

Abstract
The apoptotic effect of adenosine and its analogues was studied in fibroblast-like synoviocytes derived from rheumatoid arthritis patients (RA-FLSs). Evoked cell death was quantitatively examined by assessing DNA fragmentation using an enzyme-liked immunosorbent assay and by measuring phosphatidylserine exposure through flow cytometric analysis of annexin V binding.
Exposing cells for 24 h to 2-chloroadenosine (2-CADO), a nonspecific, adenosine deaminase (ADA)-resistant, adenosine receptor (AdoR) agonist, induced DNA fragmentation, and thus apoptosis, in RA-FLSs at concentrations ⩾50 μM. By contrast, incubation with adenosine for up to 72 h did not evoke DNA fragmentation, even in the presence of ADA inhibitor coformycin and nucleoside transporter inhibitor nitrobenzylmercaptopurin (NBMPR). Transcription of all four AdoR isoforms was detected in RA-FLSs; nevertheless selective AdoR agonists similarly failed to induce DNA fragmentation.
DNA fragmentation evoked by 2-CADO was inhibited by NBMPR and by 5′-iodotubercidin, an adenosine kinase inhibitor, but not by xanthine amine congener, an A1 and A2 receptor antagonist, or by selective AdoR antagonists.
The nonspecific caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp fluoromethyl ketone abolished the apoptotic effect of 2-CADO.
These results suggest that 2-CADO induces apoptosis in RA-FLSs independently of AdoR-mediated signalling. Instead, 2-CADO, but not adenosine, is taken up into RA-FLSs via human equilibrative nucleoside transporter-1, where it is phosphorylated by adenosine kinase. The resultant phospho-2-CADO induces DNA fragmentation by activating a caspase pathway.
Keywords: Rheumatoid arthritis, synoviocytes, 2-chloroadenosine, apoptosis, adenosine receptor, nucleoside transporter, adenosine kinase, caspase
Introduction
Rheumatoid arthritis (RA) is a chronic, autoimmune inflammatory disorder of unknown aetiology (Wicks et al., 1997); the current treatment goals are to alleviate symptoms, slow disease progression and optimize quality of life (Weinblatt, 1999). The pathophysiological characteristics of RA are infiltration of mononuclear cells and activation and proliferation of synoviocytes (Panayi et al., 1992), resulting in pannus formation and, eventually, joint destruction (Zvaifler & Firestein, 1994). There is increasing evidence, however, that early inhibition of synovial inflammation by disease-modifying antirheumatic drugs (DMARDs) can reduce disease activity and alter the clinical course of RA (Langenegger & Michel, 1999).
Methotrexate has proven to be one of the most effective DMARDs for the treatment of RA, although its mechanism of action, and indeed that of DMARDs in general, is not yet well understood. However, using the murine air pouch inflammation model, it was shown that administration of methotrexate elevates local concentrations of extracellular adenosine at sites of inflammation (Cronstein et al., 1993). It is also notable that the concentration of adenosine deaminase (ADA), an adenosine-degrading enzyme, is higher in the synovial fluid of RA patients than in that of osteoarthritis patients (Pettersson et al., 1988; Wortmann et al., 1989) and that nonselective adenosine receptor (AdoR) antagonists, theophylline and caffeine, are capable of reversing the antiinflammatory effects of methotrexate in rat adjuvant arthritis (Montesinos et al., 2000). Taken together, these findings suggest that reductions in the local concentration of adenosine may contribute to the joint inflammation of RA and raise the possibility that adenosine analogues could serve as the basis of a novel approach to the treatment of RA.
The respective roles of adenosine and adenine nucleotides in numerous physiological responses have been studied for more than six decades. These compounds are interesting in that they act both intracellularly and extracellularly, via several cell-surface receptors, with multiple regulatory pathways affecting their intracellular and extracellular concentrations (Ralevic & Burnstock, 1998). Extracellular adenosine, which accumulates in the extracellular milieu as a consequence of degradation of extracellular ATP, active membrane transport and facilitated diffusion from cells (Dalziel & Westfall, 1994), can act as an autocrine and/or paracrine factor, modulating a variety of cellular and systemic functions (Olsson & Pearson, 1990). For example, its actions via cell-surface AdoRs have potent effects on cardiovascular (Shryock & Belardinelli, 1997) and nervous system (Guieu et al., 1998) function. Four AdoRs, all of which are G-protein-coupled receptors with seven-transmembrane spanning domains (Ralevic & Burnstock, 1998), have been cloned from a variety of species including humans: two (A2A and A2B) are coupled with GS, while the others (A1 and A3) are coupled to Gi. The effects of AdoR signalling are thus primarily modulated by regulating the activities of adenylate cyclase – and hence the intracellular cyclic AMP concentration – and protein kinase A, the downstream target of cyclic AMP (Dalziel & Westfall, 1994). The A3 receptor is also coupled to Gq/11 and is able to stimulate phospholipase C, thereby elevating levels of intracellular IP3 and Ca2+ (Palmer et al., 1995).
In the present study, we investigated the direct effects of adenosine and its analogues on the regulation of apoptotic cell death in fibroblast-like synoviocytes (RA-FLSs). Our aim was to provide the basis for the development of novel strategies for the treatment of RA that are efficacious and have fewer adverse side effects than the DMARD therapy currently in use. We found that the ADA-resistant adenosine analogue 2-chloroadenosine (2-CADO) induces apoptosis in RA-FLSs, though adenosine itself and selective AdoR agonists do not. This effect of 2-CADO was attenuated by inhibition of a nucleoside transporter or adenosine kinase.
Methods
Cells
After obtaining informed consent, human RA-FLSs were isolated from synovial tissue obtained from RA patients undergoing joint replacement surgery. The diagnosis of RA had been made in these patients according to criteria established by the American College of Rheumatology (Arnett et al., 1988). The collected tissues were minced and then incubated first for 3 – 4 h at 37°C with 4 mg ml−1 collagenase and then for 30 min with 0.05% trypsin (Difco Laboratories, Detroit, MI, U.S.A.). The isolated cells were cultured in RPMI 1640 medium (Nissui, Tokyo, Japan) supplemented with 10% foetal calf serum (Life Technologies, Grand Island, NY, U.S.A.), penicillin, streptomycin and L-glutamine (complete medium). Adherent RA-FLSs were used from the second through the fifth passages, a period during which they were highly proliferative.
SW982 cells, a human synovial sarcoma line, were obtained from the American Type Tissue Culture Collection (Rockville, MD, U.S.A.) and maintained as instructed by the supplier. HeLa cells were a kind gift from Dr Riko Kitazawa (Kobe University).
