., JR, KING W., LUCHETTA G., WIEGAND J., MOERKER T., PETER H. A comparative study of the iron-clearing properties of desferrithiocin analogues with desferrioxamine B in a Cebus monkey model. Blood. 1993;81:2166–2173. [PubMed] [Google Scholar]
- BERGERON R.J., WOLLENWEBER M., WEIGAND J. An investigation of desferrithiocin metabolism. J. Med. Chem. 1994;37:2889–2895. doi: 10.1021/jm00044a009. [DOI] [PubMed] [Google Scholar]
- BERGERON R.J., WIEGAND J., WOLLENWEBER M., MCMANIS J.S., ALGEE S.E., RATIFF-THOMPSON K. Synthesis and Biological evaluation of napthyldesferrithiocin iron chelators. J. Med. Chem. 1996;39:1575–1581. doi: 10.1021/jm9508752. [DOI] [PubMed] [Google Scholar]
- BERGERON R.J., MCMANIS J.S., BUSSENIUS J., BRITTENHAM G.M., WEIGAND J. Evaluation of the desferrithiocin pharmacophore as a vector for hydroxamates. J. Med. Chem. 1999a;42:2281–2286. doi: 10.1021/jm980611q. [DOI] [PubMed] [Google Scholar]
- BERGERON R.J., WEIGAND J., MCMANIS J.S., MCCOSAR B.H., WEIMAR W.R., BRITTENHAM G.M., SMITH R.E. Effects of C-4 stereochemistry and C-4′ hydroxylation on the iron clearing efficiency and toxicity of desferrithiocin analogues. J. Med. Chem. 1999b;42:2432–2440. doi: 10.1021/jm990058s. [DOI] [PubMed] [Google Scholar]
- BERGERON R.J., WEIGAND J., WEIMAR W.R., VINSON T.J.R., BUSSENIUS J., YAO G.W., MCMANIS J.S. Desazadesmethyldesferrithocin analogues as orally effective iron chelators. J. Med. Chem. 1999c;42:95–108. doi: 10.1021/jm980340j. [DOI] [PubMed] [Google Scholar]
- BIERER B.E., NATHAN D.G. The effect of desferrithiocin, an oral iron chelator, on T-cell function. Blood. 1990;76:2502–2509. [PubMed] [Google Scholar]
- BLATT J., STITELY S. Antineuroblastoma activity of desferrioxamine in human cell lines. Cancer Res. 1987;47:1749–1750. [PubMed] [Google Scholar]
- BLATT J., TAYLOR S.R., STITELY S. Mechanisms of antinueoroblastoma activity of desferrioxamine in vitro. J. Lab. Clin. Med. 1988;112:433–436. [PubMed] [Google Scholar]
- BOMFORD A., ISAAC J., ROBERTS S., EDWARDS A., YOUNG S., WILLIAMS R. The effect of desferrioxamine on transferrin receptors, the cell cycle and growth rates of human leukaemic cells. Biochem. J. 1986;236:243–249. doi: 10.1042/bj2360243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRODIE C., SIRIWARDANA G., LUCAS J., SCHLEICHER R., TERADA N., SZAPESI A., GELFAND E., SELIGMAN P. Neuroblastoma sensitivity to growth inhibition by desferrioxamine: evidence for a block in G1 phase of the cell cycle. Cancer Res. 1993;53:3968–3975. [PubMed] [Google Scholar]
- CHENOUFI N., DRENOU B., LOREAL O., PIGEON C., BRISSOT P., LESCOAT G. Antiproliferative effect of deferiprone on the HepG2 cell line. Biochem. Pharmacol. 1998;56:431–437. doi: 10.1016/s0006-2952(98)00071-9. [DOI] [PubMed] [Google Scholar]
