

Abstract
The L-arginine-NO pathway has been implicated in the vasorelaxant effect of 17-β-oestradiol. Here we have addressed the involvement of two distinct activation steps of endothelial nitric oxide synthase (eNOS) in the 17-β-oestradiol-induced vasorelaxant effect on rat aortic rings.
Rat aortic rings contracted with phenylephrine (PE) 1 μM relaxed in a concentration related fashion to 17-β-oestradiol water soluble cyclodextrin-encapsulated (E2) only when endothelium was present. The pure anti-oestrogen of E2 receptor ICI 182,780 (20 μM) significantly inhibited E2-induced vasorelaxation.
Geldanamycin (10 μM), a specific inhibitor of heat shock protein 90 (hsp90) and Nω-nitro-L-arginine-methyl ester (L-NAME, 100 μM), a nitric oxide synthase inhibitor, significantly inhibited E2-induced vasorelaxation.
Incubation of rat aortic rings up to 6 h with LY 294002 (25 μM), a specific inhibitor of PI(3)K akt/pkb pathway reduced E2-induced vasorelaxation.
Incubation of rat isolated aorta with E2, induced prostacyclin (PGI2) release. PGI2 levels, measured as 6-keto PGF1α, were abolished by ibuprofen (10 μM), both L-NAME and GA did not influence basal or E2-stimulated PGI2 confirming the specificity of these two compounds on eNOS pathway.
In conclusion, we demonstrate that E2 interaction with its receptor is followed by a vasorelaxant effect in rat aortic rings mediated by eNOS activation through both hsp90 and akt/pkb dependent mechanisms.
Keywords: 17-β-oestradiol; eNOS; geldanamycin, hsp90; LY 294002; akt/pkb pathway
Introduction
Oestrogen is a female sex hormone that produces several biological effects through binding to the oestrogen receptors (ER) α and β (Green et al., 1986; Kuiper & Gustafsson, 1997; Paech et al., 1997). Oestrogen, as other steroids, acts through genomic and non-genomic mechanisms; in particular the non-genomic modulation of physiological and pathophysiological processes has been the object of several studies in the past few years. Oestrogen displays significant cardioprotective actions and contributes to the sex difference in the risk of cardiovascular diseases (Sullivan et al., 1988; Adams et al., 1990; Kauser et al., 1997). The beneficial effects of oestrogen on cardiovascular system is partially due to a direct vasodilatory effect of this hormone on arteries (Bush et al., 1987). Indeed, several studies showed a direct relationship between oestrogen and nitric oxide release in conductance and resistance arteries both in vitro and in vivo (Rosselli et al., 1995; Sudhir et al., 1995; Huang et al., 1998). In vitro in cultured endothelial cells, oestrogen activates the endothelial isoform of nitric oxide synthase (eNOS) (Caulin-Glaser et al., 1997) and stimulates heat shock protein 90-eNOS association in human vascular endothelial cells suggesting an involvement of hsp90 in the rapid, oestrogen receptor-mediated modulation of eNOS (Russell et al., 2000). In vivo oestrogen restores endothelium-dependent coronary vasodilatation in post-menopausal women and this action has also been ascribed to nitric oxide (Collins et al., 1995).
The ER resides in a transcriptionally inactive state with a complex of chaperone proteins within the nucleus (at equilibrium) of the target cells (Pratt & Toft, 1997; Defranco, 2000; for review see Falkenstein et al., 2000). Following the binding of the hormone, two molecules of hsp90 and the immunophillin hsp56, that maintain the receptor in an inactive form, dissociate from the receptor (Pratt & Toft, 1997) that changes its conformation initiating a cascade of events leading to the activation of specific DNA sequence regulating gene transcription (Kumar et al., 1987; Gallo & Kaufman, 1997). In bovine aortic endothelial cells, acute exposure to 17-β-oestradiol increases NO production through ERα localized in caveolae (Kim et al., 1999), a cholesterol and sphingolipid-rich microdomain of the plasmalemma (Garcia-Cardena et al., 1996) where eNOS also resides (Garcia-Cardena et al., 1997). eNOS is the nitric oxide synthase isoform responsible of NO release in vascular endothelial cells. NO is a potent vasodilator factor that plays several roles in physiological processes such as the control of basal tone of vessels, vascular remodelling (Rudic et al., 2000) and angiogenesis (Papapetropoulos et al., 1997a; Morbidelli et al., 1996). Association of hsp90 with eNOS enhances the enzyme activity, as demonstrated by the finding that geldanamycin, a specific inhibitor of hsp90, attenuates both agonist-stimulated production of nitric oxide and endothelium-dependent relaxation of isolated blood vessels (Garcia-Cardena et al., 1998). Finally, it has also been shown that the serine/threonine protein kinase (akt/pkb) is involved in the complex mechanism involving eNOS activation (Dimmeler et al., 1999; Fulton et al., 1999; Zeng & Quon, 1996; Papapetropoulos et al., 1997b) by phosphorylating the enzyme (Fulton et al., 1999; Dimmeler et al., 1999).
