

Abstract
Heat stress (HS) is known to protect the myocardium against ischaemic damage. It has been reported that reactive oxygen species (ROS) are abundantly produced during this stress. Since mechanisms triggering the HS-induced cardioprotection remain unknown, we investigated the role of ROS in the genesis of this protective phenomenon.
Rats were divided into four groups (n=8 in each group), subjected to either hyperthermia (42°C internal temperature for 15 min) or sham anaesthesia and treated or not with N-2-mercaptopropionyl glycine (MPG), a synthetic antioxidant, 10 min before HS. Twenty-four hours later, their hearts were isolated, retrogradely perfused, and subjected to a 30-min occlusion of the left coronary artery followed by 120 min of reperfusion. Myocardial Hsp 27 and 70 expression was assessed by Western blot analysis (n=4). Cardiac activities of antioxidant enzymes (superoxide dismutase and glutathione peroxidase) were also examined (n=4).
Infarct-to-risk zone ratio was significantly reduced in HS (17±1.3%) compared to Sham (34.3±1.7%) hearts. This effect was abolished by MPG pretreatment (40.6±1.9% in HS+MPG vs 39.8±2.5% in Sham+MPG hearts). This cardioprotection was associated with an enhanced Hsp 27 and 70 expression, which was not modified by MPG pretreatment. Antioxidant enzyme activities was not modified by heat stress or MPG pretreatment.
Free radical production following hyperthermia appears to play a role in the heat stress induced cardioprotection, independently of Hsp levels. Antioxidant enzyme activities do not seem to be implicated in this cardioprotective mechanism.
Keywords: Cardiovascular system, animal models, heat stress, heat shock proteins, free radicals
Introduction
Heat stress (HS) is known to increase myocardial tolerance to a subsequent sustained period of ischaemia by preserving myocardial function (Currie et al., 1988) and reducing myocardial necrosis (Donnelly et al., 1992; Yellon et al., 1992). Various mechanisms seem to initiate this cardioprotective effect (Joyeux et al., 1999). Among them, nitric oxide (NO), which constitutes the most abundant free radical in the body (Moncada et al., 1991), has been recently shown to trigger the resistance to myocardial infarction induced by HS in the isolated rat heart (Arnaud et al., 2001).
Reactive oxygen species (ROS), like superoxide anion (O2.−), are also produced during hyperthermia (Salo et al., 1991; Flanagan et al., 1998). It has been observed that these ROS play an essential role in the genesis of the delayed protection conferred by ischaemic preconditioning (IP) (Sun et al., 1996; Zhou et al., 1996). Since the cardioprotection induced 24 – 48 h following HS resembles that observed during the late phase of IP, triggers under investigation for their role in IP may provide a potential mechanism for HS-induced protection (for review, see Bolli, 2000). Hence, the initial oxidative stress occurring following hyperthermia could potentially trigger the HS-response.
Many candidates have been proposed as end-effectors of the heat stress response (for review, see Joyeux et al., 1999). Among them, the increase in endogenous cardiac antioxidant defences has been reported. Indeed, the activation of the manganese superoxide dismutase (Mn-SOD) seems to be associated with the HS-induced cardioprotection (Yamashita et al., 1998). The de novo synthesis of heat stress proteins (Hsp) seems also to mediate this protective effect. A direct correlation between the amount of Hsp 70 expression and the degree of HS-induced myocardial protection has been observed in the rat (Hutter et al., 1994) and in the rabbit (Marber et al., 1994). Furthermore, small Hsp (Hsp 27) overexpression seems to confer protection against ischaemic injury in cardiac myocytes (Martin et al., 1997).
The purpose of this study was thus to determine the role of the initial oxidative stress occurring following hyperthermia in the HS-induced cardioprotection and its potential interaction with end-effectors of this response. Therefore, we examined the effect of a synthetic antioxidant, N-2-mercaptopropionyl glycine (MPG), on the resistance to myocardial infarction conferred by heat stress in the isolated rat heart. The effect of MPG treatment on HS-induced increase in cardiac Hsp synthesis and antioxidant enzyme activities has also been investigated.
