

Abstract
We investigated the inhibitory effects of a non-acylguanidine Na+-H+ exchange (NHE) inhibitor, T-162559 ((5E,7S)-[7-(5-fluoro-2-methylphenyl)-4-methyl-7,8-dihydro-5(6H)-quinolinylideneamino] guanidine dimethanesulphonate), on NHE-1, and its cardioprotective effect against ischaemia and reperfusion injury in rats and rabbits.
T-162559 inhibited human platelet NHE-1 in a concentration-dependent manner, with an IC50 value of 13±3 nmol l−1, making it 16 and three times more potent than cariporide IC50: 209±75 nmol l−1, P<0.01) and eniporide (IC50: 40±11 nmol l−1, P=0.066), respectively. T-162559 also inhibited rat NHE-1 with an IC50 value of 14±2 nmol l−1, which was five and three times lower than that of cariporide (IC50: 75±7 nmol l−1, P<0.01) and eniporide (IC50: 44±2 nmol l−1, P<0.01), respectively.
T-162559 inhibited, in a concentration-dependent manner, the reduction in cardiac contractility, progression of cardiac contracture, and increase in lactate dehydrogenase release after global ischaemia and reperfusion in perfused rat hearts. The inhibitory effects of T-162559 were observed at a lower concentration range (10 – 100 nmol l−1) than with cariporide and eniporide. T-162559 did not alter basal cardiac contractility or coronary flow after reperfusion, suggesting that it exerts direct cardioprotective effects on the heart.
Intravenous administration of T-162559 (0.03 and 0.1 mg kg−1) significantly inhibited the progression of myocardial infarction induced by left coronary artery occlusion and reperfusion in rabbits; the infarct size normalized by area at risk was 74±6% in the vehicle group, and 47±5% and 51±7% in the T-162559-0.03 mg kg−1 and T-162559-0.1 mg kg−1 groups (both P<0.05), respectively.
These results indicate that the new structural NHE-1 inhibitor T-162559 is more potent than cariporide and eniporide and possesses a cardioprotective effect against ischaemia and reperfusion injury in rat and rabbit models.
Keywords: Na+-H+ exchanger, cariporide, eniporide, ischaemia-reperfusion, cardioprotection, myocardial infarction
Introduction
Intracellular pH is regulated by several ion transporters, including the Na-H exchanger (NHE), Na-HCO3 co-transporter (NBC), Cl-HCO3 exchanger, and Cl-OH exchanger (Reithmeier, 1994; Leem et al., 1999). One of these, NHE is a dominant ion transporter involved in the removal of protons from the cytoplasm causing Na accumulation during myocardial ischaemia, although it is quiescent around neutral pH values (Karmazyn et al., 1999; Avkiran, 2001).
In 1988, amiloride, a NHE-1 and NHE-2 inhibitor, was reported to exert protective effect against ischaemia-reperfusion injury in the rat heart, indicating the possible involvement of NHE activation in the progression of cardiac injury (Karmazyn, 1988). Although the existence of five NHE isoforms have been reported in the plasma membrane, NHE-1 has been found to be ubiquitously distributed in most tissues, and to be the primary subtype in mammalian cardiac cells (Wakabayashi et al., 1997; Klanke et al., 1995). Accordingly, inhibition of NHE-1 was speculated to be the main target of amiloride in exerting its cardioprotective effect after ischaemia and reperfusion (Satoh et al., 1994; 1995; Karmazyn et al., 1999). However, it has been well documented that amiloride possesses various pharmacological effects on ion channels, receptors and ion transporters (Kleyman & Cragoe, 1988), and thus it is difficult to exclude the possibility that the cardioprotective effect of the drug is exerted via some other pathway.
A specific NHE-1 inhibitor, cariporide, has recently been developed (Scholz et al., 1995). It protects the heart against ischaemia and reperfusion injury, limiting myocardial infarct size and suppressing ventricular fibrillation (Scholz et al., 1995; Aye et al., 1997; Miura et al., 1997). In addition, it has been reported that bolus intravenous administration of cariporide reduced the incidence of cardiac death and recurrent myocardial infraction in coronary artery bypass graft patients, based on the results of the GUARDIAN trial (Théroux et al., 2000). Since no cardioprotective agent is as yet available for clinical use, cariporide is expected to offer promise as a potentially effective new drug for the treatment of ischaemic heart disease. However, since a high dose of cariporide, 120 mg, t.i.d. is required to produce even a minimal effect in patients (Théroux et al., 2000), a new NHE-1 inhibitor having more potent inhibitory effects on NHE-1 than cariporide to provide additional benefit in patients with acute coronary syndromes is desired. Several NHE-1 inhibitors, such as EMD 85131 (hydrochloride salt of eniporide, Gumina et al., 1998), MS-31-038 (Banno et al., 1999), SM-20550 (Ito et al., 1999), BIIB513 (Gumina et al., 1999), FR183998 (Ohara et al., 1999) and TY-12533 (Aihara et al., 2000) have been reported to inhibit NHE-1 and to exert anti-ischaemic effect in animal models. However, these compounds are known to have the same basic structure, acylguanidine, which acts as a competitor of extracellular Na+. Only one imidazolylpiperadine NHE-1 inhibitor has been reported, but it remains unclear whether this drug exerts stronger cardioprotective effect than acylguanidine derivatives (Lorrain et al., 2000).
In this study, we evaluated the inhibitory effects of an aminoguanidine derivative, T-162559 (Figure 1), on NHE-1, and compared its cardioprotective effect with that of the acylguanidine NHE-1 inhibitors, cariporide and eniporide.
Figure 1.
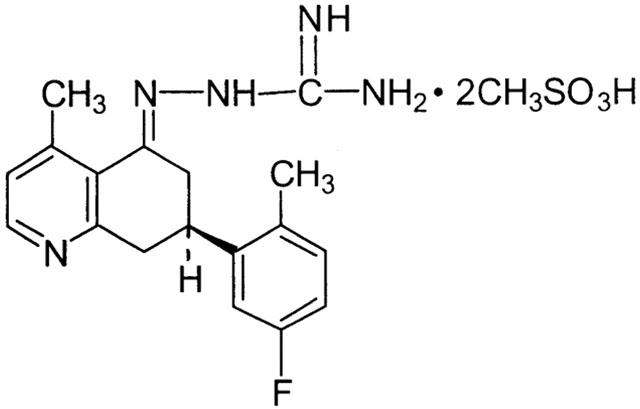
Structure of the aminoguanidine NHE-1 inhibitor T-162559.
Methods
Animal care
The following study was performed according to the recommendations of the declaration of Helsinki and internationally accepted principles for the care and use of experimental animals.
