

Abstract
We examine the sensitivity of GABAA and glycine receptors (same ionotropic superfamily) to oleamide. We address subunit-dependence/modulatory mechanisms and analogies with depressant drugs.
Oleamide modulated human GABAA currents (α1β2γ2L) in oocytes (EC50, 28.94±s.e.mean of 1.4 μM; Maximum 216%±35 of control, n=4). Modulation of human α1 glycine homo-oligomers (significant), was less marked, with a lower EC50 (P<0.05) than GABA receptors (EC50, 22.12±1.4 μM; Maximum 171%±30, n=11).
Only the hypnogenic cis geometric isomer enhanced glycine currents (without altering slope or maximal current, it reduced the glycine EC50 from 322 to 239 μM: P<0.001). Modulation was not voltage-dependent or associated with a shift in Er.
β1 containing GABAA receptors (insensitive to many depressant drugs) were positively modulated by oleamide. Oleamide efficacy was circa 2× greater at α1β1γ2L than α1β2γ2L (P=0.007). Splice variation in γ subunits did not alter oleamide sensitivity.
cis-9,10-Octadecenoamide had no effect on the equilibrium binding of [3H]-muscimol or [3H]-EBOB to mouse brain membranes. It does not directly mimic GABA, or operate as a neurosteroid-, benzodiazepine- or barbiturate-like modulator of GABAA-receptors.
The transport of [3H]-GABA into mouse brain synaptoneurosomes was unaffected by high micromolar concentrations of cis-9,10-octadecenoamide. Oleamide does not enhance GABA-ergic currents or prolong IPSCs by inhibiting GABA transport.
Oleamide is a non-selective modulator of inhibitory ionotropic receptors. The sleep lipid exerts its effects indirectly, or at a novel recognition site on the GABAA complex.
Keywords: cis-9,10-octadecenoamide/‘oleamide' recombinant GABAA receptor; recombinant glycine receptor; human/rat brain receptors and ion channels; [3H]-EBOB and [3H]-muscimol binding; synaptoneurosomes; [3H]-GABA uptake
Introduction
The fatty acid amide, cis-9,10-octadecenoamide (‘oleamide' or cOA), was isolated from the cerebrospinal fluid of sleep deprived cats, and synthetic cOA can induce sleep when injected i.p. or i.c.v. into naïve animals (Cravatt et al., 1995; Basile et al., 1999). 5HT1, 2 and 7 receptors have been cited as high-affinity targets (Boger et al., 1998a) and 5HT receptors are certainly involved in sleep (Pascoe, 1994) but the concentration of oleamide required to exert a maximal modulatory response in recombinant 5HT receptors (Huidobro Toro & Harris, 1996) is below the physiological range of oleamide reported in the CSF of even alert animals (Basile et al., 1999).
In contrast, at concentrations greater than 10 μM, oleamide can uncouple gap junctions (Boger et al., 1998b). It has been suggested that cOA blocks gap junctions by increasing the membrane homeoviscosity of neurones (Lerner, 1997). Others suggest that although oleamide (>10 μM) can fluidize membranes this is not relevant to sleep induction, but their ex vivo experiments suggest that sleep-inducing doses of the lipid result in very high brain concentrations (Gobbi et al., 1999).
Oleamide is broken down by fatty acid amide hydrolase (FAAH) enzymes, which also degrade the endocannabinoid anandamide. Exogenous oleamide's effects on locomotor activity (Cheer et al., 1999) and sleep are broadly cannabinomimetic (Mendelson & Basile, 1999). However, oleamide binds only weakly to CB1 receptors (Cheer et al., 1999) and it cannot directly alter GTPγS binding (Boring et al., 1996). A popular interpretation of the above evidence is that oleamide competes with anandamide for FAAH, causing the levels of anandamide to increase thus producing sleep (Cheer et al., 1999).
We have highlighted that GABAA receptors (see also Yost et al., 1998) and voltage-gated sodium channels are both stereoselectively modulated by oleamide (trans-oleamide was much less potent as a sleep inducer and is not active at these presumptive targets in vitro) (Laws et al., 2001). Sodium channels are blocked by oleamide in a state/voltage-dependent manner, which is an acknowledged effect of several classes of depressant drug (Verdon et al., 2000; Nicholson et al., 2001). GABAA receptor currents are enhanced in the presence of oleamide (Lees et al., 1998). The modulatory effects of oleamide on GABAA receptors, and the fact that it blocks voltage-gated sodium channels in a manner similar to anaesthetic and anticonvulsant drugs, suggests that this or related lipids may be endogenous ligands for drug recognition sites (Laws et al., 2001). Most anaesthetics (with the exception of ketamine, nitrous oxide and xenon) are demonstrably active at GABAA receptors, where their potency mirrors their anaesthetic potency in vivo (Franks & Lieb, 1994; Laws et al., 2001). Glycine receptors share sensitivity to a variety of anaesthetics and were an important tool in seeking key anaesthetic recognition domains on the GABAA receptor protein (Belelli et al., 1999b). The β subunit of the GABAA receptor and the α subunit of the glycine receptor contain sites on the M2 and M3 domains which are crucial for modulatory effects of depressant drugs. These sites confer sensitivity of the receptor not only to anaesthetics but also to the anticonvulsant loreclazole (Wafford et al., 1994) and the recently disclosed depressant effects of NSAIDS (Halliwell et al., 1999). Naturally occurring subunit combinations (isoforms) of the GABAA receptor show differing sensitivity to volatile anaesthetics, with the β1 containing receptors being insensitive. The β1 and β2 subunits contain different amino acid residues (β1, Ser-290, β2, Asn-289) in the M2 domain leading to the insensitivity of β1 to etomidate and loreclazole (Belelli et al., 1997). The M2 domain on both the α and β subunits confers absolute sensitivity to two volatile anaesthetics and ethanol (Mihic et al., 1997). A later publication by Krasowski & Harrison (2000) showed that 12 out of 13 general anaesthetics, including isoflurane and enflurane, had a reduced or no modulatory effect on GABA receptors containing mutations in the M2 domain of α and/or β subunits. This site on the M2 domain may represent a site for direct binding of oleamide to the GABAA receptor although the GABAA receptor-chloride ion channel has a remarkable capacity for allosteric regulation by both drugs and steroid hormones. In this study we examine the sensitivity of glycine receptor subunits to modulation by oleamide. Furthermore, we probe the influence of GABAA β subunits and of splice variation in γ subunits (which alter the sensitivity to ethanol) on oleamide modulation. Biochemical experiments on GABA uptake and EBOB/muscimol binding are used to seek a mechanism for potentiation of chloride currents by oleamide.