Methods
Experimental treatment groups
Male Wistar rats (280 – 340 g) were used for this study. This investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no.85-23, revised 1996).
First, rats were submitted to either heat stress (HS groups) or anaesthesia without hyperthermia (Sham groups). Prior to this procedure, the animals were treated with either MPG, a thiol compound that avidly scavenges oxidant species and especially hydroxyl radical (.OH) (Bolli et al., 1989; Sun et al., 1993), or saline, as previously described (Yamashita et al., 1998). Subsequently, all animals were allowed to recover for 24 h. Then, ischaemia – reperfusion was performed in isolated hearts. The four following experimental groups (n=8 per group) were studied: Group HS, rats were sham pretreated with saline 10 min before undergoing heat stress; Group HS+MPG, rats were pretreated with MPG (100 mg kg−1, i.p.) 10 min before undergoing heat stress; In Groups Sham and Sham+MPG, rats were similarly pretreated before being sham anaesthetized. The experimental protocols are summarized in Figure 1.
Figure 1.

Experimental protocol. MPG - N-2-mercaptopropionyl glycine.
Heat stress protocol
HS was achieved, as previously described (Joyeux et al., 1998), by placing anaesthetized (with 25 mg kg−1, i.p., sodium pentobarbitone) rats in an environmental chamber under an infrared light. Their body temperature, recorded with a rectal probe, was increased to 42±0.2°C for 15 min. Sham control animals were anaesthetized only. All rats were allowed to recover for 24 h.
Ischaemia – reperfusion protocol
Twenty-four hours after HS, rats were re-anaesthetized (60 mg kg−1, i.p., sodium pentobarbitone) and treated with heparin (500 U kg−1, iv). The heart was rapidly excised and immediately immersed in 4°C Krebs-Henseleit buffer solution (NaCl 118.0, KCl 4.7, CaCl2 1.8, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25.2 and glucose 11.0 mM). The aortic stump was then cannulated and the heart perfused retrogradely using the Langendorff technique at a constant pressure (75 mmHg) with oxygenated Krebs-Henseleit buffer. A water-filled balloon (Hugo Sachs, no.4), coupled to a pressure transducer (Statham), was inserted into the left ventricular cavity via the left atrium for pressure recording. Left ventricular end-diastolic pressure (LVEDP) was adjusted between 5 – 10 mmHg. Myocardial temperature was measured by a thermoprobe inserted into the left ventricle and was maintained constant close to 37°C. For temporary occlusion of the left coronary artery (LCA), a 3/0 silk suture (Mersilk W546, Ethicon) was placed around the artery a few millimetres distal to the aortic root. After 20 min of stabilization, regional ischaemia was induced by tightening the snare around the LCA for 30 min. Thereafter the heart was reperfused for 120 min. Coronary flow (CF) was measured throughout the ischaemia – reperfusion procedure, by collecting the effluent. Heart rate (HR) and left ventricular developed pressure (LVDP=difference between left ventricular systolic pressure and LVEDP) were continuously recorded on a polygraph (Windograph, Gould Instrument). At the end of the reperfusion period, the coronary artery ligature was re-tied and unisperse blue (Ciba-Geigy) dye was slowly infused through the aorta to delineate the myocardial risk zone. After removal of the right ventricle and connective tissues, the heart was frozen and then cut into 2 mm transverse sections from apex to base (6 – 7 slices/heart). Following defrosting, the slices were incubated at 37°C with 1% triphenyltetrazolium chloride in phosphate buffer (pH 7.4) for 10 – 20 min and fixed in 10% formaldehyde solution to distinguish stained viable tissue and unstained necrotic tissue. Left ventricular infarct zone (I) was determined using a computerized planimetric technique (Minichromax, Biolab) and expressed as a percentage of the risk zone (R) and of the left ventricle (LV).