NHE-1 in human and animal platelets
Male Wistar rats (21 – 23 weeks old, CLEA Japan Inc., Tokyo) were anaesthetized with sodium pentobarbitone (50 mg kg−1 i.p.), and blood samples (8.5 ml) were withdrawn from the abdominal aorta into syringes containing 1.5 ml of 3.8% sodium citrate (n=3 in each group). Blood samples (9/1 blood/citrate, vol/vol) were also obtained from healthy adult men (n=3, mean age: 37 years). Each sample was centrifuged at 3000 r.p.m. for 5 s, and platelet-rich plasma (PRP) was obtained. The remainder of the blood sample was then centrifuged at 3000 r.p.m. for 5 min to obtain platelet-poor plasma (PPP). Platelets were counted in an automatic blood cell counter (Sysmex K4500, Toa-iyoudenshi Co., Tokyo, Japan). The human and rat platelet counts in the PRP samples were adjusted to 4×105 cells μl−1 and 1×105 cells μl−1, respectively. Platelet NHE-1 activity was measured according to a method previously described, with minor modification (Rosskopf et al., 1991). Briefly, increases in light transmission associated with cell swelling were measured with an aggregometer (Hematracer 801, Niko Bioscience, Tokyo, Japan). PRP (200 μl) in a cuvette was stirred at 1000 r.p.m. and prewarmed for 5 min at 37°C. An increase in light transmission of PRP at 550 nm induced by platelet swelling was observed after application of Na propionate solution (600 μl, in mmol l−1: Na propionate 135, HEPES 20, CaCl2 1, MgCl2 1, glucose 10, pH 6.7). Application of Na propionate (pH 6.7) produces an acidic intracellular pH at which platelet NHE is activated, and the increase in Na+ influx associated with excretion of cytosolic H+ via NHE results in cellular swelling as a result of water accumulation in the cytoplasm (Rosskopf et al., 1991). Light transmission through PRP is increased since the density of the cellular component decreases with swelling (Rosskopf et al., 1991). We used this simple assay system to evaluate the effects of drugs on NHE-1, since NHE-1 is considered to be the dominant subtype of NHE in platelets (see Discussion).
NHE-1 inhibitors were added to the cuvette 3 min before the addition of Na propionate solution. PPP was used to correct for light transmission through the non-platelet portion of PRP. The maximum velocity of the increase in light transmission was used to evaluate platelet NHE activity. The half-maximal inhibitory concentration (IC50) value of the NHE inhibitor was obtained from the linear part of the relationship between the log concentration and NHE activity using linear regression analysis. Three to six serial concentrations at within the range in which the inhibitor induces approximately 27 – 75% reduction in enzyme activity were chosen for the analysis.
Protective effect against cardiac injury induced by global ischaemia and reperfusion in perfused rat hearts
Male Wistar rats (10 – 11 weeks old) were anaesthetizedwith sodium pentobarbitone (50 mg kg−1 i.p.. Heparin (1000 u kg−1, Shimizu Pharmaceutical Co. Ltd., Shimizu, Japan) was injected into the left femoral vein, and 1 min later, the heart was quickly removed (n=4 – 6 in each group). After dissecting out the adipose and connective tissue at 4°C, the heart was transfected to a heated organ chamber (37°C) and perfused at a constant perfusion pressure of 75 mmHg with oxygenated (95% O2 and 5% CO2) modified Krebs solution (in mmol l−1: NaCl 113.1, KCl 4.6, CaCl2 1.2, NaH2PO4 3.5, MgCl2 1.2, NaHCO3 21.9, glucose 10; pH 7.4) via a cannula inserted into the root of the aorta. The perfusion pressure was monitored with a pressure transducer (DX-312, Viggo-Spectramed, U.S.A.) connected to the pressure amplifier (AP-601G, Nihon Kohden Co. Ltd., Tokyo, Japan), and a balloon was placed inside the left ventricle to measure the left ventricular pressure. Heart rate (HR) was counted with a tachometer (AT-601G, Nihon Kohden Co. Ltd.). Coronary flow (CF) was measured using an electromagnetic flow probe (MF-27, Nihon Kohden Co. Ltd.) mounted around the perfusate tube. After a 30-min equilibration period, and 10 min before the induction of global ischaemia, infusion of vehicle or drug was started, and the infusion was continued throughout the experimental period, except for a 25-min global ischaemic period. Left ventricular developed pressure (LVDP), left ventricular end-diastolic pressure (LVEDP), HR and CF were measured before and 10 min after infusion of the drug, 10, 20 and 25 min after the induction of global ischaemia, and 10, 20, 30 and 40 min after reperfusion. The effluent perfusate was collected before the induction of global ischaemia and 40 min after reperfusion to measure the lactate dehydrogenase (LDH) activity. LDH activity in the perfusate was measured with an assay kit (LDH-UV test Wako, Wako Pure Chemical Ind. Ltd., Osaka, Japan), and normalized by the CF and wet weight of the heart. LDH release induced by global ischaemia and reperfusion in each heart was determined as the difference between the LDH activities before the drug infusion and 40 min after reperfusion.
Inhibitory effect on the extension of myocardial infarction in rabbits
Male New Zealand white rabbits (n=6 – 7 in each group, 2.5 – 4 kg, Kitayama Labs. Co. Ltd., Ina, Japan) were anaesthetized with sodium pentobarbitone (30 mg kg−1 i.v.), and a polyethylene tube was inserted into an ear vein to inject vehicle or T-162559. The lead II electrocardiogram was monitored to detect ventricular fibrillation (VF) and ventricular tachycardia (VT). The chest was opened through a left intercosal incision to expose the heart, under artificial ventilation. The pericardium was incised, and a 6-0 silk thread was placed around the first branch of the left circumflex coronary artery. After a stabilization period (at least 15 min), the coronary artery was occluded for 30 min and the snare was released. When the VF occurred during the coronary occlusion, defibrillation was attempted by gentle attaching on the surface of the heart. The chest was closed and the rabbit was left in the cage overnight with free access to water and food. The animal was anaesthetized again 24 h after the reperfusion, and the heart was quickly excised. The coronary artery was ligated again at the same site at which it had been occluded. The area at risk (AAR) was determined by injection of 1% Evans blue, and the left ventricle (LV) was sectioned into 5 – 6 slices and incubated in 1% 2,3,5,-triphenyltetrazolium chloride at 25°C for 20 min to stain intact tissue. Infarct size (IS) was expressed as a percentage of the AAR based on the wet weight. T-162559 (0.5 ml kg−1) was administered 5 min before the coronary occlusion.
Chemicals
T-162559, ((5E,7S)-[7-(5-fluoro-2-methylphenyl)-4-methyl-7,8-dihydro-5(6H)-quinolinylideneamino] guanidine dimethanesulphonate), cariporide and eniporide were synthesized by Takeda Chemical Industries, Ltd. T-162559 , cariporide and eniporide were dissolved in pure water for the in vitro experiments, and T-162559 was dissolved in saline for the in vivo experiments prior to the beginning of the study.
Statistics
All data were expressed as means±s.e.mean. If ANOVA gave a significant F value, the following analysis was performed. Dunnett's test at each time point followed by Bonferroni correction for four time points were used to evaluate the statistical significance of changes following reperfusion in the rat model. Dunnett's test was used for comparison of the IC50 values in the platelets, analysis of LDH release in the rat hearts, and the infarct size in rabbits. A P value of <0.05 was considered to denote statistical significance.
