

Abstract
In rat cerebellar astrocytes, intracellular Ca2+ store depletion by receptor agonists or sarco(endo)plasmic reticulum Ca2+ ATPase inhibitors induced a transient increase in the intracellular Ca2+ concentration ([Ca2+]i) in the absence of extracellular Ca2+ and a sustained increase in its presence.
After 10 min treatment with thapsigargin, the [Ca2+]i was unaffected by removal of thapsigargin, but fell rapidly to the basal level when extracellular Ca2+ was removed, suggesting the involvement of capacitative Ca2+ entry (CCE); this effect was not seen until cells had been exposed to thapsigargin for at least 2 min.
Using the whole cell voltage clamp technique, a 60 – 100 pA inward current was activated by store depletion, the reversal potential ranging from −5 to 0 mV.
When extracellular Na+ was isotonically replaced by Tris, the thapsigargin-induced [Ca2+]i increase was enhanced, while the inward current was reduced, indicating that store-operated Ca2+ channels were permeable to Na+; however, they were not permeable to Sr2+ or Ba2+.
Thapsigargin-induced CCE remained the same in the presence of nifedipine, La3+ or Cd2+, while it was inhibited in the presence of SK&F96365.
In cerebellar astrocytes, inhibition of protein serine/threonine phosphorylation promoted CCE.
In conclusion, in rat cerebellar astrocytes, store depletion activated a CCE via channels which were permeable to Ca2+ and Na+ and regulated by phosphorylation.
Keywords: Capacitative Ca2+ entry, intracellular Ca2+ concentration, store-operated Ca2+ channel, cerebellar astrocytes, thapsigargin, phosphorylation
Introduction
Increases in the intracellular Ca2+ concentration ([Ca2+]i) play pivotal roles in the regulation of many cellular functions, including neurotransmission, secretion, contraction, motility, adhesion, proliferation, differentiation, gene expression, and cell death, and cells have therefore evolved distinct mechanisms to control rapidly and precisely such changes (Berridge, 1997). A low resting [Ca2+]i can be maintained either by extrusion of Ca2+ across the plasma membrane or by its sequestration into intracellular organelles (Carafoli, 1987). The ATP-dependent Ca2+ pump and the Na+/Ca2+ exchanger, localized in the plasma membrane, are responsible for Ca2+ extrusion (Gill et al., 1984), while the endoplasmic reticulum (ER) is believed to be the major organelle that stores Ca2+ via a Ca2+ pump (Pozzan et al., 1994).
Ca2+ influx from the extracellular compartment and Ca2+ release from intracellular Ca2+ stores both contribute to increases in the [Ca2+]i. In excitable cells, voltage-operated Ca2+ channels (VOC) are mainly responsible for the influx of extracellular Ca2+ seen on membrane depolarization (Hess, 1990). In response to a receptor agonist, two distinct mechanisms, Ca2+ release from intracellular Ca2+ stores via IP3 generation and Ca2+ influx via receptor-operated Ca2+ channels, are responsible for the increased [Ca2+]i (Marks, 1997; Furuichi & Mikoshiba, 1995; Barnard, 1996). Thus, the intracellular Ca2+ stores have a dual function as a Ca2+ buffer, removing Ca2+ from the cytosol by an ATP-dependent process and releasing Ca2+ in response to receptor activation, thus causing an increase in the [Ca2+]i. On receptor activation, the Ca2+ permeability of the plasma membrane increases after depletion of the intracellular Ca2+ stores (Neher, 1992; Clapham, 1993); this Ca2+ influx pathway linked to a reduced store Ca2+ content is termed capacitative Ca2+ entry (CCE) and occurs via so-called store-operated Ca2+ channels (SOC) (Putney, 1990).
CCE has been identified in many cells (for review, see Parekh & Penner, 1997; Putney, 1999; Barritt, 1999), but the mechanism linking store-depletion and the increased Ca2+ permeability of the plasma membrane remains elusive. Several mechanisms have been proposed. In mast cells, an inward Ca2+ current (ICRAC, Ca2+ release-activated Ca2+ current) is seen following depletion of intracellular Ca2+ stores (Hoth & Penner, 1992), while, in mouse pancreatic acinar cells, CCE results from a ICRANC, Ca2+ release-activated nonselective cation current (Krause et al., 1996). In Drosophila, the trp genes may encode SOCs (Zhu et al., 1996; Harteneck et al., 2000). It has been suggested that a Ca2+ influx factor (CIF), generated when stores are depleted, diffuses to the plasma membrane where the SOC is located, and activates CCE (Randriamampita & Tsien, 1993). Alternatively, direct physical contact between the plasma membrane and the endoplasmic reticulum IP3 receptor, which detects depletion of the intracellular Ca2+ stores, might be responsible for activating CCE (Irvine, 1990). Recently, two studies have provided evidence to support this latter hypothesis of an exocytosis-like mechanism of CCE activation (Yao et al., 1999; Patterson et al., 1999).
In this study, using primary cultures of rat cerebellar astrocytes, we characterized store depletion-induced Ca2+ influx and found that CCE is involved in the Ca2+ signalling process in these cells and is inhibited by protein serine/threonine phosphorylation.
Methods
Cell culture
Primary cultures of rat cerebellar astrocytes were prepared from 7-day-old Sprague-Dawley rat cerebellum, as described previously (Gallo et al., 1982). Briefly, the cells were dissociated in a Ca2+-free buffer containing 0.25 mg ml−1 of trypsin and 2400 units ml−1 of DNase, and collected by centrifugation before being plated on poly-L-lysine-coated 24 mm glass coverslips (1×106 cells/coverslip) and grown in a humidified atmosphere of 5% CO2/95% air at 37°C. The freshly isolated cells from the cerebellum contained both granule neurons and glial cells. To obtain granule neuron-enriched cultures, the growth medium was supplemented with 21.5 mM KCl and 10 μM cytosine arabinoside to minimize non-neuronal cell proliferation. In the absence of high K+ and cytosine arabinoside, the number of non-neuronal cells was increased. The non-neuronal cells used in this study were identified as astrocytes by positive immunocytochemical staining for glial fibrillary acidic protein and their distinct morphology (data not shown). Astrocyte-enriched cultures were grown in basal Eagle's medium supplemented with 10% foetal bovine serum, 2 mM glutamine, 100 units ml−1 of penicillin, and 100 μg ml−1 of streptomycin. Experiments were performed on 8 – 10 day cultures.