Western blot analysis of myocardial Hsp 27 and 70
To determine Hsp 27 and 70 expression, additional animals (n=4 in each group) were submitted to either HS or sham anaesthesia, pretreated or not with MPG. Twenty-four hours later, animals were re-anaesthetized and treated with heparin as described above, before their heart was quickly excised. Hearts were minced and homogenized in a lysis buffer pH 7.4 containing (mM) Tris 50, NaCl 100, EDTA 5, NaF 1, Na3VO4 1, Na2MoO4 100 μM, protease inhibitors 5 μl ml−1, 1‰ microcystine and 1% Triton. Samples were then centrifuged for 5 min at 14,000 r.p.m., 4°C. The supernatant were diluted in sample buffer containing 2% sodium dodecyl sulphate (SDS), 10% glycerol, 62.5 mM Tris HCl (pH 6.8), 100 mM DTT, 0.1% bromophenol blue. The protein concentration in samples was determined using the BCA protein assay Kit (Pierce). Samples were loaded at 5 μg/lane and proteins were separated by electrophoresis on 12% acrylamide gels. Proteins were transfered from gel to nitrocellulose membrane, which was then incubated with antibodies to either Hsp 27 (Upstate Biotechnology) or Hsp 70 (Santa Cruz Biotechnology). The second antibody was peroxidase-conjugated anti-rabbit for Hsp 27 antisera, and peroxidase-conjugated anti-goat for Hsp 70 antisera. The membrane was developed using an enhanced chemiluminescence system (Pierce) to obtain an autoradiogram.
The relative levels of Hsp 27 and 70 were determined by densitometry of the autoradiogram (Sony video system coupled with a Minichromax digitization system and Biolab software).
Determination of myocardial antioxidant enzyme activities
To measure myocardial levels of superoxide dismutase (SOD) and glutathione peroxidase (GPx), the frozen tissue samples were homogenized on ice in (mM) Tris 10, diethylenetriaminepentaacetic acid 1 and phenylmethylsulfonyl fluoride 1, pH 7,4. SOD activity was determined using the pyrogallol assay following the procedure described by Marklund & Marklund (1974), based on the competition between pyrogallol oxidation by superoxide radicals and superoxide dismutation by SOD, photometrically read at λ=420 nm. GPx activity was measured after the rate of NADPH oxidation by t-butyl hydroperoxide at 340 nm (Turrens et al., 1992). Activities were corrected for the concentration of protein.
Statistical analysis
The data are presented as mean±s.e.mean. Comparisons in CF, HR and LVDP were performed using two-way repeated measures ANOVA. Post-hoc comparisons were done using Tukey tests. Infarct size analysis, semi-quantitative Hsp analysis and comparison in antioxidant enzyme activities were performed by a one-way ANOVA. P values ⩽0.05 were considered significant.
Materials
MPG were purchased from Sigma (France). Unisperse blue dye was obtained from Ciba-Geigy (France). Anti-Hsp 27 antibody was from Upstate biotechnology and anti-Hsp 70 from Santa Cruz Biotechnology Inc. The enhanced chemiluminescence reaction kit assay was from Pierce.
Results
Haemodynamic data
Table 1 summarizes CF, HR and LVDP data recorded in the four experimental groups during the stabilization and ischaemia-reperfusion periods. Twenty-four hours after HS or sham anaesthesia, there was no statistically significant difference in haemodynamic performance measured between the four groups.
Table 1.
Haemodynamic data
Infarct data
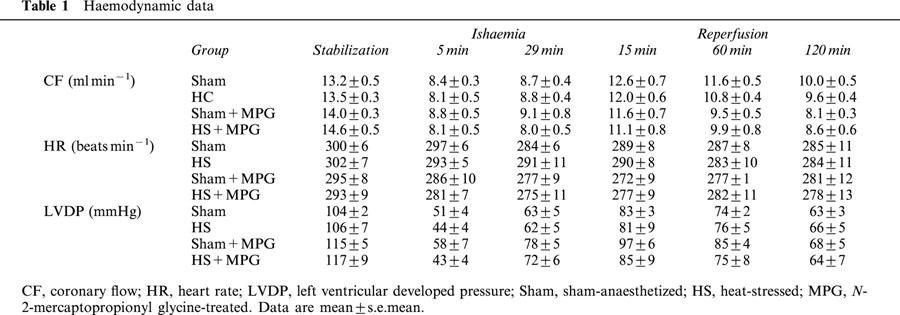
HS significantly reduced infarct size from 34.3±1.7% in Sham to 17±1.3% in HS hearts (P<0.001, one-way ANOVA). This infarct size-reducing effect of HS was abolished by MPG pretreatment (40.6±1.9%), whereas in non-heat stressed rats, MPG had no effect on infarct size (39.8±2.5%) (Figure 2). Similar results were observed concerning I/LV ratio between the four groups (data not shown). Myocardial risk zone expressed as the percentage of the left ventricle (R/LV) was similar for all groups: 45.9±2.3% in Sham, 44.7±2.6% in HS, 44.1±1.5% in Sham+MPG and 42.6±1.1% in HS+MPG groups. Therefore, differences in infarct size did not result from variability in the risk zone.