Results
Inhibitory effects of NHE-1 inhibitors on NHE-1 in human and rat platelets
The velocity of increase in light transmission through PRP induced by application of Na propionate to human platelets was inhibited by pretreatment with T-162559 (1 – 300 nmol l−1) in a concentration-dependent manner. The acylguanidine-derived NHE-1 inhibitors cariporide (10 – 3000 nmol l−1) and eniporide (3 – 1000 nmol l−1) also inhibited the velocity of light transmission through human PRP in a concentration-dependent manner (Figure 2, upper panel). The IC50 values of T-162559, cariporide and eniporide for the enzyme in human platelets were 13±3, 209±75 and 40±11 nmol l−1, respectively. The IC50 value of T-162559 for human NHE-1 was 16 and three times smaller than that of cariporide (P<0.01) and eniporide (P=0.066), respectively.
Figure 2.
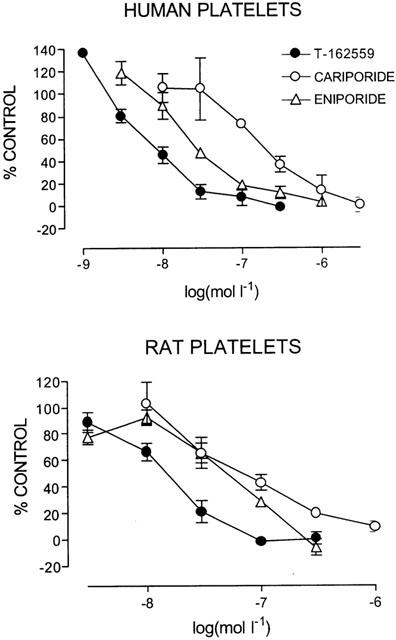
Concentration-response curves of T-162559 (closed filled circles), cariporide (open circles) and eniporide (open triangles) on the NHE-1 activity of human (upper panel) and rat (lower panel) platelets. The vertical line shows the maximum velocity of increase in light transmission induced by application of Na propionate in the presence of various concentrations of the drugs as a percentage of the value obtained in the vehicle cuvette. Values are expressed as means±s.e.mean. n=3.
Although the index of NHE activity in human platelets seemed to be slightly greater than 100% of the control in the presence of low concentrations of the inhibitors, this was not significant. In addition, in another set of experiments, neither T-162559, nor cariporide or eniporide increased the NHE activity at any concentration, and similar IC50 values to those obtained in this study were obtained.
T-162559 (3 – 300 nmol l−1) inhibited rat platelet NHE-1 in a concentration range similar to that observed for the enzyme in human platelets (Figure 2, lower panel). The IC50 value of T-162559 for NHE-1 in rat platelets was 14±2 nmol l−1. Cariporide (10 – 1000 nmol l−1) and eniporide (3 – 300 nmol l−1) also inhibited rat platelet NHE-1 with IC50 values of 75±7 and 44±2 nmol l−1, respectively, in a concentration-dependent manner (Figure 2, lower panel). Accordingly, T-162559 exerted five and three times more potent inhibitory effects on NHE-1 than cariporide (P<0.01) and eniporide (P<0.01), respectively.
Cardioprotective effect of T-162559, cariporide and eniporide against injury induced by global ischaemia and reperfusion in the perfused rat heart
In total, four hearts were excluded from the analysis, since serial arrhythmias disturbed the measurements of cardiac contraction: one heart from the vehicle group, one from the T-162559 at 30 nmol l−1 group, one from the cariporide at 1000 nmol l−1 group and one from the eniporide at 300 nmol l−1 group.
The basal LVDP value with vehicle treatment before the induction of global ischaemia was 69±3 mmHg in the rat hearts (n=15). Global ischaemia for a 25-min duration abolished the LVDP within 10 min (approximately 2 – 4 min after the start of global ischaemia) (Figures 3, 4 and 5, upper panels). Although the reduced cardiac contractility gradually recovered following reperfusion, complete recovery to the baseline value was not reached even 40 min after reperfusion (Figures 3, 4 and 5, upper panels). An increase in LVEDP, which is an indicator of cardiac contracture, was observed approximately 10 min after stopping coronary perfusion, and the pressure continued to increase during global ischaemia in the vehicle groups (Figures 3, 4 and 5, middle panels). Reperfusion of the heart was associated with further elevation of the LVEDP, suggesting the existence of reperfusion injury (Figures 3, 4 and 5, middle panels), and the LVEDP still remained high even 40 min after reperfusion (Figures 3, 4 and 5, middle panels). HR was abolished by global ischaemia, but it rapidly recovered to 85 to 95% of the pre-ischaemic value by the end of the experimental period. Reactive hyperemia was observed after the start of coronary reperfusion, and it lasted for 40 min after the reperfusion in the vehicle groups; the coronary perfusion flow was 123±5% of the baseline value (n=15, P<0.05 from baseline, paired t-test). The LDH released in the coronary perfusate was clearly increased 40 min after reperfusion as compared with the baseline level (under 0.02 IU/min/g), indicating that cardiac myocytes had been injured by the global ischaemia and reperfusion (Figures 3, 4 and 5, lower panels).
Figure 3.
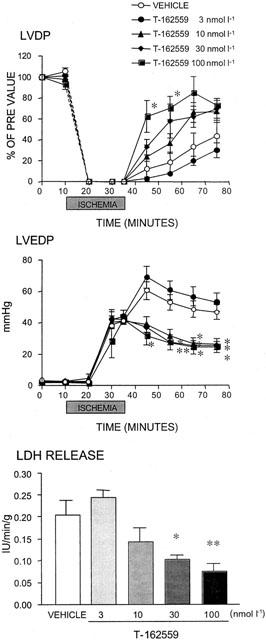
Effect of T-162559 on changes in the left ventricular developed pressure (LVDP), left ventricular end-diastolic pressure (LVEDP) and lactate dehydrogenase (LDH) release induced by global ischaemia and reperfusion in perfused rat hearts. The time-course of changes in the LVDP and LVEDP before, during and after global ischaemia is shown in the upper panels, and LDH release from the injured heart measured 40 min after reperfusion is shown in the bottom panel. The dotted line between 0 and 10 min in the top panel is an interpolation and does not accurately reflect the time course of changes in LVDP. Values are expressed as means±s.e.mean. n=5. *P⩽0.05 and **P⩽0.01 versus the vehicle group (Dunnett's test with Bonferroni correction for four time points).
Figure 4.