- CHITAMBAR C.R., SELIGMAN P.A. Effects of different transferrin forms on transferrin receptor expression, Iron uptake and cellular proliferation of human leukaemic HL60 cells. J. Clin. Invest. 1986;78:1538–1546. doi: 10.1172/JCI112746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORY J.G., CORY A.H., RAPPA G., LORICO A., LIU M.C., LIN T.S., SANTORELLI A.C. Structure-function relationships for a new series of pyridine-2-carboxaldehyde thiosemicarbazones on ribonucleotide reductase activity and tumour cell growth in culture and in vivo. Advan. Enzyme Regul. 1985;35:55–68. doi: 10.1016/0065-2571(94)00005-n. [DOI] [PubMed] [Google Scholar]
- CRAGG L., HEBBEL R.P., MILLER W., SOLOVEY A., SELBY S., ENRIGHT H. The iron chelator L1 potentiates oxidative DNA damage in iron overloaded liver cells. Blood. 1998;92:262–268. [PubMed] [Google Scholar]
- DARNELL G., RICHARDSON D.R. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents III: the effects of the ligands on molecular targets involved in proliferation. Blood. 1999;94:781–792. [PubMed] [Google Scholar]
- DEZZA L., CAZZOLA M., DANOVA M., CARLO-STELLA C., BERGAMASCHI G., BRUGNATELLI S., INVERNIZZI R., MAZZINI G., RICCARDI A., ASCARI E. Effects of desferrioxamine on normal and leukaemic human haematopoietic cell growth: in vitro and in vivo studies. Leukemia. 1989;3:104–107. [PubMed] [Google Scholar]
- DONFRANCESCO A., DEB G., DOMINICI C., PILEGGI D., CASTELLO M.A., HELSON L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990;50:4929–4930. [PubMed] [Google Scholar]
- DONFRANCESCO A., DEB G., DOMINICI C., ANGIONI A., CANIGLIA M., DE SIO L., FIDANI P., AMICI A., HELSON L. Deferoxamine, cyclophosphamide, etoposide, carboplatin, and thioteps (D-CE-CaT): A new cytoreductive chelation-chemotherapy regimen in patients with advanced neuroblastoma. Am. J. Clin. Oncol. 1992;15:319–322. doi: 10.1097/00000421-199208000-00009. [DOI] [PubMed] [Google Scholar]
- DONFRANCESCO A., DEB G., ANGIONI A., MAURIZIO C., COZZA R., JENKNER A., LANDOLFO A., BOGLINO C., HELSON L. D-CEaT: a breakthrough for patients with neuroblastoma. Anti-Cancer Drugs. 1993;4:317–321. [PubMed] [Google Scholar]
- DONFRANCESCO A., DEBERNARDI B., CARLI M., MANCINI A., NIGRO M., DE SIO L., CASALE F., BAGNULO S., HELSON L., DEB G. Deferoxamine (D) followed by cytoxan (C), etoposide (E), carboplatin (Ca), thio-TEPA (T), induction regimen in advanced neuroblastoma: Preliminary results. Eur. J. Cancer. 1995;31A:612–615. doi: 10.1016/0959-8049(95)00068-t. [DOI] [PubMed] [Google Scholar]
- HANKS J.H., WALLACE R.E. Relation of oxygenation and temperature in preservation of tissues by refrigeration. Proc. Soc. Exp. Biol. Med. 1949;71:196–200. doi: 10.3181/00379727-71-17131. [DOI] [PubMed] [Google Scholar]
- HANN H.W., STAHLHUT M.W., HANN C.L. Effect of iron and desferrioxamine on cell growth and in vitro ferritin synthesis in human hepatoma cell lines. Hepatology. 1990;11:566–569. doi: 10.1002/hep.1840110407. [DOI] [PubMed] [Google Scholar]
- HEMMAPLARDH D., MORGAN E.H. The role of endocytosis in the transferrin uptake by reticulocytes and bone marrow cells. Br. J. Haematol. 1977;36:85–96. doi: 10.1111/j.1365-2141.1977.tb05758.x. [DOI] [PubMed] [Google Scholar]