Figure 2.
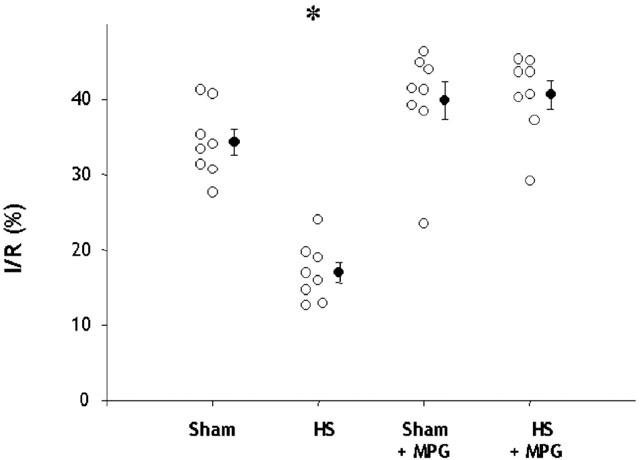
Effect of N-2-mercaptopropionyl glycine (MPG) pretreatment on myocardial infarct size assessed after 30-min coronary occlusion followed by 120-min reperfusion in isolated hearts from rats subjected to sham-anaesthesia (Sham) or heat-stress (HS). Infarct size (I) is expressed as a percentage of the risk zone (R). *P<0.001 vs the other groups (one-way ANOVA).
Western blot analysis of Hsp 27 and 70
Western blot analysis of myocardial Hsp 27 and 70 (Figures 3 and 4) expression showed a marked increase of these proteins, 24 h after HS. MPG-pretreatment did not modify the HS-induced increase in Hsp 27 and 70 expression (Figures 3 and 4).
Figure 3.
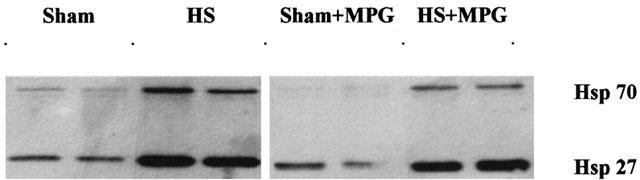
Western blot analysis of myocardial Hsp 27 and 70 in the four experimental groups. Sham, sham-anaesthetized; HS, heat-stressed; MPG, N-2-mercaptopropionyl glycine-treated.
Figure 4.
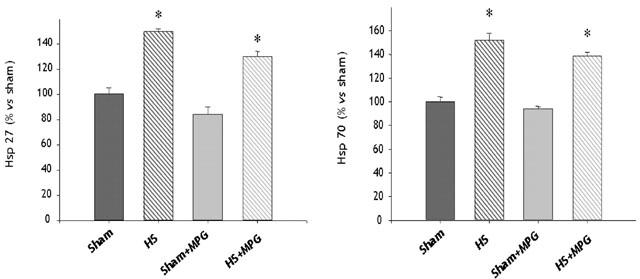
Semi-quantitative analysis of myocardial Hsp 27 and 70 levels obtained by autoradiogram in the four experimental groups, n=4 in each group). Sham, sham-anaesthetized; HS, heat-stressed; MPG, N-2-mercaptopropionyl glycine-treated. *P<0.001 vs Sham and Sham+MPG (one-way ANOVA).