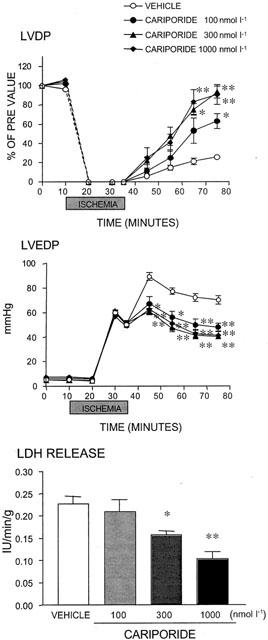
Effect of cariporide on changes in the left ventricular developed pressure (LVDP), left ventricular end-diastolic pressure (LVEDP) and lactate dehydrogenase (LDH) release induced by global ischaemia and reperfusion in perfused rat hearts. The time-course of changes in the LVDP and LVEDP before, during and after global ischaemia are shown in the upper panels, and LDH release from the injured heart measured 40 min after reperfusion is shown in the bottom panel. The dotted line between 0 and 10 min in the top panel is an interpolation and does not accurately reflect the time course of changes in LVDP. Values are expressed as means±s.e.mean. n=5. *P⩽0.05 and **P⩽0.01 versus the vehicle group (Dunnett's test with Bonferroni correction for four time points).
Figure 5.
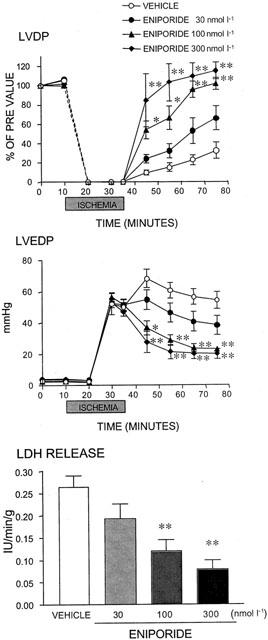
Effects of eniporide on changes in the left ventricular developed pressure (LVDP), left ventricular end-diastolic pressure (LVEDP) and lactate dehydrogenase (LDH) release induced by global ischaemia and reperfusion in perfused rat hearts. The time-course of changes in LVDP and LVEDP before, during and after global ischaemia are shown in the upper panels, and LDH release from the injured heart measured 40 min after reperfusion is shown in the bottom panel. The dotted line between 0 and 10 min in the top panel is an interpolation and does not accurately reflect the time course of changes in LVDP. Values are expressed as means±s.e.mean. n=4 – 5. *P⩽0.05 and **P⩽0.01 versus the vehicle group (Dunnett's test with Bonferroni correction for four time points).
As shown in Figure 3, T-162559 (10 – 100 nmol l−1) mitigated the reduction in LVDP during reperfusion in a concentration-dependent manner, without changing the basal contractility (Figure 3, upper panel). In particular, it accelerated the recovery of LVDP after reperfusion at higher concentrations (30 and 100 nmol l−1) (Figure 3, upper panel). Although T-162559 did not attenuate cardiac contracture during global ischaemia even at the highest concentration, it completely inhibited further progression of cardiac contracture after reperfusion at concentrations of 10 and over nmol l−1 (Figure 3, middle panel). T-162559 also reduced the release of LDH from injured cardiac cells in a concentration-dependent manner (Figure 3, lower panel).
Cariporide and eniporide exhibited similar cardioprotective effects in higher concentration ranges. Namely, cariporide (100 – 1000 nmol l−1) and eniporide (30 – 300 nmol l−1) improved the contractile function of the heart after reperfusion (Figures 4 and 5, upper panels), diminished cardiac contracture (Figures 4 and 5, middle panels), and reduced LDH release from injured cardiomyocytes (Figures 4 and 5, lower panels).
None of the drugs, T-162559, cariporide or eniporide, modulated the time-course of the changes in HR and coronary flow which increased after the start of reperfusion, at any time during the experimental period (data not shown).
The degree of improvement in contractility following the administration of these NHE-1 inhibitors in the rat model of ischaemia and reperfusion suggested that T-162559 at 10 nmol l−1, cariporide at 100 nmol l−1 and eniporide at 30 nmol l−1 exerted similar effects; the recovery rate of LVDP 40 min after reperfusion, were 42, 50 and 49%, respectively (Figures 3, 4 and 5, upper panels). The NHE-1 inhibitors also showed similar limiting effects on cardiac contracture at each of the above concentrations, with the rates of reduction in LVEDP at 40 min of 46, 36 and 33%, respectively (Figures 3, 4 and 5, middle panels). Furthermore, the minimum effective concentrations against increase in LDH were 30, 300 and 100 nmol l−1 for T-162559, cariporide and eniporide, respectively (Figures 3, 4 and 5, lower panels). Thus, the inhibitory effect of T-162559 on cardiac dysfunction and myocyte injury was approximately 10 and three times more potent than that of cariporide and eniporide, respectively, which was consistent with the drugs' inhibitory effects on rat platelet NHE-1 observed previously.
Infarct-size-limiting effect of T-162559 in the rabbit model
Myocardial infarction was established for 24 h, followed by reperfusion of the occluded coronary artery (Figure 6). VF or VT developed in 3/7, 2/7, 2/6 animals in the vehicle, T-162559- 0.03 mg kg−1 group and T-162559- 0.1 mg kg−1 group, respectively. One rabbit in the vehicle group and one in the T-162559 -0.03 mg kg−1 group died within 24 h after the start of coronary reperfusion. In the vehicle treatment group, 74±6% (n=6) of the AAR became infarcted. Intravenous administration of T-162559 (0.03 and 0.1 mg kg−1) 5 min before the coronary occlusion significantly inhibited the extension of the myocardial infarction by 24 h (Figure 6). The IS/AAR in the T-162559 treatment groups was 47±5% (n=6) and 51±7% (n=6) in the 0.03 and 0.1 mg kg−1, respectively. The AAR/LV values in the vehicle, and T-162559 – 0.03 and T-162559 – 0.1 mg kg−1 groups were not significantly different: 37±6% (n=6), 35±5% (n=6) and 45±7% (n=6), respectively.
Figure 6.
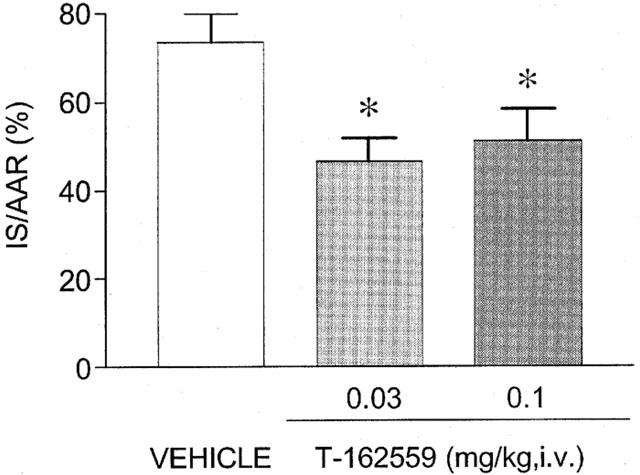
Inhibitory effects of intravenous administration of T-162559 5 min before coronary reperfusion on the extension of myocardial infarction in rabbits. Infarct size (IS) is shown as a percentage of AAR weight, and AAR was normalized by LV weight. Values are expressed as means±s.e.mean. n=6. *P⩽0.05 versus the vehicle group (Dunnett's test).
