

Abstract
The role of protein kinase C-epsilon (PKC-ε) in the development of κ-opioid receptor (κ-OR) tolerance to the effects of trans-(±)-3,4-dichloro-N-methyl-N-(2-[1-pyrrolidinyl]cyclohexyl) (U50,488H), the selective agonist of κ-OR, was determined in rat ventricular myocytes.
Incubation of ventricular myocytes with 1 μM U50,488H for 24 h significantly attenuated the inhibitory effects of 30 μM U50,488H on the electrically-induced [Ca2+]i transient and forskolin-stimulated cyclic AMP accumulation, indicating the development of tolerance to the κ-OR agonist. Chronic treatment of ventricular myocytes with U50,488H also induced translocation of PKC-ε to the particulate fraction. On the other hand, administration of 30 μM U50,488H for 10 min induced translocation of PKC-α to the particulate fraction in naïve ventricular myocytes, but not in cells pretreated with 1 μM U50,488H for 24 h.
In ventricular myocytes incubated for 24 h with 1 μM U50,488H together with 1 μM chelerythrine or 1 μM GF109203X, PKC inhibitors, or 0.1 μM εV1-2 peptide, a selective inhibitor of PKC-ε, 30 μM U50,488H still produced the inhibitory effect on the electrically-induced [Ca2+]i transient as it did in naïve ventricular myocytes. Chronic treatment of ventricular myocytes with U50,488H and chelerythrine also attenuated the development of tolerance to acute U50,488H on cyclic AMP accumulation. Cells exposed to chelerythrine, GF109203X, or εV1-2 peptide alone did not show an altered [Ca2+]i response to U50,488H.
These results indicate that activation of PKC-ε is a critical step in the development of tolerance in the κ-OR.
Keywords: Tolerance, protein kinase C-epsilon, κ-opioid receptor, ventricular myocytes
Introduction
The opioid receptor (OR), a member of the G protein-coupled receptor (GPCR) family, is widely distributed in the nervous system and other organs including the heart. In the heart, κ-OR is the predominant type (Ventura et al., 1989; Zimlichman et al., 1996). Activation of κ-OR with trans-(±)-3,4-dichloro-N-methyl-N-(2-[1-pyrrolidinyl]cyclohexyl) benzeneacetamide (U50,488H), a selective κ-OR agonist (Lahti et al., 1982), leads to inhibition of the electrically-induced intracellular Ca2+ ([Ca2+]i) transient and forskolin-stimulated cyclic AMP accumulation in rat ventricular myocytes (Bian et al., 1998; Ventura et al., 1992). In addition, it has been demonstrated that PKC mediates the inhibitory actions of κ-OR activation in ventricular myocytes (Bian et al., 1998; 2000).
It is well established that repeated or prolonged exposure to an opioid reduces the responsiveness of the receptor to that opioid. Three temporally distinct processes occur: desensitization in seconds to hours, internalization of receptors in minutes to hours, and down-regulation in hours to days (Law et al., 2000). In ventricular myocytes, exposure for 24 h to U50,488H, a κ-OR agonist, leads to development of tolerance to the effect of the agonist (Sheng & Wong, 1996; Zhang et al., 1999). The subcellular mechanisms are not fully understood. Recent studies have shown that PKC participates in the development of tolerance in the μ- and δ-ORs (Kramer & Simon, 1999; Ueda et al., 1995; Mestek et al., 1995; Xiang et al., 2000; Narita et al., 1994; Mayer et al., 1995). It is likely that PKC also plays an important role in the development of tolerance to the effects of a κ-OR agonist.
Molecular cloning and biochemical analysis has revealed that PKC is a family of at least 12 isoenzymes of closely related serine and threonine (Way et al., 2000). PKC isoforms exist in an inactive state within the cytosol but become translocated to the plasma membrane by various stimuli, and translocation of PKC is believed to be its primary mode of activation in mammalian cells (Nishizuka, 1986). In the rat heart, the predominant PKC isoforms are α, δ, ε and ζ subtypes (Rybin & Steinberg, 1994), suggesting that they may play different roles in different situations. In the study of the role of PKC, the isoform(s) involved should also be determined.
The purpose of the present study was to determine whether PKC is involved in the development of tolerance to κ-OR stimulation in rat ventricular myocytes. Using the electrically-induced intracellular [Ca2+]i transient and forskolin-stimulated cyclic AMP accumulation as measures of κ-OR sensitivity, we determined the responses to acute U50,488H administration in ventricular myocytes that had chronically exposed to a low concentration of U50,488H and a PKC inhibitor, chelerythrine or GF109203X. The response was correlated with translocation of various isoforms. To determine whether PKC-ε was involved, we made use of an isozyme-selective peptide inhibitor of PKC-ε (Csukai & Mochly-Rosen, 1999; Johnson et al., 1996), which has been shown to inhibit the effects of its activation (Johnson et al., 1996; Johnson & Mochly-Rosen, 1995). Results from the study showed that PKC-ε activation is an important step in the development of κ-OR tolerance to the effect of U50,488H.
Methods
Drugs and chemicals
U50,488H, fura-2-acetoxy-methyl ester (Fura-2/AM), type I collagenase, phenylmethylsulphonyl fluoride (PMSF) and forskolin were purchased from Sigma Chemicals Co. (U.S.A.). Chelerythrine chloride, GF109203X and leupeptin were from Calbiochem Ltd. (U.S.A.). The [3H]-cyclic AMP assay system, [8-3H]-cyclic AMP (250 μCi), molecular weight marker, and the ECL Western blotting detection reagents were from Amersham International (U.K.). PKC isoforms (-α, -δ, -ε and -ζ) antibodies were from Transduction Laboratories (U.S.A.). Both εV1-2 and control peptide were generous gift from Dr D. Mochly-Rosen of Stanford University.
Fura-2/AM, chelerythrine chloride, GF109203X and forskolin were dissolved in dimethyl sulphoxide (DMSO) and the rest in distilled water. The final concentration of DMSO was 0.1%, at which DMSO had no effect on either [Ca2+]i or cyclic AMP level.