Antioxidant enzyme activities data
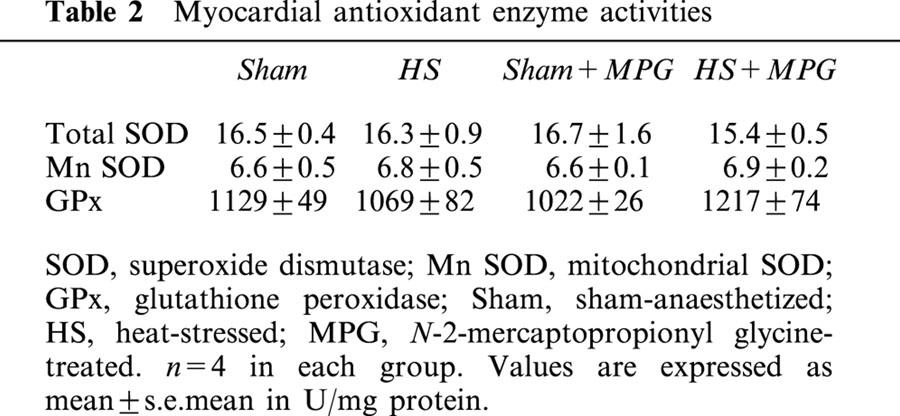
No differences in SOD and GPx activities was seen between the different groups. Results are summarized in Table 2.
Table 2.
Myocardial antioxidant enzyme activities
Discussion
In our in vitro rat model of myocardial infarction, prior HS significantly reduced infarct size performed 24 h after, in accordance with previous studies (for review, see Joyeux et al., 1999). The mechanism for this delayed protection is currently unknown. The pertinent finding of this work is the implication of ROS as triggers of this cardioprotective mechanism, since MPG, administered before HS, abolished the HS-induced-infarct size reduction, in this isolated rat heart model.
HS-induced reduction on infarct size
In our study, we investigated the effect of MPG pretreatment on the HS-induced reduction of infarct size, in the isolated rat heart. The effect of HS is well documented in the literature. Actually, it is known that hyperthermia (42°C, during 15 – 20 min) reduces the infarct size assessed 24 h later, at the end of a 30 min left coronary artery occlusion – 120 min reperfusion. This result has been observed in vivo in the rat (Donnelly et al., 1992; Hutter et al., 1994) and in the rabbit (Marber et al., 1993), as well as in the isolated rat heart (Joyeux et al., 1997; 1998). Controversially, Yamashita et al. (1998) do not observe a significant decrease in infarct size 24 h but only 48 – 72 h after HS (42°C). However, the decrease in infarct size we observed cannot be compared with this study, since reperfusion durations are different (120 min versus 48 h). Thus, the discrepancy between our study and the one by Yamashita et al. (1998) may be explained by the difference in reperfusion duration.
Delayed preconditioning mediated by ROS
ROS have yet been shown to be involved in cardioprotection. Indeed, Yamashita et al. (1998) showed, in conscious rat, that oxygen free radicals production during hyperthermia is necessary to the reduction of infarct size, as well as of ventricular fibrillation, investigated both 30 min or 48 h after HS. Moreover, it has been observed in cardiomyocytes that a burst of ROS, generated during the initial periods of brief repetitive anoxia, contributes to the late cytoprotection induced by this preconditioning (Zhou et al., 1996). In addition, anti-oxidant treatment completely blocks the development of a late protection induced by IP against stunning in conscious pigs, indicating that the initial production of ROS is one of the mechanism of the late protective response (Sun et al., 1996). ROS seem also to trigger the delayed protective effect of nitric oxide (NO) donors in conscious rabbits, since MPG coadministration abolishes the protection against myocardial stunning and infarction conferred by NO donors alone (Takano et al., 1998).
HS-induced production of ROS
Our study provides indirect demonstration that a burst of free radicals during hyperthermia is necessary for the HS-induced cardioprotection. In previous studies, hyperthermia was shown to induce production of free radicals. A sharp increase in ROS generation has been observed after hyperthermia in a cellular model, using electron paramagnetic resonance spin trapping (Flanagan et al., 1998). Moreover, oxidative stress, assessed by the efflux of both glutathione and oxidised glutathione, has been shown to be significantly increased by hyperthermia, in the rat liver (Powers et al., 1992). Zuo et al. (2000) also suggested that HS stimulates superoxide production in the mouse skeletal muscle. Finally, Salo et al. (1991) showed in the rat muscle that generation of O2.− by the mitochondria increases in a temperature-dependent way, from 37 – 45°C.