Reagents
All reagents were purchased from Sigma ImmunoChemicals (St. Louis, MO, U.S.A.) unless otherwise indicated. The selective A2A receptor agonist 2-[4-(2-carboxyethyl)phenethylamino]-5′-N-ethylcarboxamidoadenosine (CGS21680) and the A2A receptor antagonist 8-(3-chlorostyryl) caffeine (CSC) were purchased from RBI (Natick, MA, U.S.A.). The highly selective A2A receptor antagonist 4-(2-[7-amino-2-[2-furyl] [1,2,4]triazolo[2,3-a][1,3,5]triazin-5-yl]amino]ethyl)-phenol (ZM241385) was purchased from Tocris (Ballwin, MO, U.S.A.). Adenosine solution was made fresh prior to each experiment by dissolving it in complete medium. Stock solution of 2-CADO was prepared in complete medium and stored at −20°C; stock solutions of other reagents were prepared by dissolving them in dimethylsulphoxide (DMSO) and stored at −20°C. The nonspecific caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp fluoromethyl ketone (z-VAD-fmk) was purchased from Promega (Madison, WI, U.S.A.).
Flow cytometry
Flow cytometric estimation of RA – FLS apoptosis and necrosis was accomplished using annexin-V and propidium iodide (PI) staining kits (van engeland et al., 1996) (Annexin-V-FLUOS Staining Kit, Boehringer Mannheim, Mannheim, Germany) according to the manufacturer's instructions. Briefly, cells plated to a density of 0.2 – 0.3×106 cells per well in 6-well plates (Costar, Corning, NY, U.S.A.) were incubated for 24 h at 37°C under a 5% CO2 atmosphere, with or without the indicated reagents. Supernatants containing necrotic cells as well as residual attached cells were collected, washed, stained for 15 min at room temperature with Annexin-V-Fluorescein and PI, and examined using a FACSCalibur equipped with CellQuest analysis software (Becton Dickinson, San Jose, CA, U.S.A.). Cells that were negative for both annexin-V and PI were considered alive; those positive for annexin-V but negative for PI were considered apoptotic; and those positive for both annexin-V and PI were considered necrotic.
Quantitative estimation of DNA fragmentation using an enzyme-linked immunosorbent assay (ELISA)
RA – FLSs collected by brief centrifugation were plated to a density of 1×104 cells per well on flat-bottomed 96-well plates (Costar) and incubated overnight in complete medium at 37°C under a 5% CO2 atmosphere. The next day, the culture medium was exchanged for 100 μl of complete medium with or without the indicated reagent(s) and incubated for an additional 24 h. In some experiments, cells were preincubated with z-VAD-fmk (Promega) or the adenosine kinase inhibitor 5′-iodotubercidin (IT; Sigma) prior to incubation with 2-CADO. Thereafter, DNA fragmentation was quantitatively estimated in the remaining attached cells using an ELISA (Cell Detection ELISAPLUS, Boehringer Mannheim, Mannheim, Germany) according to the manufacturer's protocol. Absorbance at 405 nm (reference at 492 nm) was measured in each well, and the means±s.d. were plotted as a function of the concentration of the indicated reagent. All control incubations contained the maximal concentration of DMSO, which was typically <0.1% (v v−1); DMSO concentrations of up to 1.25% (v v−1) DMSO did not induce significant DNA fragmentation (data not shown). Each independent experiment was carried out in triplicate using RA – FLSs prepared from a different patient.
RT – PCR analysis of adenosine receptor and nucleoside transporter expression
Total RNA was prepared using the single step method of Chomczynski & Sacchi (1987) (RNA STAT-60; Tel-Test ‘B', Friendswood, TX, U.S.A.). After DNase I treatment, first-strand cDNA was synthesized using a SuperScript preamplification system (Life Technologies) according to the manufacturer's instructions. The human A1, A2A, A2B and A3 receptor sequences were amplified by RT – PCR under standard conditions with primers corresponding to nucleotides 612-631 and 978-958 of the A1 open reading frame (ORF) (GenBank X68485); 490-507 and 1001-982 of the A2A ORF (X68486); 649-669 and 989-969 of the A2B ORF (M97759); and 430-446 and 790-771 of the A3 ORF (X76981). The expected lengths of the PCR products were 367 bp, 512 bp, 341 bp and 361 bp for A1, A2A, A2B and A3, respectively. The sequences of hENT1 and hENT2, the nitrobenzylmercaptopurin (NBMPR)-sensitive and -insensitive human equilibrative nucleoside transporters, respectively, were amplified with primers corresponding to nucleotides 354-373 and 752-733 of the hENT1 ORF (GenBank U81375) and nucleotides 490-507 and 1001-982 of the hENT2 ORF (AF029358). In this case, the expected lengths of the PCR products were 399 bp and 448 bp for hENT1 and hENT2, respectively.
cyclic AMP measurements
The effects of the various treatments on cellular cyclic AMP content were assessed by incubating 0.5×106 cells with the indicated adenosine analogue(s). Levels of cyclic AMP were measured using a cyclic AMP enzyme immunoassay (EIA) system (Amersham, Arlington Heights, IL, U.S.A.) according to the manufacturer's protocol. Measurements were made in duplicate; means±s.d. were plotted as a function of stimulation time.
Statistical analysis
The Mann – Whitney U and Kruskal – Wallis rank tests were used to compare differences in DNA fragmentation and cyclic AMP accumulation. Values of P<0.05 were considered significant.
Results
Effect of 2-CADO on RA – FLS cell death
RA – FLSs and cells of the human SW982 synovial cell line were incubated with 2-CADO, a non-selective, ADA-resistant AdoR agonist (Turkozkan et al., 1996), and the percentages of live, apoptotic and necrotic cells were assessed by flow cytometric analysis of annexin-V and PI binding (Figure 1A). It was recently shown that loss of plasma membrane asymmetry is an early cell type-independent apoptotic event, resulting in the exposure of phosphatidylserine at the cell surface (Vermes et al., 1995). Annexin-V interacts strongly and specifically with phosphatidylserine, thus enabling detection of early apoptosis. Exposing the cells to 5 μM 2-CADO for 24 h had little effect on annexin-V binding, but at higher concentrations (50 and 500 μM), 2-CADO concentration-dependently increased the proportion of annexin-V-positive cells in both cell types (Figure 1A, Table 1). Nearly all annexin-V-positive RA – FLSs were PI-negative (apoptotic) (Figure 1Aa). By contrast, about 10 – 15% of SW982 cells were both annexin-V- and PI-positive (necrotic) (Figure 1Ab), which accounts for the smaller proportion of apoptotic cells and confirms that the failure to detect PI-positive RA – FLSs did not reflect a limitation of the assay, but rather the characteristics of the cells themselves (Table 1). It therefore appears that, at least under our experimental conditions, RA – FLS cell death was caused by apoptosis, not necrosis.