The concentrations of U50,488H (Bian et al., 1998; Zhang et al., 1999), εV1-2 (Chen et al., 1999; Liu et al., 1999), chelerythrine (Bian et al., 1998; Wu et al., 1999) and GF109203X (Snabaitis et al., 2000; Toullec et al., 1991) used in the present study were based on previous studies. The effects of 30 μM U50,488H on [Ca2+]i transient and cyclic AMP accumulation have been shown to be antagonized by 5 μM norbinaltorphimine, a selective κ-OR antagonist (Birch et al., 1987; Bian et al., 1998; Zhang et al., 1999).
Isolation and culture of ventricular myocytes and experimental protocol
Ventricular myocytes were isolated from adult Sprague – Dawley rats (190 – 210 g) with a collagenase method described previously (Dong et al., 1993; Sheng & Wong, 1996). The level of viable myocytes, as indicated by trypan blue exclusion, was ≈70% of total cells. The cells were incubated, under 5% CO2 atmosphere at 37°C, in culture dishes at a density of 2×105 cells/dish in 3 ml of MEM containing 0.2% bovine serum albumin, 10−8 M insulin, 100 u ml−1 of penicillin G, 100 μg ml−1 of streptomycin (Sheng & Wong, 1996). To induce the development of tolerance, 1 μM U50,488H was added to the medium bathing the ventricular myocytes for 24 h (Sheng & Wong, 1996). To determine the role of PKC, a PKC inhibitor, 1 μM chelerythrine or 1 μM GF109203X (Herbert et al., 1990; Snabaitis et al., 2000; Toullec et al., 1991), was administered 1 h and 15 min respectively before administration of 1 μM U50,488H. To determine the role of PKC-ε, 0.1 μM εV1-2, a selective PKC-ε inhibitor (Csukai & Mochly-Rosen, 1999; Johnson et al., 1996), linked to antennapedia peptide, was administered 30 min before the administration of U50,488H. Thirty minutes was required for the peptide to penetrate the sarcolemmal membrane (Chen et al., 1999; Liu et al., 1999).
Assay of cyclic AMP
Thirty μM U50,488H was added to the culture medium containing ventricular myocytes for 10 min followed by addition of 10 μM forskolin for another 5 min. The reaction was stopped with addition of 0.1 N HCl, and the sample was centrifuged at 3000×g for 5 min. The pellets were neutralized with 0.1 N NaOH for protein determination by the method of Lowry et al. (1951), using BSA as a standard. The supernatant was evaporated to dryness at 55°C under a stream of nitrogen and used for assay of cyclic AMP. The intracellular cyclic AMP level was determined by a competitive binding assay with a kit from Amersham as described previously (Bian et al., 1998). With this method a full standard curve is constructed each time an assay was performed. Briefly, 50 μl of 0.05 M Tris (hydroxymethyl) aminomethane was added to each sample on ice, followed by 50 μl of [3H]-cyclic AMP and 100 μl of binding protein. The samples were vortexed for 5 s and placed in an ice bath for 2 h. A charcoal suspension of 100 μl was then added. The samples were vortexed again for 10 s and centrifuged at 12,000 r.p.m. for 2 min at 4°C. Two hundred μl samples of supernatant were used for scintillation counting.
SDS – PAGE
Ventricular myocytes were homogenized with 20 mM Tris/HCl buffer, pH 7.5, containing 2 mM EDTA, 10 mM EGTA, 0.3% β-glycerophosphate, 50 μg ml−1 PMSF, and 50 μg ml−1 leupeptin. Both the supernatant (cytosolic, 100,000 g) and the 0.5% Triton X-100 extract of the pellet (particulate, 100,000 g) were extracted according to the method described previously (Kramer & Simon, 1999; Rybin & Steinberg, 1994). Equal amounts of cytosolic and particulate proteins (20 μg) were separated by SDS – PAGE (10% acrylamide running gel) and electroblotted to nitrocellulose membrane at 100 V for 90 min (Laemmli, 1970; Towbin et al., 1979). The membrane was rinsed three times with TBS, and incubated with 5% milk in TBS buffer for 1 h at room temperature to block non-specific reactions. Then, the membrane was incubated at 4°C overnight with primary antibody (1 : 1000) against synthetic peptides corresponding to the carboxyl-terminal variable regions of PKC-α, -ε, -δ, and -ζ. The membrane was then washed and incubated at room temperature with a peroxidase-conjugated second antibody (1 : 2000) followed by detection with an enhanced chemiluminescence kit. All gels were run in duplicate to confirm satisfactory sample separation.
Measurement [Ca2+]i in ventricular myocytes
To measure the electrically-induced [Ca2+]i transient, cells were first loaded with Fura-2/AM, and [Ca2+]i transient was determined by spectrofluorometry (Bian et al., 1998; Zhang et al., 1999). The fluorescence ratio of 340 nm (F340) over 380 nm (F380) was used as an index of [Ca2+]i because changes in the fluorescence ratio are considered to accurately reflect fluctuations in the cyotosolic [Ca2+]i (Hohl & Li, 1991).
Statistic analysis
Values presented are mean±standard error of mean (mean±s.e.). One-way ANOVA and the post hoc Tukey's test were used for multiple comparisons at a minimal significance level of P<0.05. Students's t-test was used when applicable for simple two-sample tests at the same minimal significance level.
Results
Effects of U50,488H on electrically-induced [Ca2+]i transient and forskolin-stimulated cyclic AMP accumulation in ventricular myocytes exposed to U50,488H and chelerythrine chloride or GF109203X for 24 h
In agreement with our previous observations (Bian et al., 1998; Zhang et al., 1999; Sheng & Wong, 1996), 30 μM U50,488H almost abolished the electrically-induced [Ca2+]i transient in single ventricular myocytes (Figure 1A,F). In ventricular myocytes previously exposed for 24 h to 1 μM U50,488H, which itself had no effect on the [Ca2+]i transient (Figure 1B), 30 μM U50,488H was no longer able to affect [Ca2+]i transient (Figure 1C,F), in contrast to the effect in naïve cells (Figure 1A,F). The observations in both studies indicate the development of tolerance to U50,488H in ventricular myocytes chronically exposed to the κ-OR agonist. However, in the presence of an inhibitor of PKC, 1 μM chelerythrine or 1 μM GF109203X, which itself had no effect on the electrically-induced [Ca2+]i transient, 30 μM U50,488H still inhibited the [Ca2+]i transient in ventricular myocytes previously exposed to 1 μM U50,488H (Figure 1D,E,F) as it did in naive cells (Figure 1A,F). The observation was taken to suggest that PKC was involved in the development of tolerance to a κ-opioid.
