

Abstract
The effects of ascorbate were assessed on vasodilatation mediated by endothelium-derived hyperpolarizing factor (EDHF) in the ciliary vascular bed of the bovine isolated perfused eye and in the rat isolated perfused mesenteric arterial bed.
In the bovine eye, EDHF-mediated vasodilator responses induced by acetylcholine or bradykinin were powerfully blocked when ascorbate (50 μM) was included in the perfusion medium for at least 120 min; with acetylcholine a normally-masked muscarinic vasoconstrictor response was also uncovered.
The blockade of EDHF-mediated vasodilatation by ascorbate was time-dependent (maximum blockade at 120 min) and concentration-dependent (10 – 150 μM).
Ascorbate (50 μM) also blocked acetylcholine-induced, EDHF-mediated vasodilator responses in the rat mesenteric arterial bed in a time-dependent manner (maximum blockade at 180 min).
The ability of ascorbate to block EDHF-mediated vasodilatation is likely to result from its reducing properties, since this action was mimicked in the bovine eye by two other reducing agents, namely, N-acetyl-L-cysteine (1 mM) and dithiothreitol (100 μM), but not by the redox-inactive analogue, dehydroascorbate (50 μM).
In conclusion, concentrations of ascorbate present in normal plasma block EDHF-mediated vasodilator responses in the bovine eye and rat mesentery. The mechanism and physiological consequences of this blockade remain to be determined.
Keywords: Ascorbate, antioxidant, eye, endothelium, endothelium-derived hyperpolarizing factor, EDHF, mesenteric arterial bed, vasodilatation
Introduction
The aqueous humour of the eye is formed by the epithelial cells of the ciliary body from plasma supplied via the ciliary arterial bed (Millar & Kaufman, 1995). Although the plasma concentration of ascorbate in humans is 46±8 μM (range 30 – 150 μM; Keaney & Vita, 1995; Levine et al., 1996) the epithelial cells of the ciliary body actively transport this antioxidant such that the levels found in the aqueous humour reach around 1 mM in humans and a wide range of animal species (Davson, 1980; Halliwell & Gutteridge, 1989). The precise reason for this high concentration of ascorbate in the aqueous humour is unknown, but since it avidly scavenges superoxide anion (Som et al., 1983), it may be present to compensate for the particularly low levels of superoxide dismutase found in the eye (Halliwell & Gutteridge, 1989). As a consequence of the secretion of high concentrations of ascorbate by the ciliary body, we have routinely included this antioxidant (50 μM) in the perfusate when studying aqueous humour dynamics in the bovine isolated perfused eye preparation (Shahidullah & Wilson, 1999; Wilson et al., 1993).
More recently, we have begun to study the bovine ciliary vascular bed and have found that nitric oxide exerts a small tonic vasodilator action, but it plays no part in the powerful endothelium-dependent vasodilator responses induced by acetylcholine or bradykinin in this preparation (McNeish et al., 2001). These vasodilator responses are, however, blocked by depolarizing solutions of KCl and by charybdotoxin (McNeish et al., 2001) and thus have the characteristics of the endothelium-derived hyperpolarizing factor (EDHF; for reviews see Feletou & Vanhoutte, 1999; Campbell & Harder, 2001).
Our earlier experiments on the ciliary vasculature, unlike those on aqueous humour dynamics, were conducted in the absence of ascorbate. Consequently, we had no knowledge of whether the presence of this antioxidant in the perfusate would modify vasodilator responses in the ciliary vascular bed of the bovine eye. Ascorbate does, however, have well documented effects on nitric oxide-induced vasodilator responses. Specifically, ascorbate is able to recover nitric oxide-dependent vasodilatation following its impairment by oxidant stress in isolated arterial rings (Dudgeon et al., 1998; Fontana et al., 1999). This protective action is believed to result from the ability of ascorbate to scavenge superoxide anion and so prevent it from destroying nitric oxide (Gryglewski et al., 1986; Rubanyi & Vanhoutte, 1986). An additional protective action may derive from the ability of ascorbate to elevate the levels of the cofactor, tetrahydrobiopterin, and so enhance production of nitric oxide by nitric oxide synthase (Huang et al., 2000; Heller et al., 2001). This ability of ascorbate to protect nitric oxide-dependent vasodilatation offers great potential for therapeutic intervention in a diverse range of vascular pathologies associated with oxidant stress. For example, treatment of patients with ascorbate has led to recovery of impaired nitric oxide-mediated vasodilatation in essential hypertension, chronic heart failure, atherosclerosis, hypercholesterolaemia and diabetes (for reviews see Carr et al., 2000; May, 2000).
Taking account of both the ability of ascorbate to augment nitric oxide-mediated vasodilatation, and its active transport by the ciliary body, we wished to determine if this antioxidant could affect EDHF-mediated vasodilatation in the ciliary vascular bed of the bovine isolated eye. Preliminary accounts of these findings have already been published (McNeish et al., 2002a, 2002b).
Methods
Preparation of the bovine isolated arterially perfused eye
The ciliary vascular bed of the bovine eye was perfused using the constant flow method as previously described (McNeish et al., 2001). In brief, bovine eyes obtained from a local abattoir within 1 h of killing were cannulated through a long posterior ciliary artery and perfused at 37°C with Krebs solution containing (mM): NaCl, 118; KCl, 4.7; CaCl2, 2.5; KH2PO4, 1.2; MgSO4 1.2; NaHCO3, 25; glucose, 11.5; and gassed with O2 containing 5% CO2. Flow was commenced at ∼ 0.2 – 0.5 ml min−1 and raised in 5 – 10 increments to a final constant rate of 2.5 ml min−1 over a 50 min period (increasing the flow rate too rapidly led to damage of the microvasculature in the eye). When this final flow rate was achieved, eyes were perfused for an equilibration period of at least 30 min. Perfusion pressure was measured using Gould Statham P32 ID transducers via a side arm located immediately proximal to the inflow cannula. Only eyes that had a basal perfusion pressure of 20 – 60 mmHg after the equilibration period were used for further study.
Experimental protocols with the bovine isolated arterially perfused eye
In order to observe vasodilator responses in the bovine eye, the perfusion pressure was first raised to ∼130 mmHg using the thromboxane A2-mimetic, U46619 (∼300 nM). In all experiments, responses to acetylcholine or bradykinin were elicited by adding 10 μl of the appropriate concentration with a Hamilton micro-syringe. We have previously demonstrated that vasodilator responses elicited by these two drugs are mediated solely by an EDHF-like substance and do not involve a contribution by nitric oxide or a cyclo-oxygenase product (McNeish et al., 2001). Consequently, inhibitors of nitric oxide synthase or cyclo-oxygenase were not required to study EDHF-like responses in this preparation.
