

Abstract
The effects of treatment with a number of cyclo-oxygenase inhibitors, (celecoxib, meloxicam, DuP-697 and aspirin) on ischaemia-reperfusion-induced myocardial dysfunction were examined using an in vitro perfused rabbit heart model.
Ischaemia resulted in myocardial dysfunction, as indicated by a significant increase in left ventricular end diastolic pressure and marked changes in coronary perfusion pressure and left ventricular developed pressure. In the post-ischaemic state, coronary perfusion pressure increased dramatically, left ventricular developed pressure recovered to a small degree and there were significant increases in creatinine kinase release (indicative of myocardial damage) and prostacyclin release.
Pretreatment with aspirin, or with drugs that selectively inhibit cyclo-oxygenase-2 (celecoxib, meloxicam and DuP-697), resulted in a concentration-dependent exacerbation of the myocardial dysfunction and damage. Exacerbation of myocardial dysfunction and damage was evident with 10 μM concentrations of the cyclo-oxygenase-2 inhibitors, which inhibited prostacyclin release but did not affect cyclo-oxygenase-1 activity (as measured by whole blood thromboxane synthesis).
NCX-4016, a nitric oxide-releasing aspirin derivative, significantly reduced the myocardial dysfunction and damage caused by ischaemia and reperfusion. Beneficial effects were observed even at a concentration (100 μM) that significantly inhibited prostacyclin synthesis by the heart.
The results suggest that prostacyclin released by cardiac tissue in response to ischaemia and reperfusion is derived, at least in part, from cyclo-oxygenase-2. Cyclo-oxygenase-2 plays an important protective role in a setting of ischaemia-reperfusion of the heart.
Keywords: Prostaglandin, cardiovascular, nitric oxide, reperfusion, anti-inflammatory
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are among the most commonly used drugs, primarily for their anti-inflammatory and analgesic properties. In addition to their significant toxicity in the gastrointestinal tract, NSAIDs have well documented deleterious effects on the kidney. There are conflicting reports regarding the effects of NSAIDs in various models of acute myocardial ischaemia (Ogletree & Lefer, 1976; Jugdutt et al., 1979). For example, ibuprofen has been reported to reduce infarct size in canine hearts (Kirlin et al., 1982), while indomethacin has been reported to induce coronary vasoconstriction in patients with coronary artery disease (Friedman et al., 1981). Moreover, aspirin, which inhibits platelet aggregation, does not improve exercise tolerance, change the pain threshold, or alter ischaemic electrocardiographic abnormalities in patients with angina pectoris (Frishman et al., 1976). Berti et al. (1988) and Rossoni et al. (2000) have reported that indomethacin and aspirin, respectively, aggravated ischaemia-induced ventricular dysfunction in perfused rabbit hearts and this was associated with inhibition of prostacyclin (PGI2) synthesis in the cardiac tissues. In contrast, exposure of the hearts to a nitric oxide-releasing derivative of aspirin, NCX-4016, resulted in a significant reduction of cardiac dysfunction. The cardioprotective effects of NCX-4016 were concentration-dependent and most likely mediated via the nitric oxide generated by this compound (Rossoni et al., 2000).
In recent years a new class of NSAIDs has been developed, namely the selective cyclo-oxygenase (COX)-2 inhibitors. These agents exhibit anti-inflammatory and analgesic properties with reduced toxicity in the gastrointestinal tract (Bombardier et al., 2000). However, selective COX-2 inhibitors have recently been shown to markedly reduce whole body PGI2 synthesis in healthy human volunteers (McAdam et al., 1999; Catella-Lawson et al., 1999), to significantly elevate systemic blood pressure in rats and to elicit a significant increase in leukocyte adherence to the vascular endothelium (Muscara et al., 2000). Moreover, there have been a number of recent clinical reports of selective COX-2 inhibitors increasing the incidence of myocardial infarction relative to that seen with a conventional NSAID (Bombardier et al., 2000; Mukherjee et al., 2001) or promoting thrombosis (Crofford et al., 2000).
COX-2 has been shown to be up-regulated in the myocardium of patients with congestive heart failure (Wong et al., 1998) and in the stomach of rats during ischaemia (Maricic et al., 1999). This suggests that up-regulation of COX-2 may occur as a defensive response, aimed at increasing the generation of vasodilatory prostaglandins. In the stomach, treatment of rats with a selective COX-2 inhibitor during a period of ischaemia resulted in a marked exacerbation of tissue injury (Maricic et al., 1999). It is possible that a similar phenomenon may occur in the context of myocardial ischaemia-reperfusion. Thus, in the present study, we have tested the hypothesis that selective COX-2 inhibitors will exacerbate the myocardial dysfunction that occurs as a consequence of ischaemia-reperfusion in an in vitro perfused rabbit heart model. We have compared the effects of exposure of the heart, prior to the period of ischaemia, to a number of drugs with varying degrees of selectivity for COX-2. For comparison, we have also assessed the effects of exposure of the heart to the nitric oxide-releasing aspirin derivative, NCX-4016.
Methods
The experimental procedures were approved by the Animal Care Committees of the University of Milan and the University of Calgary and the studies were performed in accordance with the principles set forth in the Italian and Canadian guidelines for the care and use of laboratory animals.