Figure 1.
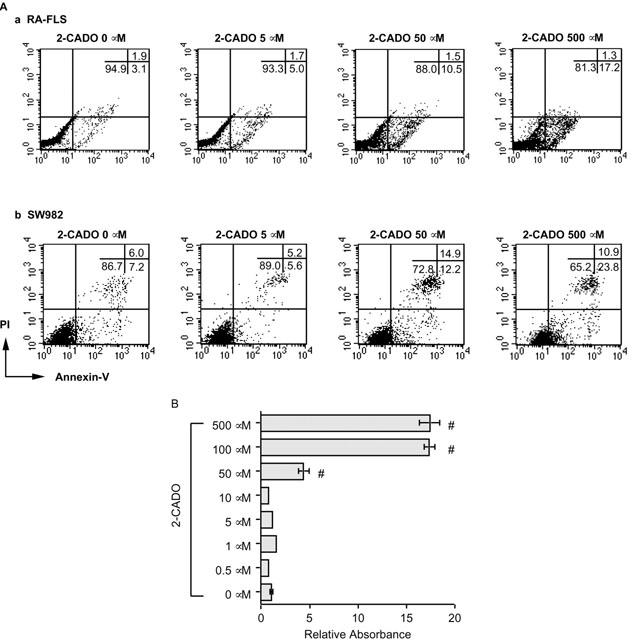
2-CADO-induced apoptosis in RA – FLSs. (A) Flow cytometric analysis of 2-CADO-induced apoptosis. RA – FLSs (Aa) and SW982 cells (Ab) were incubated for 24 h with the indicated concentration of 2-CADO, after which cell death was evaluated using annexin-V and PI staining. Data presented are representative of three independent experiments. (B) Quantitative analysis of DNA fragmentation induced by 2-CADO. RA – FLSs were incubated for 24 h in triplicate with the indicated concentration of 2-CADO, after which DNA fragmentation was estimated by ELISA. Representative data from four independent experiments are shown. The data are normalized to the value obtained in the absence of 2-CADO (control), which was arbitrarily assigned a value of 1.0; #, P<0.05 vs control incubation.
Table 1.
Relative proportion of apoptotic cells (annexin-V-positive, PI-negative) following incubation with 2-CADO

As the flow cytometric analysis revealed few PI-positive RA – FLSs at 2-CADO concentrations up to 500 μM, DNA fragmentation in attached cells was quantitated by ELISA to assess the apoptotic effect of 2-CADO in more detail. Consistent with the findings described above, 2-CADO had no effect on DNA fragmentation at concentrations less than 50 μM (Figure 1B). At higher concentrations, however, 2-CADO significantly increased DNA fragmentation, with maximum responses being elicited at concentrations of 100 μM or more. In subsequent experiments, therefore, maximum induction of apoptosis was defined as that induced by 100 or 500 μM 2-CADO, a value that served as a standard against which the effects of other adenosine analogues were compared.
Detection of AdoR expression on RA – FLSs and the effects of AdoR signalling on FLS apoptosis
Consistent with an earlier report (Boyle et al., 1996), we were able to detect expression of all four AdoR isoforms using RT – PCR (Figure 2A). Moreover, the finding that CSC, a selective A2A antagonist, did not significantly inhibit 2-CADO-induced cyclic AMP accumulation within cells, and that CGS21680, a selective A2A agonist, did not increase cyclic AMP levels (Figure 2B) suggested these cells express functional A2B receptors, which is also in agreement with the aforementioned report.
Figure 2.
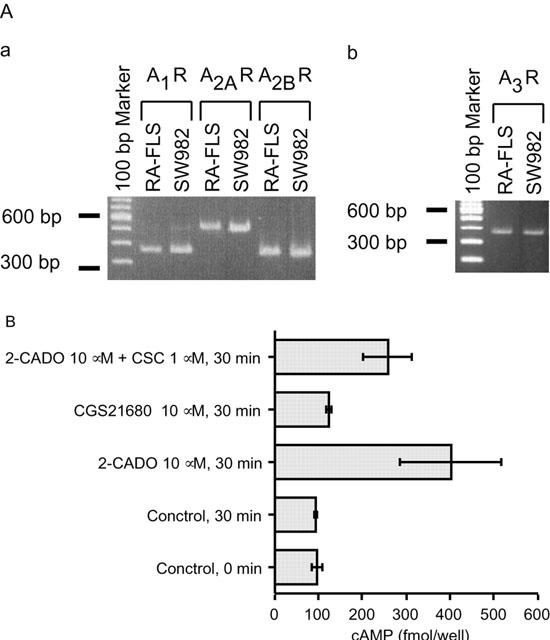
Expression of AdoR in RA – FLSs. (A) Expression of AdoR mRNA Total mRNA was extracted from RA – FLSs and SW982 cells, and each AdoR sequence was amplified by RT – PCR. Transcripts encoding A1, A2A, A2B (panel Aa) and A3 (panel Ab) AdoR were detected. Representative results from two independent experiments are shown. (B) Cyclic AMP accumulation elicited by 30 min exposures to AdoR analogues. RA – FLSs were incubated with 2-CADO (10 μM) in the presence or absence of selective A2A AdoR antagonist CSC (1 μM), or with selective A2A AdoR agonist CGS21680 (10 μM), after which the accumulated cyclic AMP was assayed. Shown are representative data from three independent experiments expressed as means±s.d.
To determine whether one or more AdoRs mediated the apoptosis induced by 2-CADO, we compared the effect of adenosine with that of 2-CADO. Because normally ADA rapidly degrades adenosine, coformycin (CF), and ADA inhibitor, was added to the incubation. To assure uniformity of conditions, the inhibitor was also added to the 2-CADO incubation, though 2-CADO is resistant to hydrolysis by ADA. In contrast to 2-CADO, adenosine did not induce DNA fragmentation, even with incubations as long as 72 h (Figure 3A and data not shown). CF itself did not induce apoptosis, nor did it affect 2-CADO-induced apoptosis (Figure 3A).
Figure 3.