Discussion
This is the first report of an aminoguanidine derivative NHE-1 inhibitor exerting cardioprotective effects against ischaemia and reperfusion in the rat heart. The results obtained in this study show that the inhibitory effect of T-162559 on human and rat NHE-1 is more potent than that of the acylguanidine derivatives, cariporide and eniporide. T-162559 was also found to have a more potent cardioprotective effect in perfused rat hearts than either of these compounds. T-162559 also showed an inhibitory effect on the extension of myocardial infarction in rabbits.
Many ion transporters have been reported to regulate platelet volume (Sarkadi & Parker, 1991). Livne et al. (1987a, 1987b) found that human platelets swell as they recover from cytoplasmic pH decrease caused by intracellular acid loading. They also clarified that the increase in platelet volume was Na+-dependent, and was abolished by replacement of extracellular Na+ by the impermeable ions K+, Rb+ and Cs+. They stated that amiloride inhibited the increase in platelet volume in a competitive manner with Na+. They, therefore, concluded that activation of NHE contributed to the platelet swelling associated with reversed transport of cytoplasmic H+ and extracellular Na+ under acidic intracellular conditions. A recent molecular analysis showed that the predominant NHE subtype in human platelets is NHE-1 (Aharonovitz & Grano, 1996), and that the selective NHE-1 inhibitor cariporide inhibits human platelet NHE activity (Weichert et al., 1997, Figure 2). These observations strongly suggested that human platelet swelling in the presence of intracellular acidosis is mediated by the activation of NHE-1. Judging from the results obtained in the present study (Figure 2), and the evidence that T-162559 has an over 100 fold lower inhibitory effect on human NHE-2 and NHE-3 as compared with that on human NHE-1 (Kawamoto et al., 2001), T-162559 is considered to be a potent and selective inhibitor of NHE-1. Although the amino acid sequence of rat platelet NHE has not been determined, it is thought to be the same as that of NHE-1, because human and rat NHE-1 show 97% homology (Noël & Pouysségur, 1995), and selective NHE-1 inhibitors, including cariporide, eniporide and T-162559, inhibit rat platelet NHE activity in concentration ranges in which they inhibit human NHE-1 (Sholtz et al., 1995; Kawamoto et al., 2001; Figure 2).
In this study, we demonstrated that T-162559 possesses a more potent inhibitory effect on NHE-1 than cariporide and eniporide in both human and rat platelets (Figure 2). Since the aromatic portion of the NHE inhibitor is considered to be important for the association with the NHE-1 protein (Benos, 1982; L'allemain et al., 1984), dihydroquinoline, which is the aromatic moiety of T-162559 has been speculated to be one of the structural candidates possessing high affinity for the NHE-1 protein. In addition, the aminoguanidine portion of T-162559 may contribute to making this compound a more potent NHE-1 inhibitor in rat and human platelets than cariporide and eniporide, for the following reason. Guanidinium ion and the protonated guanidium motif of acylguanidine derivatives have been reported to be Na+ competitors (L'allemain et al., 1984; Frelin et al., 1986; Counillon et al., 1993), and preliminary evaluation has shown that the pKa values for the acylguanidine part of cariporide and eniporide are 6.2 and 6.0, respectively (Aihara et al., 2000 and unpublished observation). By contrast, we obtained a pKa value for the aminoguanidine part of T-162559 of 8.4 (unpublished observation). This result indicates that the aminoguanidine portion of T-162559 easily associates with H+, and a sufficient number of the molecules act as an inhibitor even in the presence of a low concentration of H+, at pH 8.4. In addition, almost all of the molecules become protonated and capable of inhibiting NHE-1 at high H+ concentrations, ex. pH 6.7. In contrast, cariporide and eniporide require higher concentrations of H+ for sufficient activation to inhibit NHE-1 because of their smaller pKa values (6.2 and 6.0), and are less protonated and less active at the lower concentrations of H+, such as at pH 6.7 (Aihara et al., 2000). Since the extracellular pH was 6.7 when NHE-1 activity was measured in human and rat platelets in this study, almost all of the T-162559 molecules were able to inhibit platelet NHE-1. On the other hand, a lower percentage of acylguanidine molecules was probably in the active form capable of inhibiting NHE-1 in the mildly acidic extracellular solution. Thus, T-162559 may display a more potent inhibitory effect on NHE-1 than cariporide and eniporide, because large numbers of its molecules are in the active form at neutral pH in addition to higher affinity of the drug for the NHE-1 protein.
T-162559 inhibited global ischaemia and reperfusion-induced injury in isolated rat hearts in a concentration range similar to that in which it inhibited NHE-1 in rat platelets (Figures 2 and 3). In addition, the rank order of T-162559, cariporide and eniporide in terms of their cardioprotective efficacy against ischaemic injury in rats (Figures 3, 4 and 5) was the same as the rank order for their inhibitory effect on rat NHE-1 (Figure 2). Therefore, it was concluded that T-162559 protects the rat heart from ischaemia and reperfusion injury by inhibiting NHE-1, the same as cariporide and eniporide. Since the protection by amiloride was associated with diminished extracellular Na and Ca concentrations, it was hypothesized that NHE and Na-Ca exchange (NCX) are coupled, and that Ca overload through the reverse mode of NCX during ischaemia and the early phase of reperfusion contributed to progression of the cardiac injury (Tani & Neely, 1989; Strömer et al., 2000). Evidence has been accumulated that indicates that NBC as well as NHE is activated during ischaemia and reperfusion, and that it plays a significant role in elevating the intracellular Na concentration with concomitant increase in Ca influx through NCX (Piper et al., 1996). Thus, activation of NBC is also involved in the progression of cardiac injury (Pierce & Czubryt, 1995; Piper et al., 1996). We previously reported observing that T-162559 does not inhibit cardiac NBC and NCX-1 overexpressed in CHO cells, which were dominant subtypes of NBC and NCX, respectively, in the heart (Kawamoto et al., 2001). Therefore, it is suggested that T-162559 ameliorated cardiac injury by inhibiting NHE-1.
Although reductions in oxygen demand and increases in oxygen supply to the heart are helpful in salvaging damaged cardiac myocytes, these were not observed during the treatment with NHE-1 inhibitors. Indeed, T-162559, cariporide and eniporide did not inhibit cardiac contractility or increase coronary perfusion flow (Figures 3, 4 and 5). This is not surprising since NHE-1 is quiescent under physiological conditions, in which the neutral intracellular pH markedly reduces Na+ influx mediated by it (Takaichi et al., 1993). Thus, it is possible that NHE-1 inhibitors attenuate cardiac injury by direct protection of myocytes from factors disturbing ionic homeostasis.