Ischaemia-reperfusion in isolated rabbit heart
Male, New Zealand White rabbits (BMG-Allevamento, Cividate al Piano, BG, Italy or Reimans Fur Ranches, Calgary, AB, Canada) weighing 2.0 – 2.2 kg were used for these experiments. The hearts were excized and perfused retrogradely at 37°C through the aorta as previously described by Berti et al. (1988) and Rossoni et al. (2000). The perfusion medium (Krebs Henseleit) contained (in mM): NaCl 118, KCl 2.8, KH2PO4 1.2, CaCl2 2.5, MgSO4 1.2, NaHCO3 25 and glucose 5.5. After a period of equilibration with a 5% CO2 and 95% O2 gas mixture, the pH of the perfusate was 7.4. The rate of perfusion was maintained at 20 ml min−1 with a roller pump (Minipuls-3, Gilson, Villiers-Le Bel, France). Coronary perfusion pressure (CPP) and left ventricular pressure (LVP) were measured with two HP-1280C pressure transducers (Hewlett-Packard, Waltham, MA, U.S.A.) connected to a Hewlett-Packard dynograph (HP-7754A). LVP was recorded with a polyethylene catheter (with a small latex balloon on the top) inserted in the left ventricular cavity. The balloon was filled slowly with saline until left ventricular end-diastolic pressure (LVEDP) stabilized in the range of 4 – 6 mmHg. Left ventricular developed pressure (LVDP: peak left ventricular systolic pressure minus LVEDP) was also evaluated. The hearts were electrically paced at a frequency of 180 beats min−1 with rectangular impulses (1 ms duration, voltage 10% above threshold) by a Grass stimulator (model S-88; Grass Instruments, Quincy, MA, U.S.A.). Ischaemia was induced by reducing the flow rate from 20 to 1 ml min−1 for 40 min (ischaemic period). A normal flow rate (20 ml min−1) was then restored and the perfusion was continued for another 20 min (reperfusion period).
Each of the NSAIDs was tested at concentrations of 1, 10 and 100 μM (n=6 – 10 per group). The test drugs included three that exhibit selectivity for COX-2: celecoxib (Silverstein et al., 2000), DuP-697 (Gans et al., 1990) and meloxicam (Engelhardt et al., 1995); a non-selective COX inhibitor (aspirin) and the nitric oxide-releasing aspirin derivative, NCX 4016 (Wallace et al., 1999). One of the test drugs, or vehicle, was perfused through the hearts for 20 min before the start of the period of ischaemia.
Prostacyclin release and creatine kinase activity
The release of the PGI2 into the perfusate was measured by determining the concentrations of its stable hydration production, 6-keto-prostaglandin F1α (6-keto-PGF1α), using an enzyme-linked immunosorbent assay (ELISA) (Berti et al., 1993). The perfusates were collected for 5 min immediately before the period of ischaemia and during the first 10 min of reperfusion. Creatine kinase (CK) activity was measured in samples (taken during the ischaemia and reperfusion periods) using the spectrophotometric method of Bergmeyer et al. (1970).
Whole blood thromboxane synthesis
The synthesis of thromboxane by whole blood was determined using the method of Patrono et al. (1980). Blood was drawn from the descending aorta of rabbits into a syringe. The blood was transferred into glass tubes (1 ml per tube) to which the test drugs were added to give final concentrations of 1, 10 or 100 μM. The tubes of blood were allowed to stand at 37°C for 45 min, after which they were centrifuged (1000 g; 10 min). The serum was transferred to Eppendorf tubes then frozen at −20°C. Thromboxane B2 concentrations in the serum samples were measured using a specific enzyme-linked immunosorbent assay, according to the manufacturer's instructions (Wallace et al., 1998).
Data analysis
Multiple group comparisons were made using analysis of variance (ANOVA) and the Dunnett's multiple comparison test. Comparisons of only two groups were made using the Student's t-test with the Bonferroni correction. An associated probability (P value) of less than 5% was considered significant. In all tables and figures, results are expressed as mean±s.e.mean. Area under the curve (AUC) was estimated according to the trapezoid method (Microcal Software Inc., Northampton, MA, U.S.A.).
Materials
NCX-4016 was obtained from NicOx S.A. (Sophia Antipolis, France), celecoxib from Monsanto (St. Louis, MO, U.S.A.), DuP-697 from DuPont-Merck (Dover, DE, U.S.A.), aspirin from Sigma Chemical Co. (St. Louis, MO, U.S.A.) and meloxicam from Boehringer-Ingelheim (Danbury, CT, U.S.A.). The ELISA kits for determination of 6-keto-PGF1α and thromboxane B2 were obtained from Amersham Italia (Milan, Italy) and Medicorp (Montreal, Canada), respectively. The kits for creatine kinase determination were obtained from Boehringer-Mannheim (Milan, Italy). NCX-4016 and aspirin were initially dissolved in dimethylsulphoxide, then diluted in Krebs Henseleit solution (final concentration of dimethylsulphoxide was <1%). All other drugs were dissolved directly in the Krebs Henseleit solution.