De novo synthesis of proteins following HS
The fact that HS-induced cardioprotection requires 24 h to occur suggests that this phenomenon is related to de novo synthesis of proteins, which might mediate this response.
Hsp have been first proposed as effectors of this response, since a positive correlation between HS-induced Hsp 70 expression and infarct size reduction was described (Marber et al., 1993; 1994; Hutter et al., 1994). Our Western blot analysis of myocardial Hsp 27 and 70 expression showed a marked increase of these proteins, 24 h after HS, which was not modified by ROS scavenger pretreatment. Thus, it seems here that HS-induced cardioprotection is dissociated from induction of Hsp, since pretreatment with MPG abolished cardioprotection while having no effect on myocardial Hsp content. In the same manner, previous studies also showed that the cardioprotection conferred by HS could be abolished by blocking different mediators of this response independently of the myocardial Hsp expression (Arnaud et al., 2001; Joyeux et al., 1997; 1998). However, Yamashita et al. (1998) showed that treatment with a radical scavenger reduces the increase in Hsp 70 expression as well as inhibiting the cardioprotection induced 72 h following HS. Thus, the exact functional role of Hsp in cardioprotection is still unknown and remain to be determined. Furthermore, we have only observed here the expression of Hsp, by Western blot, and we cannot exclude possible post-translational modifications of these proteins in response to HS.
Other proteins could be upregulated in response to hyperthermia. Thus, it seems that HS also induces an increase in myocardial antioxidant activity. Indeed, it has been shown in the rat that Mn – SOD was significantly enhanced 24 h after HS in cardiomyocytes (Yamashita et al., 1997) or 48 h after HS in the heart (Yamashita et al., 1998). Maulik et al. (1995) have observed an increase in Mn – SOD activity 40 h after drug-induced HS-preconditioning in pig myocardium. However, controversy still exists on HS-enhanced antioxidant activity, which seems to be animal species and time dependant. Indeed, it has also been reported that myocardial SOD activity is not modified 24 h after HS in the rat (Currie et al., 1988), and in the mouse (Xi et al., 1998). Moreover, GPx activity seems not to be influenced by prior HS in the rat myocardium (Currie et al., 1988). Our results confirm this controversy. Indeed, we do not observe any modification in myocardial SOD or GPx activities, 24 h after HS in our rat model. Thus, antioxidant enzymes seem not to be effectors of the HS-induced cardioprotection in our rat heart model.
Conclusions
This study demonstrates that an initial burst of oxygen free radicals is necessary to initiate the HS-induced late cardioprotection, since MPG, a free radical scavenger, abolished the HS-response. This protection seems not to be related to the HS-induced increase in myocardial Hsp synthesis or antioxidant enzyme activities. Further experiments are needed to clarify the exact mechanisms occurring following ROS production and mediating the HS response.
