

Abstract
Adipocyte A1-adenosine receptors (A1 AdoR) tonically inhibit adenylyl cyclase and lipolysis. Three potential explanations for tonic activity of A1AdoR of rat epididymal adipocytes were investigated: high affinity of adenosine for the receptor, efficient coupling of receptor activation to response, and spontaneous activity of the receptor in the absence of agonist.
The affinity of adenosine for the adipocyte A1AdoR was determined as 4.6 μM by analysis of effects of an irreversible receptor antagonist on agonist concentration-response relationships. In contrast, the potency of adenosine to decrease cyclic AMP in isolated adipocytes was 1.4 nM.
Occupancy by agonist of the A1AdoR was efficiently coupled to functional response (decrease of adipocyte cyclic AMP content). Activation by adenosine of less than 1% of A1AdoRs caused a near-maximal decrease of cyclic AMP in adipocytes. Thus the receptor reserve for adenosine to decrease cyclic AMP content of adipocytes was greater than 99%.
Affinities and receptor reserves for other A1AdoR agonists were determined. Agonists appeared to differ more in their affinity for the receptor than in their intrinsic efficacy to activate it.
A1AdoRs were inactive in the absence of agonist.
It is concluded that adipocyte A1AdoR are tonically activated by endogenous adenosine at nanomolar concentrations. The expression of a high density of A1AdoR that are efficiently coupled to a functional response enables the adipocyte to respond with high sensitivity to the low-affinity agonist, adenosine. Adipocytes may be a model for cells whose functions are tonically modulated by adenosine present in the interstitium of well-oxygenated tissues.
Keywords: Adipocyte, adenosine, receptor reserve, cyclic AMP, A1-adenosine receptor, CVT-2759, FSCPX
Introduction
Activation of the A1-adenosine receptor (A1AdoR) in adipocytes reduces adenylyl cyclase activity, cyclic AMP content, and rate of lipolysis, and enhances the actions of insulin (Fain et al., 1972; Schwabe et al., 1973; 1974). Increases of cyclic AMP accumulation and lipolysis in white adipose tissue have been observed when adenosine deaminase is added to degrade endogenous adenosine (Schwabe & Ebert, 1974; Fain & Wieser, 1975; Honnor et al., 1985) and after treatments with either pertussis toxin (Moreno et al., 1983; Olansky et al., 1983; Vannucci et al., 1989) or adenosine receptor antagonists (Vannucci et al., 1989; Lanoue & Martin, 1994; Coates et al., 1994). These findings can be interpreted to indicate that adipocyte A1AdoRs are always active and mediate a tonic inhibition of adenylyl cyclase activity and lipolysis.
Tonic inhibition of adenylyl cyclase and lipolysis by the adipocyte A1AdoR could be the result of several factors. A high concentration of endogenous adenosine in adipose tissue or in preparations of isolated adipocytes may cause adenosine receptors to be tonically activated. Alternatively, a high affinity of the adipocyte A1AdoR for adenosine could explain tonic activity of the receptor at unusually low adenosine concentrations. Tonic inhibitions of adenylyl cyclase and lipolysis could also be caused by agonist-independent (i.e., spontaneous) activity of adipocyte A1AdoR. We recently demonstrated the presence of spontaneous activity of human adenosine receptors expressed at a density of 8000 fmol mg protein−1 in Chinese hamster ovary cells (Shryock et al., 1998a). Because adenosine deaminase has been reported to decrease the concentration of endogenous adenosine to undetectable levels (Honnor et al., 1985; Lohse et al., 1986), increases of adenylyl cyclase activity and lipolysis caused by adenosine receptor antagonists in the presence of ⩾1 u ml−1 of adenosine deaminase (Vannucci et al., 1989; Lanoue & Martin, 1994; Parsons et al., 1988; Coates et al., 1994) may be indicative of an action of these antagonists to reduce the spontaneous activity of adipocyte adenosine receptors. However, it is also possible that adenosine deaminase may not be effective to reduce the concentration of adenosine in the receptor compartment.
Lastly, tonic inhibition of adenylyl cyclase and lipolysis may be the result of binding of adenosine to a small number of receptors among a large population of A1AdoR that are efficiently coupled to these responses. The range of reported densities of A1AdoRs in membranes prepared from adipose tissue is high (500 – 2000 fmol mg protein−1 in rat). A small fraction of this large population of receptors would be occupied at the low adenosine concentrations presumably present endogenously in vivo and in vitro. However, activation of this small fraction of the receptor population could be sufficient to cause functional responses in adipocytes, if the receptor reserve (e.g., a measure of the efficiency of coupling of receptor activation to functional response) was high. It has been estimated that R-(−)-N6-(2-phenylisopropyl)adenosine (R-PIA) caused a half-maximal reduction of adipocyte cyclic AMP content by occupancy of only 10% of the population of adenosine receptors (Lohse et al., 1986).
The purpose of the present investigation was to quantify three factors that may contribute to an understanding of the tonic inhibition of adenylate cyclase activity by rat adipocyte A1AdoR. A fourth factor, the concentration of adenosine in the receptor compartment, was not investigated. We have estimated the affinity of the adipocyte A1AdoR for adenosine, the efficiency of coupling (receptor reserve) of A1AdoR activation to reduction of cyclic AMP accumulation, and the contribution of spontaneous activity of the receptor to an inhibition of cyclic AMP accumulation in adipocytes. Results indicated that receptor reserve for adenosine to decrease the accumulation of cyclic AMP in adipocytes is very great and tonic activity of the adipocyte A1AdoR is expected when the concentration of endogenous adenosine is 1 – 2 nM.
Methods
Drugs and chemicals
Collagenase Type I was purchased from Worthington Biochemical (Lakewood, NJ, U.S.A.). Fatty acid-free (defatted) BSA, nicotinic acid, succinyl cyclic AMP tyrosyl methyl ester, erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA), and α,β-methylene adenosine 5′-diphosphate (AOPCP) were from Sigma (St. Louis, MO, U.S.A.). Adenosine deaminase was from Boehringer – Mannheim (Indianapolis, IN, U.S.A.) and cilostamide was from Biomol (Plymouth Meeting, PA, U.S.A.). The following were from RBI (Natick, MA, U.S.A.): R-PIA, S-PIA, 2-chloro-N6-cyclopentyladenosine (CCPA), 8-cyclopentyl-1,3-dipropylxanthine (CPX), 2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine (CGS-21680), 2-phenylaminoadenosine, xanthine amine congener (XAC), N6-cyclopentyltheophylline (CPT), and isoproterenol. The A1AdoR antagonist radioligand [3H]-CPX was from New England Nuclear (Boston, MA, U.S.A.). Antibody to cyclic AMP was a gift from Dr Gary Brooker (Georgetown University), (±)-N6-endonorbornan-2-yl-9-methyladenine (N-0861) was a gift from Discovery Therapeutics (Richmond, VA, U.S.A.), {[(5-{6-[(3R)oxolan-3-yl]amino}purin-9-yl)(3S,2R,4R)-3,4-dihydroxyoxolan-2-yl]-methoxy}-N-methylcarboxamide (CVT-2759) was a gift from CV Therapeutics (Palo Alto, CA, U.S.A.). The synthesis and structure of CVT-2759 are described in United States patent number 6,258,793B1. Rolipram was a gift from Berlex Laboratories (Cedar Knolls, NJ, U.S.A.). The irreversible A1AdoR antagonist 8-cyclopentyl-3-[3-[[4-(fluorosulphonyl)benzoyl]oxy]propyl]-1-propylxanthine (FSCPX) was synthesized as described by Scammells et al. (1994). Stock solutions of adenosine receptor ligands, including FSCPX, and of rolipram and cilostamide were prepared in DMSO and stored at −20°C. Isoproterenol stock solutions were prepared in 5 mM HCl, and solutions of AOPCP, nicotinic acid, and EHNA were prepared in saline.