Based on the above evidence, here we have sought to investigate the molecular mechanism involved in 17-β-oestradiol-induced vasorelaxation, i.e eNOS phosphorylation and coupling to hsp90, in isolated rat aortic rings by using specific pharmacological tools.
Methods
Drugs
Acetylcholine (ACh), L-phenylephrine (PE), Nω-nitro-L-arginine methyl ester (L-NAME), ibuprofen, tetraethylammonium (TEA), 17-β-oestradiol water soluble cyclodextri-encapsulated (E2), LY 294002, polyethylene glycol 400 (PEG), dimethyl sulphoxide (DMSO) were purchased from Sigma Chemical Co. (Milano, Italy), ICI 182,780 was purchased from Tocris Cookson Ltd. (Bristol, U.K.), Dulbecco's modified Eagle medium (DMEM), L-glutamine, penicillin and streptomycin were purchased from Hy-Clone (Road Logan, UT, U.S.A.), geldanamycin (GA) was a generous gift of the Drug Synthesis & Chemistry Branch, Development Therapeutics program, Division of Cancer treatment at the National Cancer Institute (Bethesda, MD, U.S.A.). GA was dissolved in a solution of distilled water dH2O/PEG (1/1), ICI 182,780 was dissolved in PEG, TEA and L-NAME in dH2O. The volume added to the organ bath for each compound was 2.5 μl, except for TEA that was 25 μl.
Tissue preparation
Male Wistar rats (Charles River, 250 – 300 g) of 8 – 10 weeks of age were sacrificed and thoracic aorta was rapidly dissected and cleaned from fat and connective tissue. Rings of 2 – 3 mm length were cut and placed in organ baths (2.5 ml) filled with oxygenated (95% O2-5% CO2) Krebs solution at 37°C and mounted to isometric force transducers (type 7006, Ugo Basile, Comerio, Italy) and connected to a Graphtec linearcorder (WR3310, Japan). The composition of the Krebs solution was as follows (mol l−1): NaCl 0.118, KCl 0.0047, MgCl2 0.0012, KH2PO4 0.0012, CaCl2 0.0025, NaHCO3 0.025 and glucose 0.010. Rings were initially stretched until a resting tension of 0.5 g was reached and allowed to equilibrate for at least 30 min during which tension was adjusted, when necessary, to 0.5 g and bathing solution was periodically changed. In a preliminary study a resting tension of 0.5 g was found to develop the optimal tension to stimulation with contracting agents.
Experimental protocol
In each experiment rings were firstly challenged with PE (10−6 M) until the responses were reproducible. In order to verify the integrity of the endothelium, ACh cumulative concentration – response curve (10−8−3×10−5 M) was performed on PE-precontracted rings. Vessels that relaxed less than 75% were discarded. Selected rings were then contracted with PE (10−6 M), once plateau was reached, a cumulative concentration – response curve to E2 (10−8−3×10−5 M) was performed. In another set of experiments, rings were denuded of the endothelium and E2 cumulative concentration – response curve was performed.
Drug treatments
A preliminary study on the optimal incubation time of the drug treatments and concentration was carried out (data not shown). The experiments were then performed by using the optimal incubation time and concentrations as specified for each protocol reported below. Aortic rings were standardized with PE (10−6 M) and performing a cumulative concentration response to Ach (10−8−3×10−5 M) was performed. The experimental protocols performed were:
GA (3×10−6, 10−5 and 3×10−5 M), L-NAME (10−4 M) and ICI 182,780 (2×10−5 M) were added in the organ baths on PE-precontracted rings. After 15 min of incubation, a cumulative concentration – response curve to E2 was performed;
TEA (10−2 M) was added in the organ baths; after 60 min, the rings were contracted with PE (10−6 M) and a cumulative concentration – response curve to E2 was performed;
Ibuprofen (10−5 M) was added to the Krebs solution and ACh and a cumulative concentration – response curve to E2 was performed.