- HILETI D., PANOYIOTIDUS P., HOFFBRAND A.V. Iron chelators induce apoptosis in proliferating cells. Br. J. Haematol. 1995;89:181–187. doi: 10.1111/j.1365-2141.1995.tb08927.x. [DOI] [PubMed] [Google Scholar]
- HIRONISHI M., RADZISLAW K., YANAGIHARA R., GARRUTO R. Maltol (3-hydroxy-2-methyl-4-pyrone) toxicity in neuroblastoma cell lines and primary murine fetal hippocampal neuronal cultures. Neurodegeneration. 1996;5:325–329. doi: 10.1006/neur.1996.0044. [DOI] [PubMed] [Google Scholar]
- HOFFBRAND A.V. Prospects for oral iron chelation therapy. J. Lab. Clin. Med. 1994;123:492–494. [PubMed] [Google Scholar]
- KAPLINSKY C., ESTROV Z., FREEDMAN M.H., GELFAND E.W., COHEN A. Effect of desferrioxamine on DNA synthesis, DNA repair, cell proliferation, and differentiation of HL-60 cells. Leukemia. 1987;1:437–441. [PubMed] [Google Scholar]
- KIM W.H., CHON C.Y., MOON Y.M., KANG J.K., PARK I.S., CHOI H.J. Effect of anticancer drugs and desferrioxamine in combination with radiation on hepatoma cell lines. Yonesi Medical Journal. 1993;34:45–56. doi: 10.3349/ymj.1993.34.1.45. [DOI] [PubMed] [Google Scholar]
- KIM D.Y., KIM W.H., KANG J.K., PARK I.S., KWON O.H. The mechanism of antiproliferative effect of desferrioxamine on human hepatoma cell lines. Yonesi Medical Journal. 1994;35:62–71. doi: 10.3349/ymj.1994.35.1.62. [DOI] [PubMed] [Google Scholar]
- LAU W.Y., LEOW C.K., LI A.K.C. Hepatocellular carcinoma. Br. J. Hos. Med. 1997;57:101–104. [PubMed] [Google Scholar]
- LEDERMAN H.M., COHEN A., LEE J.W.W., FREEDMAN M.H., GELFAND E.W. Deferoxamine: A reversible S-phase inhibitor of human lymphocyte proliferation. Blood. 1984;64:748–753. [PubMed] [Google Scholar]
- LONGUEVILLE A., CRICHTON R.R. An animal model of iron overload and its application to study hepatic ferritin iron mobilization by chelators. Biochem. Pharmacol. 1986;35:3669–3678. doi: 10.1016/0006-2952(86)90650-7. [DOI] [PubMed] [Google Scholar]
- MOSMANN T. Rapid colorimetric assay for cellular growth and survival: application for proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- NAKABAYSHI H., TAKETA K., MIYANO K., YAMANE T., SATO J. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
- NASTRUZZI P., MENEGATTI E., GAMBARI R. Differential effects of liposomes-entrapped desferrioxamine on proliferation and erythroid differentiation of murine Erythroleukemia Friend cells. Biochim. Biophys. Acta. 1989;1013:36–41. doi: 10.1016/0167-4889(89)90124-9. [DOI] [PubMed] [Google Scholar]
- NOCKA K.H., PELUS L. Cell cycle specific effects of deferoxamine on human and murine haematopoietic progenitor cells. Cancer Res. 1988;48:3571–3575. [PubMed] [Google Scholar]
- PETER H.H.Industrial aspects of iron chelators: pharmaceutical applications Proteins of Iron Storage and Transport 1985Elsevier Science Publishers. Biomedical Division; 293ed Spik, G., Montreuil, J., Crichton, R.R., Mazurier, J. pp [Google Scholar]
- RENTON F., JEITNER T.M. Cell cycle-dependent inhibition of the proliferation of human neural tumour cell lines by iron chelators. Biochem. Pharmacol. 1996;51:1553–1561. doi: 10.1016/0006-2952(96)00099-8. [DOI] [PubMed] [Google Scholar]