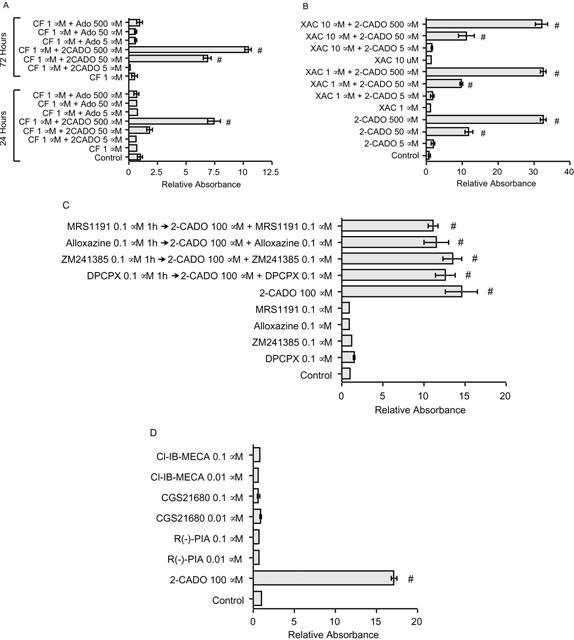
DNA fragmentation elicited by adenosine and adenosine analogues. (A) 2-CADO induced DNA fragmentation in RA – FLSs, whereas exposure to adenosine for up to 72 h did not, even in the presence of ADA inhibitor CF. CF itself did not induce DNA fragmentation, nor did it affect 2-CADO-mediated apoptosis. (B) AdoR antagonists XAC and (C) DPCPX, ZM241385, alloxazine and MRS1191 all failed to inhibit 2-CADO-induced apoptosis. MRS1191, a selective A3 receptor antagonist, tended to reduce DNA fragmentation, but the effect was not statistically significant. (D) AdoR agonists R-PIA, CGS 21680 and Cl-IB-MECA did not induce apoptosis in RA – FLSs. 2-CADO (100 μM) was included as a positive control. Data presented are representative of three (A – C) or five (D) independent experiments. In each panel, the data are normalized to the control value, which was arbitrarily assigned a value of 1.0; #, P<0.05 vs control incubation.
As mRNAs for all four AdoRs were detected in these cells (Figure 2A), it seemed possible that adenosine's failure to induce apoptosis may have been a consequence of simultaneous activation of GS- and Gi-coupled AdoRs, leading to responses that offset one another. We therefore carried out a series of experiments testing the effects of selective AdoR agonists and antagonists on RA – FLS apoptosis. As shown in Figures 3B,C, nonselective A1 and A2 receptor antagonist XAC (xanthine amine congener); A1 antagonist DPCPX [1,3-dipropyl-8-cyclopentylxanthine]; A2A antagonist ZM241385; A2B antagonist alloxazine; and A3 antagonist MRS1191 [3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate] all failed to inhibit 2-CADO-induced apoptosis. MRS1191 attenuated the effect of 2-CADO slightly, but the effect was not significant (Figure 3C). What is more, R-PIA [(R)-N6-phenylisopropyladenosine], CGS 21680 and Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)-N-methyl-5′-carbamoyladenosine] - A1, A2A and A3 receptor agonists, respectively – all failed to induce apoptosis in RA – FLS at concentrations appropriate for AdoR selectivity (Figure 3D). Direct characterization of A2B receptors was not possible because of the lack of a selective A2B receptor agonist; nonetheless, these data strongly suggest that AdoR-mediated signals are not directly responsible for 2-CADO-induced RA – FLS apoptosis.
Nucleoside transporters in 2-CADO-induced apoptosis
The observations that AdoR antagonists did not inhibit 2-CADO-induced apoptosis and that adenosine and various AdoR agonists all failed to induce apoptosis, despite expression of functional AdoRs, led us to hypothesize that 2-CADO is transported into RA – FLSs, as it is in human erythrocytes (Jarvis et al., 1985), where it activates apoptosis independently of AdoR signalling. To test this hypothesis, we first confirmed the expression of nucleoside transporters. The major equilibrative carrier of nucleoside in most cells is NBMPR-sensitive hENT1, though many cells also have a second equilibrative transporter, hENT2, which is NBMPR-insensitive (Crawford et al., 1998). Earlier pharmacological experiments showed HeLa cells to be positive for both transporters (Paterson et al., 1980); thus mRNA from those cells served as a positive control. Consistent with hENT1 being the major equilibrative nucleoside transporter, RA – FLSs from all four patients tested positive for hENT1, while only one was simultaneously hENT2-positive (Figure 4).
Figure 4.
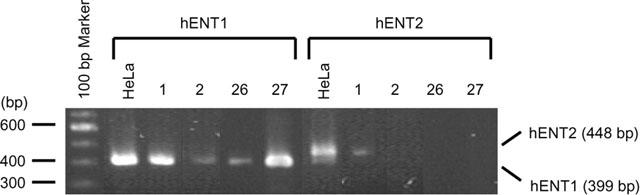
Expression of nucleoside transporter mRNAs in RA – FLSs. Total mRNA was extracted from RA – FLSs from four patients, and the expression of NBMPR-sensitive hENT1 and NBMPR-insensitive hENT2 was determined by RT – PCR; mRNA from HeLa cells served as a positive control.
Supporting our hypothesis that 2-CADO was taken up by RA – FLSs, where it acts intracellularly, NBMPR suppressed DNA fragmentation and, thus, 2-CADO-induced RA – FLS apoptosis. Adenosine again failed to promote apoptosis, even in the presence of CF and NBMPR (Figure 5). NBMPR itself had no effect on DNA fragmentation.
Figure 5.
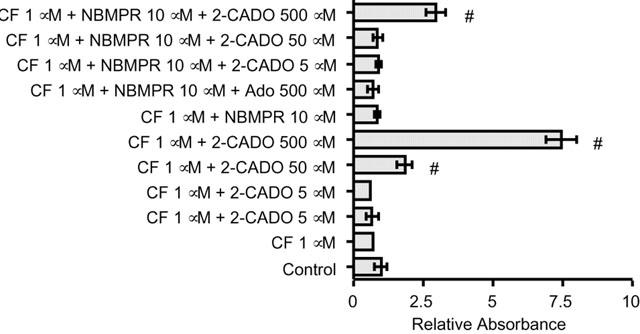
Effects of ADA inhibitor CF and nucleoside transporter inhibitor NBMPR on RA – FLSs apoptosis. RA – FLSs were incubated for 24 h in triplicate with the indicated reagents, after which DNA fragmentation was quantitated by ELISA. NBMPR partially inhibited the DNA fragmentation elicited by 2-CADO. Adenosine did not induce apoptosis even in the presence of both CF and NBMPR. Representative results from five independent experiments are shown. The data are normalized to the values obtained in the absence of 2-CADO and Ado, which was arbitrarily assigned a value of 1.0; #, P<0.05 vs control incubation.