LY 294002 treatments
Thoracic aorta was dissected, placed in sterile PBS and cleaned of fat and connective tissue. Rings, of the same size as described above, were cut and placed in a 24 multiwell plate with 980 μl DMEM solution (containing penicillin 100 U ml−1, streptomycin 100 μg ml−1 and L-glutamine 2×10−3 M) in the presence of LY 294002 (2.5×10−5 M) or vehicle (DMSO; 20 μl). This concentration of DMSO in DMEM (2%) is the maximal that can be added without affecting tissue functionality (data not shown). Thereafter, the plate was placed at 37°C in an incubator (Mod. BB6220, Heraeus Instruments, Germany) with humidified air (5% CO2/95% O2). After 1, 3, 6 and 16 h of incubation rings were washed extensively with Krebs and then mounted in organ baths, as described above (Bucci et al., 2000).
Prostacyclin (PGI2) release from rat aorta
Rat aortas were removed as described above, cut in rings, weighted and placed in a 24 multiwell plate with 500 μl of gassed Krebs solution. In each well were added GA, L-NAME or vehicles using the same concentrations used for the organ bath studies and placed in an incubator with humidified air (5% CO2/95% O2) at 37°C (Mod. BB6220, Heraeus Instruments, Germany) for 15 min. After this incubation period 100 μl of supernatant were removed in order to evaluate the basal release of PGI2 from incubation wells set for this purpose. In all the remaining wells, E2 (10−5 M) was added and left in contact for another additional 30 min. Thereafter, 100 μl aliquots were taken to assess PGI2 levels. Prostacyclin levels were measured as stable breakdown product, 6-keto-PGF1α, by enzymatic immunoassay (EIA, Cayman Chemical) and expressed as mg ml−1 per mg of tissue (wet weight) (Bucci et al., 2000). EIA was performed according to the manufacturer's directions. Data are the mean±s.e.mean of five separate experiments.
Statistical analysis
All data were expressed as mean±s.e.mean. Statistical analysis was performed by using 2-way ANOVA and paired or unpaired Student's t-test where appropriate. Differences were considered statistically significant when P was less than 0.05.
Results
17-β-oestradiol induced endothelium-dependent vasodilatation
In presence of endothelium, E2 induced a concentration-dependent vasodilatation on PE pre-contracted rings. When endothelium was removed E2 was ineffective (Figure 1).
Figure 1.

Cumulative concentration-response curves to 17-β-oestradiol (E2) in rat aortic rings with and without endothelium. Data are shown as means±s.e mean; n=6 for each group.
Effect of inhibitors on E2-induced vasodilatation
GA at the concentration of 10−5 M after 15 min of incubation significantly reduced E2-induced vasodilatation when compared to the vehicle (dH2O/PEG 1/1, Figure 2A). The lowest concentration (3×10−6 M) was inactive while the concentration of 3×10−5 M did not further inhibit E2-induced vasorelaxation (data not shown). Similarly, L-NAME at the concentration of 10−4 M, after 15 min of incubation, significantly inhibited E2-induced vasodilatation, when compared to the vehicle (dH2O), at the same extent of GA (Figure 2B). In presence of ibuprofen (10−5 M) E2-induced vasodilatation was also significantly inhibited (P<0.05, data not shown).
Figure 2.

(a) Inhibitory effect of geldanamycin (GA, 10−5 M) on E2-induced vasorelaxation. PE-precontracted rings were incubated for 15 min with GA. Vehicle was a solution of dH2O/PEG (1/1). n=6 for each group. ***P<0.001. (b) Inhibitory effect of L-NAME (10−4 M) on E2-induced vasorelaxation. PE-precontracted rings were incubated for 15 min with L-NAME. Vehicle was dH2O. n=6 to 8 for both group; ***P<0.001. (c) Incubation of the rings with TEA (10−2 M) for 60 min did not affect E2-induced vasorelaxation. Vehicle was dH2O. n=6 to 8 for each group.
In order to evaluate if calcium-activated potassium channels could be implicated in E2-induced vasorelaxation, rings were incubated with tetraethylammonium (TEA) at the concentration of 10−2 M for 60 min. This treatment did not affect E2-induced vasodilatation (Figure 2C). In these conditions TEA neither induced contraction nor enhanced significantly PE-induced contraction (data not shown).