As will be seen in the Results, acetylcholine elicited only vasodilator responses when bovine eyes were perfused with normal Krebs solution. In contrast, if eyes were perfused from the outset with Krebs solution containing ascorbate (50 μM, >120 min), acetylcholine produced vasoconstrictor responses.
Experiments were conducted to assess the time course of the ability of ascorbate to reverse acetylcholine-induced vasodilatation to vasoconstriction. In control experiments, acetylcholine (10 nmol)-induced vasodilator responses were elicited every 15 min during a 120 min study period in eyes perfused with Krebs solution in order to assess reproducibility. When the effects of ascorbate were to be assessed, control acetylcholine-induced vasodilator responses were allowed to stabilize before inclusion of ascorbate in the perfusion medium. The effects of ascorbate on acetylcholine-induced responses, elicited every 15 min, were then measured during the ensuing 120 min. During this period, the vasodilator response declined and a vasoconstrictor response developed, often resulting in a biphasic response. Consequently, vasodilator and vasoconstrictor components were measured separately and plotted at each of the 15 min time points. Similar experiments were conducted to determine if ascorbate had any effect on vasodilator responses to bradykinin in the bovine eye.
The ability of other drugs to mimic the effects of ascorbate on acetylcholine-induced responses was also assessed in similar time course experiments. These drugs were: the redox-inactive analogue, dehydroascorbate (50 μM), the thiol reducing agents, glutathione (1 mM), N-acetyl-L-cysteine (1 mM) and dithiothreitol (100 μM), and the antioxidant enzymes, superoxide dismutase (250 units ml−1) and catalase (1250 units ml−1). Experiments were also conducted to assess if washing could reverse the blocking effects of ascorbate. In such experiments, eyes were perfused from the outset with Krebs containing ascorbate (50 μM, >120 min). When reproducible acetylcholine (10 nmol)-induced control vasoconstrictor responses had been obtained, perfusion was then switched to ascorbate-free Krebs. Changes in acetylcholine-induced responses were then assessed for the ensuing 120 min.
Experiments were conducted to assess the concentration-dependence of the ability of ascorbate to reverse acetylcholine-induced vasodilatation to vasoconstriction. In control experiments, full dose – response curves to acetylcholine (1 pmol – 1 nmol) were constructed in eyes perfused with Krebs solution. These control dose – response curves were compared with those obtained following addition of ascorbate (10, 50 or 150 μM, >120 min) to the Krebs solution from the beginning of the experiment.
Preparation of the rat isolated perfused mesenteric arterial bed
Male Wistar rats (200 – 350 g) were killed by concussion followed by exsanguination. The mesenteric arterial bed was prepared using the method described by McGregor (1965). Briefly, the superior mesenteric artery was cannulated and the arterial vasculature was dissected from the intestines, suspended in a heating jacket and perfused at 37°C with Krebs solution at a flow rate of 5 ml min−1. Mesenteries were perfused for an equilibration period of 30 min before experiments were begun. Perfusion pressure was measured via a side arm proximal to the inflow cannula, as above.
Experimental protocols with the rat isolated perfused mesenteric arterial bed
Experiments were conducted with the rat isolated perfused mesenteric vascular bed preparation in order to determine if EDHF-dependent vasodilator responses to acetylcholine were also blocked by ascorbate in this preparation. In order to observe vasodilator responses, perfusion pressure was first raised using phenylephrine (3 – 10 μM) to ∼120 mmHg. All experiments investigating the actions of ascorbate were conducted on tissues treated with L-NAME (100 μM) and indomethacin (10 μM) in order to block any vasodilator contribution by nitric oxide and prostanoids. Under these conditions acetylcholine-induced vasodilatation is mediated by EDHF (McCulloch et al., 1997).
Control dose – response curves for EDHF-dependent vasodilatation to acetylcholine were constructed in mesenteries perfused with Krebs solution. These were compared with those obtained in preparations perfused with Krebs solution containing ascorbate (50 μM, >180 min). In addition, experiments were conducted to determine the time course over which ascorbate inhibits acetylcholine-induced, EDHF-dependent vasodilatation. In these experiments, control vasodilator responses to acetylcholine (10 nmol) were elicited every 15 min during a 180 min period in mesenteries perfused from the outset with Krebs solution in order to assess their reproducibility. When the effects of ascorbate were to be assessed, control acetylcholine-induced vasodilator responses were allowed to stabilize before addition of ascorbate (50 μM) to the Krebs solution. The effects of ascorbate on acetylcholine-induced vasodilatation were then assessed every 15 min during the ensuing 180 min.
Drugs and chemicals
Acetylcholine chloride, ascorbic acid, atropine sulphate, bradykinin acetate, catalase (from bovine liver), dehydroascorbate, dithiothreitol, glutathione, indomethacin, L-NAME (NG-nitro-L-arginine methyl ester), N-acetyl-L-cysteine, phenylephrine hydrochloride, superoxide dismutase (from bovine erythrocytes) and U46619 (9,11-dideoxy-11α,9α-epoxy-methanoprostaglandin F2α) were all obtained from Sigma (Poole, U.K.). Flurbiprofen was a gift from the Boots Pure Drug Company (Nottingham, U.K.). Glyceryl trinitrate (10% w w−1 in lactose) was a gift from NAPP Laboratories (Cambridge, UK). All drugs were dissolved and diluted in 0.9% saline except indomethacin (0.1 M stock), which was dissolved in ethanol and sonicated for 1 h.
Statistical analysis
Results are expressed as the mean±s.e.mean of n separate observations, each from a separate eye or mesentery. Vasoconstrictor responses are given in mmHg and vasodilator responses are expressed as percentage reduction of U46619-induced (bovine eye) or phenylephrine-induced (rat mesentery) perfusion pressure. Graphs were drawn and statistical comparisons made using Student's t-test, or one-way analysis of variance with Bonferroni's post-test, as appropriate, with the aid of a computer program, Prism (GraphPad, San Diego, U.S.A.). A probability (P) less than or equal to 0.05 was considered significant.