During global ischaemia, none of the NHE inhibitors modulated the onset and the extent of contracture in the heart (Figures 3, 4 and 5, middle panels), which is consistent with previous reports (Shipolini et al., 1997; Strömer et al., 2000). Since it has been reported that Ca overload developed under ischaemic conditions, and NHE-1 inhibition prevents its occurrence in the heart (Shipolini et al., 1997; Strömer et al., 2000), Ca overload induced by the activation of NHE-1 is not considered to be related to ischaemic contracture in the rat heart.
In the T-162559 group, the LVDP after reperfusion did not completely recover to the baseline value even at 40 min after reperfusion and at the highest concentration (Figure 3, top panel), although the other NHE inhibitors completely reversed the reduction of LVDP (Figures 4 and 5, top panels). There are two possible explanations for this phenomenon: One is that T-162559 tightly binds to NHE-1 and delays the pH recovery from the acidic side after reperfusion, which results in attenuation of cardiac contractility, since acidic pH reduces the Ca2+ sensitivity of the rat cardiac myofilaments (Ding et al., 1996). This explanation may be valid since the inhibitory effect of T-162559 was maintained even after reperfusion when the extracellular pH of cardiac myocytes quickly returned to pH 7.4, while that of cariporide and eniporide was largely lost (see previous discussion). However, the relationship between NHE-1 inhibition by T-162559 and the intracellular changes in H+ during ischaemia and reperfusion remains unclear. Another possibility is that higher concentrations of T-162559 possess a negative inotropic effect. However, this is doubtful, since the drug did not inhibit basal cardiac contractility in the rat heart (Figure 3, top panel). In addition, T-162559 is a selective NHE-1 inhibitor; it did not modulate the activities of NBC, NCX, NHE2, NHE3, L-type Ca channel, voltage-dependent Na channel, voltage-, Ca- and ATP-dependent K channels, adrenergic α1, α2, β1 and β2 receptors, adenosine A1 and A2 receptors, endothelin ETA receptor and angiotensin AT1 receptor even at the highest concentrations used in the study (Kawamoto et al., 2000 and unpublished observation). In any case, the extent of recovery in LVDP induced by T-162559 and the other NHE-1 inhibitors was not related to their cardioprotective effect, since all of the drugs induced similar reduction in LDH release at the highest concentration (Figures 3, 4 and 5, bottom panels). This was confirmed in an in vivo rat model of coronary occlusion and reperfusion; the extent of the maximal limitation of myocardial infarction induced by T-162559, cariporide and eniporide was similar (Igata et al., 2001).
T-162559 limited infarct size in both rabbit and rat models of myocardial infarction (Figure 6, Igata et al., 2001). These findings clearly indicate that the cardioprotective effect of T-162559 is not exerted in the rat alone. The amino acid sequence of NHE-1 has been demonstrated to be highly homologous in humans, rats and rabbits (>95%, Noël & Pouysségur, 1995), and for this reason T-162559 is thought to be capable of inhibiting rabbit NHE-1 and conferring on the rabbit heart tolerance against ischaemia and reperfusion. Cariporide and other NHE-1 inhibitors exert a similar cardioprotective effect in rabbits (Hendrikx et al., 1994; Miura et al., 1997; Munch-Ellingsen et al., 1998). In the rabbit model, T-162559 did not show a dose dependency between the two doses. A pharmacokinetic study of T-162559 would be necessary to determine whether the maximum inhibitory effect of the drug on NHE-1 is already obtained at the dosage of 0.03 mg kg−1 i.v.
It has been well documented that pretreatment with NHE-1 inhibitors, including cariporide and eniporide, clearly salvages cardiac myocytes from ischaemia and reperfusion injury (Karmazyn et al., 1999; Avkiran, 2001). However, it has also been reported that the protective effect of NHE-1 inhibitors is attenuated when they are administered at the time of reperfusion (Avkiran, 2001). These results indicate that NHE activity during ischaemia is the principal determinant of cardiac injury and administration during this period is important to obtain the maximal effect of NHE-1 inhibitor (Avkiran, 2001). According to the results of the GUARDIAN trial in which cariporide was used as a NHE inhibitor (Théroux et al., 2000), a significant cardioprotective effect of the drug was observed in coronary artery bypass graft patients. Since patients had the opportunity to receive cariporide before ischaemia in this subgroup, it could be judged that the maximum cardioprotection with NHE inhibitors is achieved when the drugs are administered before the onset of ischaemia in clinical therapy. However, in the early phase after reperfusion, the pH in the extracellular space of the cardiomyocytes quickly returns to neutral (Piper et al., 1996), and the inhibitory activity of cariporide (and probably eniporide) on NHE-1 is largely lost at neutral pH (Aihara et al., 2000). Thus, attenuation of cardioprotective effect of cariporide when it is administered at reperfusion may be explained by its chemical property. Therefore, T-162559 will offer confirmatory insights on the above issue since it is considered to retain its inhibitory effect on NHE-1 after reperfusion because of its higher pKa value as compared to that of cariporide and eniporide.
In conclusion, T-162559 is a structurally novel NHE-1 inhibitor that exerts a more potent cardioprotective effect against ischaemia and reperfusion injury than cariporide and eniporide. By virtue of this favourable property, this NHE inhibitor offers promise for the treatment of patients with ischaemic heart disease.
Acknowledgments
The authors are grateful to Drs Zen-ichi Terashita, Yoshimi Imura and Takehiko Naka for their helpful discussion.