Results
Effects of ischaemia-reperfusion
The reduction of the perfusion flow-rate of electrically paced isovolumic left heart preparations for 40 min resulted in a progressive increase in left ventricular end diastolic pressure (Figure 1). During reperfusion, left ventricular developed pressure was significantly reduced (Figure 2) and coronary perfusion pressure (CPP) increased consistently above baseline (Figure 3). Moreover, there was a marked increase in creatine kinase (CK) activity in the cardiac perfusates during the reperfusion period, reaching a maximum at 45 – 50 min of the reperfusion (Figure 4). In the pre-ischaemic period, 6-keto PGF1α was detectable in the perfusates (mean release of 2.7±0.2 ng min−1). During the first 10 min of the reperfusion period, the release of 6-keto PGF1α increased approximately 3 fold (mean release of 8.7±0.8 ng min−1).
Figure 1.
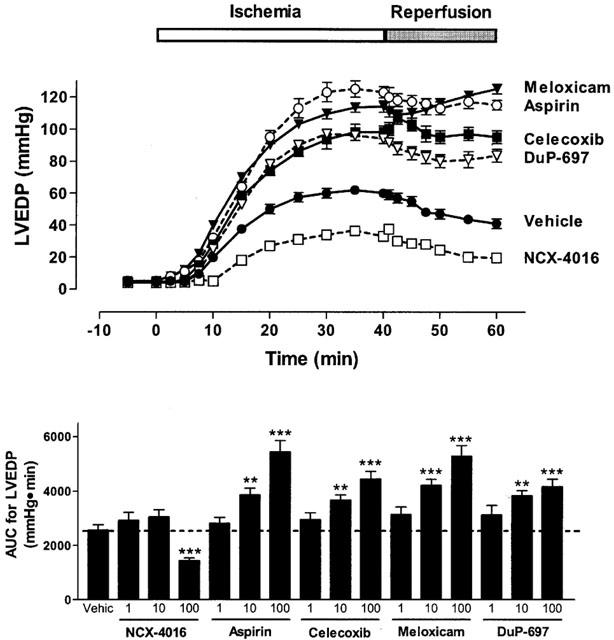
Effect of cyclo-oxygenase inhibitors on left ventricular end-diastolic pressure (LVEDP) in paced isovolumic left heart preparations subjected to low-flow ischaemia (1 ml min−1 for 40 min) and reperfusion (20 ml min−1 for 20 min). The compounds were infused for 20 min before reduction of flow. The upper panel shows the effect of the 100 μM concentration of each drug. The lower panel shows the area-under-the-curve (AUC) for all three concentrations of each test drug. *P<0.05, **P<0.01, ***P<0.001 versus the vehicle-treated group. Each point/bar represents the mean±s.e.mean of 6 – 10 experiments.
Figure 2.
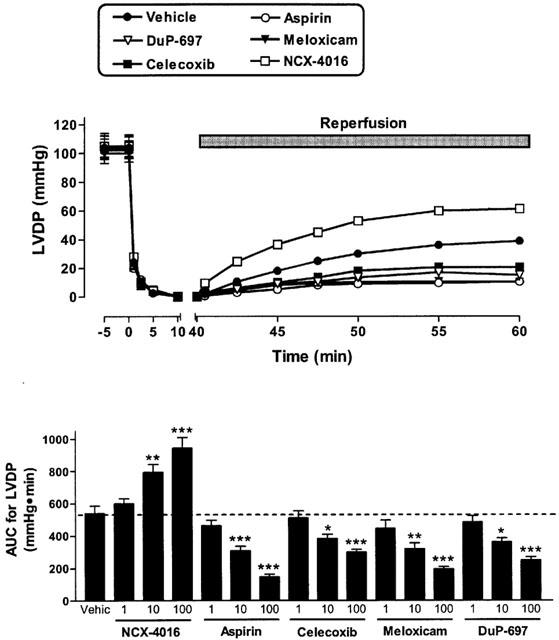
Effect of cyclo-oxygenase inhibitors on left ventricular developed pressure (LVDP) in paced isovolumic left heart preparations subjected to low-flow ischaemia (1 ml min−1 for 40 min) and reperfusion (20 ml min−1 for 20 min). The compounds were infused for 20 min before reduction of flow The upper panel shows the effect of the 100 μM concentration of each drug. The lower panel shows the area-under-the-curve (AUC) during the reperfusion period for all three concentrations of each test drug. *P<0.05, **P<0.01, ***P<0.001 versus the vehicle-treated group. Each point/bar represents the mean (±s.e.mean in the lower panel) of 6 – 10 experiments.
Figure 3.