[Ca2+]i measurement
The [Ca2+]i change in a single cell was measured using the fluorescent Ca2+ indicator, fura-2, as described previously (Grynkiewicz et al., 1985; Chin & Chueh, 1998). Briefly, cells grown on glass coverslips were loaded with fura-2 by incubation for 20 min at 37°C with 10 μM fura-2 acetoxymethyl ester and 1% pluronic acid in loading buffer (in mM: NaCl 150, KCl 5, glucose 5, CaCl2 2.2, MgCl2 1 and HEPES 10, pH 7.4). The coverslips were then mounted in a modified Cunningham chamber (Cunningham & Szenberg, 1968) attached to the stage of a Nikon diaphot inverted microscope equipped with a Nikon 40× fluor objective, and cell fluorescence monitored using a dual-excitation spectrofluorometer with a photomultiplier-based detection system (Spex Industries, Edison, NJ, U.S.A.). Only a single cell per coverslip, selected by a pinhole diaphragm placed in the image plane in front of the photomultiplier, was used in experiments; this was excited alternately with 340 and 380 nm light, and the emitted fluorescent light collected by the objective through a 510 nm long-wave pass filter. The [Ca2+]i was calculated from the ratio of the fluorescence at 340 nm and 380 nm according to the equation derived by Grykiewicz et al. (1985) using parameters obtained on our instrument for fura-2 in rats cerebellar astrocytes: Rmin=0.68; Rmax=5.42; Sf2/Sb2=4.62; Kd=135 nM. In experiments to measure Ba2+ or Sr2+ influx, the CaCl2 in the loading buffer was replaced with BaCl2 or SrCl2, respectively. Under nominally Ca2+-free conditions, extracellular Ca2+ was omitted from the loading buffer after fura-2 loading. All experiments were performed on at least 18 cells. The results of one representative experiment are illustrated graphically and the mean±s.d. values for the [Ca2+]i increase, calculated for n cells of different batches, are given in the text.
Examination of fluorescence quenching by Mn2+
Fura-2-loaded cells, grown on coverslips, were transferred to loading buffer containing 0.2 mM Ca2+ (low calcium loading buffer; LCL buffer). They were then incubated for 5 min in LCL buffer alone or LCL buffer containing 100 nM staurosporine, 100 nM phorbol 12-myristate 13-acetate (PMA), or 2 nM okadaic acid, then 1 mM Mn2+, either alone or together with 1 μM thapsigargin, was added, and Mn2+ influx monitored by the quenching of fura-2 fluorescence at the isosbestic wavelength, 360 nm. All experiments were performed at least six times, using different batches of cells. The results of one representative experiment are illustrated graphically, while the mean±s.d. for the fluorescence quenching during the initial 20 s after Mn2+ addition, calculated from the number (n) of cells, are shown in Table 1 and were analysed by Student's t-test.
Table 1.
Effect of protein phosphorylation on thapsigargin-induced Mn2+ influx

Electrophysiological recording
Thapsigargin-induced currents were measured by the whole cell voltage clamp technique using a patch clamp amplifier (Axopatch 1-D, Axon Instruments, Inc., Foster City, CA, U.S.A.) as described previously (Hamill et al., 1981). The recording chamber was superfused with bathing buffer consisting of: (in mM) NaCl 137, CsCl 4, MgCl2 0.5, CaCl2 5.4, glucose 5, HEPES 10, pH 7.4, while the pipette buffer consisted of (in mM): CsCl 110, MgATP 5, EGTA 20, tetraethylammonium chloride 20 and HEPES 10, pH 7.2. A gigaohm seal was formed with a borosilicate glass patch pipette with a tip diameter of 1 – 1.5 μm and an input resistance of 2 – 5 MΩ. Data were sampled at 2 kHz and filtered at 1 kHz (−3dB cutoff frequency) using an eight-pole Bessel-type filter (VBF/8.03, Kemo, Beckenham, Kent, U.K.). The digitised traces were stored on a magneto-optical disk unit (RMO-S550, Sony, Japan) and analysed using pCLAMP software, version 6.03 (Axon Instruments, Inc.). To record thapsigargin-induced currents, the membrane potential was held at −70 mV and step pulsed for 100 ms to −100 mV at 10 s intervals, currents being recorded before and after thapsigargin perfusion and after washout. To obtain the current-voltage relationship, ramp pulses were given at 5 s intervals from the holding potential of −40 to −100 mV, then to 100 mV, then back to the holding potential at a speed of 2 V/s. In some experiments, extracellular Na+ was isotonically replaced with Tris. All experiments were repeated six times using different batches of cells with similar results. One set of representative data is shown and the mean±s.d. values for the current change are given in the text.
Materials
Basal Eagle's medium, foetal bovine serum, glutamine, and penicillin-streptomycin were purchased from Life Technologies, Inc. (Grand Island, NY, U.S.A.). Fura-2 acetoxymethyl ester was obtained from Molecular Probes (Eugene, OR, U.S.A.). Thapsigargin, okadaic acid, staurosporine, glutamate, and PMA were purchased from Research Biochemicals International (Natick, MA, U.S.A.). ATP, poly-L-lysine, trypsin, DNase, angiotensin II, EGTA, and cytosine arabinoside were purchased from Sigma (St. Louis, MO, U.S.A.). SK&F96365 was purchased from Biomol (Plymouth Meeting, PA, U.S.A.). All other chemicals were of analytical grade and obtained from Merck (Darmstadt, Germany).
Results
To determine whether cerebellar astrocytes contained SOCs we first depleted the intracellular Ca2+ stores via IP3 generation using ATP or angiotensin II in the presence or absence of extracellular Ca2+. As shown in Figure 1 (traces a and c), in the presence of extracellular Ca2+, the [Ca2+]i increased rapidly from a basal level of 50±22 nM (n=40) to 856±60 nM (n=21) or 790±35 nM (n=19) in response to addition of 100 μM ATP or 100 nM angiotensin II, respectively, then declined to a sustained level of 213±21 nM (n=21) or 224±16 nM (n=19) 180 s after addition of ATP or angiotensin II, respectively. In the absence of extracellular Ca2+, the ATP- or angiotensin II-induced [Ca2+]i increase peaked at 682±47 nM (n=18) or 486±24 nM (n=21), respectively, 20 or 38% lower than in the presence of Ca2+, and the effect was transient, with the [Ca2+]i falling back completely to the basal level (Figure 1, traces b and d).
Figure 1.
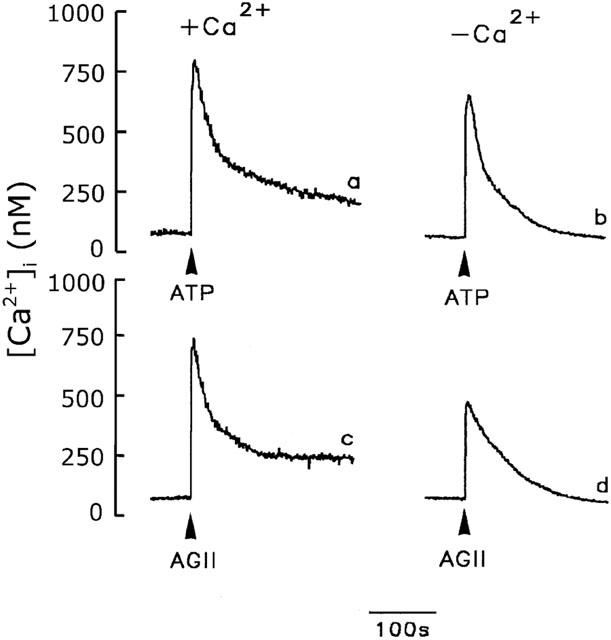
Effect of extracellular Ca2+ on the [Ca2+]i increase in cerebellar astrocytes. The [Ca2+]i increase induced by 100 μM ATP (traces a and b) or 100 nM angiotensin II (AGII) (traces c and d) were measured in the presence (traces a and c) or absence (traces b and d) of extracellular Ca2+.