Abbreviations
- AAR
area at risk
- CF
coronary flow
- HR
heart rate
- IS
infarct size
- LDH
lactate dehydrogenase
- LV
left ventricle
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end-diastolic pressure
- NHE
Na+-H+ exchanger
- PRP
platelet-rich plasma
- PPP
platelet-poor plasma
- VF
ventricular fibrillation
- VT
ventricular tachycardia
References
- AHARONOVITZ O., GRANO Y. Stimulation of mitogen-activated protein kinase and Na+/H+ exchanger in human platelets. Different effect of phorbol ester and vasopressin. J. Biol. Chem. 1996;271:16494–16499. doi: 10.1074/jbc.271.28.16494. [DOI] [PubMed] [Google Scholar]
- AIHARA K., HISA H., SATO T., YONEYAMA F., SASAMORI J., YAMAGUCHI F., YONEYAMA S., MIZUNO Y., TAKAHASHI A., NAGAI A., KIMURA T., KOGI K., SATOH S. Cardioprotective effect of TY-12533, a novel Na+/H+ exchange inhibitor, on ischemia/reperfusion injury. Eur. J. Pharmacol. 2000;404:221–229. doi: 10.1016/s0014-2999(00)00613-0. [DOI] [PubMed] [Google Scholar]
- AVKIRAN M. Protection of the ischemic myocardium by Na+/H+ exchange inhibitors: potential mechanisms of action. Basic Res. Cardiol. 2001;96:306–311. doi: 10.1007/s003950170037. [DOI] [PubMed] [Google Scholar]
- AYE N.N., XUE Y.X., HASHIMOTO K. Antiarrhythmic effects of cariporide, a novel Na+-H+ exchange inhibitor, on reperfusion ventricular arrhythmias in rat hearts. Eur. J. Pharmacol. 1997;339:121–127. doi: 10.1016/s0014-2999(97)01371-x. [DOI] [PubMed] [Google Scholar]
- BANNO H., FUJIWARA J., HOSOYA J., KITAMORI T., MORI H., YAMASHITA H., IKEDA F. Effects of MS-31-038, a novel Na+-H+ exchange inhibitor, on the myocardial infarct size in rats after postischemic administration. Arzneim.-Forsch. Drug Res. 1999;49:304–310. doi: 10.1055/s-0031-1300419. [DOI] [PubMed] [Google Scholar]
- BENOS D.J. Amiloride: a molecular probe of sodium transport in tissues and cells. Am. J. Physiol. 1982;242:C131–C145. doi: 10.1152/ajpcell.1982.242.3.C131. [DOI] [PubMed] [Google Scholar]
- COUNILLON L.T., SCHOLZ W., ALBUS U., LANG H.J., POUYSSEGUR J. Pharmacological characterization of stably transfected Na+/H+ antiporter isoforms using amiloride analogs and a new inhibitor exhibiting anti-ischemic properties. Mol. Pharmacol. 1993;44:1041–1045. [PubMed] [Google Scholar]
- DING X.L., AKELLA A.B., SONNENBLICK E.H., RAO V.G., GULATI J. Molecular basis of depression of Ca2+ sensitivity of tension by acid pH in cardiac muscles of the mouse and rat. J. Card. Fail. 1996;2:319–326. doi: 10.1016/s1071-9164(96)80019-4. [DOI] [PubMed] [Google Scholar]
- FRELIN C.P., VIGNE P., BARBRY P., LAZDUSKI M. Interaction of guanidinium and guanidinium derivatives with the Na+/H+ exchange system. FEBS Lett. 1986;154:241–245. doi: 10.1111/j.1432-1033.1986.tb09388.x. [DOI] [PubMed] [Google Scholar]
- GUMINA R.J., BUERGER E., EICKMEIER C., MOORE J., DAEMMGEN J.D., GROSS G.J. Inhibition on the Na+/H+ exchanger confers greater cardioprotection against 90 minutes of myocardial ischemia than ischemic preconditioning in dogs. Circulation. 1999;100:2519–2526. doi: 10.1161/01.cir.100.25.2519. [DOI] [PubMed] [Google Scholar]
- GUMINA R.J., MIZUMURA T., BEIER N., SCHELLING P., SCHULTZ J.J., GROSS G.J. A new sodium/hydrogen exchange inhibitor, EMD 85131, limits infarct size in dogs when administered before or after coronary artery occlusion. J. Pharmacol. Exp. Ther. 1998;286:175–183. [PubMed] [Google Scholar]
- HENDRIKX M., MUBAGWA K., VERDONCK F., OVERLOOP K., HECKE P.V., VANSTAPEL F., LOMMEL A.V., VERBEKEN E., LAUWERYNS J., FLAMENG W. New Na+-H+ exchange inhibitor HOE694 improves postischemic function and high-energy phosphate resynthesis and reduces Ca2+ overload in isolated perfused rabbit heart. Circulation. 1994;89:2787–2798. doi: 10.1161/01.cir.89.6.2787. [DOI] [PubMed] [Google Scholar]
- IGATA H., FUJIWARA S., KANEKO M., ABE A., KUSUMOTO K., FUKUMOTO S., SHIRAISHI M., WATANABE T. Jpn. J. Pharmacol. 2001. p. 78P.
- ITO Y., IMAI S., UI G., NAKANO M., IMAI K., KAMIYAMA H., NAGANUMA F., MATSUI K., OHASHI N., NAGAI R. A Na+-H+ exchange inhibitor (SM-20550) protects from microvascular deterioration and myocardial injury after reperfusion. Eur. J. Pharmacol. 1999;374:355–366. doi: 10.1016/s0014-2999(99)00283-6. [DOI] [PubMed] [Google Scholar]
- KARMAZYN M. Amiloride enhances postischemic ventricular recovery: possible role of Na+-H+ exchange. Am. J. Physiol. 1988;255:H608–H615. doi: 10.1152/ajpheart.1988.255.3.H608. [DOI] [PubMed] [Google Scholar]
- KARMAZYN M., GAN T.G., HUMPHREYS R.A., YOSHIDA H., KUSUMOTO K. The myocardial Na+-/H+ exchange. Structure, regulation, and its role in heart disease. Circ. Res. 1999;85:777–786. doi: 10.1161/01.res.85.9.777. [DOI] [PubMed] [Google Scholar]
- KAWAMOTO T., KIMURA H., KUSUMOTO K., FUKUMOTO S., SHIRISHI M., WATANABE T., SAWADA H. Potent and selective inhibition of the human Na+/H+ exchanger isoform NHE-1 by a novel aminoguanidine derivative T-162559. Eur. J. Pharmacol. 2001;420:1–8. doi: 10.1016/s0014-2999(01)00991-8. [DOI] [PubMed] [Google Scholar]
- KLANKE R.A., YAN R.S., CALLEN J., WANG Z., MENETON P., BAIRD N., KANDASAMY R.A., ORLOWSKI J., OTTERUD B.E., LEPPERT M., SHULL G.E., MENON A.G. Molecular cloning and physical mapping of a novel human Na+/H+ exchanger (NHE5/SLC9A5) to chromosome 16q22.1. Genomics. 1995;25:615–622. doi: 10.1016/0888-7543(95)80002-4. [DOI] [PubMed] [Google Scholar]
- KLEYMAN T.R., CRAGOE E.J. , JR Amiloride and its analogs as tools in the study of ion transport. J. Membrane Biol. 1988;105:1–21. doi: 10.1007/BF01871102. [DOI] [PubMed] [Google Scholar]
- L'ALLEMAIN G., FRANCHI A., CRAGOE E., POUYSSÉGUR Blockade of the Na+/H+ antiport abolishes growth factor-induced DNA synthesis in fibroblasts. Structure-activity relationships in the amiloride series. J. Biol. Chem. 1984;259:4313–4319. [PubMed] [Google Scholar]