Isolation of adipocytes
Adipocytes were isolated from the epididymal fat pads of male Sprague – Dawley rats (380 – 420 g) fed ad libitum and maintained on a 12-h light-dark cycle for 1 – 2 weeks. Adipocytes from rats of the size used are mature and express A1 – but not A2-AdoR (Vassaux et al., 1993). Rats were anaesthetized with halothane and killed by transection of the aorta. Epididymal fat tissue was isolated and placed into Krebs – Ringer – HEPES (KRH) buffer. Buffer composition (in mM) was: NaCl 100, KCl 4.7, CaCl2 2.5, NaHCO3 3.6, MgSO4 1.19, KH2PO4 1.18, dextrose 5, pyruvic acid 5, ascorbic acid 1, and HEPES 5 (titrated to pH 7.4). Visible blood vessels were trimmed away, and adipose tissue was minced with scissors. Minced tissue from one rat was placed into 25 ml of fresh KRH buffer containing Type I collagenase (1 mg ml−1) and 1% (wt v−1) defatted BSA in a plastic 50-ml tube. Nicotinic acid (2 μM) was added to inhibit lipolysis without causing activation of adenosine receptors or desensitization of A1AdoR-mediated responses (Green et al., 1992). The tissue was digested for 1 h at 34 – 35°C in an orbital shaker bath and the digest was poured through a nylon-mesh filter (210 μm). The cell filtrate was placed in a 50-ml plastic tube. Adipocytes floated to the top of the column of buffer; the infranatent solution (42 – 45 ml) was removed. Fresh KRH buffer at 36°C containing 1% defatted BSA was added to the adipocyte suspension and the wash procedure was repeated. After three washes, 2 ml of adipocyte suspension was either diluted in 20 ml of fresh KRH buffer with 1% defatted BSA for use in experiments, used to prepare membranes for binding assays, or pretreated to inactivate A1AdoRs. A small volume of the cell suspension was aspirated into a capillary tube for determination of the fractional occupation (the lipocrit) of the suspension by the fat cells as described by Honnor et al. (1985). The range of values of lipocrit was 3 – 6% in the adipocyte suspensions.
Pretreatment of adipocytes with FSCPX
Isolated adipocytes from two fat pads were suspended in 20 ml of KRH buffer containing 0.1% defatted BSA, 2 μM nicotinic acid, adenosine deaminase (2 u ml−1) and either DMSO (vehicle, final content 0.1%) or FSCPX (1 – 10 μM) and incubated for 1 h at 36°C without shaking. Cells were then washed four times as described above in 40 ml of KRH buffer containing 1% defatted BSA and 2 μM nicotinic acid, and twice in 40 ml of KRH buffer containing only 1% defatted BSA. The time for washing cells was approximately 70 min. Cells were resuspended in 20 ml of KRH buffer with 1% defatted BSA and adenosine deaminase (2 u ml−1) for use in experiments.
Assay of actions of A1AdoR agonists on cyclic AMP content of isolated adipocytes
Aliquots (100 μl, 45,000 – 90,000 cells) of the freshly prepared adipocyte cell suspension were pipetted into wells of 24-well cell culture clusters (Costar, Corning, NY, U.S.A.) containing 0.4 ml of KRH medium and the appropriate concentration of A1AdoR agonist to be tested. Next, 0.5 ml of KRH medium with 60 nM isoproterenol was added to each well. All KRH media contained 1% defatted BSA, 1 mM ascorbic acid (to prevent oxidation of isoproterenol), 10 μM rolipram and 1 μM cilostamide to inhibit cyclic AMP phosphodiesterases, and adenosine deaminase (2 u ml−1). Inhibition of cyclic AMP phosphodiesterases has been shown to reduce the release of nucleotides from adipocytes, and thereby the extracellular formation of adenosine (Kather, 1990). Culture clusters were placed in an orbital shaker bath maintained at 34 – 36°C during incubations of cells with drugs. Incubations were terminated after 4 min by addition of 200 μl of 300 mM HCl to each well to lyse the cells. Cell lysates were stored at 2°C overnight and cyclic AMP content of lysates was determined the following day.
The protocol was modified to assay actions of adenosine. Adenosine is formed by adipocytes and released into the medium (Schwabe et al., 1973), and therefore adenosine deaminase (2 u ml−1) was added to the medium used for final suspension of isolated adipocytes. To inhibit adenosine deaminase during the subsequent incubation of cells with exogenous adenosine, EHNA (10 μM) was added to the adipocyte suspension immediately before cells were aliquoted for incubations, and was present in the incubation media added to cells. The 5′-nucleotidase inhibitor AOPCP (50 μM) was added to the medium to decrease formation of adenosine from AMP. A relatively dilute suspension of cells (20,000 cells ml−1) was used in these experiments to further reduce the rate of accumulation of adenosine.
Assay of cyclic AMP content of cell lysates
Cyclic AMP content was determined by RIA. Cyclic AMP in samples and standards was acetylated to increase the binding of antibody and sensitivity of the assay. Acetylation was done in glass tubes by addition of 4.5 μl of a 3.5 to 1 (v v−1) mixture of triethylamine and acetic anhydride to 100 μl of sample. Antibody to cyclic AMP and 125I-labelled succinyl cyclic AMP tyrosyl methyl ester (20,000 d.p.m.) were added to each sample and the combination (215 μl total volume) was incubated at room temperature for 2 h. Hydroxyapatite (75 μl of an aqueous suspension) was added to each sample to bind antibody and antibody-bound cyclic AMP. Samples with hydroxyapatite were incubated at 2°C for 10 min. Incubations were terminated by collection of hydroxyapatite with its adsorbed antibody-bound cyclic AMP on glass fibre filters by vacuum filtration using a cell harvester (Brandel, Gaithersburg, MD, U.S.A.). The radioactivity of antibody-bound 125I-labelled cyclic AMP ester on filter paper was quantified by gamma counting. Cyclic AMP content of adipocyte extracts was estimated by comparison with results of parallel assays of standards of known cyclic AMP content.
Radioligand binding assays of A1AdoR density
A crude membrane fraction was prepared from isolated adipocytes for estimation of A1AdoR density by assay of saturation binding of [3H]-CPX. Adipocytes were suspended in 30 ml of a chilled solution containing 0.25 M sucrose, 1 mM EDTA, and 10 mM Tris-HCl (pH 7.4). The suspension was placed in a chilled Potter – Elvehjem tissue grinder and homogenized with 10 up-and-down strokes of a motor-driven pestle. The homogenate was cooled on ice and the infranate under the fat cake was aspirated, transferred to a 50-ml centrifuge tube, and centrifuged at 500×g for 10 min at 4°C. The infranate under the fat cake was again aspirated, resuspended in fresh buffer, and homogenized a second time using six strokes of the tissue grinder. Cell membranes were collected by centrifugation of the homogenate at 15,000×g for 15 min. The final membrane pellet was resuspended in a solution containing 0.25 M sucrose, 0.1 mM phenylmethylsulphonyl fluoride, leupeptin and aprotinin (5 μg ml−1 each), 2 u ml−1 of adenosine deaminase, and 10 mM Tris-HCl buffer, pH 7.4. The membrane suspension was frozen and stored in liquid nitrogen. For saturation binding assays, 10-μl aliquots (10 μg protein) of membrane suspension were incubated in glass tubes with 0.15 – 10 nM [3H]-CPX and adenosine deaminase (2 u ml−1), with or without 10 μM CPT, for 2 h at room temperature in a total volume of 200 μl of 50 mM Tris-HCl buffer. Incubations were terminated by dilution of samples with 4 ml of ice-cold 50 mM Tris-HCl buffer and immediate collection of membranes onto glass fibre filters by vacuum filtration using a cell harvester. Filters were quickly washed three times with ice-cold buffer to remove unbound radioligand. Filter discs containing trapped membranes and bound radioligand were placed in plastic tubes with 4 ml of scintillation cocktail. Radioactivity was quantified by scintillation counting. Nonspecific binding of [3H]-CPX was defined as [3H]-CPX bound in the presence of 10 μM CPT. Specific binding of [3H]-CPX was determined by subtracting nonspecific from total binding, and was plotted as a function of radioligand concentration by use of the Prism computer program (GraphPad, San Diego, CA, U.S.A.). Triplicate determinations were made at each concentration of radioligand in each assay.