- RICHARDSON D.R., PONKA P., BAKER E. The effects of the iron (III) chelator, desferrioxamine, on iron and transferrin uptake by the human malignant melanoma cell. Cancer Res. 1994;54:685–689. [PubMed] [Google Scholar]
- RICHARDSON D.R., TRAN E.H., PONKA P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood. 1995;86:4295–4306. [PubMed] [Google Scholar]
- RICHARDSON D.R., MILNES K. The potential of iron chelators of the pyridoxal isonicotonoyl hydrazone class as effective iron chelators II: the mechanism of action of ligands derived from salicylaldehyde benzoyl hydrazone and 2-hydroxy-1-napththylaldehyde benzoyl hydrazone. Blood. 1997;89:3025–3038. [PubMed] [Google Scholar]
- RUSTGI V.K. Epidemiology of hepatocellular carcinoma. Ann. Intern. Med. 1998;108:390–401. doi: 10.7326/0003-4819-108-3-390. [DOI] [PubMed] [Google Scholar]
- SCHAFER D.F., SORRELL M.F. Hepatocellular carcinoma. The Lancet. 1999;353:1253–1257. doi: 10.1016/S0140-6736(98)09148-X. [DOI] [PubMed] [Google Scholar]
- SELIG R.A., MADAFIGLIO J., HABER M., NORRIS M.D., WHITE L., STEWART B.W. Ferritin production and desferrioxamine cytotoxicity in human neuroblastoma cell lines. Anticancer Res. 1993;13:721–726. [PubMed] [Google Scholar]
- STAHEL E., MAZIER D., GUILLOUZO A., MILTGEN F., LANDAU I., MELLOUK S., BEAUDOIN R.L., LANGLOIS P., GENTILINI M. Iron chelators in vitro inhibitory effect on the liver stage of rodent and human malaria. Am. J. Trop. Med. Hyg. 1988;39:236–240. doi: 10.4269/ajtmh.1988.39.236. [DOI] [PubMed] [Google Scholar]
- TIMEUS F., VALLE P., CRESCENZIO N., RUGGIERI L., ROSSO P., PAGLIARDI G.L., CORDERO DI MONTEZEMOLO L., GABUTTI V., RAMENGHI U. Effect of desferrioxamine and hydroxypyridones on haemopoietic progenitors and neuroectodermal tumour cells. Am. J. Hematol. 1994;47:183–186. doi: 10.1002/ajh.2830470307. [DOI] [PubMed] [Google Scholar]
- TORTI S., TORTI F., WHITMAN S., BRECHBIEL M., PARK G., PLANALP R. Tumour cell cytotoxicity of a novel metal chelator. Blood. 1998;92:1384–1389. [PubMed] [Google Scholar]
- VOEST E.E., ROOTH H., NEIJT J.P., VAN ASBECK B.S., MARX J.J.M. The in vitro response of human tumour cells to desferrioxamine is growth medium dependent. Cell. Prolif. 1993;26:77–88. doi: 10.1111/j.1365-2184.1993.tb00008.x. [DOI] [PubMed] [Google Scholar]
- WEBER L. ENZYMOLOGY OF CANCER CELL. N. Eng. J. Med. 1977;293:486–492. doi: 10.1056/NEJM197703032960905. [DOI] [PubMed] [Google Scholar]
- WITT L., YAP T., BLAKLEY R.L. Regulation of ribonucleotide reductase activity and its possible exploitation in chemotherapy. Advan. Enzyme Regul. 1979;17:157–171. doi: 10.1016/0065-2571(79)90012-8. [DOI] [PubMed] [Google Scholar]
- WOLFE L.C., NICOLOSI R.J., RENAUD M.M., FINGER J., PETER H.H., NATHAN D.G. A non human primate model for the study of oral iron chelators. Br. J. Haematol. 1989;72:456–461. doi: 10.1111/j.1365-2141.1989.tb07732.x. [DOI] [PubMed] [Google Scholar]