Adenosine kinase in 2-CADO-induced RA – FLS apoptosis
The metabolic pathway activated by intracellular 2-CADO remains largely unknown, though it is known that, once inside cells, 2-CADO is phosphorylated by adenosine kinase (Yamanaka et al., 1986). Furthermore, the cytotoxicity of 2-CADO in human B lymphoid cells is dependent on this phosphorylation (Yamanaka et al., 1984). To test whether 2-CADO-induced RA – FLS apoptosis is dependent on phosphorylation by adenosine kinase, the cells were initially incubated for 3 h, with or without adenosine kinase inhibitor IT, after which they were cultured for an additional 24 h with various concentrations of 2-CADO. As shown in Figure 6, preincubation with IT almost completely blocked the apoptotic effect of 2-CADO, while IT itself had no effect on DNA fragmentation. This suggests that, when taken up by RA – FLSs , 2-CADO is phosphorylated by adenosine kinase, and that it is the resultant phospho-2-CADO that activates the apoptosis pathway.
Figure 6.
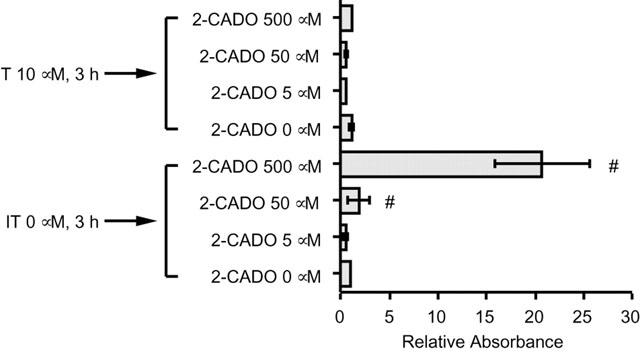
Effect of adenosine kinase inhibitor IT on 2-CADO-induced apoptosis. RA – FLSs were preincubated for 3 h, with or without 10 μM IT. After washing, the cells were incubated with 2-CADO for an additional 24 h, after which DNA fragmentation was assayed. Representative results from three independent experiments are shown. The data are normalized to the value obtained in the absence of 2-CADO, which was arbitrarily assigned a value of 1.0; #, P<0.05 vs control incubation.
Effect of caspases on 2-CADO induced apoptosis
To examine the contribution made by the caspase pathway to 2-CADO-induced RA – FLS apoptosis, the cells were incubated with caspase inhibitor z-VAD-fmk. As shown in Figure 7, apoptosis was completely blocked when 20 μM z-VAD-fmk was present during the incubation with 500 μM 2-CADO, regardless of whether or not the cells were preincubated with the inhibitor prior to addition of 2-CADO. Moreover, the apoptosis was partially inhibited even when the cells were only preincubated for 60 min with z-VAD-fmk, which was then removed before incubation with 2-CADO was started. It thus appears that phospho-2-CADO activates a caspase pathway leading to apoptosis in RA – FLSs.
Figure 7.
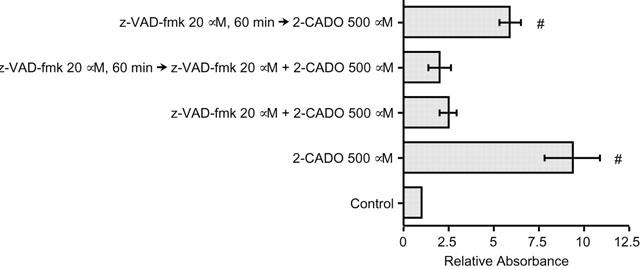
Inhibition of 2-CADO-induced RA – FLSs apoptosis by caspase inhibitor z-VAD-fmk. Cells were preincubated for 60 min or without 20 μM z-VAD-fmk, washed and then incubated for an additional 24 h with 2-CADO with or without z-VAD-fmk. Representative results from three independent experiments using RA – FLSs derived from two different patients are shown. The data are normalized to the control value, which was arbitrarily assigned a value of 1.0; #, P<0.05 vs control incubation.
Discussion
Adenosine is a ubiquitous endogenous nucleoside that modulates a variety of physiological processes, including immune responses and inflammation (Abbracchio & Burnstock, 1998). The first evidence that adenosine affects the immune system came from a finding in patients with severe combined immunodeficiency (SCID). The ADA deficiency resulted in the accumulation of both intracellular and extracellular adenosine, which led to markedly impaired immunity (Giblett et al., 1972). This immunosuppressive effect of adenosine is currently explained by the direct lymphotoxicity of adenosine catabolites as well as by the inhibition of TCR-triggered T-cell proliferation and effector functions through A2A receptor-mediated signalling (Koshiba et al., 1997).
The majority of adenosine's effects are mediated via interaction with specific cell membrane receptors (Fredholm et al., 1994), and four AdoR isoforms (A1, A2A, A2B, and A3) have been cloned from humans and other species. Signalling via AdoR is carried mainly as changes in the intracellular cyclic AMP concentration (Fredholm et al., 1994, as A2 receptors are coupled with GS, and A1 and A3 receptors are coupled to Gi. RA – FLSs apparently express all four AdoR mRNAs (Figure 2A), which is consistent with the earlier report that synoviocyte collagenase gene expression is downregulated by A2B receptor stimulation (Boyle et al., 1996). 2-CADO, a non-hydrolysable adenosine analogue, was used in this study because of the very short life of adenosine (Dawicki et al., 1988). Although 2-CADO was able to signal via functional A2 receptors, resulting in accumulation of cyclic AMP (Figure 2B), adenosine was unable to induce DNA fragmentation, even in the presence of CF, an ADA inhibitor, and NBMPR, a nucleoside transporter inhibitor, which suggests the apoptotic effect of 2-CADO is independent of AdoR-mediated signalling. This was confirmed by our observation that 2-CADO was taken up into cells by the hENT nucleoside transporter, where it was phosphorylated by adenosine kinase.