Calculation of receptor reserves for A1AdoR agonists to decrease cyclic AMP content of intact adipocytes
Concentration-response data for each agonist to decrease cyclic AMP content of adipocytes pretreated with either 0.1% DMSO (vehicle control) or FSCPX (1 – 10 μM) were imported into Table Curve (Jandel Scientific, Sausalito, CA, U.S.A.) and fit to a dose-response logistic function (equation 1 in Morey et al., 1998) with a non-linear regression technique. Concentrations of agonist causing equal percentage reductions of isoproterenol-stimulated cyclic AMP content of adipocytes pretreated with vehicle and FSCPX were determined from the computer-fitted concentration-response relationships as previously described (Morey et al., 1998). These pairs of agonist concentrations were entered into the user-defined equation, A=A′qKA/[KA+(1−q)A′], wherein A and A′ are concentrations of agonist that caused equal levels of functional response in control and FSCPX-treated cells, respectively. The equation was solved for values of q and KA, the fraction of receptors that is functional after treatment of cells with FSCPX, and the agonist equilibrium dissociation constant, respectively. The fractional receptor occupancy (ρ) at any given agonist concentration [A] was then calculated as ρ=[A]/([A]+KA), and a plot of fractional receptor occupancy as a function of agonist concentration was made. Using data expressing both fractional receptor occupancy and cyclic AMP response as functions of agonist concentration, a plot of cyclic AMP response as a function of fractional receptor occupancy was made. The percentage of receptors occupied by each agonist to cause responses that were 50 and 90% of maximal was determined from the plots.
Effects of the A1AdoR agonist CVT-2759 on heart rate and serum non-esterified fatty acid (NEFA) concentration of awake male rats
Two sets of six male Sprague – Dawley rats (350 – 450 g) were used. Heart rate was measured by telemetry. For transmitter implantation, a rat was anaesthetized, a midline abdominal incision was made using sterile technique, and a transmitter was sutured to the abdominal wall. The two transmitter leads were tunnelled through the wall, passed subcutaneously, one to the left shoulder, the other to the right thigh, and secured in place with sutures. For experiments, heart rate of the awake rat was recorded using a Dataquest ART Gold System (Data Sciences International, St. Paul MN, U.S.A.). Cardiac electrical activity was recorded for 10-s periods and used to calculate heart rate in beats per min. After recording of a control (zero time) heart rate, either vehicle (0.5 ml 31% DMSO in saline) or CVT-2759 (0.5 or 2 mg kg−1 on different days) was injected into the intraperitoneal cavity of each rat, and heart rate was monitored continuously for an additional 3 h.
The effect of CVT-2759 (0.5 mg kg−1) to reduce serum NEFA concentration was determined. Using aseptic conditions and sterile technique, a catheter (0.25-mm outer diameter) was implanted in the left common carotid artery of each rat 3 days before an experiment. The catheter was tunnelled up through the back and out of the skin. Following recovery, rats were placed in metabolic cages to facilitate blood sampling. Blood samples (0.2 ml) were obtained before and at 20, 60, 120, and 180 min after intraperitoneal injection of either CVT-2759 (0.5 mg kg−1) or vehicle (0.9% DMSO in saline). A 0.4-ml volume of 1% sodium citrate in saline was administered following withdrawal of each blood sample to replace blood volume and prevent clotting in the carotid artery catheter. Serum was collected from each sample after centrifugation of the clotted blood. Serum samples were stored at −80°C until analysis. Serum NEFA concentration was determined using an enzymatic colorimetric assay kit (Wako Chemicals USA, Richmond, VA, U.S.A.).
Data analysis
Decreases of cyclic AMP content of adipocytes caused by A1AdoR agonists in the presence of isoproterenol were normalized to the value of cyclic AMP content in the presence of isoproterenol alone (control). Concentration – response data for reduction of cyclic AMP content in each experiment were analysed to provide a mean value of response at each concentration of agonist. Mean values of responses from several similar experiments with each agonist were then analysed using Prism (variable slope) to determine the mean and s.e.mean of responses and values of curve-fitting parameters including the concentration of agonist causing a half-maximal response (EC50), log EC50, Hill slope, and KA. These values are given together with 95% confidence intervals. Differences between values of EC50 and KA among different agonists were considered to be significant when their 95% confidence intervals did not overlap. Results of all other experiments were analysed as described in figure legends and data are presented graphically as means with s.e.mean.
Results
The potency of adenosine and other A1AdoR agonists to decrease the cyclic AMP content of rat epididymal adipocytes in the presence of 30 nM isoproterenol was very high (Figure 1). Values of EC50 for the A1AdoR agonists R-PIA, CCPA, and adenosine to reduce cyclic AMP content of adipocytes were 0.20, 0.30, and 1.4 nM, respectively (Table 1). The agonist rank order of potency was R-PIA>CCPA>adenosine>S-PIA>CVT-2759>2-phenylaminoadenosine=CGS-21680. The compound CVT-2759, which is a partial agonist at cardiac A1AdoRs and causes minimal change of heart rate and a modest slowing of atrioventricular conduction (Wu et al., 2001), was a full agonist to decrease cyclic AMP content of rat adipocytes (Figure 1). Actions of A1AdoR agonists to decrease cyclic AMP content of adipocytes were antagonized by 0.1 μM CPX (not shown).
Figure 1.
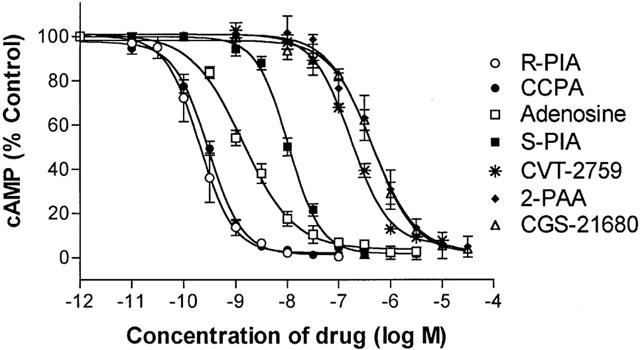
Concentration – response relationships for A1AdoR agonists to decrease cyclic AMP content of rat isolated adipocytes in the presence of 30 nM isoproterenol. Isoproterenol caused an increase of cyclic AMP content by 20 fold above basal, and A1AdoR agonists attenuated this increase by 95%. Symbols indicate the mean and s.e.of mean values from 4 – 8 experiments. In each experiment, six repeats were done at each concentration of ligand. Data were fit using a four-parameter logistic equation describing a sigmoidal dose-response relationship with variable Hill slope (Prism, GraphPad). Values of Hill slopes of curves for R-PIA, CCPA, S-PIA, CVT-2759, and CGS-21680 were significantly greater than 1.0 (mean value of 1.33). Values of Hill slopes for adenosine and 2-phenylaminoadenosine (2-PAA) were not significantly different from 1.0. See Methods for details of experimental conditions.
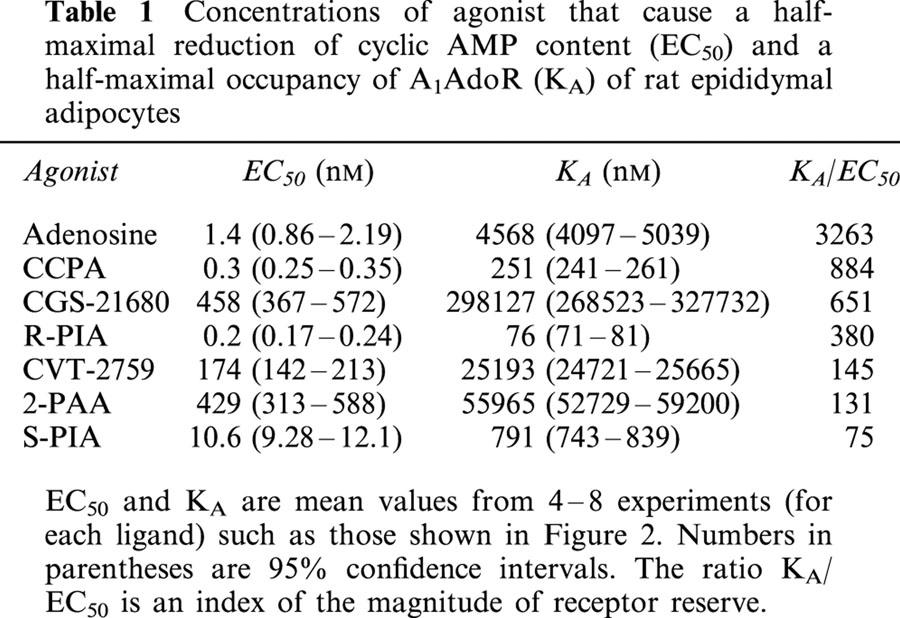
Table 1.