Results
Effects of ascorbate on acetylcholine-induced, EDHF-dependent vasodilatation in the ciliary vascular bed of the bovine isolated perfused eye
When bovine isolated eyes were perfused with normal Krebs solution, acetylcholine (10 nmol) induced powerful falls (59.5±4.5%, n=8, Figure 1a) in the U46619-induced perfusion pressure. We have previously shown that these vasodilator responses are mediated solely by an EDHF-like factor (McNeish et al., 2001). In contrast, in eyes perfused with Krebs solution containing ascorbate (50 μM, >120 min) from the outset, acetylcholine produced a powerful vasoconstrictor response (87.0±16.8 mmHg, n=6) followed by a small vasodilator response (17.3±6.3%, n=6, Figure 1). This acetylcholine-induced vasoconstriction was unaffected by the cyclo-oxygenase inhibitor, flurbiprofen (30 μM, 20 min), but was reduced to −7.6±5.3 mmHg (n=14, P<0.001) by atropine (100 nM, 20 min). The perfusion pressure generated by U46619 (300 nM) was not, however, different in control eyes (123.3±10, n=7) and in eyes treated with ascorbate (122.7±12.0 mmHg, n=8).
Figure 1.
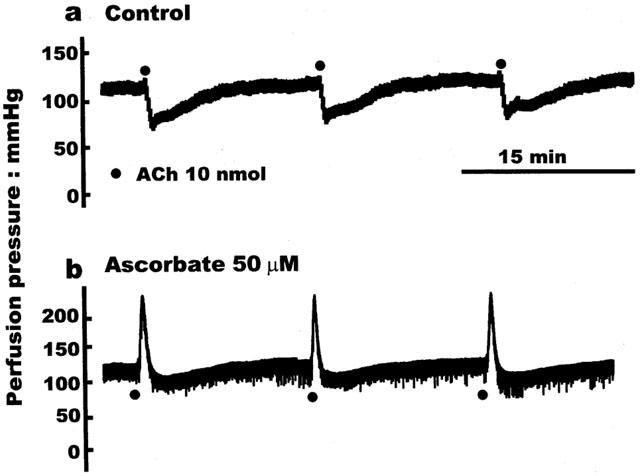
Original traces showing the difference in the responses produced by acetylcholine (ACh; 10 nmol) in the bovine isolated eye following perfusion with: (a) Control Krebs solution, in which acetylcholine elicited only vasodilator responses; or (b) Krebs containing ascorbate (50 μM, 120 min), in which acetylcholine produced powerful vasoconstriction, followed by weak residual vasodilatation.
Time course of ascorbate-induced reversal of acetylcholine-induced vasodilatation to vasoconstriction
The time course of the ability of ascorbate to inhibit acetylcholine-induced vasodilatation and uncover a vasoconstrictor response was studied over a 120 min period. In control experiments, acetylcholine (10 nmol) induced only vasodilatation and these responses, elicited every 15 min remained stable over the 120 min period (Figure 2a). In contrast, inclusion of ascorbate (50 μM) into the perfusion medium led to a time-dependent attenuation of the acetylcholine (10 nmol)-induced vasodilatation and development of vasoconstriction; vasodilator responses first began to decline after 30 min and were virtually abolished by 120 min (Figure 2a). Vasoconstrictor responses (Figure 2b), first observed 60 min after treatment with ascorbate, were fully developed and remained stable after 90 min (Figure 2b). Once the effects of ascorbate were established, washing out the antioxidant for up to 120 min failed to restore the vasodilator responses to acetylcholine or inhibit acetylcholine-induced vasoconstriction (data not shown).
Figure 2.
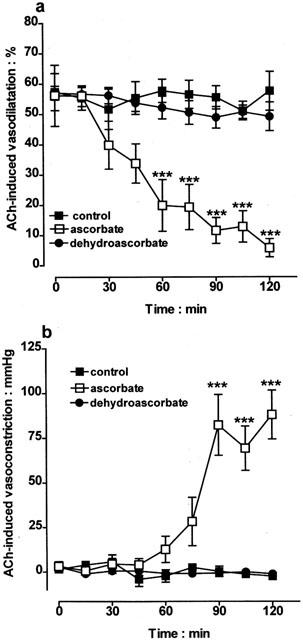
Control experiments showing that vasodilator responses to acetylcholine (ACh; 10 nmol) elicited in the bovine isolated perfused eye every 15 min remained stable for a 120 min study period. In contrast, inclusion of ascorbate (50 μM) in the perfusion medium led to a time-dependent loss of vasodilatation (a) and development of vasoconstriction (b). The redox-inactive analogue, dehydroascorbate (50 μM) lacked the activity of ascorbate. Data represent mean±s.e.mean of 6 – 7 observations, each from a separate eye. ***P<0.001 indicates a difference from control.
In contrast to the effects with acetylcholine, vasodilator responses to 10 nmol glyceryl trinitrate (51.4±4.3%, n=7) were unaffected following treatment with ascorbate (50 μM; 48.3±4.2%, n=7).
Concentration-dependence of ascorbate-induced reversal of acetylcholine-induced vasodilatation to vasoconstriction
The concentration-dependence of the ascorbate-induced reversal of acetylcholine-induced vasodilatation to vasoconstriction was examined. In these experiments bovine eyes were perfused with a range of concentrations of ascorbate (10, 50 and 150 μM) for at least 120 min before constructing dose – response curves to acetylcholine (1 pmol – 1 μmol). Figure 3 shows that the ability of ascorbate to inhibit the vasodilator response and uncover a vasoconstrictor response to acetylcholine was indeed concentration-dependent. Nevertheless, even the lowest concentration of ascorbate tested (10 μM), resulted in significant loss of acetylcholine-induced vasodilatation and development of vasoconstriction.
Figure 3.
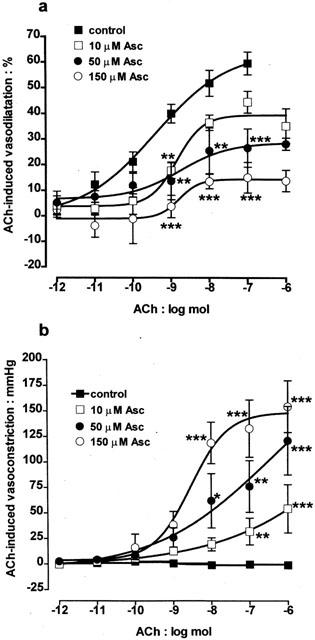
Dose-response curves showing acetylcholine (ACh; 1 pmol-1 μmol)-induced responses in the bovine isolated perfused eye. Under control conditions, acetylcholine produced only vasodilator responses. In contrast, following perfusion for 120 min with ascorbate (Asc; 10, 50 or 150 μM), a concentration-dependent loss of vasodilatation (a) and development of vasoconstriction (b) was seen. Responses in the presence of ascorbate were therefore biphasic, consisting of an initial vasoconstriction followed by a residual vasodilatation. Data represent the mean±s.e.mean of 5 – 10 observations, each from a separate eye. *P<0.05, **P<0.01 and ***P<0.001 indicate a difference from control.