The sustained [Ca2+]i phase seen in the presence of extracellular Ca2+ represented the influx of Ca2+, probably via SOCs, since intracellular Ca2+ stores are depleted by ATP- or angiotensin II-induced IP3 generation. When the intracellular Ca2+ stores were depleted using the intracellular Ca2+ pump inhibitors, thapsigargin and cyclopiazonic acid, similar results were seen. As shown in Figure 2A, a slow but significant [Ca2+]i increase was induced by 1 μM thapsigargin (Chin & Chueh, 1998) and reached the peak of 321±40 nM (n=15) at 210 s in the presence (trace a) or absence (trace b) of extracellular Ca2+. The [Ca2+]i returned to the basal level in the absence of extracellular Ca2+, while it remained high in its presence. This sustained [Ca2+]i increase, about 463±41 nM (n=15) 15 min after thapsigargin addition, represents extracellular Ca2+ entry, rather than intracellular Ca2+ release, since the [Ca2+]i was not affected by thapsigargin removal after the cells had been exposed to thapsigargin for 10 min, but immediately declined to the basal level when extracellular Ca2+ was removed and returned to the previous sustained level when 2.2 mM Ca2+ was added back to the bathing solution (Figure 2B). Identical results were seen using cyclopiazonic acid (10 μM) (Krishna et al., 2001) (Figure 3). The basal and sustained [Ca2+]i was 77±12 nM (n=15) and 291±35 nM (n=15), respectively.
Figure 2.
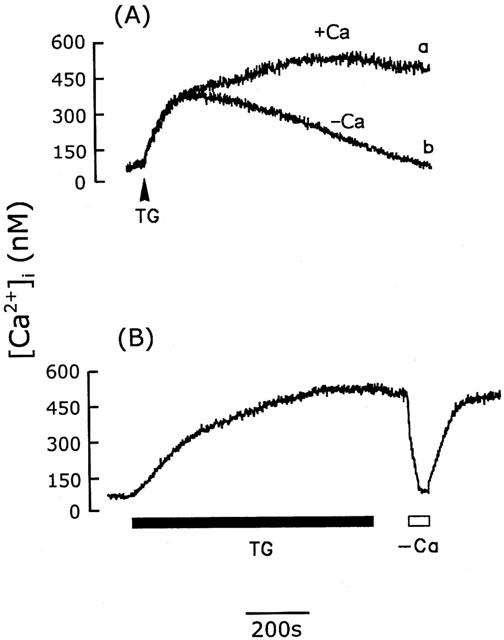
Activation of CCE by thapsigargin in cerebellar astrocytes. (A) [Ca2+]i increases induced by 1 μM thapsigargin (TG) in the presence (trace a) or absence (trace b) of extracellular Ca2+. (B) After 10 min exposure to thapsigargin (1 μM) (TG), the [Ca2+]i remained elevated after removal of thapsigargin, but fell to the basal level when extracellular Ca2+ was removed and returned to the elevated level when Ca2+ was added back to the bathing buffer. Similar results were seen using five different batches of cells (n=15).
Figure 3.
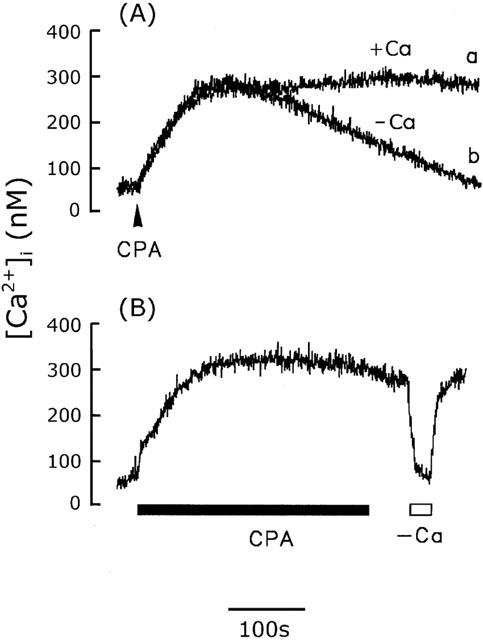
Activation of CCE by cyclopiazonic acid in cerebellar astrocytes. (A) [Ca2+]i increase induced by 10 μM cyclopiazonic acid (CPA) in the presence (trace a) or absence (trace b) of extracellular Ca2+. (B) After 10 min exposure to CPA, the [Ca2+]i was unaffected by removal of CPA, but fell to the basal level when extracellular Ca2+ was removed. The experiment was repeated five times using different batches of cells with similar results (n=15).
Our data indicate that, in rat cerebellar astrocytes, CCE occurs following depletion of the intracellular Ca2+ stores. Figure 4 shows the relationship between CCE and the duration of thapsigargin treatment. To detect CCE, we determined whether the [Ca2+]i decreased after the simultaneous removal of thapsigargin and extracellular Ca2+. As shown in Figure 4 (trace a), when extracellular Ca2+ and thapsigargin were removed 1.5 min after exposure of cells to 1 μM thapsigargin, the [Ca2+]i was unaffected, indicating that Ca2+ influx via SOCs was not activated and that the [Ca2+]i increase was attributable to intracellular Ca2+ release. However, in another cell after 2 min of thapsigargin treatment, CCE was detected, since the [Ca2+]i was reduced when thapsigargin and extracellular Ca2+ were removed (trace b), and this effect became progressively greater the longer the cells were exposed to thapsigargin (traces c – f). In this experiment, each trace used a fresh individual cell to expose thapsigargin for indicated time. These results indicate that longer exposure to thapsigargin resulted in a lower amount of releasable Ca2+ within the Ca2+ stores and a greater contribution of extracellular Ca2+ influx to the thapsigargin-induced increased [Ca2+]i. To confirm that the degree of availability of the releasable Ca2+ within the Ca2+ pools fell with increased time of exposure to thapsigargin, [Ca2+]i increase induced by 100 μM ATP was measured 1 min after extracellular Ca2+ was removed following exposure of individual cell to thapsigargin for indicated time. As shown in Figure 4 (trace f), virtually no ATP-induced [Ca2+]i increase was seen after cells were exposed to thapsigargin for 10 min, while a rapid and significant [Ca2+]i increase was evoked if cells were exposed to thapsigargin for a shorter time period (traces a – c). Figure 5 summarizes the statistical data measured in Figure 4. The dependence of Ca2+ influx via CCE on thapsigargin exposure time period contrasts with the amplitude of 100 μM ATP-induced [Ca2+]i increase, an indication of the releasable Ca2+ within the Ca2+ stores.
Figure 4.