Concentrations of agonist that cause a half-maximal reduction of cyclic AMP content (EC50) and a half-maximal occupancy of A1AdoR (KA) of rat epididymal adipocytes
Pretreatment of adipocytes with the irreversible A1AdoR antagonist FSCPX caused a marked reduction of sensitivity of adipocytes to adenosine (Figure 2). Similar effects of FSCPX treatment to reduce the responsiveness of adipocytes to all seven tested agonists were observed. Analysis of the effects of FSCPX on agonist concentration – response relationships (see Methods) indicated that 98 – 99.9% of receptors were inactivated by 1 – 10 μM FSCPX in the various experiments. Pretreatment of adipocytes with FSCPX did not decrease the action of nicotinic acid to reduce the cyclic AMP content of adipocytes in the presence of isoproterenol (Figure 2). Nicotinic acid has been shown to cause a G protein-mediated inhibition of adipocyte adenylyl cyclase activity (Aktories et al., 1983; Lorenzen et al., 2001), but does not stimulate A1AdoR. The results suggest that FSCPX selectively reduced the function of adenosine receptors, which is consistent with previous reports (Srinivas et al., 1996; 1997; Morey et al., 1998; Baker et al., 2000).
Figure 2.
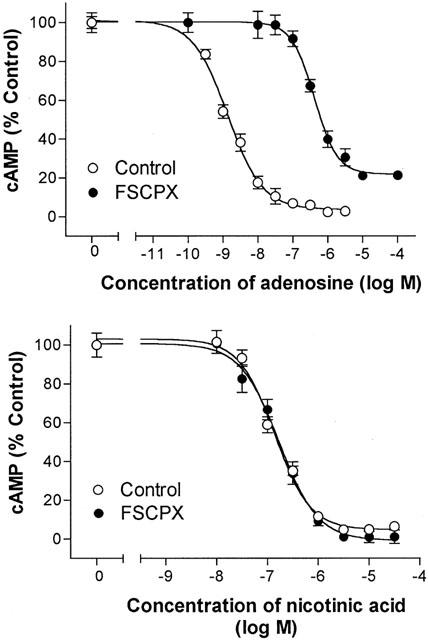
Concentration – response relationships for adenosine (top panel) and nicotinic acid (bottom panel) to decrease cyclic AMP content of control and FSCPX (10 μM)-treated rat adipocytes in the presence of 30 nM isoproterenol. Symbols indicate mean and s.e. of mean values of six repeats from each of 4 – 8 experiments. Data were fit as described in Methods to derive a value of KA for each A1AdoR agonist.
To confirm that treatment with FSCPX irreversibly reduced the number of adipocyte A1AdoR that could be bound by a receptor ligand, adipocytes were pretreated with FSCPX, washed to remove unbound drug, homogenized, and used to prepare cell membranes for assay of A1AdoRs. In membranes prepared from control cells the density of A1AdoR detected by [3H]-CPX was 690±50 fmol mg protein−1 (n=4). In membranes from cells pretreated with 0.3 μM FSCPX for 1 h, the maximal specific binding of [3H]-CPX was reduced to 341±33 fmol mg protein−1 (n=4, P<0.05 vs control). The equilibrium dissociation constant of binding of [3H]-CPX was not changed by pretreatment with 0.3 μM FSCPX (values of pKD for [3H]-CPX were 8.85±0.09 and 8.99±0.07 for control and treated membranes, respectively). In membranes from cells pretreated with 10 μM FSCPX, no specific binding of the antagonist [3H]-CPX was detected. The results indicate that treatment of adipocytes with 10 μM FSCPX causes a loss of A1AdoR as detected by both radioligand binding and functional assays.
Values of KA for A1AdoR agonists were calculated from data such as those shown in Figure 2, using the procedure of Furchgott & Bursztyn (1967) as described in Methods, and are listed in Table 1. These values are much higher than the values of EC50 for each agonist (Table 1). The affinity of adenosine for the adipocyte A1AdoR, 4.6 μM, was 3263 fold greater than the EC50 value of 1.4 nM for adenosine to decrease cyclic AMP content of adipocytes.
The fraction of total receptors occupied at selected concentrations of each agonist was determined by application of the law of mass action (see Methods). Plots of both receptor occupancy and response (reduction of adipocyte cyclic AMP content) as functions of adenosine concentration are shown in Figure 3. A comparison of the occupancy and response plots indicates that cyclic AMP content of adipocytes was reduced in the presence of nanomolar concentrations of adenosine when very few adenosine receptors were occupied. In fact, the occupancy of adenosine receptors was <1% at adenosine concentrations below 50 nM. However, activation by adenosine of only 0.03 and 0.44% of adipocyte A1AdoRs caused responses that were 50 and 90% of maximal, respectively. The relationships between adipocyte response and occupancy of A1AdoRs by each of seven agonists are shown in Figure 4. They are markedly non-linear, indicating the presence of substantial receptor reserve for each agonist tested.
Figure 3.
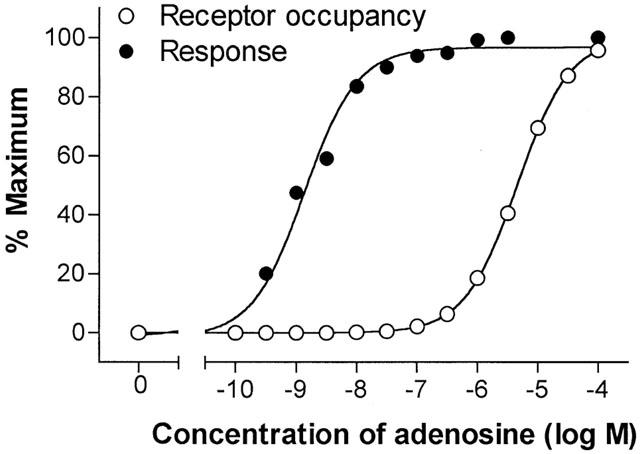
Plots of relationships between adenosine concentration and response (decrease of cyclic AMP in adipocytes) and adenosine concentration and occupancy of A1AdoR. The relationship between adenosine concentration and receptor occupancy was calculated using an equation for the law of mass action as stated in Methods. The adenosine concentration – response relationship was plotted using data shown in Figures 1 and 2 (control).
Figure 4.
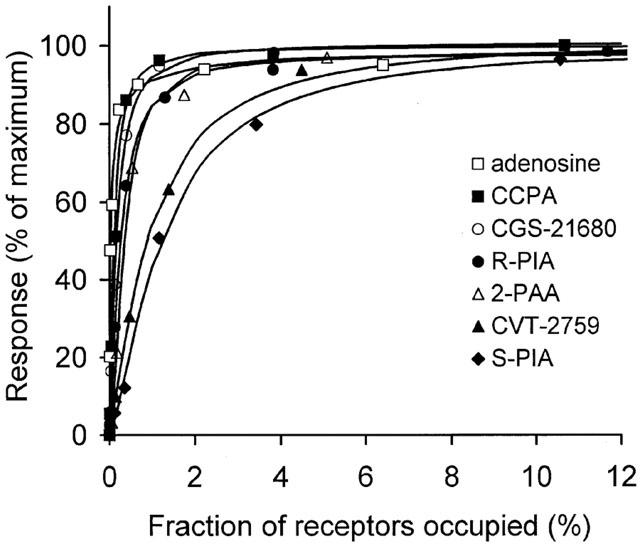
The relationship between fractional occupancy of A1AdoR in adipocytes and response (decrease of cyclic AMP content in the presence of 30 nM isoproterenol), for each of seven agonists. Please see Methods regarding calculation of the occupancy – response relationship.
Two experiments were designed to determine if adipocyte A1AdoR were active when not bound to an agonist. In the first experiment (Figure 5) aliquots of isolated adipocytes were incubated with 30 nM isoproterenol in the absence and presence of either XAC (1 nM – 1 μM) or N-0861 (0.3 – 50 μM). It was previously shown that XAC is an inverse agonist whereas N-0861 is a neutral antagonist of A1AdoR (Shryock et al., 1998a). We reasoned that if spontaneous activity of A1AdoR in the absence of adenosine inhibited the cyclic AMP response to isoproterenol, then an inverse agonist of A1AdoR would decrease this spontaneous activity and thereby increase cyclic AMP formation in the presence of isoproterenol. Adenosine deaminase (2 u ml−1) and 50 μM AOPCP (an inhibitor of 5′-nucleotidase-catalyzed formation of adenosine from 5′-AMP) were used to degrade endogenous adenosine and to reduce adenosine formation, respectively. As shown in Figure 5, adenosine deaminase and AOPCP significantly increased adipocyte cyclic AMP content in the presence of 30 nM isoproterenol (P<0.01). However, the A1AdoR antagonists XAC (1 nM – 1 μM) and N-0861 (0.3 – 50 μM) did not cause a further increase of cyclic AMP content of adipocytes in the presence of isoproterenol, adenosine deaminase, and AOPCP (Figure 5). The results indicated that endogenous adenosine, and not spontaneous receptor activity, was responsible for inhibition by adipocyte A1AdoR of adenylyl cyclase activity in the presence of isoproterenol.