Time course of ascorbate-induced blockade of bradykinin-induced vasodilatation
Experiments were conducted to determine if bradykinin-induced, EDHF-dependent vasodilator responses were blocked by ascorbate in the bovine eye. In control experiments bradykinin (10 pmol) induced vasodilator responses (43.8±3.9%, n=8), and these remained stable over the 120 min study period (Figure 4). In contrast, inclusion of ascorbate (50 μM) into the perfusion medium led to a time-dependent attenuation of bradykinin-induced vasodilatation: vasodilator responses began to decline after 60 min and were virtually abolished by 120 min. In contrast to acetylcholine, vasoconstrictor responses to bradykinin were never observed, even when the vasodilator blockade by ascorbate was fully established.
Figure 4.
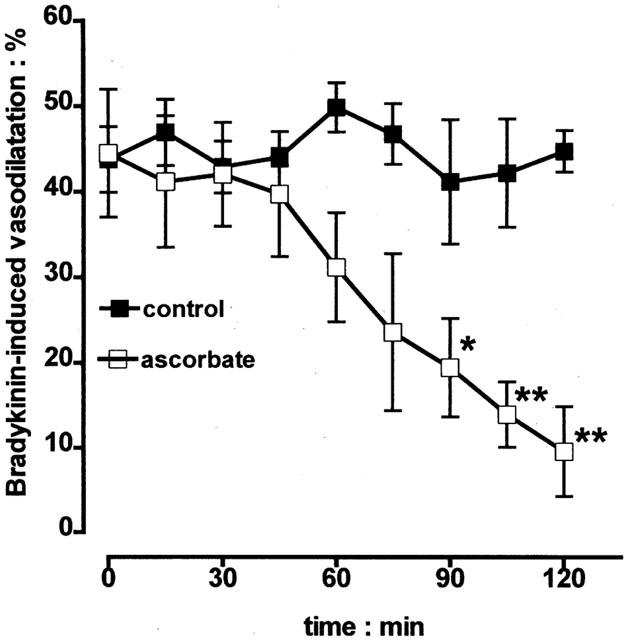
Time course experiments showing that vasodilator responses to bradykinin (10 pmol), elicited in the bovine isolated perfused eye every 15 min, remained stable for a 120 min study period. In contrast, inclusion of ascorbate (50 μM) in the perfusion medium led to a time-dependent loss of vasodilatation. Data represent the mean±s.e.mean of 5 – 9 observations, each from a separate eye. *P<0.05 and **P<0.01 indicate a difference from control.
Effects of antioxidants on acetylcholine-induced vasodilatation
Experiments were conducted to determine if the ability of ascorbate to reverse acetylcholine-induced vasodilatation to vasoconstriction resulted from an antioxidant action. Control vasodilator responses to acetylcholine (10 nmol) were obtained before examining the effects of a range of agents over the ensuing 120 min. Inclusion of the redox-inactive analogue, dehydroascorbate (50 μM) in the perfusion medium failed to reproduce the reversal of acetylcholine-induced vasodilatation to vasoconstriction seen with ascorbate (Figure 2). Moreover, superoxide dismutase (250 units ml−1) and catalase (1250 units ml−1), which scavenge superoxide anion and hydrogen peroxide, respectively, also had no effect on acetylcholine-induced vasodilatation (Figure 5).
Figure 5.
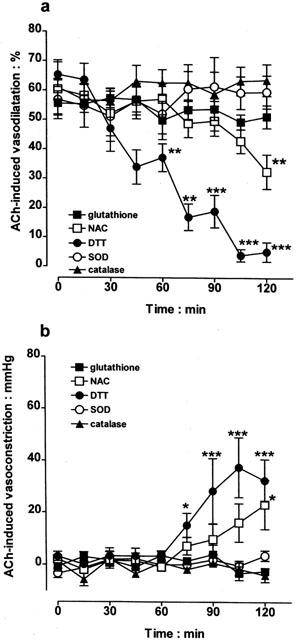
After having obtained control vasodilator responses to acetylcholine (ACh; 10 nmol) in the bovine isolated perfused eye, the effects of infusion of a number of antioxidants, namely, glutathione (1 mM), N-acetyl-L-cysteine (NAC; 1 mM), dithiothreitol (DTT; 100 μM), superoxide dismutase (SOD; 250 units ml−1) and catalase (1250 units ml−1), were examined on these vasodilator responses over the ensuing 120 min. Loss of acetylcholine-induced vasodilatation and any development of vasoconstriction are shown in (a) and (b), respectively. Data represent the mean±s.e.mean of 5 – 10 observations, each from a separate eye. *P<0.05, **P<0.01 and ***P<0.001 indicate a difference from control.
Perfusion with glutathione (1 mM) also had no effect on acetylcholine-induced vasodilatation (Figure 5). In contrast, perfusion with two other thiol-reducing agents, N-acetyl-L-cysteine (1 mM) and dithiothreitol (100 μM), did inhibit vasodilatation and uncover a vasoconstrictor response to acetylcholine, with dithiothreitol being the more effective.
Effects of ascorbate on acetylcholine-induced,EDHF-dependent vasodilatation in the rat isolated perfused mesenteric arterial bed
Experiments were conducted to determine if the ability of ascorbate to attenuate EDHF-dependent vasodilator responses in the bovine eye was also seen in the rat perfused mesenteric arterial bed preparation. Following perfusion with Krebs solution and vasoconstriction with phenylephrine (3 – 10 μM), acetylcholine (1 pmol – 100 nmol) elicited vasodilatations which were dose-dependent (Figure 6a). The EDHF-dependent component of vasodilatation was revealed by inclusion of the nitric oxide synthase inhibitor, L-NAME (100 μM) and the cyclo-oxygenase inhibitor, indomethacin (10 μM), in the perfusion medium (McCulloch et al., 1997). This EDHF-dependent component of acetylcholine-induced vasodilatation was inhibited when ascorbate (50 μM) was present in the perfusion medium for 180 min (Figure 6a). In contrast to the bovine eye, vasoconstrictor responses to acetylcholine were never observed in the rat mesentery even when vasodilator blockade by ascorbate was fully established.