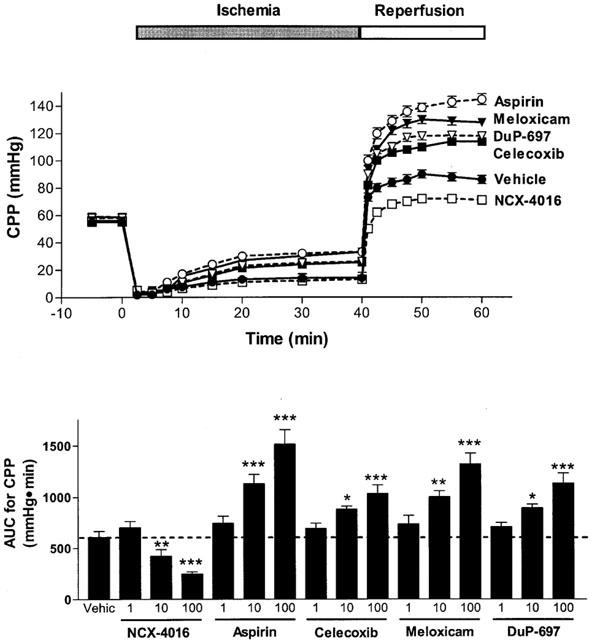
Effect of cyclo-oxygenase inhibitors on coronary perfusion pressure (CPP) in paced isovolumic heart preparations subjected to low-flow ischaemia (1 ml min−1 for 40 min) and reperfusion (20 ml min−1 for 20 min). The compounds were infused for 20 min before the reduction of flow. The upper panel shows the effect of the 100 μM concentration of each drug. The lower panel shows the area-under-the-curve (AUC) during the reperfusion period for all three concentrations of each test drug. *P<0.05, **P<0.01, ***P<0.001 versus the vehicle-treated group. Each point/bar represents the mean (±s.e.mean in the lower panel) of 6 – 10 experiments.
Figure 4.
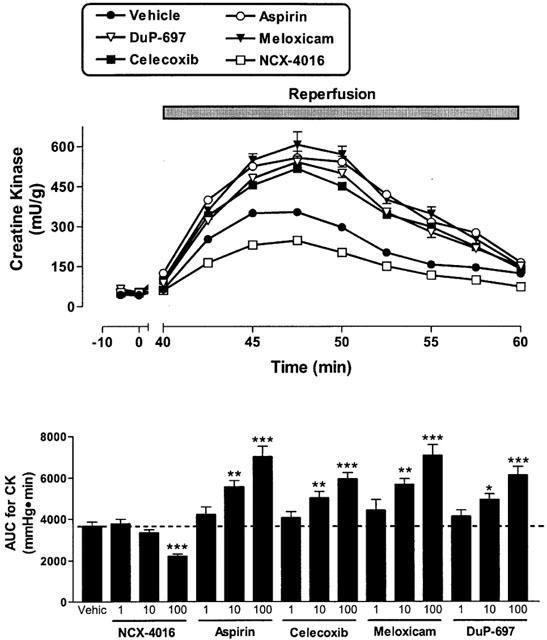
Effect of cyclo-oxygenase inhibitors on creatine kinase (CK) activity in perfusates of rabbit heart preparations subjected to low-flow ischaemia (1 ml min−1 for 40 min) and reperfusion (20 ml min−1 for 20 min). The compounds were infused for 20 min before reduction of flow. The upper panel shows the effect of the 100 μM concentration of each drug. The lower panel shows the area-under-the-curve (AUC) for all three concentrations of each test drug. *P<0.05, **P<0.01, ***P<0.001 versus the vehicle-treated group. Each point/bar represents the mean (±s.e.mean in the lower panel) of 6 – 10 experiments.
Effects of COX-2 inhibitors
When the hearts were perfused with celecoxib, DuP-697 or meloxicam for 20 min before the ischaemia period, a worsening of the cardiac mechanics was observed. This aggravation, which occurred in a concentration-dependent manner, consisted not only of an increase in ventricular contraction (LVEDP; Figure 1), but also in the strength of contractility (LVDP) during reperfusion (Figure 2). The exacerbation of the ischaemic damage was also evident from the concentration-dependent augmentation of CPP and an increase in CK activity during reperfusion (Figures 3 and 4). Each of the COX-2 inhibitors dose-dependently inhibited the release of 6-keto-PGF1α into the perfusate (Figure 5). Whole blood thromboxane synthesis was measured as an index of COX-1 activity (Wallace et al., 2000). Each of the selective COX-2 inhibitors spared thromboxane synthesis at the lower two concentrations, but inhibited thromboxane synthesis (by ∼50%) at the highest concentration tested (Figure 5).
Figure 5.
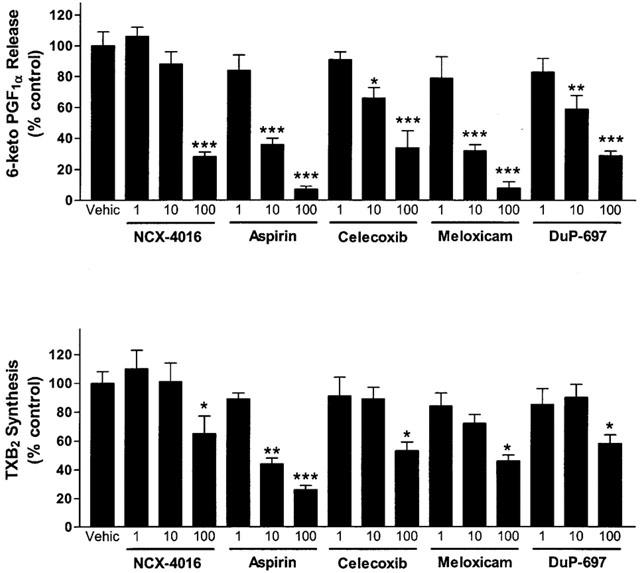
Upper panel: effect of cyclo-oxygenase inhibitors on release of 6-keto PGF1α in perfusates of rabbit heart preparations subjected to low-flow ischaemia (1 ml min−1 for 40 min) and reperfusion (20 ml min−1 for 20 min). The data shown are from samples collected during the first 10 min of the reperfusion period. Lower panel: effects of cyclo-oxygenase inhibitors on whole blood thromboxane synthesis in vitro. *P<0.05, **P<0.01, ***P<0.001 versus the vehicle-treated group. The results are expressed as a percentage of the levels of release of these prostanoids in the vehicle-treated group. Each bar represents the mean±s.e.mean of 5 – 10 experiments.