Figure 5.
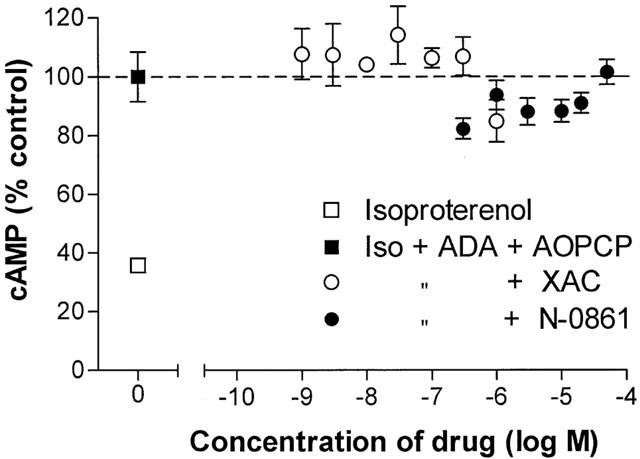
Absence of effects of the A1AdoR inverse agonist XAC and the neutral antagonist N-0861 on cyclic AMP content of rat isolated adipocytes in the presence of adenosine deaminase (2 u ml−1) and 50 μM AOPCP. Cells (approximately 20,000) were incubated for 4 min at 36°C in KRH buffer containing 4% fatty acid-free albumin with 30 nM isoproterenol alone or isoproterenol, ADA, and AOPCP in the absence or presence of either XAC or N-0861 as indicated. Symbols represent the mean and s.e.mean of six determinations in each of five experiments.
In a second experiment (Figure 6), the responses of adipocytes to a neutral antagonist (N-0861) and a different inverse agonist of A1AdoR (CPX) were compared. The neutral antagonist N-0861 would not be expected to reduce the spontaneous activity of A1AdoR, although it antagonizes agonist-induced receptor activation (Shryock et al., 1998a). In contrast, the inverse agonist CPX has been shown to reduce both agonist-dependent and agonist-independent (spontaneous) activity of A1AdoR (Shryock et al., 1998a). Thus if the activity of adipocyte A1AdoR to inhibit cyclic AMP formation is caused only by the binding of endogenous adenosine to the receptor, then both CPX and N-0861 should attenuate this activity and raise adipocyte cyclic AMP content. If, however, cyclic AMP formation in adipocytes is restrained by A1AdoR that are active spontaneously, then CPX but not N-0861 will increase cyclic AMP content in the presence of isoproterenol. Results indicated that both CPX (1 nM – 1 μM) and N-0861 (3 nM – 50 μM) caused concentration-dependent and equivalent 10 fold increases of adipocyte cyclic AMP content in the presence of 100 nM isoproterenol (without adenosine deaminase and AOPCP) (Figure 6). This suggests that the activity of adipocyte A1AdoR is mediated by endogenous adenosine and not by spontaneous changes in receptor conformation.
Figure 6.
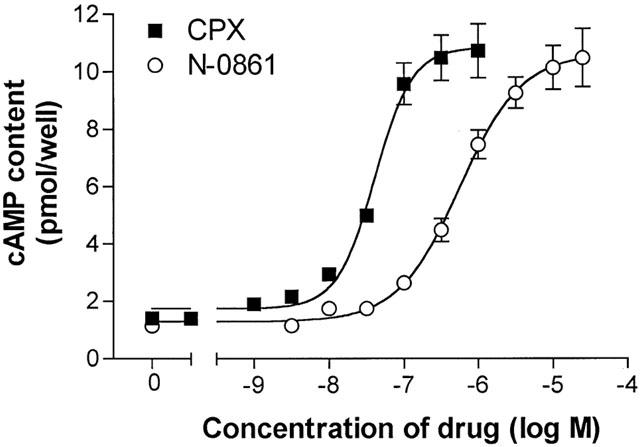
Concentration – response relationships for the A1AdoR inverse agonist CPX and the neutral antagonist N-0861 to increase cyclic AMP content of rat isolated adipocytes in the presence of 100 nM isoproterenol. Adenosine deaminase (2 u ml−1) and 50 μM AOPCP increased adipocyte cyclic AMP content to 8.25±0.86 pmol well−1 (not shown). Points indicate the mean and s.e.mean of six determinations in each of 4 – 7 experiments.
Because inhibitory G proteins mediate the action of adenosine to inhibit adenylyl cyclase activity in adipocytes, we attempted to determine if there was an excess (‘reserve') of G proteins for mediating the response to A1AdoR activation. Isolated adipocytes were treated in the absence and presence of pertussis toxin (0.05 and 1 μg ml−1) for 2 h, then washed and incubated with the A1AdoR agonist CCPA. If pertussis toxin could shift the concentration – response relationship for CCPA to the right without reducing efficacy (maximal response), in the manner of FSCPX (Figure 2), then there would appear to be a reserve of G protein. On the other hand, if there were no reserve of G protein for coupling to adipocyte A1AdoR, then pertussis toxin would reduce both the efficacy and potency of an A1AdoR agonist at the same time. The data shown in Figure 7 indicate that pertussis toxin decreased both the efficacy and potency of CCPA concurrently, thus suggesting that there is no reserve of G protein to mediate responses to A1AdoR activation.
Figure 7.
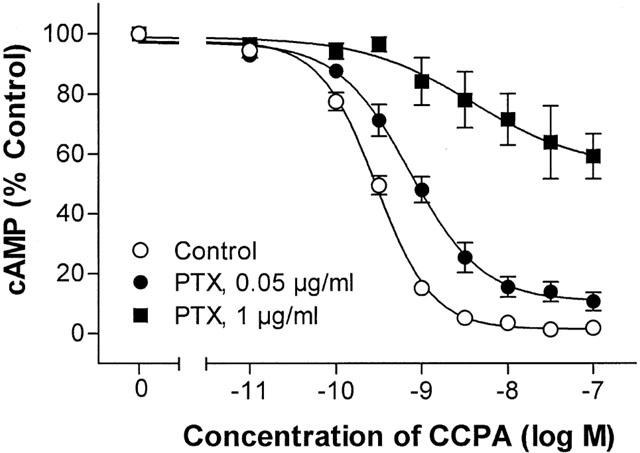
Attenuation by pertussis toxin of the action of CCPA to reduce adipocyte cyclic AMP content. Rat isolated adipocytes (45,000 – 90,000 cells) were incubated without (control) or with pertussis toxin at the indicated doses for 2 h at 35°C (without shaking) in KRH buffer with 2 μM nicotinic acid and 1% fatty acid-free BSA. Cells were washed and incubated for 4 min with 100 nM isoproterenol, adenosine deaminase (2 u ml−1), and the indicated concentrations of CCPA. Symbols indicate the mean and s.e.mean of six determinations in each of 10 (control) or five (0.05 and 1 μg toxin) experiments.
Given the presence of many ‘spare' A1AdoRs for agonists to reduce cyclic AMP formation in adipocytes but few ‘spare' receptors for actions of A1AdoR agonists on heart cells (Srinivas et al., 1997), it is predictable that binding of a weak or ‘partial' agonist to A1AdoR in adipocytes would cause a greater functional response than binding of such agonist to A1AdoR in heart. Weak agonists of A1AdoR have previously been shown to cause a greater reduction of lipolysis than of heart rate (van schaick et al., 1998). To confirm this assumption, we measured heart rate and plasma NEFA concentration before and after an intraperitoneal injection of the weak A1AdoR agonist CVT-2759 into awake rats. Injection of rats with CVT-2759 (0.5 or 2 mg kg−1, n=6 each) caused no significant change in heart rate. In contrast, 0.5 mg kg−1 of CVT-2759 significantly reduced the concentration of NEFA in rat blood serum from 0.70±0.04 to 0.24±0.02 and 0.39±0.04 mM (P<0.05, one-way ANOVA and Student – Newman – Keuls test, n=6 animals) at 20 and 60 min post-injection, respectively (not shown).