Effect of oestrogens receptor pure antagonist ICI 182,780 on E2-induced vasodilatation in rat aorta
To demonstrate that the E2-induced vasorelaxation on aortic rings was mediated through the interaction of E2 with its receptor, ICI 182,780, a pure anti-oestrogen of E2 receptor was used. When aortic rings were incubated with ICI 182,780 at the concentration of 2×10−5 M for 15 min, inhibition of E2-induced vasodilatation occurred (Figure 3).
Figure 3.
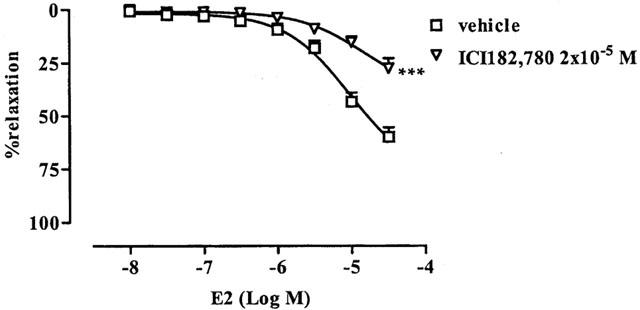
Inhibitory effect of pure anti-oestrogen IC I182,780 (2×10−5 M) on E2-induced vasorelaxation. PE-precontracted rings were incubated for 15 min with ICI 182,780. n=5 for both groups. Vehicle was PEG. ***P<0.001.
Effect of LY 294002 on E2-induced vasodilatation
Incubation of the rings with LY 294002 at concentration of 2.5×10−5 M for 1 and 3 h did not affect E2-induced vasorelaxation. Following 6 and 16 h of incubation LY 294002 significantly inhibited E2-induced vasodilatation (Figure 4). At 6 and 16 h LY 294002 reduced also Ach-induced vasorelaxation (data not shown).
Figure 4.

Time course of LY 294002 on E2-induced vasorelaxation. Rings were treated for 1, 3, 6 and 16 h with LY 294002 (2.5×10−5 M) or vehicle (DMSO) as described in conditioned medium. After incubation rings were washed repeatedly and then mounted in organ baths to perform the experiments. n=5 for each group; **P<0.01, ***P<0.001 vs vehicle group.
E2 induces prostacyclin (PGI2) release from rat aorta
PGI2 levels, measured as 6-keto-PGF1α, were significantly increased following 30 min of incubation with E2 (10−5 M, Figure 5). GA and L-NAME did not influence basal or E2-stimulated PGI2 levels produced by rat aortic rings. On the other hand, ibuprofen treatment completely blocked E2-induced PGI2 release (Figure 5). LY 294002 was not tested since DMSO at the lowest amount needed to dissolve the drug interfered with the assay.
Figure 5.

Levels of 6-keto-PGF1α release under basal and E2-stimulated conditions. Rings were treated with L-NAME (10−4 M for 15 min), GA (10−5 M for 15 min) or vehicles dH2O or dH2O/PEG 1/1, respectively. In a separate set of experiments Ibuprofen 10−5 M was dissolved in Krebs solution and used as positive control n=6 for each group.
Discussion
In this study we have examined the involvement of L-arginine NO pathway in the vasodilatory effect of 17-β-oestradiol on rat aortic rings. To perform this study we used water soluble E2 cyclodestrin-encapsulated, since 17-β-oestradiol is soluble only in organic solvents that usually interfere in tissue bath experiments. The first experiments we performed was the endothelium removal to confirm that E2-induced vasodilatation is endothelium-dependent. This finding is in agreement with another study where following endothelium removal, rat arteriole lost their ability to modify their diameter following E2 stimulation (Huang et al., 2000). To further gain insights into the mechanisms involved we used a pharmacological approach. For this purpose we used L-NAME that acts as false substrate for nitric oxide synthase blunting NO release in endothelium (Southan & Szabo, 1996), geldanamycin, a specific inhibitor of eNOS activation through its binding to hsp90 (Garcia-Cardena et al., 1998) and LY 294002, a specific inhibitor of phosphatidylinositol-3-OH kinase (PI(3)K), that regulates through akt, NO release in endothelial cells by eNOS phosphorylation (Fulton et al., 1999).
L-NAME strongly reduced E2-induced vasorelaxation suggesting that nitric oxide is the main vasorelaxing factor responsible for E2-induced vasodilatation in this condition. However, this inhibitor widely used to show NO involvement, being a false substrate, does not give information on the possible molecular mechanism(s) involved.