Effects of aspirin and NCX-4016
Aspirin produced effects similar to those of the selective COX-2 inhibitors; that is, an augmentation of the myocardial dysfunction induced by ischaemia and reperfusion (Figures 1, 2, 3 and 4). Aspirin also caused significant inhibition of the generation of 6-keto PGF1α, of a similar magnitude to that seen with the COX-2 inhibitors. However, the inhibitory effects of aspirin on thromboxane synthesis were observed at a lower concentration (10 μM) than that required for inhibition by the selective COX-2 inhibitors (Figure 5).
As previously observed (Rossoni et al., 2000), perfusion of the hearts with NCX-4016 resulted in a dose-dependent protection against ischaemia-reperfusion-associated cardiac dysfunction (Figures 1, 2, 3 and 4). This beneficial effect was obtained even with the highest concentration of NCX-4016, which significantly inhibited 6-keto-PGF1α generation (Figure 5). At the highest concentration tested, NCX-4016 also significantly inhibited thromboxane synthesis (Figure 5).
Discussion
This study demonstrates that COX inhibitors, such as aspirin, celecoxib, meloxicam and DuP-697, aggravate the ischaemic damage and dysfunction in electrically paced, left heart preparations of the rabbit. These compounds aggravate the decreased ventricular contractility during perfusion flow restriction and markedly increase the post-ischaemic ventricular dysfunction. A common feature of these drugs, at the concentrations that caused significant exacerbation of myocardial dysfunction and damage, is the significant suppression of prostacyclin generation by the heart tissue. Previous studies have documented that prostacyclin synthesis increases during myocardial ischaemia (Berti et al., 1993), and this was confirmed in the present study. This increase in prostacyclin synthesis reduces the vasospasm that occurs as a result of ischaemia, in part by reducing the vasoconstrictive effects of endothelin-1 (Berti et al., 1993), which has been shown to be released into plasma during acute myocardial ischaemia (Miyauchi et al., 1989).
The results of this study also suggest that COX-2 is the source of a significant proportion of the prostacyclin that was released in response to ischaemia and reperfusion. COX-2 has been shown to be expressed in the vascular endothelium (Habib et al., 1993; Busija et al., 1996; Schmedtje et al., 1997; Caughey et al., 2001) and in vascular smooth muscle (Vinals et al., 1997). McAdam et al. (1999) demonstrated that in healthy, young volunteers, selective suppression of COX-2 resulted in profound inhibition of total body excretion of prostacyclin metabolites. Recently Schrör et al. (1998) demonstrated that subjecting perfused rabbit hearts to ischaemia resulted in induction of COX-2 and a parallel enhancement of prostacyclin generation. Schmedtje et al. (1997) demonstrated that exposure of cultured human endothelial cells to hypoxia resulted in rapid up-regulation of COX-2, while Hennan et al. (2001) reported that COX-2-derived prostacyclin played a key role in the maintenance of blood flow in a dog thrombosis model. These findings are consistent with our own observations that prostacyclin synthesis increased significantly following ischaemia, and that three NSAIDs with selectivity for COX-2 each caused an augmentation of myocardial dysfunction and damage at concentrations at which they did not significantly inhibit COX-1 (as measured by the whole blood thromboxane synthesis assay). It is noteworthy that there appeared to be a parallel relationship between the ability of the COX-2 inhibitors to suppress prostacyclin synthesis and the degree of augmentation of myocardial dysfunction and damage. This was also true in the case of aspirin, which suppressed COX-1 (thromboxane synthesis) in parallel with the suppression of prostacyclin synthesis. Of course, it is important to bear in mind that the present study involved the acute administration of selective COX-2 inhibitors. In a clinical setting, these agents are generally used on a chronic basis. Whether or not chronic administration of selective COX-2 inhibitors would similarly affect susceptibility to myocardial dysfunction is not clear.
COX-2-derived prostaglandins, particularly prostacyclin, appear to play an important protective role during ischaemia-reperfusion or other types of tissue stress. These findings are consistent with a number of recent studies. For example, COX-2 was shown to be induced in the stomach by ischaemia, and treatment with a COX-2 inhibitor resulted in a marked exacerbation of damage (Maricic et al., 1999). Dowd et al. (2001) reported that doxorubicin administration to rats resulted in induction of COX-2, elevated prostacyclin synthesis and cardiac injury. Administration of a selective COX-2 inhibitor resulted in a significant exacerbation of cardiac injury. COX-2 also appears to play a role in ischaemic preconditioning, in both the rabbit (Shinmura et al., 2000) and mouse (Guo et al., 2000). However, COX-2 does not appear to play a role in the preconditioning induced in rabbit by adenosine A1 or A3 agonists (Kodani et al., 2001).