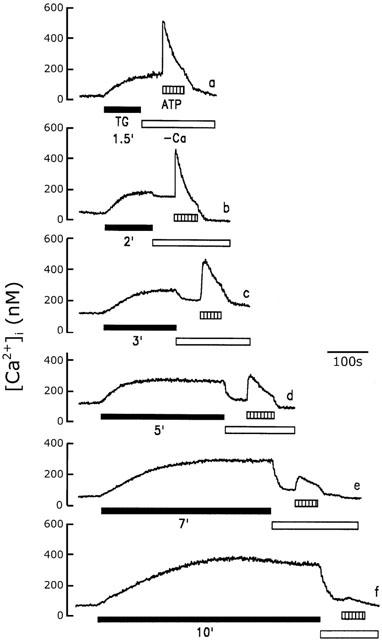
Dependence of CCE on the thapsigargin exposure time. After cells were treated with 1 μM thapsigargin (TG) for 1.5 min (trace a), 2 min (trace b), 3 min (trace c), 5 min (trace d), 7 min (trace e), or 10 min (trace f), as indicated, extracellular Ca2+ was removed, then, 1 min later, the ATP (100 μM) was added. [Ca2+]i increase was measured. The experiment was repeated six times using different batches of cells with similar results.
Figure 5.
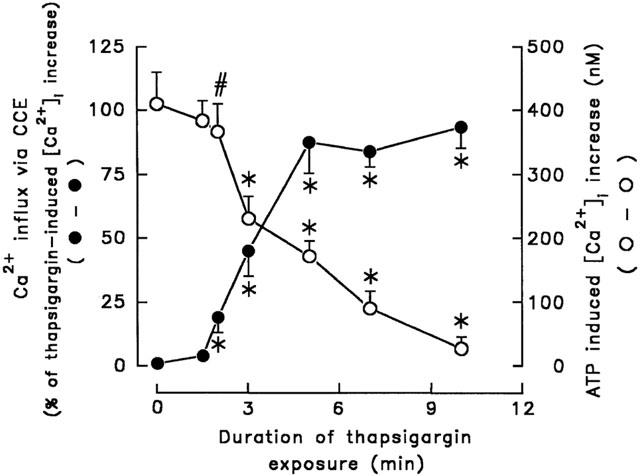
Effect of thapsigargin exposure time on CCE and ATP-induced [Ca2+]i increase. Data were obtained from Figure 4. Thapsigargin exposure time was plotted versus thapsigargin-induced CCE or ATP-induced [Ca2+]i increase. The former was expressed as the per cent of [Ca2+]i decrease in response to the removal of extracellular Ca2+ in thapsigargin-induced [Ca2+]i increase and the latter was the subsequent ATP (100 μM)-induced [Ca2+]i increase in the absence of extracellular Ca2+. Data were the mean±s.d. (n=18). *P<0.001 and #P<0.01, significant different from control cells without treatment with thapsigargin, respectively.
In many cells, the SOC is also permeable to Na+ (Krause et al., 1996). We therefore determined the effect of Na+ on SOC-mediated Ca2+ influx in rat cerebellar astrocytes. Figure 6 shows thapsigargin-induced [Ca2+]i increases measured in Na+-containing (trace a) and Na+-free buffer (trace b). As shown in trace b, when extracellular Na+ was isotonically replaced with Tris and the cells exposed to 1 μM thapsigargin for 5 min, the sustained [Ca2+]i increase, 310±20 nM (n=5), was completely attributable to extracellular Ca2+ influx, since the [Ca2+]i rapidly declined to the basal level, 44±12 nM (n=5), when extracellular Ca2+ was removed. Subsequent addition of 100 μM ATP did not induce a [Ca2+]i increase, indicating that the intracellular Ca2+ stores were indeed depleted; this contrasts with the situation in Na+-containing buffer in which 10 min treatment with 1 μM thapsigargin was required to completely deplete the intracellular Ca2+ stores (Figure 4, trace f). These results indicate that, under Na+-free conditions, depletion of the intracellular Ca2+ stores was readily achieved, which, in turn, activated CCE.
Figure 6.
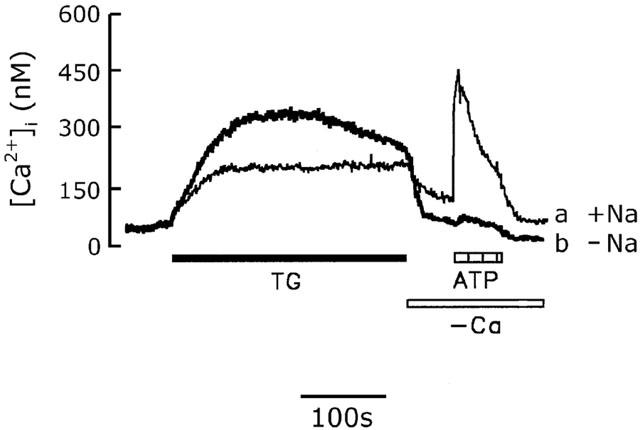
Effect of extracellular Na+ on the thapsigargin-induced CCE. Cells were bathed in either Na+-containing buffer (trace a) or Na+-free loading buffer in which Na+ was isotonically replaced by Tris (trace b) and the [Ca2+]i changes induced by 1 μM thapsigargin (TG), Ca2+ removal, or 100 μM ATP were measured. The experiment was repeated five times with similar results.
To further characterize SOC-mediated Ca2+ entry and the effect of Na+ on SOCs, we used the whole cell voltage clamp technique to measure the thapsigargin-induced current by step pulses. The membrane potential was held at −70 mV and step hyperpolarized to −100 mV for 100 ms at 10 s intervals, and the amplitudes of the currents measured at −100 mV before, and after, perfusion with thapsigargin, and after thapsigargin washout were plotted versus time. As shown in Figure 7, following exposure of cells to 1 μM thapsigargin, an average of 80±13 pA (n=10) inward current was evoked and was still present after thapsigargin washout (Figure 7B), while no current change was seen after perfusion with buffer alone (Figure 7A). To obtain the current-voltage curve for the thapsigargin-activated current, the current was recorded by voltage ramps spanning from −100 mV to +100 mV in 100 ms at 5 s intervals with a holding potential of −40 mV. As shown in Figure 8, the current-voltage curve for the thapsigargin-activated current showed an outward rectification relationship, with the reversal potential ranging from −5 to 0 mV (left panel, middle). The amplitude of the thapsigargin-activated current was reduced when the Na+ in the bathing solution was isotonically replaced with Tris (right panel, middle), while the reversal potential remained the same. The reduction in current at −100 mV was 24±6% (n=10) and the reversal potentials were −2.4±0.3 mV (n=10) in Na+ containing and 1.6±0.2 mV (n=10) in Tris containing buffer. Similar current-voltage curves were seen after thapsigargin washout in both Na+- and Tris-containing buffer (bottom panels).
Figure 7.