Discussion
The important finding of this study is that the receptor reserves for adenosine and other A1AdoR agonists to decrease adipocyte cyclic AMP content were extremely high. Activation of 0.03% of the total population of adipocyte A1AdoRs by adenosine caused a reduction of cyclic AMP content equivalent to 50% of that caused by activation of all of the A1AdoR (Figure 4). Because the adenosine concentration required to activate 0.03% of A1AdoR is much lower than that required to activate 50% of the A1AdoR, the EC50 for adenosine to decrease cyclic AMP content of adipocytes (1.4 nM) was 3263 fold lower than the calculated affinity (KA value) of adenosine for A1AdoR in the intact adipocyte (4.6 μM). Thus, adipocyte adenosine receptors are very efficiently coupled to inhibition of adenylyl cyclase. The expression of a high density (500 – 2000 fmol mg protein−1) of receptors efficiently coupled to a functional response enables the adipocyte to respond with high sensitivity to the low-affinity endogenous ligand, adenosine. Our data indicate that adipocyte A1AdoR were not spontaneously active. Therefore, tonic inhibition by A1AdoR of adenylyl cyclase activity and lipolysis in vitro and in vivo is probably caused by the presence of a nanomolar concentration of endogenous extracellular adenosine, sufficient to occupy <0.1% of adipocyte A1AdoR.
The affinity (KA value) of adenosine for the adipocyte A1AdoR was estimated to be 4.6 μM in this study. The values of KA for adenosine binding to A1AdoR coupled to activation of IKAdo and inhibition of β-adrenergic-stimulated ICa,L in guinea-pig atrial myocytes (Srinivas et al., 1997) were 2.7 and 5.6 μM, respectively. The value of KA for adenosine to bind to A2AAdoR in guinea-pig coronary artery was 1.8 μM (Shryock et al., 1998b). Thus AdoR in rat adipocytes and guinea-pig heart have similar, micromolar affinities for adenosine. It should be noted that values of KA represent apparent equilibrium dissociation constants for agonists to bind to adipocyte A1AdoR in intact cells (in the presence of G proteins and guanine nucleotides). They cannot be equated to values of Ki determined in membrane binding assays, although some agreement between values of KA (intact cell assay) and Ki (membrane binding assay in presence of GTP or guanylylimidodiphosphate) has been noted (Morey et al., 1998).
The receptor reserve for adenosine to activate A1AdoR, as judged by the value of the ratio of KA to EC50 (Table 1), was the highest of any of the A1AdoR agonists tested in this study. Agonists that were more potent than adenosine (e.g., R-PIA, CCPA) had a much greater affinity for the A1AdoR than adenosine, but were apparently less able to activate a functional response when bound to the receptor (i.e., had a lower intrinsic efficacy), in comparison to adenosine. On the other hand, CGS-21680, which is known as a highly selective A2AAdoR agonist, appeared to be able to activate the A1AdoR as efficiently as the potent A1AdoR agonist CCPA, but had a very low affinity for the receptor. Because of the high sensitivity of adipocytes to activation of A1AdoRs, however, CGS-21680 caused a half-maximal decrease of adipocyte cyclic AMP content at a concentration of only 0.5 μM (Table 1). The micromolar potency of CGS-21680 to decrease adipocyte cyclic AMP content could not be predicted from its affinity (0.3 mM, Table 1) alone. The finding serves to illustrate the principle that the potency of an agonist to cause a response is dependent both on the affinity of the agonist for the receptor and the receptor reserve (which itself is a function of receptor density and the efficiency of coupling of receptor activation to response).
Because pertussis toxin treatment of adipocytes concurrently reduced both the potency and efficacy of CCPA to reduce adipocyte cyclic AMP content, we conclude that inactivation of G proteins cannot be remedied by activation of a greater number of adenosine receptors. Thus there is no reserve of G proteins for an adenosine-induced inhibition of adenylyl cyclase. Recently, Baker et al. (2000) similarly concluded that there was no reserve of G protein for activation by A1AdoR in DDT1MF-2 cells. Adipocyte A1AdoR are known to be tightly associated with inhibitory G proteins (Londos et al., 1978; Ukena et al., 1984; Ramkumar & Stiles, 1988) and down-regulation of adipocyte content of inhibitory G proteins occurs upon excessive stimulation of A1AdoR (Parsons & Stiles, 1987; Green, 1987). It might be concluded that adenosine receptor and inhibitory G protein activities and regulation are intimately coupled in adipocytes, and that one is not likely to be affected independently of the other.
Tonic receptor activation is known to cause desensitization of cellular responsiveness to agonists. However, it is possible that tonic inhibition by adenosine of cyclic AMP accumulation can occur with minimal down-regulation of A1AdoR and inhibitory G proteins, given that activation of a small fraction (<0.1%) of adipocyte A1AdoR is sufficient to cause the tonic response. Results of studies by Green (1987) and Green et al. (1990) indicated that >1 nM of R-PIA was needed to cause down-regulation of adipocyte A1AdoR and inhibitory G proteins. Using a KA value of 76 nM for R-PIA (this study, Table 1), the per cent occupancy of adipocyte A1AdoR in the presence of 1 nM R-PIA can be calculated to be 1.3% (see Methods, ‘Calculation of receptor reserves for . . .'). It would therefore appear that occupancy of 0.1% of A1AdoR will cause no or minimal receptor and G protein down-regulation. If, however, the reserve of adipocyte A1AdoR were reduced, then higher concentrations of adenosine and occupancy of a greater percentage of the population of A1AdoR would be required to elicit a tonic response. This higher receptor occupancy might be associated with increased receptor down-regulation and desensitization. Thus, we speculate that the association of a high A1AdoR reserve and a tonic responsiveness of the adipocyte to adenosine may not be coincidental; rather, the former may be important to maintain the latter.
The receptor reserves for adenosine and A1AdoR agonists to reduce cyclic AMP content of adipocytes were much higher than the reported receptor reserves for actions of A1AdoR agonists on atrial myocytes, atrioventricular conduction, or the coronary vasculature (Srinivas et al., 1997; Morey et al., 1998; Dennis et al., 1992; Shryock et al., 1998b). The density of A1AdoR is ⩾10 fold higher in adipose than in cardiac tissue. The A1-adenosine receptor-effector systems in heart and adipose tissue are different but appropriate to serve the physiologic roles for adenosine in the two tissues. Cardiac AdoR are inactive under normal conditions of oxygen balance, and become active when oxygen consumption for cellular work exceeds the supply of oxygen by the coronary circulation, causing an increased local formation of adenosine (Bardenheuer & Schrader, 1986). Increased adenosine formation by myocytes and nodal cells causes activation of their own A1AdoR and the A2AAdoR of coronary arterioles, to reduce heart rate and cardiac work, and to increase coronary blood flow, respectively. This is the paradigm for a role of adenosine as a ‘retaliatory metabolite' (Belardinelli & Shryock, 1992). In this paradigm a high sensitivity of cells to adenosine is unnecessary because the local concentration of adenosine rises rapidly to activate adenosine receptors and retaliatory responses when tissue oxygenation is less than optimal. There is no evidence that adenosine is a retaliatory metabolite in adipose tissue. Catecholamines increase lipolysis and ATP utilization in adipocytes but do not appear to increase adenosine formation (Schwabe et al., 1973; Fain, 1979; Kather, 1988). The major pathway for AMP degradation in adipocytes is reported to be via IMP to inosine and hypoxanthine (Kather, 1988), neither of which are agonists of adenosine receptors. In adipose tissue adenosine appears to be a tonic modulator of function, inhibiting adenylyl cyclase and lipolysis, and increasing the responsiveness of the adipocyte to actions of insulin. Adenosine's role of tonic modulator is served by a high density of A1AdoR, efficiently coupled to adenylyl cyclase, and sensitive to concentrations of adenosine likely to be present in the interstitium of a tissue with relatively modest and fixed energy demands.