It is important to bear in mind that the present study involved the acute administration of selective COX-2 inhibitors. In a clinical setting, these agents are generally used on a chronic basis. Whether or not chronic administration of selective COX-2 inhibitors would similarly affect susceptibility to myocardial dysfunction is not clear. It is noteworthy, however, that a role of COX-2 in preservation of myocardial function in a clinical setting has recently been suggested. Chronic use of selective COX-2 inhibitors has been suggested to increase the risk of serious cardiovascular events, including myocardial infarction (Bombardier et al., 2000; Mukherjee et al., 2001). In one study, which excluded patients at risk of acute myocardial infarction, use of the selective COX-2 inhibitor rofecoxib was found to be associated with ∼5-times as many myocardial infarctions as observed in a group of similar patients taking naproxen, a conventional NSAID (Bombardier et al., 2000). It has been suggested that naproxen exerted a beneficial effect, due to inhibition of platelet aggregation, but there is little direct evidence to support this hypothesis (Mukherjee et al., 2001). A recent meta-analysis of trials with selective COX-2 inhibitors further suggested an association between use of selective COX-2 inhibitors and myocardial infarction (Mukherjee et al., 2001).
As has been reported previously, NCX-4016 significantly reduced the myocardial dysfunction and damage caused by ischaemia-reperfusion. This was in sharp contrast to the other NSAIDs, despite the fact that NCX-4016 produced significant suppression of prostacyclin at the highest concentration tested (comparable to the level of inhibition observed with the highest concentratons of celecoxib and DuP-697). Rossoni et al. (2001) have suggested that these beneficial effects of NCX-4016 are attributable to the release of nitric oxide from this compound, and the subsequent beneficial effect that mediator would exert in terms of preventing vasoconstriction of myocardial blood vessels. They also reported that in perfused rabbit hearts, the blockade of nitric oxide synthase with NG-monomethyl-L-arginine worsened the ischaemia-reperfusion damage, an event prevented by prior treatment with NCX-4016 but not by aspirin. Inhibition of nitric oxide synthesis caused an even greater release of prostacyclin from the perfused myocardial tissue (Berti et al., 1993), further suggesting that the release of prostacyclin occurs as a consequence of the reduced flow, as has been demonstrated previously (Rubanyi et al., 1986). More recently, it has been reported that NCX-4016, but not aspirin, dose-dependently reduced infarct size due to myocardial ischaemia and reperfusion in anaesthetized rats (Rossoni et al., 2001). These authors claimed that the beneficial effects of NCX-4016 appeared to be due primarily to the nitric oxide release from this compound, which could modulate a number of cellular events leading to inflammation, coronary microcirculation obstruction, arrhythmias and myocardial tissue necrosis.
Taken together, these findings suggest that NSAIDs that selectively inhibit COX-2 display negative effects in experimental myocardial ischaemia-reperfusion similar to those observed with conventional NSAIDs, such as aspirin. COX-2-derived prostaglandins appear to perform an important protective role in the heart during ischaemia-reperfusion.
Acknowledgments
Dr. Wallace is an Alberta Heritage Foundation for Medical Research Senior Scientist. This work was supported by a grant from the Heart and Stroke Foundation of Canada.
Abbreviations
- ANOVA
analysis of variance
- AUC
area-under-the-curve
- CK
creatine kinase
- COX
cyclo-oxygenase
- CPP
coronary perfusion pressure
- ELISA
enzyme-linked immunosorbent assay
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end-diastolic pressure
- LVP
left ventricular pressure
- NSAID
nonsteroidal anti-inflammatory drug
- PGI2
prostacyclin
References
- BERGMEYER H.U., RICH W., BUTTER H., SCHMIDT E., HILLMAN G., KREUZ F.H., STAMM D., LANG H., SZASZ G., LANE D. Standardization of methods for estimation of enzyme activity in biological fluids. Z. Klin. Chem. Klin. Bioch. 1970;8:658–660. [Google Scholar]
- BERTI F., ROSSONI G., DELLA BELLA D., VILLA L.M., BUSCHI A., TRENTO F., BERTI C. Nitric oxide and prostacyclin influence coronary vasomotor tone in perfused rabbit heart and modulate endothelin-1 activity. J. Cardiovasc. Pharmacol. 1993;22:321–326. doi: 10.1097/00005344-199308000-00023. [DOI] [PubMed] [Google Scholar]