Figure 1.
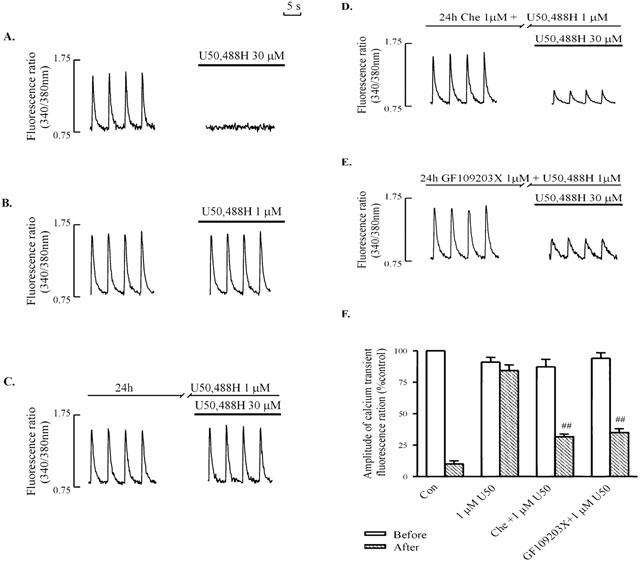
Effects of U50,488H on electrically-stimulated [Ca2+]i transient in naive ventricular myocytes (A, B), and in myocytes pretreated with 1 μM U50,488H for 24 h in the absence of PKC inhibitors (C) or presence of 1 μM chelerythrine (D) or 1 μM GF109203X (E). [Ca2+]i transient was induced by electrical stimulation at 0.2 Hz. (A – E): Representative tracings. (F) Group results showing effects of 30 μM U50,488H transient in naive ventricular myocytes, and in myocytes pretreated with 1 μM U50,488H for 24 h in the presence of a PKC inhibitor. Values are mean±s.e. of 6 – 12 cells in 4 – 6 rats. All measurements were recorded at ∼15 min after administration of U50,488H. The amplitude of the [Ca2+]i transient without U50,488H is 100%. chelerythrine was added 60 min before addition of U50,488H (1 μM) for 24 h. One μM GF109203X was added 15 min before addition of U50,488H (1 μM). □num;□num;P<0.01 vs corresponding value in U50,488H group. Before: before addition of 30 μM U50,488H; after: after addition of 30 μM U50,488H. U50: U50,488H. Con: control. Che: chelerythrine.
Similarly, forskolin-stimulated cyclic AMP accumulation in ventricular myocytes was significantly attenuated by 30 μM U50,488H (Figure 2). The attenuating effect of 30 μM U50,488H was completely abolished in myocytes previously exposed to 1 μM U50,488H for 24 h (Figure 2). In the presence of 1 μM chelerythrine, however, 30 μM U50,488H still inhibited the forskolin-stimulated cyclic AMP accumulation as it did in naïve myocytes (Figure 2).
Figure 2.
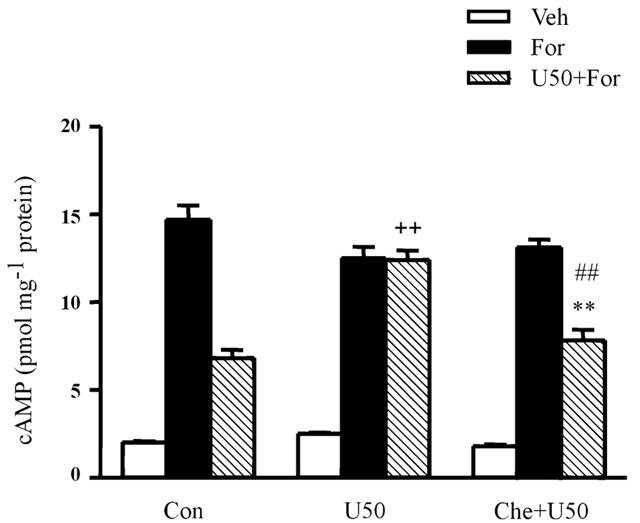
Effect of U50,488H on forskolin-stimulated cyclic AMP accumulation in rat ventricular myocytes. Values are mean±s.e. n=8 – 10. **P<0.01 vs corresponding value in U50 group. ##P<0.01 vs corresponding values in For group. ++P<0.01 compared with the corresponding values in control group. For Abbreviations, refer to Figure 1.
Immunoblot analysis of translocation of individual PKC isoforms in ventricular myocytes treated with U50,488H
Following exposure of the myocytes to 1 μM U50,488H for 24 h, which induced tolerance to its effects, the expression of the particulate fraction of PKC-ε increased while that of the cytosolic fraction decreased (Figure 3), indicating translocation of the isoform. No significant translocation of PKC-α, -δ or -ζ was observed (Figure 3)
Figure 3.
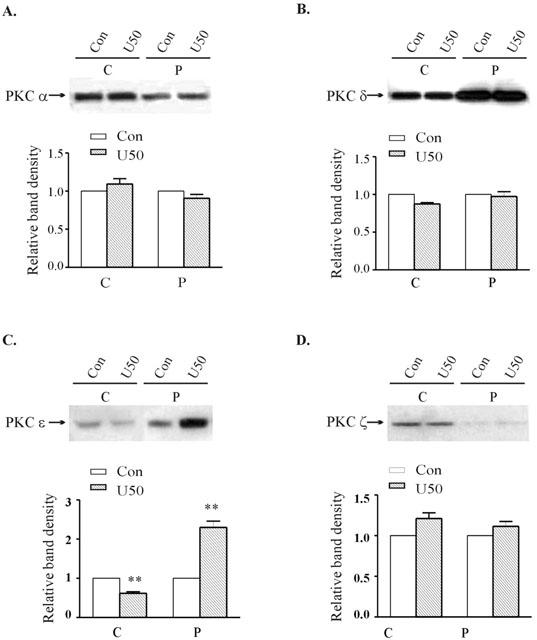
Effect of 24 h treatment with 1 μM U50,488H on the subcellular distribution of PKC isozymes in ventricular myocytes. Upper panel: Representative Western blots. Lanes 1 and 2 represent, respectively to cytosolic fractions isolated from untreated and 1 μM U50,488H-treated cells; Lanes 3 and 4 represent, respectively to particulate fractions isolated from untreated and 1 μM U50,488H-treated group. Lower panel: Group results showing expression of different isoforms as assessed by densitometric analysis. Data are expressed as percentage changes in the density of autoradiographic bands of cytosolic (C) or particulate (P) fractions from U50,488H-treated ventricular myocytes (hatched bars) relative to those from untreated cells (white bars, 100%). The data are expressed as mean±s.e.mean. (n=6). **P<0.01, vs corresponding control group.