Figure 6.
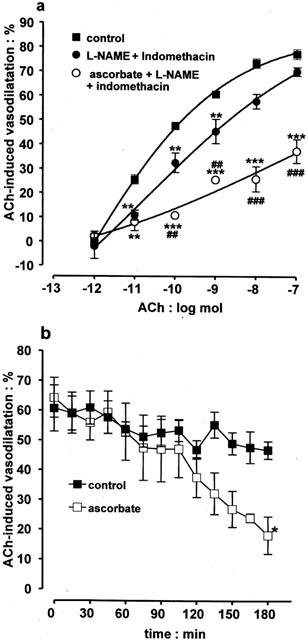
Graphs showing the ability of ascorbate to inhibit acetylcholine (ACh)-induced, EDHF-dependent vasodilatation in the rat perfused mesenteric arterial bed. (a) Control dose-response curves to acetylcholine (1 pmol – 100 nmol) and the residual EDHF-dependent vasodilatation seen following treatment with the combination of L-NAME (100 μM) and indomethacin (10 μM). This residual EDHF-dependent vasodilatation was inhibited following inclusion of ascorbate (50 μM,) in the perfusion medium for 180 min. (b) Time course experiments showing that the EDHF-dependent vasodilatations produced by acetylcholine (10 nmol) in preparations treated with L-NAME and indomethacin did not change significantly during a 180 min study period. In contrast, inclusion of ascorbate (50 μM) in the perfusion medium led to a time-dependent loss of vasodilatation. Data represent the mean±s.e.mean of 5 – 11 observations, each from a separate mesentery. *P<0.05, **P<0.01 and ***P<0.001 indicate a difference from control. In (a) ##P<0.01 and ###P<0.001 indicate a significant blockade by ascorbate of EDHF-induced vasodilator responses, obtained in the presence of L-NAME and indomethacin.
Experiments were conducted to determine the time course over which ascorbate (50 μM) attenuated acetylcholine-induced vasodilatation in the rat mesentery. In control experiments acetylcholine (10 nmol)-induced vasodilatation, did not change significantly over a 180 min study period (Figure 6b). In contrast, inclusion of ascorbate (50 μM) in the perfusion medium led to a time-dependent attenuation of acetylcholine-induced vasodilatation: responses first began to decline at 120 min and this intensified during the remaining 60 min.
Discussion
Surprisingly, the findings of the present study demonstrate that ascorbate, at concentrations occurring in human plasma (mean 46±8, range 30 – 150 μM; Keaney & Vita, 1995; Levine et al., 1996), blocks agonist-induced vasodilator responses mediated by EDHF. Specifically, when the ciliary vascular bed of the bovine isolated eye is perfused with Krebs solution containing ascorbate (50 μM, >120 min), acetylcholine induces powerful vasoconstrictor responses. In contrast, when perfused with standard Krebs solution, it induces vasodilatation that is blocked by depolarizing solutions of potassium chloride or by charybdotoxin (McNeish et al., 2001), and thus has the characteristics of the EDHF (Edwards et al., 1998; Feletou & Vanhoutte, 1999). This acetylcholine-induced vasoconstriction was unaffected by the cyclo-oxygenase inhibitor, flurbiprofen (10 μM), precluding the involvement of a vasoconstrictor prostanoid. It was, however, abolished by atropine (100 nM), indicating that it was muscarinic in nature and probably resulted from the well-characterized direct contractile action of acetylcholine on vascular smooth muscle (Furchgott & Zawadzki, 1980).
We have previously reported a similar ‘reversal' of vasodilatation to vasoconstriction in the bovine eye when charybdotoxin was used to attenuate acetylcholine-induced, EDHF-dependent vasodilatation (McNeish et al., 2001). It was therefore likely that ascorbate was blocking the EDHF pathway at some point, thus uncovering a normally-masked muscarinic vasoconstrictor response. Indeed, this conclusion was strengthened by the results of time course experiments. These showed that although acetylcholine-induced, EDHF-mediated vasodilator responses were stable during a 120 min study period, inclusion of ascorbate (50 μM) in the perfusion medium led to both a time-dependent fall in vasodilatation and development of vasoconstriction. By the end of the 120 min period vasodilatation was almost abolished and vasoconstriction had developed fully, while at intermediate time points a biphasic response, consisting of an initial vasoconstriction followed by weak residual vasodilatation was observed.
Further investigation revealed that the ability of ascorbate to reverse acetylcholine-induced vasodilatation to vasoconstriction was concentration-dependent, with even the lowest concentration studied (10 μM), producing a significant effect. Moreover, at the highest concentration of ascorbate tested (150 μM), vasodilator responses were virtually abolished, with acetylcholine now producing almost exclusively vasoconstriction. Thus, the inhibitory action of ascorbate was seen over the full range of concentrations found in human plasma (30 – 150 μM; Keaney & Vita, 1995) and even lower than this.
Bradykinin also elicits EDHF-dependent vasodilatation in the bovine isolated perfused eye (McNeish et al., 2001), so it was important to determine if the blocking action of ascorbate extended to this vasodilator. We found that bradykinin-induced, EDHF-mediated vasodilator responses were stable during a 120 min study period when the eye was perfused with standard Krebs solution. Moreover, in common with experiments when acetylcholine was used as the vasodilator, inclusion of ascorbate (50 μM) in the perfusion medium led to a time-dependent fall in bradykinin-induced vasodilatation, such that at 120 min the response was virtually abolished. In contrast to acetylcholine, vasoconstrictor responses to bradykinin were never observed, even when the blocking action of ascorbate was fully established. This observation therefore strengthens our conclusion that the effect of ascorbate in the bovine eye is to block EDHF-mediated vasodilatation and not to invoke a normally-absent vasoconstrictor response. Moreover, these findings indicate that the blocking action of ascorbate is mediated by an action on an element of the EDHF signal transduction cascade common to both acetylcholine and bradykinin. This effect is likely to be highly selective since the vasodilator actions of glyceryl trinitrate were entirely unaffected by ascorbate.