It is known that hsp90 coordinates the trafficking and the regulation of several signalling proteins, in particular it has been shown that hsp90 associates with eNOS enhancing the activation of this enzyme (Garcia-Cardena et al., 1998). Incubation of aortic rings with GA reduced ACh-induced vasorelaxation indicating the involvement of hsp90 in agonist-stimulated production of nitric oxide (Garcia-Cardena et al., 1998). In our experiments GA, at the same concentration and incubation time that inhibits ACh-induced vasorelaxation, significantly reduced E2-induced vasorelaxation. This effect was already maximal at the concentration of 10−5 M and did not further increase at the concentration of GA to 3×10−5 M. A lower concentration, such as 3×10−6 M, had no effect on E2-induced vasorelaxation. This phenomenon could be explained considering how GA acts on eNOS activation. GA specifically binds to hsp90, hindering the interaction between eNOS and hsp90 (Garcia-Cardena et al., 1998). It has been also shown that GA 10−5 M reduces ACh-induced vasorelaxation in rat aortic rings (Garcia-Cardena et al., 1998). Most likely, GA 10−5 M is the concentration that subtracts an amount of hsp90 molecules sufficient to interfere with the activation mechanism of the enzyme and this effect is not further increased with GA 3×10−5 M. For the opposite reason GA 3×10−6 M did not affect E2-induced vasodilatation e.g. there is still enough free hsp90 able to interact with eNOS. Thus, it is feasible that when E2 binds to its receptor liberation of two molecules of hsp90 occurs that in turn could activate eNOS.
Another mechanism through which eNOS is activated and that has been recently described is its activation through phosphorylation. Indeed, recently Fulton et al. (1999) showed that the serine/threonine protein kinase akt (protein kinase B) can directly phosphorylate eNOS on serine 1179 and activate the enzyme, leading to NO production. They have shown that incubating endothelial cells with LY 294002, a specific inhibitor of PI(3)K, there was a partial block of NO release. For this purpose LY 294002, a specific inhibitor of PI(3)K was used to further characterize the mechanism of the activation of eNOS as responsible of E2-induced vasorelaxation. In our experiments by incubating rat aortic rings with LY 294002 (2.5×10−5 M) there was a remarkable inhibition of E2-induced vasorelaxation. In particular we performed a time-course of LY 294002 effect on E2-induced vasorelaxation. Following 1 and 3 h of incubation LY 294002 did not affect E2-induced vasorelaxation, after 6 h of incubation instead LY 294002 strongly inhibited E2-induced vasorelaxation and this effect increased up to 16 h of incubation. Thus these results, taken all together, indicate that eNOS activation, through both mechanisms, is essential in E2-induced vasorelaxation.
It has been shown that E2 activates the arachidonic acid cascade and that E2 stimulates cyclo-oxygenase derived products in cultured rat aortic smooth muscle cells and in isolated rat aorta (Chang et al., 1981, 1983). Conversely, in male rat aortic endothelial cells (RAEC) oestrogen increases PGI2 release but not through COX activation (Myers et al., 1996) suggesting that E2 stimulation can increase endogenous PGI2 release in RAEC. Our data also confirm an involvement of PGI2 in E2-mediated vasorelaxation since ibuprofen reduced significantly, E2-induced vasorelaxation, as also shown by others (Chang et al., 1983). GA and L-NAME did not modify PGI2 release while ibuprofen completely abolished basal and E2-stimulated PGI2 release implying that the inhibitory effect is not mediated through COX inhibition. These results further stress the specificity of these compounds on two critical steps of the L-arginine-NO pathway.
Involvement of potassium channels in E2-induced vasodilatory effect can be ruled out since pre-treatment of the rings with the unselective calcium-activated potassium channels blocker TEA was ineffective on E2-induced vasorelaxation. TEA has been used at the concentration described to block Ach-induced vasorelaxation in rat isolated renal arteries (Jiang et al., 2000) and at this same concentration, in our experimental conditions, inhibited ACh-induced vasodilatory effect (data not shown). Concerning this particular effect it has been already shown that TEA dose-dependently inhibited E2-induced uterine vasodilation without affecting mean arterial pressure, heart rate and uterine blood flow in ovariectomized nonpregnant ewes (Rosenfeld et al., 2000) implying a different action of E2 on the vascular system compared to uterus.