On the other hand, acute exposure of naïve ventricular myocytes to 30 μM U50,488H for 10 min increased the particulate fraction, and decreased the cytosolic fraction of the PKC-α isoform (Figure 4), indicating translocation of the α isoform. The expression of the other PKC isoforms, -δ, -ε and -ζ was unaffected (Figure 4).
Figure 4.
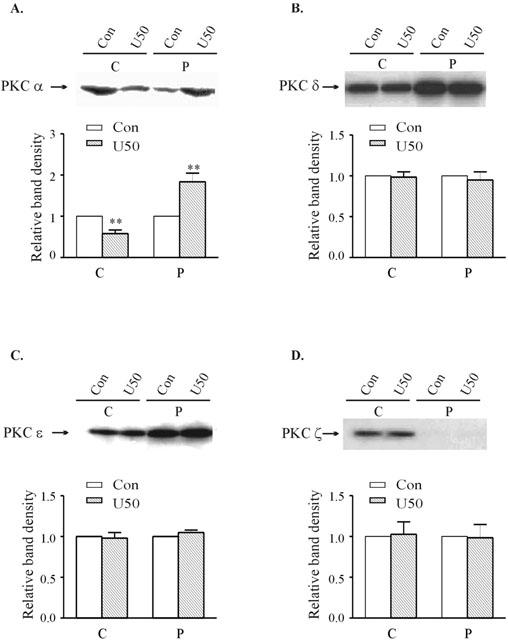
Effects of 10 min treatment with 30 μM U50,488H on the subcellular distribution of PKC-α (A), -δ (B), -ε (C) and -ζ (D) in naïve ventricular myocytes. Upper panel: Representative Western blots. Lanes 1 and 2 represent, respectively, cytosolic fractions isolated from untreated and U50,488H-treated cells; Lanes 3 and 4 represent, respectively, particulate fractions isolated from untreated and U50,488H-treated cells. Lower panel: Group results showing expression of different isoenzymes as assessed by densitometric analysis. Data are expressed as percentage changes in the density of autoradiographic bands of cytosolic (C) or particulate (P) fractions from U50,488H-treated ventricular myocytes (hatched bars) relative to those from untreated cells (white bars, 100%). The data are expressed as mean±s.e.mean. (n=5). **P<0.01, vs corresponding control value.
Effect of 30 μM U50,488H on electrically-induced [Ca2+]i transient in ventricular myocytes exposed to U50,488H and εV1-2 peptide for 24 h
In order to delineate the role of PKC-ε in the development of tolerance to U50,488H in ventricular myocytes, we made use of a selective PKC-ε inhibitor, εV1-2 peptide. In ventricular myocytes previously exposed to 0.1 μM εV1-2 peptide and 1 μM U50,488H for 24 h, U50,488H inhibited the electrically-induced [Ca2+]i transient (Figure 5B,D). The effect was similar to that in ventricular myocytes previously exposed to 1 μM U50,488H and a PKC inhibitor (Figure 1D,E,F), but in contrast to the absence of inhibitory effect in ventricular myocytes pretreated only with U50,488H (Figure 1C,F) or with U50,488H and the control peptide for 24 h (Figure 5C,D). Thirty μM U50,488H also increased the cytosolic fraction of PKC-α, but decreased the particulate fraction of the isoform (Figure 6), a response just the opposite to that in naïve ventricular myocytes. On the other hand, the cytosolic fraction of PKC-ε decreased while the particulate fraction increased slightly (Figure 6), which is also different to the lack of response to 30 μM U50,488H in naïve ventricular myocytes (Figure 4).
Figure 5.
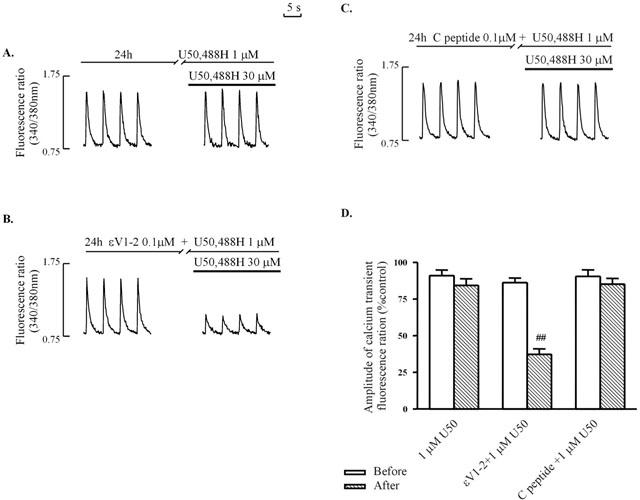
Effects of 1 μM U50,488H pretreatment on inhibition of electrically-stimulated [Ca2+]i transient in ventricular myocytes by 30 μM U50,488H in the absence (A) or presence (B) of 0.1 μM εV1-2 peptide and 0.1 μM control peptide. (A – C): Representative tracing. (D): Group results. Values are mean±s.e.mean of 6 – 12 cells in 4 – 6 rats. The amplitude of the [Ca2+]i transient without U50,488H is 100%. 0.1 μM εV1-2 peptide was added 30 min before addition of U50,488H (1 μM) for 24 h. □num;□num;P<0.01 corresponding value in U50,488H group. For experimental protocol, and abbreviations, refer to Figure 1.
Figure 6.