Nucleoside transporters are classified into two major types based on their mechanism of action: equilibrative bidirectional processes driven by chemical gradients and inwardly directed concentrative processes driven by the Na+ electrochemical gradient (Thorn & Jarvis, 1996). The former are found in most mammalian cell types and can be divided into two subclasses – NBMPR-sensitive hENT1 and NBMPR-insensitive hENT2 – depending upon their sensitivity to NBMPR at concentrations in the nanomolar range. hENT2 is present primarily in specialized epithelia (Cass et al., 1998), while hENT1 more widely expressed. Recent molecular cloning of hENT1 (Griffiths et al., 1997) and hENT2 (Crawford et al., 1998) enabled us to access transcription of these transporters in RA – FLSs using RT – PCR. Consistent with earlier findings (Crawford et al., 1998), we observed hENT1 to be the major equilibrative nucleoside transporter in these cells. Nevertheless, cells from one patient did express both hENT1 and hENT2 mRNAs. For that reason, we used 10 μM NBMPR in our experiments, which is a relatively high concentration capable of inhibiting both hENT1 and hENT2: the IC50 for hENT2 with respect to inhibition of uridine transport in HeLa cells is 6 μM; the residual hENT2 activity at that concentration is about one-third of maximum (Crawford et al., 1998). In RA – FLSs, 10 μM NBMPR completely abolished the DNA fragmentation evoked by 50 μM 2-CADO and partially inhibited that evoked by 500 μM 2-CADO (Figure 5), confirming that intracellular 2-CADO is responsible for the observed RA – FLS apoptosis. Jarvis et al. (1985) reported that the apparent Km for 2-CADO uptake is 23 μM in human erythrocytes, and that 2-CADO competitively inhibits high-affinity binding of NBMPR with an apparent Ki of 180 μM. It is therefore not surprising that NBMPR does not completely inhibit the DNA fragmentation evoked by 500 μM 2-CADO.
Once taken up into cells, phosphorylation of the intracellular 2-CADO was necessary for induction of cell death, which is consistent with earlier observations in human B lymphoid cells (Yamanaka et al., 1984). As such, in the presence of 10 μM IT, a concentration at which 95% of intracellular adenosine is unphosphorylated (Dawicki et al., 1988), 2-CADO-induced DNA fragmentation in RA – FLSs was almost completely blocked. Thus, phosphorylation of intracellular 2-CADO by adenosine kinase was a requirement for activation of the pathway(s) leading to the apoptosis.
By activating a specific DNase that cleaves chromosomal DNA, proteases of the caspase family play a crucial role in the degradation of nuclear DNA into nucleosomal units, which is one of the hallmarks of apoptotic cell death (Nagata, 2000). The nonspecific caspase inhibitor z-VAD-fmk (20 μM) is known to inhibit apoptosis in T-cells (Atkinson et al., 1998). That z-VAD-fmk completely blocked the apoptotic effect of 2-CADO in the present study confirms that the caspase cascade is activated by 2-CADO – most likely phospho-2-CADO – and is solely responsible for the DNA fragmentation seen in RA – FLSs exposed to 2-CADO.
Our observation that extracellular adenosine and AdoR agonists failed to induce apoptosis does not preclude the possibility that adenosine acts as an antirheumatic agent via AdoR-mediated signalling (Cronstein et al., 1993; Montesinos et al., 2000). It is known that the concentrations of such proinflammatory cytokines as TNF-alpha and IL-6 are significantly elevated in RA joints (Lettesjo et al., 1998), and that blockade of cytokine signalling by specific antibodies is effective in the treatment of RA (Moreland et al., 1997; Yoshizaki et al., 1998). In that regard, AdoR-mediated signalling in RA – FLSs may serve to interfere with cytokine-activated signalling pathways rather than to directly induce apoptosis. Consistent with that hypothesis, our preliminary findings indicate that, in at least some RA – FLS preparations, DNA fragmentation occurs in cells exposed to adenosine in the presence of TNF-alpha (data not shown). Alternatively or in addition, extracellular adenosine may affect the function of cells other than FLSs. Several types of inflammatory and immune cells, including neutrophils and lymphocyte, accumulate in RA joints. Consequently, the in vivo biological effects of exogenous adenosine and its analogues should reflect the sum of the individual effects on each of the cell types present within the joint. It is thus notable that activation of A2A receptors reportedly inhibits oxidative bursting, degranulation and adhesion by neutrophils (Cronstein et al., 1992); IL-12 production by monocytes (Link et al., 2000); and antigen-specific cytotoxicity and cytokine production by T-cells (Koshiba et al., 1997). Moreover, A3 receptor signalling mediates production of cytokines MIP-1alpha, IL-6 and IL-12 in neutrophils and macrophages, and inhibits neutrophil infiltration in mouse collagen-induced arthritis (Szabo et al., 1998). A 2B receptor signalling also stimulates IL-8 secretion from human mast cells (Feoktistov & Biaggioni, 1995) and inhibits M-CSF-dependent macrophage proliferation in murine bone marrow-derived macrophages (Xaus et al., 1999). It is therefore interesting to us that whereas administration of the nonselective AdoR antagonist (theophylline or caffeine) reverses the antiinflammatory effects of methotrexate in rat adjuvant arthritis, administration of a selective A1, A2A or A2B receptor antagonist does not (Montesinos et al., 2000). This suggests a necessity for activation of multiple AdoRs in the treatment of RA. It remains to be clarified whether AdoR-mediated signalling is truly antirheumatic and, if so, which cell(s) is responsible.
Finally, the results presented suggest that the use of 2-CADO may represent a new and effective approach to the treatment of RA. Local administration of 2-CADO into RA joints may be useful in controlling pathophysiological FLS proliferation by directly inducing apoptosis and by affecting the function of infiltrating inflammatory and immune cells, while minimizing systemic side effects. Also advantageous is the ADA-resistant nature of 2-CADO (Turkozkan et al., 1996), since levels of ADA are elevated in the synovial fluid of RA patients (Pettersson et al., 1988). Additional in vivo analysis aimed at establishing the safety and efficacy of 2-CADO in the treatment of RA would thus seem warranted.
Acknowledgments
The authors thank Ms Kyoko Tanaka, Ms Maimi Kanamori and Ms Sugayo Kanagawa (Kobe University) for their technical assistance and Dr William F. Goldman (MST Editing Company, Baltimore MD, U.S.A.) for editorial assistance and preparation of the manuscript. This work is supported partly by Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science, and by Sapporo Bioscience Foundation.