The ciliary body actively transports ascorbate, such that the concentration found in the aqueous humour is normally around 1 mM in a wide range of species, including humans (Millar & Kaufman, 1995). We therefore wished to determine if the ability of ascorbate to block EDHF-mediated vasodilatation was peculiar to the eye, or was a more general phenomenon. Accordingly, we extended our studies to a different preparation where the role of EDHF has been characterized extensively, i.e. the rat isolated perfused mesenteric arterial bed (Adeagbo & Triggle, 1993; McCulloch et al., 1997). In this preparation, the vasodilatation induced by acetylcholine in the presence of inhibitors of nitric oxide synthase and cyclo-oxygenase is mediated solely by EDHF. Our experiments revealed that acetylcholine-induced, EDHF-dependent vasodilator responses in the perfused mesentery were indeed blocked in a time-dependent manner when ascorbate (50 μM) was included in the perfusion medium.
The time required for maximal blockade (180 min) was somewhat longer than in the bovine eye (120 min), but time-matched experiments conducted in the absence of ascorbate showed that acetylcholine-induced vasodilatation was well maintained during this period. Thus, the ability of ascorbate to block EDHF-mediated vasodilatation is not limited to the bovine eye, but is a more widespread phenomenon. Whether or not ascorbate blocks EDHF-dependent vasodilatation at all vascular sites remains to be determined.
We investigated the possibility that the ascorbate-induced blockade of EDHF-mediated vasodilatation was due to an antioxidant action. The failure of the redox-inactive analogue, dehydroascorbate (50 μM), to mimic the effects of ascorbate on acetylcholine-induced vasodilatation supports this view. It also eliminates the possibility that the breakdown products of dehydroascorbate, i.e. oxalic and L-threonic acids (Halliwell & Gutteridge, 1989), could account for the ability of ascorbate to block EDHF-mediated vasodilatation.
Neither superoxide dismutase (250 units ml−1) nor catalase (1250 units ml−1), which scavenge superoxide and hydrogen peroxide, respectively, shared the ability of ascorbate to block acetylcholine-induced, EDHF-mediated vasodilatation. Thus, the blocking action cannot be attributed to the scavenging of superoxide anion or hydrogen peroxide. The lack of effect of catalase is of additional significance, since it would counter the recent suggestion that hydrogen peroxide is the EDHF (Matoba et al., 2000).
Glutathione (1 mM) also failed to mimic the ability of ascorbate to block acetylcholine-induced, EDHF-mediated vasodilatation. In contrast, two other more powerful thiol-reducing agents, N-acetyl-L-cysteine (1 mM) and dithiothreitol (100 μM), each promoted a time-dependent fall in acetylcholine-induced vasodilatation and uncovered a vasoconstrictor response. These results may simply be explained by the general antioxidant action of the two compounds, but they raise the possibility that they (and ascorbate) might block EDHF-mediated vasodilatation by reducing a critical disulphide group at some point on its signal transduction cascade. Indeed, redox modification of thiol groups has been reported to modulate the gating of the intermediate conductance, calcium-activated potassium channel (Cai & Sauve, 1997), which, together with the small conductance calcium-activated potassium channel, is generally involved in EDHF-mediated vasodilatation (Edwards et al., 1998; Feletou & Vanhoutte, 1999). An alternative locus for the inhibitory action of ascorbate might be cytochrome P450. It has been suggested that EDHF-dependent vasodilatation may be mediated by a product of a cytochrome P450 enzyme (Bauersachs et al., 1994; Fisslthaler et al., 1999). Cytochrome P450s are redox sensitive, and ascorbate is known to inhibit their activity (Anderson & Kappas, 1991; Ghosh et al., 1997). Furthermore, there is growing evidence that communication via myoendothelial gap junctions plays a critical role in the EDHF response (Chaytor et al., 2001). It would therefore be of interest to determine if inhibition of cytochrome P450 enzymes or gap junctional communication underlies the ability of ascorbate to block EDHF-mediated vasodilatation.
The blockade of EDHF-mediated vasodilatation in the bovine eye and rat mesentery by ascorbate at concentrations well within the normal plasma range raises several important issues. For example, if our in vitro experiments reflect the behaviour of the vascular beds in vivo, they would suggest that EDHF activity is normally greatly suppressed. Such a situation seems almost inconceivable, since numerous reports describe vasodilator responses attributed to EDHF in living animals (Nishikawa et al., 1999; Welsh & Segal, 2000) and humans (Honing et al., 2000; Katz & Krum, 2001). Furthermore, the blocking action of ascorbate on EDHF contrasts markedly with its actions on nitric oxide, where enhancement of vasodilator activity is widely reported. For example, ascorbate is able to restore nitric oxide-dependent vasodilatation following its impairment by oxidant stress in isolated arterial rings (Dudgeon et al., 1998; Fontana et al., 1999). Moreover, treatment of patients with ascorbate has led to restoration of impaired nitric oxide-mediated vasodilatation in essential hypertension (Taddei et al., 1998; Natali et al., 2000), atherosclerosis (Levine et al., 1996), hypercholesterolaemia (Ting et al., 1997), insulin-dependent (Timimi et al., 1998) and non-insulin-dependent diabetes (Ting et al., 1996) and chronic heart failure (Ellis et al., 2001; Hornig et al., 1998).
Although highly speculative, it is possible that ascorbate may act in vivo as a redox switch to activate EDHF under conditions of oxidant stress. Under such conditions nitric oxide is destroyed by the superoxide anion (Gryglewski et al., 1986; Rubanyi & Vanhoutte, 1986) and low molecular weight antioxidants such as ascorbate are rapidly consumed (for review see Frei, 1994). The depletion of ascorbate could then potentially activate the normally suppressed EDHF-dependent vasodilator mechanism. Indeed, support for this concept comes from a recent report showing that in heart failure, where nitric oxide-dependent vasodilatation is impaired by oxidant stress in the forearm circulation, EDHF becomes the dominant vasodilator mechanism (Katz & Krum, 2001). Further evidence of a reciprocal interaction between these two vasodilators is suggested by the findings that nitric oxide inactivates EDHF in the porcine coronary circulation both in vitro (Bauersachs et al., 1996) and in vivo (Nishikawa et al., 2000). Furthermore, it has been suggested that the EDHF-mediated component of vasodilatation becomes fully established in porcine isolated coronary arteries only when the nitric oxide-dependent component is inhibited (Kilpatrick & Cocks, 1994). Whatever the precise interaction between these two important vasodilators, our new findings with ascorbate may have important consequences for cardiovascular pathologies where nitric oxide-mediated vasodilatation is impaired by oxidant stress.
In summary, ascorbate at normal plasma concentrations blocks EDHF-dependent vasodilatation in the ciliary vascular bed of the bovine eye and the mesenteric arterial bed of the rat. The mechanism and physiological consequences of this action of ascorbate remain to be determined.