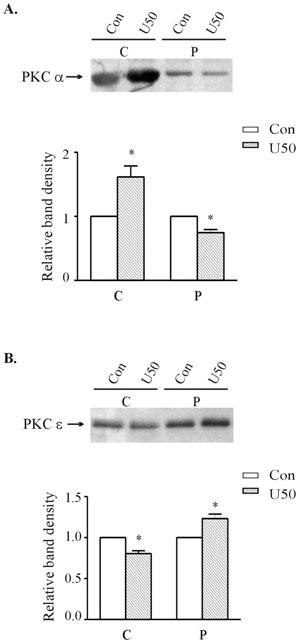
Effect of 10 min treatment with 30 μM U50,488H on the subcellular distribution of PKC-α (A) and -ε (B) in ventricular myocytes pretreated with 1 μM U50,488H for 24 h. Upper panel: Representative Western blots. Lanes 1 and 2 represent, respectively to cytosolic fractions isolated from untreated and U50,488H-treated cells. Lanes 3 and 4 represent, respectively to particulate fractions isolated from untreated and U50,488H-treated cells. Lower panel: Group results showing expression of different isoforms as assessed by densitometric analysis. Data are expressed as percentage changes in the density of autoradiographic bands of cytosolic (C) or particulate (P) fractions from cells subjected to 30 μM U50,488H (hatched bars) relative to those from untreated cells (white bars, 100%). The data are expressed as mean±s.e. (n=6). *P<0.05 vs corresponding control.
It should be noted that exposure of myocytes to εV1-2 peptide for 24 h alone did not alter the electrically-induced [Ca2+]i transient (Figure 5B). Nor was the inhibitory effect of 30 μM U50,488H on the electrically-induced [Ca2+]i transient altered in naïve ventricular myocytes by the presence of 0.1 μM εV1-2 peptide for 30 min (Figure 7).
Figure 7.
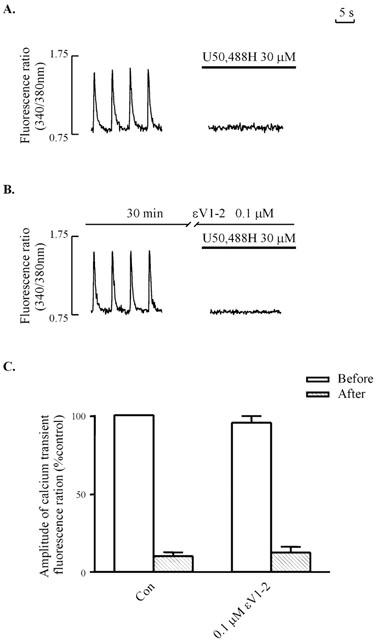
Thirty μM U50,488H-induced inhibition of electrically-stimulated [Ca2+]i transient in naive ventricular myocytes in the absence (A) or presence (B) of 0.1 μM εV1-2 peptide. (A,B): Representative tracings. (C): Group results. Values are mean±s.e.mean of 6 – 12 cells in 4 – 6 rats. The amplitude of the [Ca2+]i transient without U50,488H is 100%. 0.1 μM εV1-2 peptide was added 30 min before addition of U50,488H. For experimental protocol, and abbreviations, refer to Figure 1.
Discussion
There are two important observations in the present study. Firstly, chronic exposure to of ventricular myocytes to U50,488H, which induced tolerance to the effects of the agonist, was accompanied by translocation of PKC-ε. Secondly, blockade of PKC-ε with a selective inhibitor attenuated the development of tolerance. The observations are evidence that PKC-ε mediates the development of tolerance in the κ-OR following chronic exposure to a κ-OR agonist. In previous studies, PKC-ε has been shown to mediate delayed cardioprotection of various forms of preconditioning (Qiu et al., 1998; Ping et al., 1999; Wang et al., 2001). It seems that this particular PKC-isoform plays an important role in the heart in more than one situation.
Individual PKCs are classified into the following groups based on their common structural characteristics, and dependencies on Ca2+, phospholipids, and diacylglycerol (DAG) for activity: conventional (α, βI, βII, and γ; Ca2+- and DAG-regulated); novel (δ, ε, ζ, and θ; DAG-regulated), and atypical (ζ and ι/λ; regulated by neither DAG nor Ca2+) (Way et al., 2000). A previous study showed that, in SH-SY5Y cells, which express PKC-α, -ε, and -ζ (Turner et al., 1996), μ-OR down-regulation induced by its agonist was directly correlated with the binding of [3H]-phorbol 12,13-dibutyrate ([3H]-PDBu) in the membrane fraction, an index of total PKC activity, and translocation of PKC-α and ε, which are DAG-regulated (Kramer & Simon, 1999). It was also shown that prior treatment with phorbol ester did not affect the agonist-induced μ-OR down-regulation (Kramer & Simon, 1999). Since phorbol ester depletes PKC isoforms, which are DAG-regulated and Ca2+ sensitive, but not PKC isoforms of the atypical group (-ζ), which are not affected by either DAG or Ca2+ (Kramer & Simon, 1999), the evidence was taken by the authors to suggest that the conventional (α), novel (ε) and atypical (ζ) PKC isoforms are involved in the agonist-induced μ-OR down-regulation. In the present study, we correlated the development of tolerance in the κ-OR of the heart with translocation of PKC-isoforms, which suggested that PKC-ε is involved in development of tolerance. More importantly, we found that blockade of PKC-ε with a selective inhibitor attenuated the development of tolerance. The result indicates that PKC-ε mediates the development of tolerance in the κ-OR of the heart.
In the present study we found that the tolerance to the effect of U50,488H following 24 h exposure to the agonist was abolished by chelerythrine or GF109203X, suggesting that PKC plays a mediatory role in the development of tolerance in the κ-OR. A mediatory role of PKC in the development of tolerance/receptor down regulation is not unique in κ-OR. In previous studies, it has been reported that PKC was involved in agonist-induced δ-OR desensitization in Xenopus oocytes (Ueda et al., 1995) and μ-OR down-regulation in SH-SY5Y neuroblastoma cells (Kramer & Simon, 1999). It has also been shown that PKC mediates the agonist-independent (heterologous) receptor down regulation in δ-OR in NG108-15 cells (Fan et al., 1998). It appears that PKC is an important secondary messenger mediating desensitization/receptor down-regulation/tolerance in the ORs. There are, however, observations suggesting that PKC does not play any role in receptor down-regulation/tolerance. After prolonged exposure to phorbol ester, known to deplete the total cellular PKC protein in HEK-293, the δ-OR was still capable of developing tolerance to the δ-OR agonist (Pei et al., 1995). It cannot be ruled out, however, that a small amount of PKC-ε may still remain after chronic PMA exposure and exert an influence on agonist-induced opioid receptor tolerance.