- BERTI F., ROSSONI G., MAGNI F., CARUSO D., OMINI C., PUGLISI L., GALLI G. Non steroidal anti-inflammatory drugs aggravate acute myocardial ischemia in the perfused rabbit heart: a role for prostacyclin. J. Cardiovasc. Pharmacol. 1988;12:438–444. doi: 10.1097/00005344-198810000-00009. [DOI] [PubMed] [Google Scholar]
- BOMBARDIER C., LAINE L., REICIN A., SHAPIRO D., BURGOS-VARGAS R, DAVIS B., DAY R, FERRAZ M.B., HAWKEY C.J., HOCHBERG M.C., KVIEN T.K., SCHNITZER T.J. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- BUSIJA D.W., THORE C., BEASLEY T., BARI F. Induction of cyclooxygenase-2 following anoxic stress in piglet cerebral arteries. Microcirculation. 1996;3:379–386. doi: 10.3109/10739689609148310. [DOI] [PubMed] [Google Scholar]
- CATELLA-LAWSON F., MCADAM B., MORRISON B.W., KAPOOR S., KUJUBU D., ANTES L., LASSETER K.C., QUAN H., GERTZ B.J., FITZGERALD G.A. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J. Pharmacol. Exp. Ther. 1999;289:735–741. [PubMed] [Google Scholar]
- CAUGHEY G.E., CLELAND L.G., PENGLIS P.S., GAMBLE J.R., JAMES M.J. Roles of cyclooxygenase (Cox)-1 and Cox-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by cox-2. J. Immunol. 2001;167:2831–2838. doi: 10.4049/jimmunol.167.5.2831. [DOI] [PubMed] [Google Scholar]
- CROFFORD L.J., OATES J.C., MCCUNE W.J., GUPTA S., KAPLAN M.J., CATELLA-LAWSON F., MORROW J.D., MCDONAGH K.T., SCHMAIER A.H. Thrombosis in patients with connective tissue diseases treated with specific cyclooxygenase 2 inhibitors. A report of four cases. Arthritis Rheum. 2000;43:1891–1896. doi: 10.1002/1529-0131(200008)43:8<1891::AID-ANR28>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- DOWD N.P., SCULLY M., ADDERLEY S.R., CUNNINGHAM A.J., FITZGERALD D.J. Inhibition of cyclooxygenase-2 aggravates doxorubicin-mediated cardiac injury in vivo. J. Clin. Invest. 2001;108:585–590. doi: 10.1172/JCI11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGELHARDT G., HOMMA D., SCHLEGEL K., UTZMANN R., SCHNITZLER C. Anti-inflammatory, analgesic, antipyretic and related properties of meloxicam, a new non-steroidal anti-inflammatory agent with favourable gastrointestinal tolerance. Inflamm. Res. 1995;44:423–433. doi: 10.1007/BF01757699. [DOI] [PubMed] [Google Scholar]
- FRIEDMAN P.L., BROWN E.J., GUNTHER S., ALEXANDER R.W., BARRY W.H., MUDGE G.H., GROSSMAN W. Coronary vasoconstriction effect of indomethacin in patients with coronary artery disease. N. Engl. J. Med. 1981;305:1171–1175. doi: 10.1056/NEJM198111123052002. [DOI] [PubMed] [Google Scholar]
- FRISHMAN W.H., CHRISTODOULOU J., WEKSLER B., SMITHEN C., KILLIP T., SCHEIDT S. Aspirin therapy in angina pectoris: effect on platelet aggregation, exercise tolerance, and electrocardiographic manifestation of ischemia. Am. Heart J. 1976;92:3–10. doi: 10.1016/s0002-8703(76)80397-3. [DOI] [PubMed] [Google Scholar]
- GANS K.R., GALBRAITH W., ROMAN R.J., HABER S.B., KERR J.S., SCHMIDT W.K., SMITH C., HEWES W.E., ACKERMAN N.R. Antiinflammatory and safety profile of DuP 697, a novel orally effective prostaglandin synthesis inhibitor. J. Pharmacol. Exp. Ther. 1990;254:180–187. [PubMed] [Google Scholar]
- GUO Y., BAO W., WU W.J., SHINMURA K., TANG X.L., BOLLI R. Evidence for an essential role of cyclooxygenase-2 as a mediator of the late phase of ischemic preconditioning in mice. Basic Res. Cardio. 2000;95:479–484. doi: 10.1007/s003950070024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HABIB A., CREMINON C., FROBERT Y., GRASSI J., PRADELLES P., MACLOUF J. Demonstration of an inducible cyclooxygenase in human endothelial cells using antibodies raised against the carboxyl-terminal region of the cyclooxygenase-2. J. Biol. Chem. 1993;268:23448–23454. [PubMed] [Google Scholar]
- HENNAN J.K., HUANG J., BARRETT T.D., DRISCOLL E.M., WILLENS D.E., PARK A.M., CROFFORD L.J., LUCCHESI B.R. Effects of selective cyclooxygenase-2 inhibition on vascular responses and thrombosis in canine coronary arteries. Circulation. 2001;104:820–825. doi: 10.1161/hc3301.092790. [DOI] [PubMed] [Google Scholar]
- JUGDUTT B.I., HUTCHINS G.M., BULKLEY B.J., PITT B., BECKER L.C. Effect of indomethacin on collateral blood flow and infarct size in the conscious dog. Circulation. 1979;59:734–743. doi: 10.1161/01.cir.59.4.734. [DOI] [PubMed] [Google Scholar]
- KIRLIN P.C., ROMSON J.L., PITT B., ABRAMS G.D., SCHORK M.A., LUCCHESI B.R. Ibuprofen-mediated infarct size reduction: effects on regional myocardial function in canine myocardial infarction. Am. J. Cardiol. 1982;50:846–849. doi: 10.1016/0002-9149(82)91244-9. [DOI] [PubMed] [Google Scholar]
- KODANI E., SHINMURA K., XUAN Y.T., TAKANO H., AUCHAMPACH J.A., TANG X.L., BOLLI R. Cyclooxygenase-2 does not mediate late preconditioning induced by activation of adenosine A1 or A3 receptors. Am. J. Physiol. 2001;281:H959–H968. doi: 10.1152/ajpheart.2001.281.2.H959. [DOI] [PubMed] [Google Scholar]