We have further investigated if the effect of E2 is due to the interaction with its receptor(s). Our data show that in aortic rings treated with ICI 182,780, an oestrogen receptor pure antagonist, E2-induced vasorelaxation was inhibited. If the incubation was increased up to 1 h, when the compound acts by blocking the E2 genomic effect (Wakeling et al., 1991), ICI 182,780 was ineffective. It is known that the complex ER – ICI 182,780 has a shorter half-life when compared to ER – E2 complex due to an increased ER turn over-provoked by ICI 182,780 (Dauvais et al., 1992). This particular mechanism could explain why, when incubated for a longer time, ICI 182,780 did not affect E2-induced vasorelaxation. On the other hand, using shorter incubation times e.g. up to 15 min there was a significant inhibition of E2-induced vasorelaxation due to the fact that ICI 182,780, in this condition, exerts mainly its effect by preventing E2 – ER interaction.
In conclusion, we have shown that once E2 binds to its receptor, eNOS is stimulated to release NO. eNOS activation process involves its coupling to hsp90 and phosphorylation. In fact, by using LY 294002, a specific inhibitor of PI(3)K pathway, and GA, an hsp90 binding substance, a marked reduction of E2-induced vasodilatation occurred. In addition we have also shown that this effect is also linked to E2 – ER interaction in a non-genomic fashion. To our knowledge, this is the first demonstration that the L-arginine-NO pathway component of E2-induced vasorelaxation is linked to both eNOS phosphorylation through akt/pkb pathway and coupling to hsp90.
Abbreviations
- ACh
acetylcholine
- akt/pkb
serine/threonine protein kinase
- COX
cyclo-oxygenase
- DMEM
Dulbecco's modified Eagle medium
- DMSO
dimethylsulphoxide
- E2
17-β-oestradiol
- eNOS
endothelial nitric oxide synthase
- ER
oestrogen receptor
- GA
geldanamycin
- hsp90
heat shock protein 90
- L-NAME
Nω-nitro-L-arginine methyl ester
- NO
nitric oxide
- PBS
Phosphate Buffer Solution
- PE
phenylephrine
- PEG
polyethylene glycol 400
- PGI2
prostacyclin
- PI(3)K
phosphatidylinositol-3-OH kinase
- TEA
tetraethylammonium
References
- ADAMS M.R., KAPLAN J.F., MANUCK S.B., KORITNIK D.R., PARKS J.S., WOLFE M.S., CLARKSON T. Inhibition of coronary artery atherosclerosis by 17-beta estradiol in ovariectomized monkeys. Arteriosclerosis. 1990;10:1051–1057. doi: 10.1161/01.atv.10.6.1051. [DOI] [PubMed] [Google Scholar]
- BUCCI M., GRATTON J.P., RUDIC R.D., ACEVEDO L., ROVIEZZO F., CIRINO G., SESSA W.C. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 2000;6:1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- BUSH T.L., BARRETT-CONNOR E., COWAN L.D., CRIQUI M.H., WALLACE R.B., SUCHINDDRAN C.M., TYROLER H.A., RIFKIND B.M. Cardiovascular mortality and noncontraceptive use of oestrogen in women: results from the Lipid Research Clinics Program follow-up study. Circulation. 1987;75:1102–1109. doi: 10.1161/01.cir.75.6.1102. [DOI] [PubMed] [Google Scholar]
- CAULIN-GLASER T., GARCIA-CARDENA G., SARREL P., SESSA W.C., BENDER J.R. 17-β-estradiol regulation of human endothelial cell basal nitric oxide release, indipendent of cytosolic Ca2+ mobilization. Circ.Res. 1997;81:885–892. doi: 10.1161/01.res.81.5.885. [DOI] [PubMed] [Google Scholar]
- CHANG W.C., NAKAO J., MUROTA S., TAI H.H. Induction of fatty acid cyclooxygenase in rat aortic smooth muscle cells by estradiol. Prostaglandins Leukot. Med. 1983;10:33–37. doi: 10.1016/s0262-1746(83)80018-3. [DOI] [PubMed] [Google Scholar]
- CHANG W.C., NAKAO J., NEICHI T., ORIMO H., MUROTA S. Effects of estradiol on the metabolism of arachidonic acid by aortas and platelets in rats. Biochim. Biophys. Acta. 1981;664:291–297. [PubMed] [Google Scholar]