Acknowledgments
This work was supported by the Wellcome Trust and the British Heart Foundation.
Abbreviations
- DTT
dithiothreitol
- EDHF
endothelium-derived hyperpolarizing factor
- NAC
N-acetyl-L-cysteine
References
- ADEAGBO A.S.O., TRIGGLE C.R. Varying extracellular [K+]: a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J. Cardiovasc. Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- ANDERSON K.E., KAPPAS A. Dietary-regulation of cytochrome P450. Ann. Rev. Nutr. 1991;11:141–167. doi: 10.1146/annurev.nu.11.070191.001041. [DOI] [PubMed] [Google Scholar]
- BAUERSACHS J., HECKER M., BUSSE R. Display of the characteristics of endothelium-derived hyperpolarizing factor by a cytochrome P450-derived arachidonic acid metabolite in the coronary microcirculation. Br. J. Pharmacol. 1994;113:1548–1553. doi: 10.1111/j.1476-5381.1994.tb17172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUERSACHS J., POPP R., HECKER M., SAUER E., FLEMING I., BUSSE R. Nitric oxide attenuates the release of endothelium-derived hyperpolarising factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- CAI S., SAUVE R. Effects of thiol-modifying agents on a K(Ca2+) channel of intermediate conductance in bovine aortic endothelial cells. J. Membr. Biol. 1997;158:147–158. doi: 10.1007/s002329900252. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., HARDER D.R. Prologue: EDHF - what is it. Am. J. Physiol. 2001;280:H2413–H2416. doi: 10.1152/ajpheart.2001.280.6.H2413. [DOI] [PubMed] [Google Scholar]
- CARR A.C., ZHU B.Z., FREI B. Potential antiatherogenic mechanisms of ascorbate (vitamin C) and alpha- tocopherol (vitamin E) Circ. Res. 2000;87:349–354. doi: 10.1161/01.res.87.5.349. [DOI] [PubMed] [Google Scholar]
- CHAYTOR A.T., MARTIN P.E.M., EDWARDS D.H., GRIFFITH T.M. Gap junctional communication underpins EDHF-type relaxations evoked by Ach in the rat hepatic artery. Am. J. Physiol. 2001;280:H2441–H2450. doi: 10.1152/ajpheart.2001.280.6.H2441. [DOI] [PubMed] [Google Scholar]
- DAVSON H. The aqueous humour and the intraocular pressure Physiology of the Eye 1980London: Churchill Livingstone; ed. Davson, H., p 26 [Google Scholar]
- DUDGEON S., BENSON D.P., MACKENZIE A., PAISLEY-ZYSKIEWICZ K., MARTIN W. Recovery by ascorbate of impaired nitric oxide-dependent relaxation resulting from oxidant stress in rat aorta. Br. J. Pharmacol. 1998;125:782–786. doi: 10.1038/sj.bjp.0702120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- ELLIS G.R., ANDERSON R.A., CHIRKOV Y.Y., MORRIS-THURGOOD J., JACKSON S.K., LEWIS M.J., HOROWITZ J.D., FRENNEAUX M.P. Acute effects of vitamin C on platelet responsiveness to nitric oxide donors and endothelial function in patients with chronic heart failure. J. Cardiovasc. Pharmacol. 2001;37:564–570. doi: 10.1097/00005344-200105000-00008. [DOI] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. The third pathway: endothelium-dependent hyperpolarization. J. Physiol. Pharmacol. 1999;50:525–534. [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- FONTANA L., MCNEILL K.L., RITTER J.M., CHOWIENCZYK P.J. Effects of vitamin C and of a cell permeable superoxide dismutase mimetic on acute lipoprotein-induced endothelial dysfunction in rabbit aortic rings. Br. J. Pharmacol. 1999;126:730–734. doi: 10.1038/sj.bjp.0702331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREI B. Reactive oxygen species and antioxidant vitamins: mechanisms of action. Am. J. Med. 1994;97:5S–13S. doi: 10.1016/0002-9343(94)90292-5. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GHOSH M.K., MUKHOPADHYAY M., CHATTERJEE I.B. NADPH-initiated cytochrome P450-dependent free iron-independent microsomal lipid peroxidation: Specific prevention by ascorbic acid. Mol. Cell. Biochem. 1997;166:35–44. doi: 10.1023/a:1006841228483. [DOI] [PubMed] [Google Scholar]
- GRYGLEWSKI R.J., PALMER R.M., MONCADA S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- HALLIWELL B., GUTTERIDGE J.M.C. Free Radicals in Biology and Medicine 1989Oxford: Clarendon Press; ed. Halliwell, B. & Gutteridge, J.M.C [Google Scholar]
- HELLER R., UNBEHAUN A., SCHELLENBERG B., MAYER B., WERNER-FELMAYER G., WERNER E.R. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J. Biol. Chem. 2001;276:40–47. doi: 10.1074/jbc.M004392200. [DOI] [PubMed] [Google Scholar]
- HONING M.L.H., SMITS P., MORRISON P.J., RABELINK T.J. Bradykinin-induced vasodilation of human forearm resistance vessels is primarily mediated by endothelium-dependent hyperpolarization. Hypertension. 2000;35:1314–1318. doi: 10.1161/01.hyp.35.6.1314. [DOI] [PubMed] [Google Scholar]
- HORNIG B., ARAKAWA N., KOHLER C., DREXLER H. Vitamin C improves endothelial function of conduit arteries in patients with chronic heart failure. Circulation. 1998;97:363–368. doi: 10.1161/01.cir.97.4.363. [DOI] [PubMed] [Google Scholar]
- HUANG A., VITA J.A., VENEMA R.C., KEANEY J.F. Ascorbic acid enhances endothelial nitric-oxide synthase activity by increasing intracellular tetrahydrobiopterin. J. Biol. Chem. 2000;275:17399–17406. doi: 10.1074/jbc.M002248200. [DOI] [PubMed] [Google Scholar]
- KATZ S.D., KRUM H. Acetylcholine-mediated vasodilation in the forearm circulation of patients with heart failure: indirect evidence for the role of endothelium-derived hyperpolarizing factor. Am. J. Cardiol. 2001;87:1089–1092. doi: 10.1016/s0002-9149(01)01466-7. [DOI] [PubMed] [Google Scholar]