An implication of our findings is that the concentration of adenosine in the vicinity of adipocyte A1AdoR in vivo must be much lower than in blood plasma. The concentration of adenosine in rat blood plasma was reported to be 100 – 200 nM (Yamada et al., 1992). Because A1AdoR agonists reduce (van schaick et al., 1998; this study) and A1AdoR antagonists increase (Lanoue & Martin, 1994) the plasma concentration of NEFA, endogenous adenosine appears to be causing a submaximal inhibition of adenylyl cyclase and lipolysis in vivo. This suggests that adenosine concentration in the receptor compartment is close to the EC50 value of 1.4 nM for adenosine to reduce cyclic AMP in this study. If this assumption is correct, then proper function of the vascular endothelium may be important to maintain a low concentration of adenosine in the extravascular space of adipose tissue. The endothelium is reported to be a barrier to adenosine and is active in adenosine metabolism. We speculate that damage to the endothelium may cause an increase of adenosine concentration in the interstitium of adipose tissue. This may explain the findings that adenosine receptors in adipose tissue appear to be overactive as inhibitors of lipolysis in obese individuals (Lanoue & Martin, 1994), in spite of a reduction of receptor density (Kaartinen et al., 1991). Chronic stimulation of adipocyte A1AdoR by R-PIA has been shown to cause receptor down-regulation and insulin resistance (Parsons & Stiles, 1987; Green, 1987; Green et al., 1992). Insulin resistance and altered endothelial function are characteristic of diabetes and obesity.
Our finding of receptor reserve for A1AdoR agonists to decrease cyclic AMP content in adipocytes is supported by a report (Hoffman et al., 1989) that adipocytes from rats treated chronically with R-PIA showed a reduced sensitivity but no reduction of the maximal effect of R-PIA to inhibit isoproterenol-stimulated cyclic AMP accumulation. Green et al. (1992) reported a similar reduction of sensitivity without a change of the maximal response to R-PIA, in spite of a 50 – 60% loss of A1AdoR, in a study of isolated adipocytes chronically exposed to R-PIA. The finding by Kather (1988) that the EC50 for adenosine to reduce lipolysis of isolated human adipocytes was 6 nM suggests that a reserve of A1AdoR for reduction of lipolysis by adenosine is likely to be present in human fat cells.
In conclusion, the binding of adenosine to a small fraction of the large population of adipocyte A1AdoR was sufficient to cause a decrease of cell cyclic AMP content. Because adipocytes have a high density of A1AdoR, and activation by adenosine of a small fraction of adipocyte A1AdoR can mediate a functional response, adipocytes have a high sensitivity to adenosine. Tonic activity of the receptor will occur when the concentration of adenosine in the receptor compartment is 1 – 2 nM. Adipocyte A1AdoR were not active in the absence of agonist and their affinity for adenosine was low relative to the EC50 for adenosine to decrease cyclic AMP. It is suggested that the interstitial adenosine concentration in adipose tissue must normally be much lower than the adenosine concentration in blood plasma.
Acknowledgments
We gratefully acknowledge the support of National Institutes of Health awards HL56785 and DK56747. Additional support for experiments on awake rats was generously provided by CV Therapeutics, Inc.
Abbreviations
- A1AdoR
A1-adenosine receptor
- AOPCP
α,β-methylene adenosine 5′-diphosphate
- CCPA
2-chloro-N6-cyclopentyladenosine
- CGS-21680
2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine
- CPT
N6-cyclopentyltheophylline
- CPX
8-cyclopentyl-1,3-dipropylxanthine
- CVT-2759
{[(5-{6-[(3R)oxolan-3-yl]amino}purin-9-yl)(3S,2R,4R)-3,4-dihydroxyoxolan-2-yl]-methoxy}-N-methylcarboxamide
- EHNA
erythro-9-(2-hydroxy-3-nonyl)-adenine
- FSCPX
8-cyclopentyl-3-[3-[[4-(fluorosulphonyl)benzoyl]oxy]propyl]-1-propylxanthine
- KA
agonist equilibrium dissociation constant
- KRH
Krebs – Ringer – HEPES
- N-0861
(±)-N6-endonorbornan-2-yl-9-methyladenine
- NEFA
non-esterified fatty acid
- ρ
fractional receptor occupancy
- R-PIA
R-(−)-N6-(2-phenylisopropyl)adenosine
- XAC
xanthine amine congener
References
- AKTORIES K., SCHULTZ G., JAKOBS K.H. Inhibition of adenylate cyclase and stimulation of a high affinity GTPase by the antilipolytic agents, nicotinic acid, acipimox and various related compounds. Arzneimittelforschung. 1983;33:1525–1527. [PubMed] [Google Scholar]
- BAKER S.P., SCAMMELLS P.J., BELARDINELLI L. Differential A1-adenosine receptor reserve for inhibition of cyclic AMP accumulation and G-protein activation in DDT1MF-2 cells. Br. J. Pharmacol. 2000;130:1156–1164. doi: 10.1038/sj.bjp.0703405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARDENHEUER H., SCHRADER J. Supply-to-demand ratio for oxygen determines formation of adenosine by the heart. Am. J. Physiol. 1986;250:H173–H180. doi: 10.1152/ajpheart.1986.250.2.H173. [DOI] [PubMed] [Google Scholar]
- BELARDINELLI L., SHRYOCK J.C. Does adenosine function as a retaliatory metabolite in the heart. News Physiol. Sci. 1992;7:52–56. [Google Scholar]
- COATES J., SHEEHAN M.J., STRONG P. 1,3-Dipropyl-8-cyclopentyl xanthine (DPCPX): a useful tool for pharmacologists and physiologists. Gen. Pharmacol. 1994;25:387–394. doi: 10.1016/0306-3623(94)90185-6. [DOI] [PubMed] [Google Scholar]
- DENNIS D., JACOBSON K., BELARDINELLI L. Evidence of spare A1-adenosine receptors in guinea pig atrioventricular node. Am. J. Physiol. 1992;262:H661–H671. doi: 10.1152/ajpheart.1992.262.3.H661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAIN J.N. Effect of lipolytic agents on adenosine and AMP formation by fat cells. Biochim. Biophys. Acta. 1979;573:510–520. doi: 10.1016/0005-2760(79)90225-x. [DOI] [PubMed] [Google Scholar]
- FAIN J.N., POINTER R.H., WARD W.F. Effects of adenosine nucleosides on adenylate cyclase, phosphodiesterase, cyclic adenosine monophosphate accumulation, and lipolysis in fat cells. J. Biol. Chem. 1972;247:6866–6872. [PubMed] [Google Scholar]
- FAIN J.N., WIESER P.B. Effects of adenosine deaminase on cyclic adenosine monophosphate accumulation, lipolysis, and glucose metabolism of fat cells. J. Biol. Chem. 1975;250:1027–1034. [PubMed] [Google Scholar]
- FURCHGOTT R.F., BURSZTYN P. Comparison of dissociation constants and of relative efficacies of selected agonists acting on parasympathetic receptors. Ann. NY Acad. Sci. 1967;144:882–899. [Google Scholar]
- GREEN A. Adenosine receptor down-regulation and insulin resistance following prolonged incubation of adipocytes with an A1 adenosine receptor agonist. J. Biol. Chem. 1987;262:15702–15707. [PubMed] [Google Scholar]
- GREEN A., JOHNSON J.L., MILLIGAN G. Down-regulation of Gi sub-types by prolonged incubation of adipocytes with an A1 adenosine receptor agonist. J. Biol. Chem. 1990;265:5206–5210. [PubMed] [Google Scholar]
- GREEN A., MILLIGAN G., DOBIAS S.B. Gi down-regulation as a mechanism for heterologous desensitization in adipocytes. J. Biol. Chem. 1992;267:3223–3229. [PubMed] [Google Scholar]