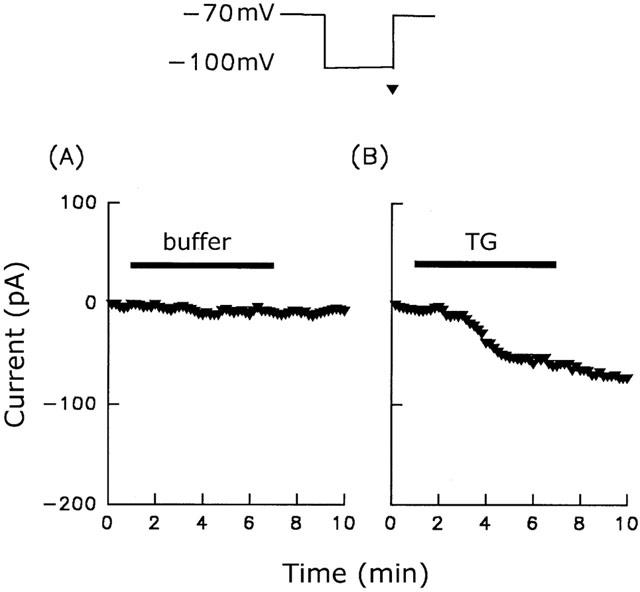
ISOC activation by thapsigargin. The current was measured by step-pulse recording from a holding potential of −70 mV to a test potential of −100 mV over a period of 100 ms executed every 10 s. From these pulses, the amplitudes of the currents measured at −100 mV (indicated by the triangle) before, and after, perfusion with, and after washout of, buffer (A) or 1 μM thapsigargin (B) were plotted versus time. The perfusion period is indicated by the horizontal bars. Similar results were seen using six different batches of cells (n=10).
Figure 8.
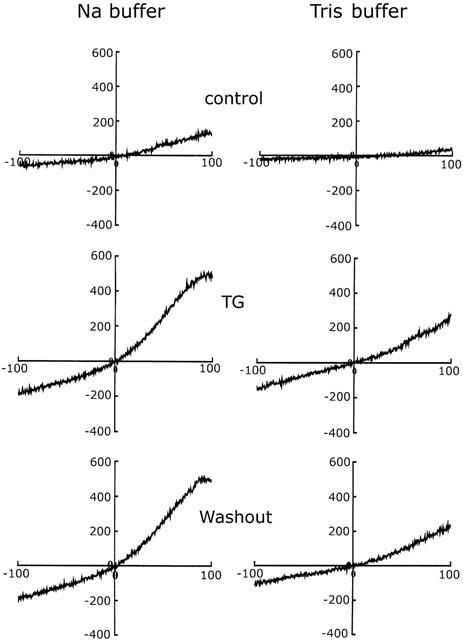
Membrane currents in response to extracellular application of thapsigargin. The holding potential was −40 mV and ramp pulses were given every 5 s from −100 mV to 100 mV at a speed of 2 V/s. I-V curves for control cells (top traces), cells showing the maximal response to thapsigargin (middle traces), and cells after thapsigargin washout (bottom traces) in Na+-containing (left traces) and Na+-free (right traces) bathing buffer are shown. Similar results were seen using six different batches of cells (n=10).
Our data suggested that the SOC in rat cerebellar astrocytes is permeable to both Ca2+ and Na+. We next determined whether it was permeable to Ba2+ and Sr2+. If this was the case, Ba2+ and Sr2+ were not the substrate for Ca2+ pump and would accumulate in the cytosol, resulting in a progressive increase in fura-2 fluorescence. As shown in Figure 9 (trace a), in the presence of 2.2 mM BaCl2, the fluorescence change induced by thapsigargin was indistinguishable from that seen in Ca2+-free medium (Figure 2A, trace b). Similar results were obtained when extracellular Ca2+ was replaced by Sr2+ (Figure 9, trace b).
Figure 9.
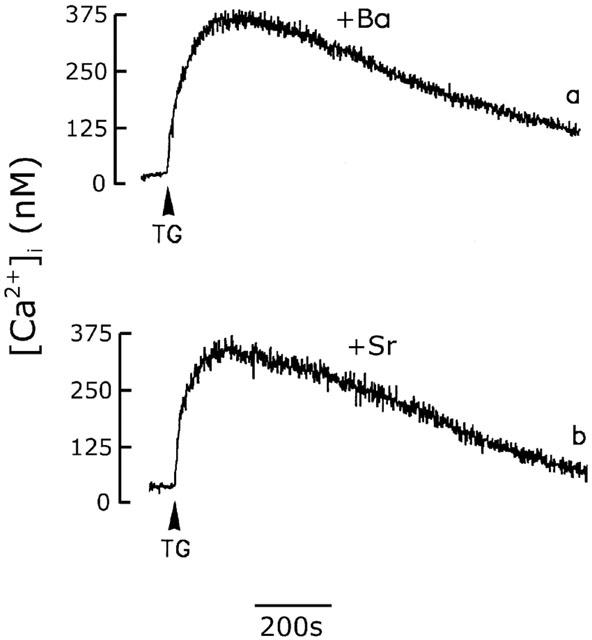
Effect of Ba2+ or Sr2+ on the thapsigargin-induced [Ca2+]i increase. Cells were bathed in Ca2+-free loading buffer in which Ca2+ was replaced by equimolar concentrations of Ba2+ (trace a) or Sr2+ (trace b) and the thapsigargin (1 μM)-induced [Ca2+]i increase was measured. The experiment was repeated three times with similar results.
To further confirm that Ca2+ influx following intracellular Ca2+ stores depletion we observed in cerebellar astrocytes is due to the activation of SOC rather than VOC, we next determined the sensitivity of this Ca2+ influx pathway to nifedipine, Cd2+, and La3+, agents known to block VOC, and SK&F96365, a specific SOC inhibitor. As shown in Figure 10, cerebellar astrocyte was individually treated with buffer, 10 μM nifedipine, 100 μM Cd2+, 100 μM La3+, and 10 μM SK&F96365 for 5 min, 1 μM thapsigargin was then added to each group. Except for cells treated with SK&F96365, a prolonged [Ca2+]i increase was seen in all cells; the sustained [Ca2+]i being 306±38 nM (n=8), 293±32 nM (n=8), 321±41 nM (n=8) and 298±34 nM (n=8) in cells treated with buffer, nifedipine, Cd2+ and La3+, respectively. This prolonged [Ca2+]i increase was due to Ca2+ influx, since it immediately declined to the basal level when extracellular Ca2+ was removed and returned to the same sustained level when 2.2 mM Ca2+ was added back to the bathing solution. In the presence of SK&F96365, thapsigargin-induced [Ca2+]i increase was transient, and the [Ca2+]i returned to the basal level even in the presence of extracellular Ca2+. This is indistinguishable from that seen in Ca2+-free medium (Figure 2A, trace b).
Figure 10.
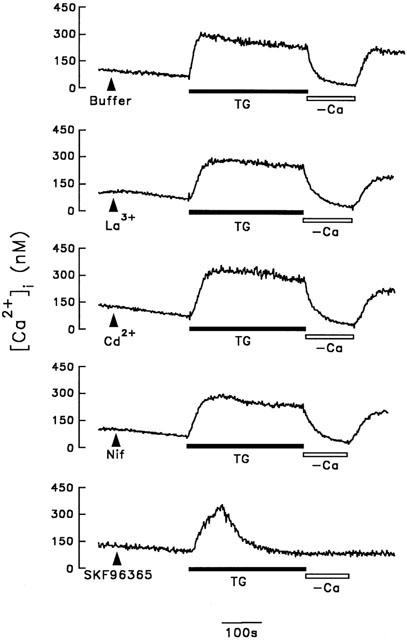
Effect of La3+, Cd2+, nifedipine or SK&F96365 on thapsigargin-induced CCE. After cells were initially treated with buffer, 100 μM La3+, 100 μM Cd2+, 10 μM nifedipine (NIF) or 10 μM SK&F96365 as indicated, thapsigargin (TG) (1 μM) was then added in each group. Five min after thapsigargin addition, extracellular Ca2+ was removed from and added back to the bathing buffer. [Ca2+]i changes were measured. Similar results were seen using three different batches of cells (n=8).