Available evidence on chelerythrine being a selective PKC inhibitor is controversial. Chelerythrine was first reported to be a selective PKC inhibitor in 1990 (Herbert et al., 1990). However, two recent studies showed that chelerythrine produces effects independent of PKC (Lee et al., 1998; Yu et al., 2000). On the other hand recent studies also showed that both chelerythrine and calphostin C, another PKC inhibitor, activate JNKs and p38MAPKs (Heidkamp et al., 2001) and induce apoptosis in cardiac myocytes through generation of reactive oxygen species (Clerk, 2001; Yamamoto et al., 2001). In the present study we found that another PKC inhibitor, GF109203X, blocked the development of tolerance to the effect of U50 as chelerythrine did. It is likely that the effect of chelerythrine observed in the present study was mainly via PKC.
The internalization of human κ-OR expressed in Chinese hamster ovary cells exposed to U50,488H for 30 min involves both β-arrestin and dynamin I (Li et al., 1999), similarly to that of GPCRs. In the present study, we found that PKC-ε mediated the development of κ-OR tolerance in ventricular myocytes previously exposed to U50,488H for 24 h. So different mechanisms may be in operation at different times after exposure to the agonist. Alternatively, these mechanisms are related to each other. Further studies are needed.
A third important observation of the present study is that translocation of PKC-α occurred in response to acute exposure to 30 μM U50,488H, an observation reported previously (Ventura & Pintus, 1997). On the other hand there was no translocation of other PKC isoforms. The observations in the previous and present studies suggest that the isoform may be involved in the acute effect of U-50,488H. In support of this suggestion, we also observed that there was no translocation of this isoform in response to acute 30 μM U50,488H in ventricular myocytes previously exposed to 1 μM U50,488H, when the agonist did not elicit a significant response in the myocytes. However, the exact role of PKC-α in the acute response to κ-OR stimulation can only be elucidated when studies with a selective inhibitor of PKC-α have been conducted.
In the present study, we found that exposure to U50,488H abolished the inhibition of forskolin-stimulated cyclic AMP accumulation of rat ventricular myocytes, a finding also observed in human κ-OR (Zhu et al., 1998). However, Joseph & Bidlack (1995) observed no desensitization of U50,488H-elicited inhibition of forskolin-stimulated adenylate cyclase in R1.1 cells pretreated with 0.1 μM U50,488H for 24 – 48 h. The discrepancy may be the result of the different cells used in different studies.
One limitation of the present study is that we cannot rule out the possibility that other PKC-isoforms are also important in the development of tolerance. This is limited by the fact that selective inhibitors for PKC-α and PKC-ζ are not available yet. In a preliminary study we tried to determine the development of tolerance to U50,488H in the presence of rottlerin, PKC-δ inhibitor. We were not able to keep the ventricular myocytes alive for 24 h because of the toxic effect of the inhibitor (Majumder et al., 2000; Soltoff, 2001).
In conclusion, the present study has demonstrated for the first time that PKC-ε translocation occurs following chronic exposure to U50,488H, that induced development of tolerance, and that blockade of PKC-ε attenuated the tolerance. Modulation of PKC-ε translocation and activation may prove useful for the management of pain and opiate addiction.
Acknowledgments
The study was supported by a grant from the Committee of Research and Conference Grants, The University of Hong Kong. We thank Dr I. Bruce for advice on the use of English, Dr G.R. Li and Dr N.S. Wong for helpful discussion, and Mr C.P. Mok and Mr H. Yang for technical assistance. J.-J. Zhou was on leave from the Department of Physiology, the Fourth Military Medical University of Xi'an, P. R. of China.
Abbreviations
- Che
Chelerythrine
- DAG
diacylglycerol
- GPCR
G protein-coupled receptor
- OR
opioid receptor
- PKC
protein kinase C
- U50,488H
trans-(±)-3,4-dichloro-N-methyl-N-(2-[1-pyrrolidinyl]cyclohexyl) benzeneacetamide
References
- BIAN J.S., PEI J.M., CHEUNG C.S., ZHANG W.M., WONG T.M. kappa-opioid receptor stimulation induces arrhythmia in the isolated rat heart via the protein kinase C/Na(+)-H(+)exchange pathway. J. Mol. Cell Cardiol. 2000;32:1415–1427. doi: 10.1006/jmcc.2000.1175. [DOI] [PubMed] [Google Scholar]
- BIAN J.S., WANG H.X., ZHANG W.M., WONG T.M. Effects of kappa-opioid receptor stimulation in the heart and the involvement of protein kinase C. Br. J. Pharmacol. 1998;124:600–606. doi: 10.1038/sj.bjp.0701857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIRCH P.J., HAYES A.G., SHEEHAN M.J., TYERS M.B. Norbinaltorphimine: antagonist profile at kappa opioid receptors. Eur. J. Pharmacol. 1987;144:405–408. doi: 10.1016/0014-2999(87)90397-9. [DOI] [PubMed] [Google Scholar]
- CHEN C.H., GRAY M.O., MOCHLY-ROSEN D. Cardioprotection from ischemia by a brief exposure to physiological levels of ethanol: role of epsilon protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 1999;96:12784–12789. doi: 10.1073/pnas.96.22.12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLERK A. Death by protein kinase c inhibitor: a stressful event. J. Mol. Cell Cardiol. 2001;33:1773–1776. doi: 10.1006/jmcc.2001.1458. [DOI] [PubMed] [Google Scholar]