- COLLINS P., ROSANO G.M.C. , SARREL P.M., ULRICH L., ADAMOPOULOS S., BEALE C.M., MCNEILL J.G., POOLE-WILSON P.A. 17-β-estradiol attenuates acetylcholine-induced coronary arterial constriction in women but not men with coronary heart disease. Circulation. 1995;92:24–30. doi: 10.1161/01.cir.92.1.24. [DOI] [PubMed] [Google Scholar]
- DAUVAIS S., DANIELIAN P.S., WHITE R., PARKER M.G. Antiestrogen ICI 164,384 reduces cellular oestrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. USA. 1992;89:4037–4041. doi: 10.1073/pnas.89.9.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEFRANCO D.B. Role of molecular chaperones in subnuclear trafficking of glucocorticoid receptors. Kidney Int. 2000;57:1241–1249. doi: 10.1046/j.1523-1755.2000.00957.x. [DOI] [PubMed] [Google Scholar]
- DIMMELER S., FLEMING I., FISSLTHALER B., HERMANN C., BUSSE R., ZELHER A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorilation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- FALKENSTEIN E., TILLMANN H.C., CHRIST M., FEURING M., WEHLING M. Multiple actions of steroid hormones a focus on rapid non-genomic effects. Pharmacol. Rev. 2000;52:513–556. [PubMed] [Google Scholar]
- FULTON D., GRATTON J.P., MCCABE T.J., FONTANA J., FUJIO Y., WALSH K., FRANKE T.F., PAPAPETROPOULOS A., SESSA W.C. Regulation of endothelium derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALLO M.A., KAUFMAN D. Antagonistic and agonistic effects of tamoxifen significance in human cancer. Sem. Oncol. 1997;24:1.71–1.80. [PubMed] [Google Scholar]
- GARCIA-CARDENA G., FAN R., SHAH V., SORRENTINO R., CIRINO G., PAPAPETROPOULOS A., SESSA W.C. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- GARCIA-CARDENA G., OH P., LIU J., SCHNITZER J.E., SESSA W.C. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signalling. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARCIA-CARDENA G., MARTASEK P., SILER MASTERS B.S., SKIDD P.M., COUET J., LI S., LISANTI M.P., SESSA W.C. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. J. Biol. Chem. 1997;272:25437–25440. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- GREEN S., WALTER P., KUMAR V., KRUST A., BORNERT J.M., ARGOS P., CHAMBON P. Human estrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. 1986;320:134–139. doi: 10.1038/320134a0. [DOI] [PubMed] [Google Scholar]
- HUANG A., SUN D., KOLLER A., KALEY G. Gender difference in flow dependent dilation and regulation of shear stress: role of estrogen and nitric oxide. Am. J. Physiol. 1998;275:R1571–R1577. doi: 10.1152/ajpregu.1998.275.5.R1571. [DOI] [PubMed] [Google Scholar]
- HUANG A., SUN D., KOLLER A., KALEY G. 17-β-estradiol restores endothelial nitric oxide release to shear stress in arterioles of male hypertensive rats. Circulation. 2000;101:94–100. doi: 10.1161/01.cir.101.1.94. [DOI] [PubMed] [Google Scholar]
- JIANG F., GUANG, LI C., RAND M.J. Mechanism of nitric oxide-indipendent relaxations induced by carbachol and acetylcholine in rat isolated renal arteries. Br. J. Pharmacol. 2000;130:1191–1200. doi: 10.1038/sj.bjp.0703408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAUSER K., RUBANYI G.M. Potential cellular signalling mechanism mediating upregulation of endothelial nitric oxide production by oestrogen. J. Vasc. Res. 1997;34:229–236. doi: 10.1159/000159227. [DOI] [PubMed] [Google Scholar]
- KIM H.P., LEE J.Y., JEONG J.K., BAE S.W., LEE H.K., JO I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor α localized in caveolae. Biochem. Biophys. Res. Commun. 1999;263:257–262. doi: 10.1006/bbrc.1999.1348. [DOI] [PubMed] [Google Scholar]
- KUIPER G.G.J.M., GUSTAFSSON J.A. The novel oestrogen receptor-β-subtype: potential role in the cell and promoter specific actions of estrogens and anti-estrogens. FEBS Lett. 1997;410:87–90. doi: 10.1016/s0014-5793(97)00413-4. [DOI] [PubMed] [Google Scholar]