We finally determined whether CCE activity was regulated by protein phosphorylation by examining the effects of staurosporine, a protein kinase inhibitor, PMA, a protein kinase C activator, and okadaic acid, a phosphatase inhibitor, on SOC-mediated Mn2+ influx after store depletion. As with Sr2+ or Ba2+, once Mn2+ enters the cell, it is trapped in the cytosol, and the fluorescence of fura-2 is quenched after it binds to Mn2+. As shown in Figure 11, addition of 1 μM thapsigargin to cerebellar astrocytes caused slight, but significant, fluorescence quenching (trace b) compared to control cells (trace a) and this quenching was slightly reduced following 5 min pretreatment of cells with 1 μM PMA or 2.5 nM okadaic acid (traces f and h, respectively), but significantly potentiated by 1 μM staurosporine (trace d). In control cells, the basal fluorescence quenching was not affected by any of these three drugs (compare trace a with traces c, e, and g). The statistical data are summarized in Table 1.
Figure 11.
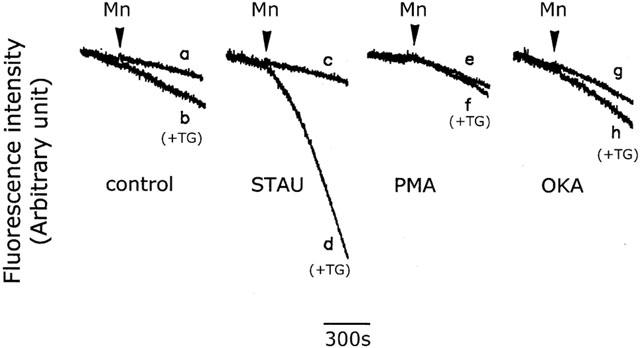
Effect of protein phosphorylation on the thapsigargin-induced CCE. Fura-2-loaded cells were bathed in loading buffer containing 0.2 mM Ca2+ alone (control) (traces a and b), or plus 100 nM staurosporine (STAU) (traces c and d), 100 nM PMA (traces e and f), or 2 nM okadaic acid (OKA) (traces g and h). Five min later, 1 mM Mn2+ (traces a, c, e and g) or 1 mM Mn2+ plus 1 μM thapsigargin (TG) (traces b, d, f and h) was added, and the fluorescence quenching due to Mn2+ influx measured. The experiment was repeated six times using different batches of cells with similar results (n=15).
Discussion
Under physiological conditions, depletion of the intracellular Ca2+ stores occurs following receptor occupancy as a result of IP3 generation, and Ca2+ influx via the SOC is then activated, resulting in the so-called CCE (Putney, 1990; Berridge, 1997; Parekh & Penner, 1997; Barritt, 1999). In the present study, in rat cerebellar astrocytes, a sustained [Ca2+]i increase was seen in response to ATP or angiotensin II stimulation in the presence of extracellular Ca2+, but not in its absence (Figure 1). A slow [Ca2+]i increase was seen following inhibition of sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) by thapsigargin or cyclopiazonic acid (Figures 2 and 3), indicating that, under basal physiological condition, there was a dynamic equilibrium between Ca2+ sequestration into, and Ca2+ leakage from, the intracellular Ca2+ stores, and that no second messenger was required for Ca2+ leakage. Thus, by inhibiting Ca2+ sequestration, the Ca2+ stores could be depleted as a result of Ca2+ leakage. In the present study, two structurally distinct SERCA inhibitors, thapsigargin and cyclopiazonic acid, were equally effective in causing Ca2+ store depletion and increasing the Ca2+ permeability of the plasma membrane (Figures 2 and 3). This Ca2+ influx following store depletion was due to the activation of SOC since it was blocked by SK&F96365 (Figure 10).
Our results further showed that Ca2+ influx following store depletion was resistant to nifedipine, La3+ and Cd2+ (Figure 10). It has been previously shown that store depletion-operated current was resistant to La3+ in mouse anococcygeus smooth muscle cells (Wayman et al., 1997) and pancreatic acinar cells (Krause et al., 1996), but attenuated in A7r5 vascular smooth muscle cells (Gardner & Benoit, 2000) and guinea-pig enteric glial cells (Zhang et al., 1998). Similarly, it was resistant to Cd2+ in mouse pancreatic acinar cells (Krause et al., 1996), while being inhibited in anococcygeus smooth muscle cells, (Wayman et al., 1997). Thus, different cell types may explain the variety of the cation specificity of SOC.
The K+-driven current in response to store depletion was negligible, since KCl was replaced by CsCl in both the bathing and pipette buffers and 20 mM tetraethylammonium chloride was present. The outward rectification at the positive potential range is more prominent in the left middle panel of Figure 8. Since there was no K+ present, the outward current induced by thapsigargin might be driven by Cl−. In contrast, the limb of I-V curve at the negative membrane potential range is linear. Therefore, we monitored the progression of the inward current at membrane potential of −100 mV in the absence and presence of thapsigargin and expressed the time course of changes in Figure 7. When Na+ in the bathing buffer was isotonically substituted with Tris, the thapsigargin-induced current at −100 mV was reduced (Figure 8), indicating that the contribution of Na+ to the store depletion-induced current is profound in normal Na+-containing physiological buffer. Thus, in rat cerebellar astrocytes, the SOC is permeable to Na+. In contrast, it is not permeable to Sr2+ and Ba2+ (Figure 9). In mast cells (Hoth & Penner, 1992) and Xenopus oocytes (Parekh et al., 1993), the SOC shows high selectivity for Ca2+, while, in mouse pancreatic acinar cells, it does not discriminate between monovalent cations and possesses significant conductance for Ca2+ and Ba2+ (Krause et al., 1996). Different subtypes of SOC may therefore exist in different animal species.
In contrast, when the thapsigargin-induced Ca2+ influx was measured to reflect CCE activity, the [Ca2+]i increase was higher in Na+-free buffer than in Na+-containing buffer (Figure 6). This finding is consistent with the hypothesis that Ca2+ enters through a nonselective cation channel. In the absence of Na+, more Ca2+ enters cells via the SOC. The Na+/Ca2+ exchanger is involved in regulating [Ca2+]i and removal of extracellular Na+ inhibits the activity of the exchanger, causing an increase in the [Ca2+]i. Thus, in our study, it is also possible that the greater thapsigargin-induced [Ca2+]i increases seen in Na+-free buffer were due to inhibition of the Na+/Ca2+ exchanger. Alternatively, Na+ may inhibit the action of thapsigargin or SOC activity.