- LEEM C.H., LAGADIC-GROSSMAN D., VAUGHAN-JONES R.D. Characterization of intracellular pH in the guinea-pig ventricular myocyte. J. Physiol. 1999;517:159–180. doi: 10.1111/j.1469-7793.1999.0159z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIVNE A., GRINSTEIN S., ROTHSTEIN A. Volume-regulating behavior of human platelets. J. Cell. Physiol. 1987a;131:354–363. doi: 10.1002/jcp.1041310307. [DOI] [PubMed] [Google Scholar]
- LIVNE A., GRINSTEIN S., ROTHSTEIN A. Characterization of Na+-H+ exchange in platelets. Thromb. Haemost. 1987b;58:971–977. [PubMed] [Google Scholar]
- LORRAIN J., BRIAND V., FAVENNEC E., DUVAL N., GROSSET A., JANIAK P., HOORNAERT C., CREMER G., LATHAM C., O'CONNOR S.E. Pharmacological profile of SL 59.1227, a novel inhibitor of the sodium/hydrogen exchanger. Br. J. Pharmacol. 2000;131:1188–1194. doi: 10.1038/sj.bjp.0703671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIURA T., OGAWA T., SUZUKI K., GOTO M., SHIMAMOTO K. Infarct size limitation by a new Na+-H+ exchange inhibitor, hoe642: difference from preconditioning in the role of protein kinase C. J. Am. Coll. Cardiol. 1997;29:693–701. doi: 10.1016/s0735-1097(96)00522-0. [DOI] [PubMed] [Google Scholar]
- MUNCH-ELLINGSEN J., LØKEBØ J.E., BUGGE E., YTREHUS K. Equal reduction in infarct size by ethylisopropyl-amiloride pretreatment and ischemic preconditioning in the in situ rabbit heart. Mol. Cell. Biochem. 1998;186:13–18. [PubMed] [Google Scholar]
- NOËL J., POUYSSÉGUR J. Hormonal regulation, pharmacology, and membrane sorting of vertebrate Na+/H+ exchanger isoforms. Am. J. Physiol. 1995;268:C283–C296. doi: 10.1152/ajpcell.1995.268.2.C283. [DOI] [PubMed] [Google Scholar]
- OHARA F., SUGIMOTO T., YAMAMOTO N., OHKUBO K., MAEDA K., OZAKI T., SEKI J., GOTO T. Preischemic and postischemic treatment with a new Na+/H+-exchange inhibitor, FR 183998, shows cardioprotective effects in rats with cardiac ischaemia and reperfusion. J. Cardiovasc. Pharmacol. 1999;34:848–856. doi: 10.1097/00005344-199912000-00012. [DOI] [PubMed] [Google Scholar]
- PIERCE G.N., CZUBRYT M.P. The contribution of ionic imbalance to ischemia/reperfusion-induced injury. J. Mol. Cell. Cardiol. 1995;27:53–63. doi: 10.1016/s0022-2828(08)80007-7. [DOI] [PubMed] [Google Scholar]
- PIPER H.M., BALSER C., LADILOV Y.V., SCHÄFER M., SIEGMUND B., RUIZ-MEANA M., GARCIA-DORADO D. The role of Na+/H+ exchange in ischemia-reperfusion. Basic. Res. Cardiol. 1996;91:191–202. doi: 10.1007/BF00788905. [DOI] [PubMed] [Google Scholar]
- REITHMEIER R.A.F. Mammalian exchangers and co-transporters. Current Opin. Cell Biol. 1994;6:583–594. doi: 10.1016/0955-0674(94)90080-9. [DOI] [PubMed] [Google Scholar]
- ROSSKOPF D., MORGENSTERN E., SCHOLZ W., OSSWALD U., SIFFERT W. Rapid determination of the elevated Na+-H+ exchange in platelets of patients with essential hypertension using an optical swelling assay. J. Hypertens. 1991;9:231–238. [PubMed] [Google Scholar]
- SARKADI B., PARKER J.C. Activation of ion transport pathways by changes in cell volume. Biochim. Biophys. Acta. 1991;1071:407–427. doi: 10.1016/0304-4157(91)90005-h. [DOI] [PubMed] [Google Scholar]
- SATOH H., HAYASHI H., KATOH H., TERADA H., KOBAYASHI A. Na+/H+ and Na+/Ca2+ exchange in regulation of Na+(i) and Ca2+(I) during metabolic inhibition. Am. J. Physiol. 1995;268:H1239–H1248. doi: 10.1152/ajpheart.1995.268.3.H1239. [DOI] [PubMed] [Google Scholar]
- SATOH H., HAYASHI H., NODA N., TERADA H., KOBAYASHI A., HIRONO M., YAMASHITA Y., YAMAZAKI N. Regulation of Na+(i) and Ca2+(i) in guinea pig myocytes: Dual loading of fluorescent indicators SBFI and fluo 3. Am. J. Physiol. 1994;266:H568–H576. doi: 10.1152/ajpheart.1994.266.2.H568. [DOI] [PubMed] [Google Scholar]
- SCHOLZ W., ALBUS U., COUNILON L., GOGELEIN H., LANG H.-J., LINZ W., WEICHERT A., SCHOLKENS B.A. Protective effects of HOE642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischemia and reperfusion. Cardiovasc. Res. 1995;29:260–268. [PubMed] [Google Scholar]
- SHIPOLINI A.R., YOKOYAMA H., GALIÑANESM M., EDMONDSON S.J., HEARSE D.J., AVKIRAN M. Na+/H+ exchanger activity does not contribute to protection by ischemic preconditioning in the isolated rat heart. Circulation. 1997;96:3617–3625. doi: 10.1161/01.cir.96.10.3617. [DOI] [PubMed] [Google Scholar]
- STRÖMER H., DE GROOT M.C.H., HORN M., FAUL C., LEUPOLD A., MORGAN J.P., SCHOLTZ W., NEUBAUER S. Na+/H+ exchange inhibition with HOE642 improves postischemic recovery due to attenuation of Ca2+ overload and prolonged acidosis on reperfusion. Circulation. 2000;29:693–701. doi: 10.1161/01.cir.101.23.2749. [DOI] [PubMed] [Google Scholar]
- TANI M., NEELY J.R. Role of intracellular Na+ in Ca2+ overload and depressed recovery of ventricular function of reperfused ischemic rat hearts. Circ. Res. 1989;65:1045–1056. doi: 10.1161/01.res.65.4.1045. [DOI] [PubMed] [Google Scholar]
- TAKAICHI K., BALKOVETZ D.F., VAN MEIR E., WARNOCK D.G. Cytosolic pH sensitivity of an expressed human NHE-1 Na+-H+ exchanger. Am. J. Physiol. 1993;264:C944–C950. doi: 10.1152/ajpcell.1993.264.4.C944. [DOI] [PubMed] [Google Scholar]
- THÉROUX P., CHAITMAN B.R., DANCHIN N., ERHARDT L., MEINERTZ T., SCHROEDER J.S., TOGNONI G., WHITE H.D., WILLERSON J.T., JESSEL A. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Circulation. 2000;102:3032–3038. doi: 10.1161/01.cir.102.25.3032. [DOI] [PubMed] [Google Scholar]
- WAKABAYASHI S., SHIGEKAWA M., POUYSSEGUR J. Molecular physiology of vertebrate Na+/H+ exchangers. Physiol. Rev. 1997;77:51–74. doi: 10.1152/physrev.1997.77.1.51. [DOI] [PubMed] [Google Scholar]
- WEICHERT A., FABER S., JANSEN H.W., SCHOLZ W., LANG H.J. Synthesis of the highly selective Na+/H+ exchange inhibitors cariporide mesilate and (3-methanesulfonyl-4-piperidino-benzyl) guanidine. Arzneim.-Forsch. Drug Res. 1997;47:1204–1207. [PubMed] [Google Scholar]