- KUMAR V., GREEN S., STACK G., BERRY M., JIN J.R., CHAMBON P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–951. doi: 10.1016/0092-8674(87)90581-2. [DOI] [PubMed] [Google Scholar]
- MORBIDELLI L., CHANG C.H., DOUGLAS J.G. , GRANGER H.J., LEDDA F., ZICHE M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am. J. Physiol. 1996;270:H411–H415. doi: 10.1152/ajpheart.1996.270.1.H411. [DOI] [PubMed] [Google Scholar]
- MYERS S.I., TURNAGE R.H., BARTULA L., KALLEY B., MENG Y. Prostagl. Leukot. Essent. Fatty Acid. 1996;54:403–409. doi: 10.1016/s0952-3278(96)90023-x. [DOI] [PubMed] [Google Scholar]
- PAPAPETROPOULOS A., DESAI K.M., RUDIC R.D., MAYER B., ZHANG R., RUIZ-TORRES M.P., GARCIA-CARDENA G., MADRI J.A., SESSA W.C. Nitric oxide synthase inhibitors attenuate transforming-growth factor beta-1 stimulated capillary organization in vitro. Am. J. Phatol. 1997a;150:1835–1844. [PMC free article] [PubMed] [Google Scholar]
- PAPAPETROPOULOS A., GARCIA-CARDENA G., MADRI J.A., SESSA W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest. 1997b;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAECH K., WEBB P., KUIPER G.G.J.M., NILSSON S., GUSTAFSSON J.A., KUSHNER P.J., SCANLAN ThS. Differential ligand activation of estrogen receptors ERα and ERβ at AP1-sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- PRATT W.B., TOFT D.O. Steroid receptor interactions with heat shock protein and immunophillin chaperones. Endocrinol. Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- ROSENFELD C.R., WHITE R.E., ROY T., COX B.E. Calcium activated potassium channels and nitric oxide coregulate estrogen-induced vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H319–H328. doi: 10.1152/ajpheart.2000.279.1.H319. [DOI] [PubMed] [Google Scholar]
- ROSSELLI M., IMTHURN B., KELLER P.J., JACSON E.K., DUBEY P.K. Circulating nitric oxide (nitrite/nitrate) levels in postmenopausal women substituted with 17-β-estradiol and norethisterone acetate: a two year follow-up study. Hypertension. 1995;25:848–853. doi: 10.1161/01.hyp.25.4.848. [DOI] [PubMed] [Google Scholar]
- RUDIC R.D., BUCCI M., FULTON D., SEGAL S.S., SESSA W.C. Temporal events underlyng arterial remodeling after chronic flow reduction in mice. Circ. Res. 2000;86:1160–1166. doi: 10.1161/01.res.86.11.1160. [DOI] [PubMed] [Google Scholar]
- RUSSELL K.S., HAYNES M.P., CAULIN-GLASER T., ROSNECK J., SESSA W.C., BENDER J.R. Estrogen stimulates heat shock protein 90 binding to endothelial nitric oxide synthase in human vascular endothelial cells. Effects on calcium sensitivity and NO release. J. Biol. Chem. 2000;275:5026–5030. doi: 10.1074/jbc.275.7.5026. [DOI] [PubMed] [Google Scholar]
- SOUTHAN G.J., SZABO C. Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem. Pharmacol. 1996;51:383–394. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- SUDHIR K., CHOU T.M., MULLEN W.L., HAUSMANN D., COLLINS P., YOCK P.G., CHATTERJEE K. Mechanism of estrogen-induced vasodilation: in vivo studies in canine coronary conductance and resistance arteries. J. Am. Coll. Cardiol. 1995;26:807–814. doi: 10.1016/0735-1097(95)00248-3. [DOI] [PubMed] [Google Scholar]
- SULLIVAN J.M., VANDER ZWAAG R., LEMP G.F., HUGHES J.P., MADDOCK V., KROETZ F.W., RAMANATHAN K.B., MIRVIS D.M. Postmenopausal estrogen use and coronary atherosclerosis. Ann. Intern. Med. 1988;108:358–363. doi: 10.7326/0003-4819-108-3-358. [DOI] [PubMed] [Google Scholar]
- WAKELING A.E., DUKES M., BOWLER J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- ZENG G., QUON M.J. Insulin-stimulated production of nitric oxide is inhibited by Wortmannin. Direct measurement in vascular endothelial cells. J. Clin. Invest. 1996;98:894–898. doi: 10.1172/JCI118871. [DOI] [PMC free article] [PubMed] [Google Scholar]