- KEANEY J.F., VITA J.A. Atherosclerosis, oxidative stress, and antioxidant protection in endothelium-derived relaxing factor action. Prog. Cardiovasc. Dis. 1995;38:129–154. doi: 10.1016/s0033-0620(05)80003-9. [DOI] [PubMed] [Google Scholar]
- KILPATRICK E.V., COCKS T.M. Evidence for differential roles of nitric oxide (NO) and hyperpolarisation in endothelium-dependent relaxation of pig isolated coronary artery. Br. J. Pharmacol. 1994;112:557–565. doi: 10.1111/j.1476-5381.1994.tb13110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVINE G.N., FREI B., KOULOURIS S.N., GERHARD M.D., KEANEY J.F., VITA J.A. Ascorbic acid reverses endothelial vasomotor dysfunction in patients with coronary artery disease. Circulation. 1996;93:1107–1113. doi: 10.1161/01.cir.93.6.1107. [DOI] [PubMed] [Google Scholar]
- MATOBA T., SHIMOKAWA H., NAKASHIMA M., HIRAKAWA Y., MUKAI Y., HIRANO K., KANAIDE H., TAKESHITA A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J. Clin. Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAY J.M. How does ascorbic acid prevent endothelial dysfunction. Free Radic. Biol. Med. 2000;28:1421–1429. doi: 10.1016/s0891-5849(00)00269-0. [DOI] [PubMed] [Google Scholar]
- MCCULLOCH A.I., BOTTRILL F.E., RANDALL M.D., HILEY C.R. Characterization and modulation of EDHF-mediated relaxations in the rat isolated superior mesenteric arterial bed. Br. J. Pharmacol. 1997;120:1431–1438. doi: 10.1038/sj.bjp.0701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGREGOR D.D. The effect of sympathetic nerve stimulation on vasoconstrictor responses in perfused mesenteric blood vessels of the rat. J. Physiol. 1965;177:21–30. doi: 10.1113/jphysiol.1965.sp007572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCNEISH A.J., WILSON W.S., MARTIN W. Dominant role of an endothelium-derived hyperpolarizing factor (EDHF)-like vasodilator in the ciliary vascular bed of the bovine isolated perfused eye. Br. J. Pharmacol. 2001;134:912–920. doi: 10.1038/sj.bjp.0704332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCNEISH A.J., WILSON W.S., MARTIN W. Ascorbic acid attenuates EDHF-mediated vasodilatation in the bovine isolated perfused eye Br. J. Pharmacol. 2002aBPS meeting Imperial College London; (abstract C018) [Google Scholar]
- MCNEISH A.J., WILSON W.S., MARTIN W. Antioxidants attenuate EDHF-mediated vasodilatation in the bovine isolated perfused eye Br. J. Pharmacol. 2002bBPS meeting Imperial College London; abstract P091 [Google Scholar]
- MILLAR C., KAUFMAN P.L. Aqueous humor: secretion and dynamics Duane's Foundations of Clinical Ophthalmology 1995London and Philadelphia. Harper & Row; 1–52.ed. Tasmin, W., pp [Google Scholar]
- NATALI A., SIRONI A.M., TOSCHI E., CAMASTRA S., SANNA G., PERISSINOTTO A., TADDEI S., FERRANNINI E. Effect of vitamin C on forearm blood flow and glucose metabolism in essential hypertension. Arterioscler. Thromb. Vasc. Biol. 2000;20:2401–2406. doi: 10.1161/01.atv.20.11.2401. [DOI] [PubMed] [Google Scholar]
- NISHIKAWA Y., STEPP D.W., CHILIAN W.M. In vivo location and mechanism of EDHF-mediated vasodilation in canine coronary microcirculation. Am. J. Physiol. 1999;277:H1252–H1259. doi: 10.1152/ajpheart.1999.277.3.H1252. [DOI] [PubMed] [Google Scholar]
- NISHIKAWA Y., STEPP D.W., CHILIAN W.M. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. Am. J. Physiol. 2000;279:H459–H465. doi: 10.1152/ajpheart.2000.279.2.H459. [DOI] [PubMed] [Google Scholar]
- RUBANYI G.M., VANHOUTTE P.M. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am. J. Physiol. 1986;250:H822–H827. doi: 10.1152/ajpheart.1986.250.5.H822. [DOI] [PubMed] [Google Scholar]
- SHAHIDULLAH M., WILSON W.S. Atriopeptin, sodium azide and cGMP reduce secretion of aqueous humour and inhibit intracellular Ca2+ release in bovine cultured ciliary epithelium. Br. J. Pharmacol. 1999;127:1438–1446. doi: 10.1038/sj.bjp.0702681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOM S., RAHA C., CHATTERJEE I.B. Ascorbic acid: a scavenger of superoxide radical. Acta Vitaminol. Enzymol. 1983;5:243–250. [PubMed] [Google Scholar]
- TADDEI S., VIRDIS A., GHIADONI L., MAGAGNA A., SALVETTI A. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation. 1998;97:2222–2229. doi: 10.1161/01.cir.97.22.2222. [DOI] [PubMed] [Google Scholar]
- TIMIMI F.K., TING H.H., HALEY E.A., RODDY M.A., GANZ P., CREAGER M.A. Vitamin C improves endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1998;31:552–557. doi: 10.1016/s0735-1097(97)00536-6. [DOI] [PubMed] [Google Scholar]
- TING H.H., TIMIMI F.K., BOLES K.S., CREAGER S.J., GANZ P., CREAGER M.A. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Clin. Invest. 1996;97:22–28. doi: 10.1172/JCI118394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TING H.H., TIMIMI F.K., HALEY E.A., RODDY M.A., GANZ P., CREAGER M.A. Vitamin C improves endothelium-dependent vasodilation in forearm resistance vessels of humans with hypercholesterolemia. Circulation. 1997;95:2617–2622. doi: 10.1161/01.cir.95.12.2617. [DOI] [PubMed] [Google Scholar]
- WELSH D.G., SEGAL S.S. Role of EDHF in conduction of vasodilation along hamster cheek pouch arterioles in vivo. Am. J. Physiol. 2000;278:H1832–H1839. doi: 10.1152/ajpheart.2000.278.6.H1832. [DOI] [PubMed] [Google Scholar]
- WILSON W.S., SHAHIDULLAH M., MILLAR C. The bovine arterially perfused eye: an in vitro method for the study of drug mechanisms on IOP, aqueous humour formation and uveal vasclature. Curr. Eye Res. 1993;12:609–620. doi: 10.3109/02713689309001840. [DOI] [PubMed] [Google Scholar]