Abbreviations
- CF
coronary flow
- Cu
Zn SOD, copper, zinc superoxide dismutase
- GPx
glutathione peroxidase
- HR
heart rate
- HS
heat stress
- Hsp
heat stress protein
- I
infarct zone
- IP
ischaemic preconditioning
- LV
left ventricle
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end-diastolic pressure
- Mn SOD
manganese superoxide dismutase
- MPG
N-2-mercaptopropionyl glycine
- NO
nitric oxide
- O2.−
superoxide anion
- .OH
hydroxyl radical
- R
risk zone
- ROS
reactive oxygen species
- VF
ventricular fibrillation
References
- ARNAUD C., LAUBRIET A., JOYEUX M., GODIN-RIBUOT D., ROCHETTE L., DEMENGE P., RIBUOT C. Role of nitric oxide synthases in the infarct size-reducing effect conferred by heat stress in isolated rat hearts. Br. J. Pharmacol. 2001;132:1845–1851. doi: 10.1038/sj.bjp.0703942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLLI R. The late phase of preconditioning. Circ. Res. 2000;87:972–983. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- BOLLI R., JEROUDI M.O., PATEL B.S., ARUOMA O.I., HALLIWELL B., LAI E.K., MCCAY P.B. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial ‘stunning' is a manifestation of reperfusion injury. Circ. Res. 1989;65:607–622. doi: 10.1161/01.res.65.3.607. [DOI] [PubMed] [Google Scholar]
- CURRIE R.W., KARMAZYN M., KLOC M., MAILER K. Heat-shock response is associated with enhanced postischemic ventricular recovery. Circ. Res. 1988;63:543–549. doi: 10.1161/01.res.63.3.543. [DOI] [PubMed] [Google Scholar]
- DONNELLY T.J., SIEVERS R.E., VISSERN F.L., WELCH W.J., WOLFE C.L. Heat shock protein induction in rat hearts. A role for improved myocardial salvage after ischemia and reperfusion. Circulation. 1992;85:769–778. doi: 10.1161/01.cir.85.2.769. [DOI] [PubMed] [Google Scholar]
- FLANAGAN S.W., MOSELEY P.L., BUETTNER G.R. Increased flux of free radicals in cells subjected to hyperthermia: detection by electron paramagnetic resonance spin trapping. FEBS Lett. 1998;431:285–286. doi: 10.1016/s0014-5793(98)00779-0. [DOI] [PubMed] [Google Scholar]
- HUTTER M.M., SIEVERS R.E., BARBOSA V., WOLFE C.L. Heat-shock protein induction in rat hearts. A direct correlation between the amount of heat-shock protein induced and the degree of myocardial protection. Circulation. 1994;89:355–360. doi: 10.1161/01.cir.89.1.355. [DOI] [PubMed] [Google Scholar]
- JOYEUX M., BAXTER G.F., THOMAS D.L., RIBUOT C., YELLON D.M. Protein kinase C is involved in resistance to myocardial infarction induced by heat stress. J. Mol. Cell. Cardiol. 1997;29:3311–3319. doi: 10.1006/jmcc.1997.0556. [DOI] [PubMed] [Google Scholar]
- JOYEUX M., GODIN-RIBUOT D., PATEL A., DEMENGE P., YELLON D.M., RIBUOT C. Infarct size-reducing effect of heat stress and alphal adrenoceptors in rats. Br. J. Pharmacol. 1998;125:645–650. doi: 10.1038/sj.bjp.0702137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOYEUX M., GODIN-RIBUOT D., YELLON D.M., DEMENGE P., RIBUOT C. Heat stress response and myocardial protection. Fundam. Clin. Pharmacol. 1999;13:1–10. doi: 10.1111/j.1472-8206.1999.tb00314.x. [DOI] [PubMed] [Google Scholar]
- MARBER M.S., LATCHMAN D.S., WALKER J.M., YELLON D.M. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- MARBER M.S., WALKER J.M., LATCHMAN D.S., YELLON D.M. Myocardial protection after whole body heat stress in the rabbit is dependent on metabolic substrate and is related to the amount of the inducible 70-kD heat stress protein. J. Clin. Invest. 1994;93:1087–1094. doi: 10.1172/JCI117059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARKLUND S., MARKLUND G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- MARTIN J.L., MESTRIL R., HILAL-DANDAN R., BRUNTON L.L., DILLMANN W.H. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation. 1997;96:4343–4348. doi: 10.1161/01.cir.96.12.4343. [DOI] [PubMed] [Google Scholar]