- HOFFMAN B.B., PROKOCIMER P., THOMAS J.M., VAGELOS R, CHANG H., REAVEN G.M. Cellular tolerance to adenosine receptor-mediated inhibition of lipolysis: altered adenosine 3′,5′-monophosphate metabolism and protein kinase activation. Endocrinology. 1989;124:2434–2442. doi: 10.1210/endo-124-5-2434. [DOI] [PubMed] [Google Scholar]
- HONNOR R.C., DHILLON G.S., LONDOS C. cAMP-dependent protein kinase and lipolysis in rat adipocytes. I. Cell preparation, manipulation, and predictability in behaviour. J. Biol. Chem. 1985;260:15122–15129. [PubMed] [Google Scholar]
- KAARTINEN J.M., HRENIUK S.P., MARTIN L.F., RANTA S., LANOUE K.F., OHISALO J.J. Attenuated adenosine-sensitivity and decreased adenosine-receptor number in adipocyte plasma membranes in human obesity. Biochem. J. 1991;279:17–22. doi: 10.1042/bj2790017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATHER H. Purine accumulation in human fat cell suspensions. Evidence that human adipocytes release inosine and hypoxanthine rather than adenosine. J. Biol. Chem. 1988;263:8803–8809. [PubMed] [Google Scholar]
- KATHER H. Beta-adrenergic stimulation of adenine nucleotide catabolism and purine release in human adipocytes. J. Clin. Invest. 1990;85:106–114. doi: 10.1172/JCI114399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANOUE K.F., MARTIN L.F. Abnormal A1 adenosine receptor function in genetic obesity. FASEB J. 1994;8:72–80. doi: 10.1096/fasebj.8.1.8299893. [DOI] [PubMed] [Google Scholar]
- LOHSE M.J., KLOTZ K.-N., SCHWABE U. Agonist photoaffinity labeling of A1 adenosine receptors: persistent activation reveals spare receptors. Mol. Pharmacol. 1986;30:403–409. [PubMed] [Google Scholar]
- LONDOS C., COOPER D.M.F., SCHLEGEL W., RODBELL M. Adenosine analogs inhibit adipocyte adenylate cyclase by a GTP-dependent process: basis for actions of adenosine and methylxanthines on cyclic AMP production and lipolysis. Proc. Natl. Acad. Sci. U.S.A. 1978;75:5362–5366. doi: 10.1073/pnas.75.11.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LORENZEN A., STANNEK C., LANG H., ANDRIANOV V., KALVINSH I., SCHWABE U. Characterization of a G protein-coupled receptor for nicotinic acid. Mol. Pharmacol. 2001;59:349–357. doi: 10.1124/mol.59.2.349. [DOI] [PubMed] [Google Scholar]
- MORENO F.J., MILLS I., GARCIA-SAINZ J.A., FAIN J.N. Effects of pertussis toxin treatment on the metabolism of rat adipocytes. J. Biol. Chem. 1983;258:10938–10943. [PubMed] [Google Scholar]
- MOREY T.E., BELARDINELLI L., DENNIS D.M. Validation of Furchgott's method to determine agonist-dependent A1-adenosine receptor reserve in guinea-pig atrium. Br. J. Pharmacol. 1998;123:1425–1433. doi: 10.1038/sj.bjp.0701747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLANSKY L., MYERS G.A., POHL S.L., HEWLETT E.L. Promotion of lipolysis in rat adipocytes by pertussis toxin: reversal of endogenous inhibition. Proc. Natl. Acad. Sci. U.S.A. 1983;80:6547–6551. doi: 10.1073/pnas.80.21.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARSONS W.J., STILES G.L. Heterologous desensitization of the inhibitory A1 adenosine receptor-adenylate cyclase system in rat adipocytes. J. Biol. Chem. 1987;262:841–847. [PubMed] [Google Scholar]
- PARSONS W.J., RAMKUMAR V., STILES G.L. Isobutylmethylxanthine stimulates adenylate cyclase by blocking the inhibitory regulatory protein, Gi. Mol. Pharmacol. 1988;34:37–41. [PubMed] [Google Scholar]
- RAMKUMAR V., STILES G.L. Reciprocal modulation of agonist and antagonist binding to A1 adenosine receptors by guanine nucleotides is mediated via a pertussis toxin-sensitive G protein. J. Pharmacol. Exp. Ther. 1988;246:1194–1200. [PubMed] [Google Scholar]
- SCAMMELLS P.J., BAKER S.P., BELARDINELLI L., OLSSON R.A. Substituted 1,3-dipropylxanthines as irreversible antagonists of A1 adenosine receptors. J. Med. Chem. 1994;37:2704–2712. doi: 10.1021/jm00043a010. [DOI] [PubMed] [Google Scholar]
- SCHWABE U., EBERT R. Stimulation of cyclic adenosine 3′,5′-monophosphate accumulation and lipolysis in fat cells by adenosine deaminase. Naunyn-Schmiedeberg's Arch. Pharmacol. 1974;282:33–44. doi: 10.1007/BF00647401. [DOI] [PubMed] [Google Scholar]
- SCHWABE U., EBERT R., ERBLER H.C. Adenosine release from isolated fat cells and its significance for the effects of hormones on cyclic 3′,5′-AMP levels and lipolysis. Naunyn-Schmiedeberg's Arch. Pharmacol. 1973;276:133–148. doi: 10.1007/BF00501186. [DOI] [PubMed] [Google Scholar]
- SCHWABE U., SCHÖNHÖFER P.S., EBERT R. Facilitation by adenosine of the action of insulin on the accumulation of adenosine 3′ : 5′-monophosphate, lipolysis, and glucose oxidation in isolated fat cells. Eur. J. Biochem. 1974;46:537–545. doi: 10.1111/j.1432-1033.1974.tb03647.x. [DOI] [PubMed] [Google Scholar]
- SHRYOCK J.C., OZECK M.J., BELARDINELLI L. Inverse agonists and neutral antagonists of recombinant human A1 adenosine receptors stably expressed in Chinese hamster ovary cells. Mol. Pharmacol. 1998a;53:886–893. [PubMed] [Google Scholar]
- SHRYOCK J.C., SNOWDY S., BARALDI P.G., CACCIARI B., SPALLUTO G., MONOPOLI A., ONGINI E., BAKER S.P., BELARDINELLI L. A2A-adenosine receptor reserve for coronary vasodilation. Circulation. 1998b;98:711–718. doi: 10.1161/01.cir.98.7.711. [DOI] [PubMed] [Google Scholar]
- SRINIVAS M., SHRYOCK J.C., DENNIS D.M., BAKER S.P., BELARDINELLI L. Differential A1 adenosine receptor reserve for two actions of adenosine on guinea pig atrial myocytes. Mol. Pharmacol. 1997;52:683–691. doi: 10.1124/mol.52.4.683. [DOI] [PubMed] [Google Scholar]
- SRINIVAS M., SHRYOCK J.C., SCAMMELLS P.J., RUBLE J., BAKER S.P., BELARDINELLI L. A novel irreversible antagonist of the A1-adenosine receptor. Mol. Pharmacol. 1996;50:196–205. [PubMed] [Google Scholar]
- UKENA D., POESCHLA E., SCHWABE U. Guanine nucleotide and cation regulation of radioligand binding to Ri adenosine receptors of rat fat cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1984;326:241–247. doi: 10.1007/BF00505325. [DOI] [PubMed] [Google Scholar]
- VANNUCCI S.J., KLIM C.M., MARTIN L.F., LANOUE K.F. A1-Adenosine receptor-mediated inhibition of adipocyte adenylate cyclase and lipolysis in Zucker rats. Am. J. Physiol. 1989;257:E871–E878. doi: 10.1152/ajpendo.1989.257.6.E871. [DOI] [PubMed] [Google Scholar]
- VAN SCHAICK E.A., TUKKER H.E., ROELEN H.C.P.F., IJZERMAN A.P., DANHOF M. Selectivity of action of 8-alkylamino analogues of N6-cyclopentyladenosine in vivo: haemodynamic versus anti-lipolytic responses in rats. Br. J. Pharmacol. 1998;124:607–618. doi: 10.1038/sj.bjp.0701868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VASSAUX G., GAILLARD D., MARI B., AILHAUD G., NEGREL R. Differential expression of adenosine A1 and A2 receptors in preadipocytes and adipocytes. Biochem. Biophys. Res. Commun. 1993;193:1123–1130. doi: 10.1006/bbrc.1993.1742. [DOI] [PubMed] [Google Scholar]
- WU L., BELARDINELLI L., ZABLOCKI J.A., PALLE V., SHRYOCK J.C. A partial agonist of the A1-adenosine receptor selectively slows AV conduction in guinea pig hearts. Am. J. Physiol. 2001;280:H334–H343. doi: 10.1152/ajpheart.2001.280.1.H334. [DOI] [PubMed] [Google Scholar]
- YAMADA K., GOTO A., ISHII M., YOSHIOKA M., MATSUOKA H., SUGIMOTO T. Plasma adenosine concentrations are elevated in conscious spontaneously hypertensive rats. Clin. Exp. Pharmacol. Physiol. 1992;19:563–567. doi: 10.1111/j.1440-1681.1992.tb00505.x. [DOI] [PubMed] [Google Scholar]