- CSUKAI M., MOCHLY-ROSEN D. Pharmacologic modulation of protein kinase C isozymes: the role of RACKs and subcellular localisation. Pharmacol. Res. 1999;39:253–259. doi: 10.1006/phrs.1998.0418. [DOI] [PubMed] [Google Scholar]
- DONG H., SHENG J.Z., LEE C.M., WONG T.M. Calcium antagonistic and antiarrhythmic actions of CPU-23, a substituted tetrahydroisoquinoline. Br. J. Pharmacol. 1993;109:113–119. doi: 10.1111/j.1476-5381.1993.tb13539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAN G.H., ZHAO J., WU Y.L., LOU L.G., ZHANG Z., JING Q., MA L., PEI G. N-Methyl-D-aspartate attenuates opioid receptor-mediated G protein activation and this process involves protein kinase C. Mol. Pharmacol. 1998;53:684–690. doi: 10.1124/mol.53.4.684. [DOI] [PubMed] [Google Scholar]
- HEIDKAMP M.C., BAYER A.L., MARTIN J.L., SAMAREL A.M. Differential activation of mitogen-activated protein kinase cascades and apoptosis by protein kinase C epsilon and delta in neonatal rat ventricular myocytes. Circ. Res. 2001;89:882–890. doi: 10.1161/hh2201.099434. [DOI] [PubMed] [Google Scholar]
- HERBERT J.M., AUGEREAU J.M., GLEYE J., MAFFRAND J.P. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- HOHL C.M., LI Q.A. Compartmentation of cAMP in adult canine ventricular myocytes. Relation to single-cell free Ca2+ transients. Circ. Res. 1991;69:1369–1379. doi: 10.1161/01.res.69.5.1369. [DOI] [PubMed] [Google Scholar]
- JOHNSON J.A., GRAY M.O., CHEN C.H., MOCHLY-ROSEN D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J. Biol. Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- JOHNSON J.A., MOCHLY-ROSEN D. Inhibition of the spontaneous rate of contraction of neonatal cardiac myocytes by protein kinase C isozymes. A putative role for the epsilon isozyme. Circ. Res. 1995;76:654–663. doi: 10.1161/01.res.76.4.654. [DOI] [PubMed] [Google Scholar]
- JOSEPH D.B., BIDLACK J.M. The kappa opioid receptor expressed on the mouse R1.1 thymoma cell line down-regulates without desensitizing during chronic opioid exposure. J. Pharmacol. Exp. Ther. 1995;272:970–976. [PubMed] [Google Scholar]
- KRAMER H.K., SIMON E.J. Role of protein kinase C (PKC) in agonist-induced mu-opioid receptor down-regulation: II. Activation and involvement of the alpha, epsilon, and zeta isoforms of PKC. J. Neurochem. 1999;72:594–604. doi: 10.1046/j.1471-4159.1999.0720594.x. [DOI] [PubMed] [Google Scholar]
- LAEMMLI U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LAHTI R.A., VONVOIGTLANDER P.F., BARSUHN C. Properties of a selective kappa agonist, U-50,488H. Life Sci. 1982;31:2257–2260. doi: 10.1016/0024-3205(82)90132-1. [DOI] [PubMed] [Google Scholar]
- LAW P.Y., WONG Y.H., LOH H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- LEE S.K., QING W.G., MAR W., LUYENGI L., MEHTA R.G., KAWANISHI K., FONG H.H., BEECHER C.W., KINGHORN A.D., PEZZUTO J.M. Angoline and chelerythrine, benzophenanthridine alkaloids that do not inhibit protein kinase C. J. Biol. Chem. 1998;273:19829–19833. doi: 10.1074/jbc.273.31.19829. [DOI] [PubMed] [Google Scholar]
- LI J.G., LUO L.Y., KRUPNICK J.G., BENOVIC J.L., LIU-CHEN L.Y. U50,488H-induced internalization of the human kappa opioid receptor involves a beta-arrestin- and dynamin-dependent mechanism. Kappa receptor internalization is not required for mitogen-activated protein kinase activation. J. Biol. Chem. 1999;274:12087–12094. doi: 10.1074/jbc.274.17.12087. [DOI] [PubMed] [Google Scholar]
- LIU G.S., COHEN M.V., MOCHLY-ROSEN D., DOWNEY J.M. Protein kinase C-epsilon is responsible for the protection of preconditioning in rabbit cardiomyocytes. J. Mol. Cell Cardiol. 1999;31:1937–1948. doi: 10.1006/jmcc.1999.1026. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MAJUMDER P.K., PANDEY P., SUN X., CHENG K., DATTA R., SAXENA S., KHARBANDA S., KUFE D. Mitochondrial translocation of protein kinase C delta in phorbol ester-induced cytochrome c release and apoptosis. J. Biol. Chem. 2000;275:21793–21796. doi: 10.1074/jbc.C000048200. [DOI] [PubMed] [Google Scholar]
- MAYER D.J., MAO J., PRICE D.D. The development of morphine tolerance and dependence is associated with translocation of protein kinase C. Pain. 1995;61:365–374. doi: 10.1016/0304-3959(95)00023-L. [DOI] [PubMed] [Google Scholar]
- MESTEK A., HURLEY J.H., BYE L.S., CAMPBELL A.D., CHEN Y., TIAN M., LIU J., SCHULMAN H., YU L. The human mu opioid receptor: modulation of functional desensitization by calcium/calmodulin-dependent protein kinase and protein kinase C. J. Neurosci. 1995;15:2396–2406. doi: 10.1523/JNEUROSCI.15-03-02396.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARITA M., FENG Y., MAKIMURA M., HOSKINS B., HO I.K. A protein kinase inhibitor, H-7, inhibits the development of tolerance to opioid antinociception. Eur. J. Pharmacol. 1994;271:543–545. doi: 10.1016/0014-2999(94)90817-6. [DOI] [PubMed] [Google Scholar]
- NISHIZUKA Y. Studies and perspectives of protein kinase C. Science. 1986;233:305–312. doi: 10.1126/science.3014651. [DOI] [PubMed] [Google Scholar]
- PEI G., KIEFFER B.L., LEFKOWITZ R.J., FREEDMAN N.J. Agonist-dependent phosphorylation of the mouse delta-opioid receptor: involvement of G protein-coupled receptor kinases but not protein kinase C. Mol. Pharmacol. 1995;48:173–177. [PubMed] [Google Scholar]