Protein phosphorylation plays a role in CCE regulation. Firstly, protein tyrosine phosphorylation is involved in CCE. In human platelets, depletion of intracellular Ca2+ stores stimulates protein tyrosine phosphorylation (Vostal et al., 1991; Jenner et al., 1994), while, in human foreskin fibroblasts, bradykinin- or thapsigargin-induced Ca2+ entry is inhibited by tyrosine kinase inhibitors (Lee et al., 1993). In addition, there is increasing evidence that serine/threonine phosphorylation potentiates CCE. In Xenopus oocytes, okadaic acid potentiates Ca2+ entry once the stores have been emptied (Parekh et al., 1993), while, in lymphocytes and astrocytoma cells, it also prevents CIF degradation and enhances CCE (Randriamampita & Tsien, 1995). However, opposing effects of serine/threonine phosphorylation have been reported in CCE regulation in human neutrophils (Montero et al., 1994), platelets (Murphy et al., 1996), HeLa cells (Berlin & Preston, 1993), and rat basophilic leukaemia cells (BRL-2H3) (Parekh & Penner, 1995). In the present study, we showed that CCE in rat cerebellar astrocytes was regulated by phosphorylation, since inhibition of protein serine/threonine phosphorylation by staurosporine potentiated CCE, while promotion of protein serine/threonine phosphorylation by activation of protein kinase C or inhibition of protein phosphatase inhibited CCE activity (Figure 11). Many studies have shown that various Ca2+ transporters, including plasma membrane Ca2+ pump, SERCA, IP3 receptor, SOC and VOC, are regulated by phosphorylation/dephosphorylation. In this study, we compared the [Ca2+]i responses with and without thapsigargin treatment. Measurements of change of [Ca2+]i reflect the final [Ca2+]i after the summation of the operation of all Ca2+ transporters within the cell. The drugs used in this study, including staurosporine, PMA and okadaic acid may affect other Ca2+ transporters and result in the change of [Ca2+]i. However, if there would be any, the influence is the same in control and thapsigargin-treated cells.
As shown in Figure 2B, in the presence of extracellular Ca2+, removal of thapsigargin had no effect on the thapsigargin-induced [Ca2+]i increase once the cells had been exposed to thapsigargin for 10 min. Similarly, the thapsigargin-induced inward current was unaffected by thapsigargin washout (Figure 7). Thus, once the intracellular Ca2+ stores were depleted, Ca2+ entered via SOC, regardless of whether thapsigargin was present, suggesting that thapsigargin is an irreversible inhibitor. In the presence of SERCA inhibitors, the intracellular Ca2+ stores were maintained in a depleted state, resulting in CCE activation and the [Ca2+]i remaining at a plateau (Figures 2 and 3). This result also indicates that, in rat cerebellar astrocytes, the SOC is not inhibited by a high [Ca2+]i. In Jurkat leukaemic T lymphocytes and rat basophilic leukaemia cells (RBL-2H3), SOCs are regulated by high [Ca2+]i feedback inhibition (Zweifach & Lewis, 1995; Parekh, 1998), while, in RBL-1 cells, they are activated by a low [Ca2+]i independently of global Ca2+ store depletion (Krause et al., 1999).
The SOC-induced CCE plays a crucial role in the homeostasis of [Ca2+]i, which in turn controls many cellular functions. In the disease states, the delicate balance of diverse signal transduction pathways might be injured and phosphorylation regulation changed. Consequently, dysregulation of [Ca2+]i resulted from the disturbances of CCE will be linked to cell death or degeneration. CCE may therefore represent an important therapeutic target for the design of drugs. In cerebellar astrocytes, CCE has not been fully characterized. Whether depolarization occurs following Na+ influx mediated by CCE requires further investigation. Similarly, the effect of this increased cytosolic Na+ concentration on mitochondrial Ca2+ regulation is not clear. In addition to refilling the intracellular Ca2+ stores, the increased Ca2+ via SOC may also have other cellular functions. For example, Ca2+ in the microdomain of channel mouth may activate enzymes or target proteins. The possibility that second messengers may modulate SOCs in cerebellar astrocytes is of interest. The cerebellar astrocytes are the major arachidonic acid production site in the brain (Moore et al., 1991), the interrelationship of SOC and second messenger-operated Ca2+ entry also needs further study.
In conclusion, in rat cerebellar astrocytes, store depletion activates CCE via SOCs, which are permeable to Na+, but not to Sr2+ and Ba2+. In addition, SOC activity is regulated by serine/threonine phosphorylation.
Acknowledgments
We thank Dr Thomas Barkas for helpful discussion. This work was supported by grants from the National Science Council (NSC90-2316-B016-001) and the National Defense Medical Center (DOD-90-33).
Abbreviations
- [Ca2+]i
intracellular Ca2+ concentration
- CCE
capacitative Ca2+ entry
- CIF
calcium influx factor
- PMA
phorbol 12-myristate 13-acetate
- SERCA
sarco(endo)plasmic reticulum Ca2+ ATPase
- SOC
store-operated Ca2+ channel
- VOC
voltage-operated Ca2+ channel
References
- BARNARD E.A. The transmitter-gated channels: a range of receptor types and structures. Trends Pharmacol. Sci. 1996;17:305–309. [PubMed] [Google Scholar]
- BARRITT G.J. Receptor-activated Ca2+ inflow in animal cells: a variety of pathways tailored to meet different intracellular Ca2+ signaling requirements. Biochem. J. 1999;337:153–169. [PMC free article] [PubMed] [Google Scholar]
- BERLIN R.D., PRESTON S.F. Okadaic acid uncouples calcium entry from depletion of intracellular stores. Cell Calcium. 1993;14:379–386. doi: 10.1016/0143-4160(93)90042-5. [DOI] [PubMed] [Google Scholar]
- BERRIDGE M.J. Elementary and global aspects of calcium signaling. J. Physiol. 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARAFOLI E. Intracellular calcium homeostasis. Annu. Rev. Biochem. 1987;56:395–433. doi: 10.1146/annurev.bi.56.070187.002143. [DOI] [PubMed] [Google Scholar]
- CHIN T.Y., CHUEH S.H. Sphingosylphosphorylcholine stimulates mitogen-activated protein kinase via a Ca2+-dependent pathway. Am. J. Physiol. 1998;275:C1255–C1263. doi: 10.1152/ajpcell.1998.275.5.C1255. [DOI] [PubMed] [Google Scholar]
- CLAPHAM D.E. Cellular calcium. A mysterious new influx factor. Nature. 1993;364:763–764. doi: 10.1038/364763a0. [DOI] [PubMed] [Google Scholar]
- CUNNINGHAM A.J., SZENBERG A. Further improvements in the plaque technique for detecting single antibody-forming cells. Immunology. 1968;14:599–600. [PMC free article] [PubMed] [Google Scholar]