- MARICIC N., EHRLICH K., GRETZER B., SCHULIGOI R, RESPONDEK M., PESKAR B.M. Selective cyclooxygenase-2 inhibitors aggravate ischaemia-reperfusion injury in the rat stomach. Br. J. Pharmacol. 1999;128:1659–1666. doi: 10.1038/sj.bjp.0702966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCADAM B.F., CATELLA-LAWSON F., MARDINI I.A., KAPOOR S., LAWSON J.A., FITZGERALD G.A. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Pharmacology. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIYAUCHI T., YANAGISAWA M., TOMIZAWA T., SUGISHITA Y., SUZUKI N., FUJINO M., AJISAKA R, GOTO K., MASAKI T. Increased plasma concentrations of endothelin-1 and big endothelin-1 in acute myocardial infarction. Lancet. 1989;2:53–54. doi: 10.1016/s0140-6736(89)90303-6. [DOI] [PubMed] [Google Scholar]
- MUKHERJEE D., NISSEN S.E., TOPOL E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- MUSCARA M.N., VERGNOLLE N., LOVREN F., TRIGGLE C.R., ELLIOTT S.N., ASFAHA S., WALLACE J.L. Selective cyclo-oxygenase-2 inhibition with celecoxib elevates blood pressure and promotes leukocyte adherence. Br. J. Pharmacol. 2000;129:1423–1430. doi: 10.1038/sj.bjp.0703232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGLETREE M.L., LEFER A.M. Influence of non-steroidal anti-inflammatory agents on myocardial ischemia. J. Pharmacol. Exp. Ther. 1976;197:582–593. [PubMed] [Google Scholar]
- PATRONO C., CIABATTONI G., PINCA E., PUGLIESE F., CATRUCCI G., DE SALVO A., SATTA M.A., PESKAR B.A. Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb. Res. 1980;17:317–327. doi: 10.1016/0049-3848(80)90066-3. [DOI] [PubMed] [Google Scholar]
- ROSSONI G., BERTI M., DE GENNARO COLONNA V., DEL SOLDATO P., BERTI F. Myocardial protection by the nitroderivative of aspirin, NCX 4016: in vitro and in vivo experiments in the rabbit. Ital. Heart J. 2000;1:146–155. [PubMed] [Google Scholar]
- ROSSONI G., MANFREDI B., DE GENNARO COLONNA V., BERNAREGGI M., BERTI F. The nitroderivative of aspirin, NCX 4016, reduces infarct size caused by myocardial ischemia-reperfusion in the anesthetized rat. J. Pharmacol. Exp. Ther. 2001;297:380–387. [PubMed] [Google Scholar]
- RUBANYI G.M., ROMERO J.C., VANHOUTTE P.M. Flow-induced release of endothelium-derived relaxing factor. Am. J. Physiol. 1986;250:H1145–H1149. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- SCHMEDTJE J.F., JI Y.S., LIU W.L., DUBOIS R.N., RUNGE M.S. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 1997;272:601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- SCHRÖR K., ZIMMERMANN K.C., TANNHÄUSER R. Augmented myocardial ischaemia by nicotine: mechanisms and their possible significance. Br. J. Pharmacol. 1998;125:79–86. doi: 10.1038/sj.bjp.0702061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHINMURA K., TANG X-L., WANG Y., XUAN Y-T., LIU S-Q., TAKANO H., BHATNAGAR A., BOLLI R. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc. Nat. Acad. Sci. U.S.A. 2000;97:10197–10202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SILVERSTEIN F.E., FAICH G., GOLDSTEIN J.L., SIMON L.S., PINCUS T., WHELTON A., MAKUCH R, EISEN G., AGRAWAL N.M., STENSON W.F., BURR A.M., ZHAO W.W., KENT J.D., LEFKOWITH J.B., VERBURG K.M., GEIS G.S. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284:1247–1255. doi: 10.1001/jama.284.10.1247. [DOI] [PubMed] [Google Scholar]
- VINALS M., MARTINEZ-GONZALEZ J., BADIMON J.J., BADIMON L. HDL-induced prostacyclin release in smooth muscle cells is dependent on cyclooxygenase-2 (Cox-2) Arterioscler. Thromb. Vasc. Biol. 1997;17:3481–3488. doi: 10.1161/01.atv.17.12.3481. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., BAK A., MCKNIGHT W., ASFAHA S., SHARKEY K.A., MACNAUGHTON W.K. Cyclooxygenase-1 contributes to inflammatory responses in rats and mice: implications for GI toxicity. Gastroenterology. 1998;115:101–109. doi: 10.1016/s0016-5085(98)70370-1. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., MCKNIGHT W., REUTER B.K., VERGNOLLE N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., MUSCARA M.N., MCKNIGHT W., DICAY M., DEL SOLDATO P., CIRINO G. In vivo antithrombotic effects of a nitric oxide-releasing aspirin derivative, NCX-4016. Thromb. Res. 1999;93:43–50. doi: 10.1016/s0049-3848(98)00134-0. [DOI] [PubMed] [Google Scholar]
- WONG S.C., FUKUCHI M., MELNYK P., RODGER I., GIAID A. Induction of cyclooxygenase-2 and activation of nuclear factor KB in myocardium of patients with congestive heart failure. Circulation. 1998;98:100–103. doi: 10.1161/01.cir.98.2.100. [DOI] [PubMed] [Google Scholar]