- MAULIK N., ENGELMAN R.M., WEI Z., LIU X., ROUSOU J.A., FLACK J.E., DEATON D.W., DAS D.K. Drug-induced heat-shock preconditioning improves postischemic ventricular recovery after cardiopulmonary bypass. Circulation. 1995;92:II381–II388. doi: 10.1161/01.cir.92.9.381. [DOI] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M., HIGGS E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- POWERS R.H., STADNICKA A., KALBFLEISH J.H., SKIBBA J.L. Involvement of xanthine oxidase in oxidative stress and iron release during hyperthermic rat liver perfusion. Cancer Res. 1992;52:1699–1703. [PubMed] [Google Scholar]
- SALO D.C., DONOVAN C.M., DAVIES K.J. HSP70 and other possible heat shock or oxidative stress proteins are induced in skeletal muscle, heart, and liver during exercise. Free Radic. Biol. Med. 1991;11:239–2346. doi: 10.1016/0891-5849(91)90119-n. [DOI] [PubMed] [Google Scholar]
- SUN J.Z., KAUR H., HALLIWELL B., LI X.Y., BOLLI R. Use of aromatic hydroxylation of phenylalanine to measure production of hydroxyl radicals after myocardial ischemia in vivo. Direct evidence for a pathogenetic role of the hydroxyl radical in myocardial stunning. Circ. Res. 1993;73:534–549. doi: 10.1161/01.res.73.3.534. [DOI] [PubMed] [Google Scholar]
- SUN J.Z., TANG X.L., PARK S.W., QIU Y., TURRENS J.F., BOLLI R. Evidence for an essential role of reactive oxygen species in the genesis of late preconditioning against myocardial stunning in conscious pigs. J. Clin. Invest. 1996;97:562–576. doi: 10.1172/JCI118449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKANO H., TANG X.L., QIU Y., GUO Y., FRENCH B.A., BOLLI R. Nitric oxide donors induce late preconditioning against myocardial stunning and infarction in conscious rabbits via an antioxidant-sensitive mechanism. Circ. Res. 1998;83:73–84. doi: 10.1161/01.res.83.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURRENS J.F., THORNTON J., BARNARD M.L., SNYDER S., LIU G., DOWNEY J.M. Protection from reperfusion injury by preconditioning hearts does not involve increased antioxidant defenses. Am. J. Physiol. 1992;262:H585–H589. doi: 10.1152/ajpheart.1992.262.2.H585. [DOI] [PubMed] [Google Scholar]
- XI L., CHELLIAH J., NAYEEM M.A., LEVASSEUR J.E., HESS M.L., KUKREJA R.C. Whole body heat shock fails to protect mouse heart against ischemia/reperfusion injury: role of 72 kDa heat shock protein and antioxidant enzymes. J. Mol. Cell. Cardiol. 1998;30:2213–2227. doi: 10.1006/jmcc.1998.0781. [DOI] [PubMed] [Google Scholar]
- YAMASHITA N., HOSHIDA S., NISHIDA M., IGARASHI J., TANIGUCHI N., TADA M., KUZUYA T., HORI M. Heat shock-induced manganese superoxide dismutase enhances the tolerance of cardiac myocytes to hypoxia-reoxygenation injury. J. Mol. Cell. Cardiol. 1997;29:1805–1813. doi: 10.1006/jmcc.1997.0415. [DOI] [PubMed] [Google Scholar]
- YAMASHITA N., HOSHIDA S., TANIGUCHI N., KUZUYA T., HORI M. Whole-body hyperthermia provides biphasic cardioprotection against ischemia/reperfusion injury in the rat. Circulation. 1998;98:1414–1421. doi: 10.1161/01.cir.98.14.1414. [DOI] [PubMed] [Google Scholar]
- YELLON D.M., PASINI E., CARGNONI A., MARBER M.S., LATCHMAN D.S., FERRARI R. The protective role of heat stress in the ischaemic and reperfused rabbit myocardium. J. Mol. Cell. Cardiol. 1992;24:895–907. doi: 10.1016/0022-2828(92)91102-b. [DOI] [PubMed] [Google Scholar]
- ZHOU X., ZHAI X., ASHRAF M. Direct evidence that initial oxidative stress triggered by preconditioning contributes to second window of protection by endogenous antioxidant enzyme in myocytes. Circulation. 1996;93:1177–1184. doi: 10.1161/01.cir.93.6.1177. [DOI] [PubMed] [Google Scholar]
- ZUO L., CHRISTOFI F.L., WRIGHT V.P., LIU C.Y., MEROLA A.J., BERLINER L.J., CLANTON T.L. Intra- and extracellular measurement of reactive oxygen species produced during heat stress in diaphragm muscle. Am. J. Physiol. Cell Physiol. 2000;279:C1058–C1066. doi: 10.1152/ajpcell.2000.279.4.C1058. [DOI] [PubMed] [Google Scholar]