- FURUICHI T., MIKOSHIBA D. Inositol 1,4,5-trisphosphate receptor-mediated Ca2+ signaling in the brain. J. Neurochem. 1995;64:953–960. doi: 10.1046/j.1471-4159.1995.64030953.x. [DOI] [PubMed] [Google Scholar]
- GALLO V., CIOTTI M.T., COLETTI A., ALOISI F., LEVI G. Selective release of glutamate from cerebellar granule cells differentiating in culture. Proc. Natl. Acad. Sci. U.S.A. 1982;79:7919–7923. doi: 10.1073/pnas.79.24.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARDNER J.D., BENOIT J.N. Effect of capacitative calcium entry on agonist-induced calcium transients in A7r5 vascular smooth muscle cells. J. Biomed. Sci. 2000;7:304–310. doi: 10.1007/BF02253249. [DOI] [PubMed] [Google Scholar]
- GILL D.L., CHUEH S.H., WHITLOW C.L. Functional importance of the synaptic plasma membrane calcium pump and sodium-calcium exchanger. J. Biol. Chem. 1984;259:10807–10813. [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HARTENECK C., PLANT T.D., SCHULTZ G. From worm to man: three subfamilies of TRP channels. Trends Neurosci. 2000;23:159–166. doi: 10.1016/s0166-2236(99)01532-5. [DOI] [PubMed] [Google Scholar]
- HESS P. Calcium channels in vertebrate cells. Annu. Rev. Neurosci. 1990;13:337–356. doi: 10.1146/annurev.ne.13.030190.002005. [DOI] [PubMed] [Google Scholar]
- HOTH M., PENNER R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- IRVINE R.F. ‘Quantal' Ca2+ release and the control of Ca2+ entry by inositol phosphates–a possible mechanism. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- JENNER S., FARNDALE R.W., SAGE S.O. The effect of calcium-store depletion and refilling with various bivalent cations on tyrosine phosphorylation and Mn2+ entry in fura-2-loaded human platelets. Biochem. J. 1994;303:337–339. doi: 10.1042/bj3030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRAUSE E., PFEIFFER F., SCHMID A., SCHULZ I. Depletion of intracellular calcium stores activates a calcium conducting nonselective cation current in mouse pancreatic acinar cells. J. Biol. Chem. 1996;271:32523–32528. doi: 10.1074/jbc.271.51.32523. [DOI] [PubMed] [Google Scholar]
- KRAUSE E., SCHMID A., GONZALEZ A., SCHULZ I. Low cytoplasmic [Ca2+] activates ICRAC independently of global Ca2+ store depletion in RBL-1 cells. J. Biol. Chem. 1999;274:36957–36962. doi: 10.1074/jbc.274.52.36957. [DOI] [PubMed] [Google Scholar]
- KRISHNA S., WOODROW C., WEBB R, PENNY J., TAKEYASU K., KIMURA M., EAST J.M. Expression and functional characterization of a Plasmodium falciparum Ca2+-ATPase (PfATP4) belonging to a subclass unique to apicomplexan organisms. J. Biol. Chem. 2001;276:10782–10787. doi: 10.1074/jbc.M010554200. [DOI] [PubMed] [Google Scholar]
- LEE K.-M., TOSCAS K., VILLEREAL M.L. Inhibition of bradykinin- and thapsigargin-induced Ca2+ entry by tyrosine kinase inhibitors. J. Biol. Chem. 1993;268:9945–9948. [PubMed] [Google Scholar]
- MARKS A.R. Intracellular calcium-release channels: regulators of cell life and death. Am. J. Physiol. 1997;272:H597–H605. doi: 10.1152/ajpheart.1997.272.2.H597. [DOI] [PubMed] [Google Scholar]
- MONTERO M., GARCIA-SANCHO J., ALVAREZ J. Phosphorylation down-regulates the store-operated Ca2+ entry pathway of human neutrophils. J. Biol. Chem. 1994;269:3963–3967. [PubMed] [Google Scholar]
- MOORE S.A., YODER E., MURPHY S., DUTTON G.R., SPECTOR A.A. Astrocytes, not neurons, produce decosahexaenoic acid (22, 6ω-3) and arachidonic acid (20, 4ω-6) J. Neurochem. 1991;56:518–524. doi: 10.1111/j.1471-4159.1991.tb08180.x. [DOI] [PubMed] [Google Scholar]
- MURPHY C.T., BULLOCK A.J., WESTWICK J. A role for protein phosphorylation in modulating Ca2+ elevation in rabbit platelets treated with thapsigargin. Biochem. J. 1996;313:83–89. doi: 10.1042/bj3130083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEHER E. Controls on calcium influx. Nature. 1992;355:298–299. doi: 10.1038/355298a0. [DOI] [PubMed] [Google Scholar]
- PAREKH A.B. Slow feedback inhibition of calcium release-activated calcium current by calcium entry. J. Biol. Chem. 1998;273:14925–14932. doi: 10.1074/jbc.273.24.14925. [DOI] [PubMed] [Google Scholar]
- PAREKH A.B., PENNER R. Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAREKH A.B., PENNER R. Store depletion and calcium influx. Physiol. Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- PAREKH A.B., TERLAU H., STUHMER W. Depletion of InsP3 stores activates a Ca2+ and K+ current by means of a phosphatase and a diffusible messenger. Nature. 1993;364:814–818. doi: 10.1038/364814a0. [DOI] [PubMed] [Google Scholar]
- PATTERSON R.L., VAN ROSSUM D.B., GILL D.L. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- POZZAN T., RIZZUTO R, VOLPE P., MELDOLESI J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- PUTNEY J.W., JR Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- PUTNEY J.W., JR “Kissin' cousins”: intimate plasma membrane-ER interactions underlie capacitative calcium entry. Cell. 1999;99:5–8. doi: 10.1016/s0092-8674(00)80056-2. [DOI] [PubMed] [Google Scholar]
- RANDRIAMAMPITA C., TSIEN R.Y. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- RANDRIAMAMPITA C., TSIEN R.Y. Degradation of a calcium influx factor (CIF) can be blocked by phosphatase inhibitors or chelation of Ca2+ J. Biol. Chem. 1995;270:29–32. doi: 10.1074/jbc.270.1.29. [DOI] [PubMed] [Google Scholar]
- VOSTAL J.G., JACKSON W.L., SHULMAN N.R. Cytosolic and stored calcium antagonistically control tyrosine phosphorylation of specific platelet proteins. J. Biol. Chem. 1991;266:16911–16916. [PubMed] [Google Scholar]
- WAYMAN C.P., MCFADZEAN I., GIBSON A., TUCKER J.F. Cellular mechanisms underlying carbachol-induced oscillations of calcium-dependent membrane current in smooth muscle cells from mouse anococcygeus. Br. J. Pharmacol. 1997;121:1301–1308. doi: 10.1038/sj.bjp.0701279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAO Y., FERRER-MONTIEL A.V., MONTAL M., TSIEN R.Y. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- ZHANG W., SAROSI G.A., JR, BARNHART D.C., MULHOLLAND M.W. Endothelin-stimulated capacitative calcium entry in enteric glial cells: synergistic effects of protein kinase C activity and nitric oxide. J. Neurochem. 1998;71:205–212. doi: 10.1046/j.1471-4159.1998.71010205.x. [DOI] [PubMed] [Google Scholar]
- ZHU X., JIANG M., PEYTON M., BOULAY G., HURST R, STEFANI E., BIRNBAUMER L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]
- ZWEIFACH A., LEWIS R.S. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. J. Biol. Chem. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]