- PING P., TAKANO H., ZHANG J., TANG X.L., QIU Y., LI R.C., BANERJEE S., DAWN B., BALAFONOVA Z., BOLLI R. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ. Res. 1999;84:587–604. doi: 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- QIU Y., PING P., TANG X.L., MANCHIKALAPUDI S., RIZVI A., ZHANG J., TAKANO H., WU W.J., TESCHNER S., BOLLI R. Direct evidence that protein kinase C plays an essential role in the development of late preconditioning against myocardial stunning in conscious rabbits and that epsilon is the isoform involved. J. Clin. Invest. 1998;101:2182–2198. doi: 10.1172/JCI1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RYBIN V.O., STEINBERG S.F. Protein kinase C isoform expression and regulation in the developing rat heart. Circ. Res. 1994;74:299–309. doi: 10.1161/01.res.74.2.299. [DOI] [PubMed] [Google Scholar]
- SHENG J.Z., WONG T.M. Chronic U50,488H abolishes inositol 1,4,5-trisphosphate and intracellular Ca2+ elevations evoked by kappa-opioid receptor in rat myocytes. Eur. J. Pharmacol. 1996;307:323–329. doi: 10.1016/0014-2999(96)00280-4. [DOI] [PubMed] [Google Scholar]
- SNABAITIS A.K., YOKOYAMA H., AVKIRAN M. Roles of mitogen-activated protein kinases and protein kinase C in alpha(1A)-adrenoceptor-mediated stimulation of the sarcolemmal Na(+)-H(+) exchanger. Circ. Res. 2000;86:214–220. doi: 10.1161/01.res.86.2.214. [DOI] [PubMed] [Google Scholar]
- SOLTOFF S.P. Rottlerin is a mitochondrial uncoupler that decreases cellular atp levels and indirectly blocks protein kinase cdelta tyrosine phosphorylation. J. Biol. Chem. 2001;276:37986–37992. doi: 10.1074/jbc.M105073200. [DOI] [PubMed] [Google Scholar]
- TOULLEC D., PIANETTI P., COSTE H., BELLEVERGUE P., GRAND-PERRET T., AJAKANE M., BAUDET V., BOISSIN P., BOURSIER E., LORIOLLE F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- TOWBIN H., STAEHELIN T., GORDON J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURNER N.A., WALKER J.H., BALL S.G., VAUGHAN P.F. Down-regulation or long-term inhibition of protein kinase C (PKC) reduces noradrenaline release evoked via either PKC-dependent or PKC-independent pathways in human SH-SY5Y neuroblastoma cells. Neurosci. Lett. 1996;220:37–40. doi: 10.1016/s0304-3940(96)13245-6. [DOI] [PubMed] [Google Scholar]
- UEDA H., MIYAMAE T., HAYASHI C., WATANABE S., FUKUSHIMA N., SASAKI Y., IWAMURA T., MISU Y. Protein kinase C involvement in homologous desensitization of delta-opioid receptor coupled to Gi1-phospholipase C activation in Xenopus oocytes. J. Neurosci. 1995;15:7485–7499. doi: 10.1523/JNEUROSCI.15-11-07485.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENTURA C., BASTAGLI L., BERNARDI P., CALDARERA C.M., GUARNIERI C. Opioid receptors in rat cardiac sarcolemma: effect of phenylephrine and isoproterenol. Biochim. Biophys. Acta. 1989;987:69–74. doi: 10.1016/0005-2736(89)90456-2. [DOI] [PubMed] [Google Scholar]
- VENTURA C., PINTUS G. Opioid peptide gene expression in the primary hereditary cardiomyopathy of the Syrian hamster. III. Autocrine stimulation of prodynorphin gene expression by dynorphin B. J. Biol. Chem. 1997;272:6699–6705. doi: 10.1074/jbc.272.10.6699. [DOI] [PubMed] [Google Scholar]
- VENTURA C., SPURGEON H., LAKATTA E.G., GUARNIERI C., CAPOGROSSI M.C. Kappa and delta opioid receptor stimulation affects cardiac myocyte function and Ca2+ release from an intracellular pool in myocytes and neurons. Circ. Res. 1992;70:66–81. doi: 10.1161/01.res.70.1.66. [DOI] [PubMed] [Google Scholar]
- WANG G.Y., ZHOU J.J., SHAN J., WONG T.M. Protein Kinase C-epsilon is a trigger of delayed cardioprotection against myocardial ischemia of kappa-opioid receptor stimulation in rat ventricular myocytes. J. Pharmacol. Exp. Ther. 2001;299:603–610. [PubMed] [Google Scholar]
- WAY K.J., CHOU E., KING G.L. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends. Pharmacol. Sci. 2000;21:181–187. doi: 10.1016/s0165-6147(00)01468-1. [DOI] [PubMed] [Google Scholar]
- WU S., LI H.Y., WONG T.M. Cardioprotection of preconditioning by metabolic inhibition in the rat ventricular myocyte. Involvement of kappa-opioid receptor. Circ. Res. 1999;84:1388–1395. doi: 10.1161/01.res.84.12.1388. [DOI] [PubMed] [Google Scholar]
- XIANG B., YU G.H., GUO J., CHEN L., HU W., PEI G., MA L. Heterologous activation of protein kinase C stimulates phosphorylation of delta-opioid receptor at serine 344 and this results in beta-arrestin- and clathrin-mediated receptor internalization. J. Biol. Chem. 2000;276:4709–4716. doi: 10.1074/jbc.M006187200. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO S., SETA K., MORISCO C., VATNER S.F., SADOSHIMA J. Chelerythrine rapidly induces apoptosis through generation of reactive oxygen species in cardiac myocytes. J. Mol. Cell Cardiol. 2001;33:1829–1848. doi: 10.1006/jmcc.2001.1446. [DOI] [PubMed] [Google Scholar]
- YU R., MANDLEKAR S., TAN T.H., KONG A.N. Activation of p38 and c-Jun N-terminal kinase pathways and induction of apoptosis by chelerythrine do not require inhibition of protein kinase C. J. Biol. Chem. 2000;275:9612–9619. doi: 10.1074/jbc.275.13.9612. [DOI] [PubMed] [Google Scholar]
- ZHANG W.M., WU S., YU X.C., WANG H.X., BIAN J.S., WONG T.M. Effects of U50488 and bremazocine on [Ca2+]i and cAMP in naive and tolerant rat ventricular myocytes: evidence of kappa opioid receptor multiplicity in the heart. J. Mol. Cell Cardiol. 1999;31:355–362. doi: 10.1006/jmcc.1998.9998. [DOI] [PubMed] [Google Scholar]
- ZHU J., LUO L.Y., MAO G.F., ASHBY B., LIU-CHEN L.Y. Agonist-induced desensitization and down-regulation of the human kappa opioid receptor expressed in Chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 1998;285:28–36. [PubMed] [Google Scholar]
- ZIMLICHMAN R., GEFEL D., ELIAHOU H., MATAS Z., ROSEN B., GASS S., ELA C., EILAM Y., VOGEL Z., BARG J. Expression of opioid receptors during heart ontogeny in normotensive and hypertensive rats. Circulation. 1996;93:1020–1025. doi: 10.1161/01.cir.93.5.1020. [DOI] [PubMed] [Google Scholar]