Abbreviations
- ADA
adenosine deaminase
- AdoR
cell surface adenosine receptors
- 2-CADO
2-chloroadenosine
- CF
coformycin
- CGS21680
2-[4-(2-carboxyethyl)phenethylamino]-5′-N-ethylcarboxamidoadenosine
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-N-methyl-5′-carbamoyladenosine
- CSC
8-(3-chlorostyryl) caffeine
- DMARDs
disease modifying antirheumatic drugs
- DMSO
dimethylsulphoxide
- DPCPX
1,3-dipropyl-8-cyclopentylxanthine
- EIA
enzyme immunoassay
- ELISA
enzyme-liked immunosorbent assay
- hENT
human equilibrative nucleoside transporter
- IT
5′-iodotubercidin
- MRS1191
3-ethyl-5-benzyl-2-methyl-4-phenylethynyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate
- NBMPR
nitrobenzylmercaptopurin
- ORF
open reading frame
- PI
propidium iodide
- RA
rheumatoid arthritis
- RA-FLSs
fibroblast-like synoviocytes from rheumatoid arthritis patients
- R-PIA
(R)-N6-phenylisopropyladenosine
- XAC
xanthine amine congener
- z-VAD-fmk
benzyloxycarbonyl-Val-Ala-Asp fluoromethyl ketone
- ZM241385
4-(2-[7-amino-2-[2-furyl][1,2,4]triazolo[2,3-a][1,3,5]triazin-5-yl]amino]ethyl)-phenol
References
- ABBRACCHIO M.P., BURNSTOCK G. Purinergic signalling: pathophysiological roles. Jpn. J. Pharmacol. 1998;78:113–145. doi: 10.1254/jjp.78.113. [DOI] [PubMed] [Google Scholar]
- ARNETT F.C., EDWORTHY S.M., BLOCH D.A., MCSHANE D.J., FRIES J.F., COOPER N.S., HEALEY L.A., KAPLAN S.R., LIANG M.H., LUTHRA H.S., AL E. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- ATKINSON E.A., BARRY M., DARMON A.J., SHOSTAK I., TURNER P.C., MOYER R.W., BLEACKLEY R.C. Cytotoxic T lymphocyte-assisted suicide. Caspase 3 activation is primarily the result of the direct activation of granzyme B. J. Biol. Chem. 1998;273:21261–21266. doi: 10.1074/jbc.273.33.21261. [DOI] [PubMed] [Google Scholar]
- BOYLE D.L., SAJJADI F.G., FIRESTEIN G.S. Inhibition of synoviocyte collagenase gene expression by adenosine receptor stimulation. Arthritis Rheum. 1996;39:923–930. doi: 10.1002/art.1780390608. [DOI] [PubMed] [Google Scholar]
- CASS C.E., YOUNG J.D., BALDWIN S.A. Recent advances in the molecular biology of nucleoside transporters of mammalian cells. Biochem. Cell Biol. 1998;76:761–770. doi: 10.1139/bcb-76-5-761. [DOI] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate- phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- CRAWFORD C.R., PATEL D.H., NAEVE C., BELT J.A. Cloning of the human equilibrative, nitrobenzylmercaptopurine riboside (NBMPR)-insensitive nucleoside transporter ei by functional expression in a transport-deficient cell line. J. Biol. Chem. 1998;273:5288–5293. doi: 10.1074/jbc.273.9.5288. [DOI] [PubMed] [Google Scholar]
- CRONSTEIN B.N., LEVIN R.I., PHILIPS M., HIRSCHHORN R., ABRAMSON S.B., WEISSMANN G. Neutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptors. J. Immunol. 1992;148:2201–2206. [PubMed] [Google Scholar]
- CRONSTEIN B.N., NAIME D., OSTAD E. The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Invest. 1993;92:2675–2682. doi: 10.1172/JCI116884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DALZIEL H.H., WESTFALL D.P. Receptors for adenine nucleotides and nucleosides: subclassification, distribution, and molecular characterization. Pharmacol. Rev. 1994;46:449–466. [PubMed] [Google Scholar]
- DAWICKI D.D., AGARWAL K.C., PARKS R.J. Adenosine metabolism in human whole blood. Effects of nucleoside transport inhibitors and phosphate concentration. Biochem. Pharmacol. 1988;37:621–626. doi: 10.1016/0006-2952(88)90134-7. [DOI] [PubMed] [Google Scholar]
- FEOKTISTOV I., BIAGGIONI I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J. Clin. Invest. 1995;96:1979–1986. doi: 10.1172/JCI118245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREDHOLM B.B., ABBRACCHIO M.P., BURNSTOCK G., DALY J.W., HARDEN T.K., JACOBSON K.A., LEFF P., WILLIAMS M. Nomenclature and classification of purinoceptors. Pharmacol. Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- GIBLETT E.R., ANDERSON J.E., COHEN F., POLLARA B., MEUWISSEN H.J. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972;2:1067–1069. doi: 10.1016/s0140-6736(72)92345-8. [DOI] [PubMed] [Google Scholar]
- GRIFFITHS M., BEAUMONT N., YAO S.Y., SUNDARAM M., BOUMAH C.E., DAVIES A., KWONG F.Y., COE I., CASS C.E., YOUNG J.D., BALDWIN S.A. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat. Med. 1997;3:89–93. doi: 10.1038/nm0197-89. [DOI] [PubMed] [Google Scholar]
- GUIEU R., DUSSOL B., HALIMI G., BECHIS G., SAMPIERI F., BERLAND Y., SAMPOL J., COURAUD F., ROCHAT H. Adenosine and the nervous system: pharmacological data and therapeutic perspectives. Gen. Pharmacol. 1998;31:553–561. doi: 10.1016/s0306-3623(98)00071-8. [DOI] [PubMed] [Google Scholar]
- JARVIS S.M., MARTIN B.W., NG A.S. 2-Chloroadenosine, a permeant for the nucleoside transporter. Biochem. Pharmacol. 1985;34:3237–3241. doi: 10.1016/0006-2952(85)90340-5. [DOI] [PubMed] [Google Scholar]
- KOSHIBA M., KOJIMA H., HUANG S., APASOV S., SITKOVSKY M.V. Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. J. Biol. Chem. 1997;272:25881–25889. doi: 10.1074/jbc.272.41.25881. [DOI] [PubMed] [Google Scholar]
- LANGENEGGER T., MICHEL B.A. Drug treatment for rheumatoid arthritis. Clin. Orthop. 1999. pp. 22–30. [DOI] [PubMed]
- LETTESJO H., NORDSTROM E., STROM H., NILSSON B., GLINGHAMMAR B., DAHLSTEDT L., MOLLER E. Synovial fluid cytokines in patients with rheumatoid arthritis or other arthritic lesions. Scand. J. Immunol. 1998;48:286–292. doi: 10.1046/j.1365-3083.1998.00399.x. [DOI] [PubMed] [Google Scholar]
- LINK A.A., KINO T., WORTH J.A., MCGUIRE J.L., CRANE M.L., CHROUSOS G.P., WILDER R.L., ELENKOV I.J. Ligand-activation of the adenosine A2a receptors inhibits IL-12 production by human monocytes. J. Immunol. 2000;164:436–442. doi: 10.4049/jimmunol.164.1.436. [DOI] [PubMed] [Google Scholar]
- MONTESINOS M.C., YAP J.S., DESAI A., POSADAS I., MCCRARY C.T., CRONSTEIN B.N. Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine: evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis. Rheum. 2000;43:656–663. doi: 10.1002/1529-0131(200003)43:3<656::AID-ANR23>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- MORELAND L.W., BAUMGARTNER S.W., SCHIFF M.H., TINDALL E.A., FLEISCHMANN R.M., WEAVER A.L., ETTLINGER R.E., COHEN S., KOOPMAN W.J., MOHLER K., WIDMER M.B., BLOSCH C.M. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein [see comments] N. Engl. J. Med. 1997;337:141–147. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- NAGATA S. Apoptotic DNA fragmentation. Exp. Cell. Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- OLSSON R.A., PEARSON J.D. Cardiovascular purinoceptors. Physiol. Rev. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- PALMER T.M., GETTYS T.W., STILES G.L. Differential interaction with and regulation of multiple G-proteins by the rat A3 adenosine receptor. J. Biol. Chem. 1995;270:16895–16902. doi: 10.1074/jbc.270.28.16895. [DOI] [PubMed] [Google Scholar]
- PANAYI G.S., LANCHBURY J.S., KINGSLEY G.H. The importance of the T cell in initiating and maintaining the chronic synovitis of rheumatoid arthritis. Arthritis Rheum. 1992;35:729–735. doi: 10.1002/art.1780350702. [DOI] [PubMed] [Google Scholar]
- PATERSON A.R., LAU E.Y., DAHLIG E., CASS C.E. A common basis for inhibition of nucleoside transport by dipyridamole and nitrobenzylthioinosine. Mol. Pharmacol. 1980;18:40–44. [PubMed] [Google Scholar]
- PETTERSSON T., KLOCKARS M., WEBER T.H., VON E.R. Adenosine deaminase activity in joint effusions. Scand. J. Rheumatol. 1988;17:365–369. doi: 10.3109/03009748809105272. [DOI] [PubMed] [Google Scholar]
- RALEVIC V., BURNSTOCK G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- SHRYOCK J.C., BELARDINELLI L. Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am. J. Cardiol. 1997;79:2–10. doi: 10.1016/s0002-9149(97)00256-7. [DOI] [PubMed] [Google Scholar]
- SZABO C., SCOTT G.S., VIRAG L., EGNACZYK G., SALZMAN A.L., SHANLEY T.P., HASKO G. Suppression of macrophage inflammatory protein (MIP)-1alpha production and collagen-induced arthritis by adenosine receptor agonists. Br. J. Pharmacol. 1998;125:379–387. doi: 10.1038/sj.bjp.0702040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THORN J.A., JARVIS S.M. Adenosine transporters. Gen. Pharmacol. 1996;27:613–620. doi: 10.1016/0306-3623(95)02053-5. [DOI] [PubMed] [Google Scholar]
- TURKOZKAN N., BILIGHAN A., CAYCI B., DOGULU F., AYKOL S. The effects of 2-chloroadenosine and deoxycoformycin on the ATP level, Na-K ATPase activity in experimental brain ischemia of gerbil. Neurol. Res. 1996;18:345–348. doi: 10.1080/01616412.1996.11740434. [DOI] [PubMed] [Google Scholar]
- VAN ENGELAND M., RAMAEKERS F.C., SCHUTTE B., REUTELINGSPERGER C.P. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry. 1996;24:131–139. doi: 10.1002/(SICI)1097-0320(19960601)24:2<131::AID-CYTO5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- VERMES I., HAANEN C., STEFFENS N.H., REUTELINGSPERGER C. A novel assay for apoptosis. Flow cytometry detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Meth. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- WEINBLATT M.E. The role of current strategies in the future treatment of rheumatoid arthritis. Rheumatology (Oxford) 1999;2:19–23. [PubMed] [Google Scholar]
- WICKS I., COOLEY H., SZER J. Autologous hemopietic stem cell transplantation: a possible cure for rheumatoid arthritis. Arthritis Rheum. 1997;40:1005–1011. doi: 10.1002/art.1780400603. [DOI] [PubMed] [Google Scholar]
- WORTMANN R.L., VEUM J.A., RACHOW J.W. Purine catabolic enzymes in human synovial fluids. Adv. Exp. Med. Biol. 1989. pp. 393–398. [DOI] [PubMed]
- XAUS J., VALLEDOR A.F., CARDO M., MARQUES L., BELETA J., PALACIOS J.M., CELADA A. Adenosine inhibits macrophage colony-stimulating factor-dependent proliferation of macrophages through the induction of p27kip-1 expression. J. Immunol. 1999;163:4140–4149. [PubMed] [Google Scholar]
- YAMANAKA H., KAMATANI N., NISHIDA Y., NISHIOKA K., MIKANAGI K. Relationship between phosphorylation and cytotoxicity of 2-chloroadenosine and 6-methylmercaptopurine riboside in human cells. Biochim. Biophys. Acta. 1984;798:291–294. doi: 10.1016/0304-4165(84)90318-0. [DOI] [PubMed] [Google Scholar]
- YAMANAKA H., KAMATANI N., NOBORI T., NISHIOKA K., NISHIDA Y., MIKANAGI K.2-Chloroadenosine is phosphorylated and increases the production of hypoxanthine in human cells Adv. Exp. Med. Biol. 1986195583–587.Pt B [DOI] [PubMed] [Google Scholar]
- YOSHIZAKI K., NISHIMOTO N., MIHARA M., KISHIMOTO T. Therapy of rheumatoid arthritis by blocking IL-6 signal transduction with a humanized anti-IL-6 receptor antibody. Springer Semin. Immunopathol. 1998;20:247–259. doi: 10.1007/BF00832010. [DOI] [PubMed] [Google Scholar]
- ZVAIFLER N.J., FIRESTEIN G.S. Pannus and pannocytes. Alternative models of joint destruction in rheumatoid arthritis. Arthritis Rheum. 1994;37:783–789. doi: 10.1002/art.1780370601. [DOI] [PubMed] [Google Scholar]