Methods
Recombinant receptors in oocytes
Stage V – VI oocytes were isolated from female xenopus laevis and stripped of their follicular layer (Edwards & Lees, 1997). cRNA for the human glycine α1 homo-oligomer (kindly supplied by Dr S. Daniels, University of Cardiff) was injected into the vegetal pole. cDNA for human subunits (specified combinations in text: all kindly supplied by Dr Paul Whiting, MSD, Harlow) were injected blind into the animal pole in sterile buffer (0.2 – 0.3 ng of each cDNA subunit mixed to 20 nl per oocyte). Injected oocytes were incubated at 18 – 22°C for 1 – 5 days prior to electrophysiological studies. All data reported here was obtained from a minimum of two independent batches of oocytes.
Electrophysiology
Oocyte electrodes were bevelled (Narishige diamond wheel Model no. EG-40) to give a resistance of 0.5 – 1.5 MΩ and filled with 2 M KCl. Frog ringer saline contained (mM): NaCl 115, KCl 2.5, HEPES 10, CaCl2 1.8, pH 7.2 (NaOH) and was perfused at approximately 10 ml min−1. Cells were voltage clamped at −60 mV using the two electrode voltage clamp technique (GeneClamp 500, Axon Instruments, CA, U.S.A.). Results were measured as a current shift. Recordings were made on a chart recorder, and digitized using Polyview Software (Polyview, Grass Instruments, U.S.A.). GABA and glycine were applied for long enough to produce peak responses (5 – 30 s). All experiments were conducted at 22 – 24°C.
Oocyte pharmacology
Mefenamic Acid (courtesy of Dr Bob Halliwell, University of Durham, U.K.) was dissolved in DMSO and diluted 1 in 1000 with extracellular saline. Oleamide was dissolved in DMSO and diluted 1 in 1000 with extracellular saline. All experimental salines contained 0.1% DMSO: to study oleamide, 0.033% BSA was also routinely added (to facilitate the dissolution of oleamide). Oleamide and mefenamic acid were formulated daily and perfused from glass containers via Teflon lines. GABA and glycine were dissolved in saline and applied via a rapid (solenoid based) agonist perfusion system. Drugs or receptor modulators were superfused at 10 ml min−1 between and during agonist pulses (to achieve/maintain equilibrium).
Radioligand binding
cis-9,10-Octadecenoamide and the corresponding trans isomer were synthesized and purified by Professor C.R. Ganellin (University College London). The radiochemicals [3H]-EBOB (30 Ci mmol−1)); [methylene-3H]-muscimol (11.8 Ci mmol−1) and [3H]-GABA (36.2 Ci mmol−1) were obtained from NEN Life Science Products (Boston, MA, U.S.A.). Lindane, picrotoxin, bicuculline, bovine serum albumin (BSA) and nipecotic acid were obtained from Sigma-Aldrich Canada Ltd (Oakville, Ontario, Canada). Unlabelled GABA was purchased from Calbiochem-Neurobiochem Corporation (La Jolla, CA, U.S.A.). Whatman GF/C filters were purchased from Fisher Scientific (Nepean, Ontario, Canada), 12,14-dichloro-dehydroabietic acid was purchased from Helix Biotech Corporation (Richmond, BC, Canada). Binding and transmitter uptake experiments were conducted using male CD1 mice (20 – 30 g) obtained from Charles River Laboratories (St. Constance, Quebec, Canada). Mice were maintained on a 12 h light : 12 h dark photoperiod and given ad libitum access to food and water. Animal husbandry and all experimental procedures involving mice complied with the Canadian Council on Animal Care guidelines.
[3H]-muscimol binding assay
Synaptic membranes were routinely prepared from the brains of four mice, as described by Beaumont et al. (1978), and binding assays were conducted using the procedure of Negro et al. (1995). Briefly, membranes (circa 0.5 mg protein) were incubated with [3H]-muscimol (20 nM final concentration) and oleamide isomers, drugs or solvent (DMSO) controls, in darkness, at 4°C for 30 min. Incubations were terminated by adding 4 ml ice-cold Tris-citrate buffer and rapid mixing. Membranes were promptly filtered on Whatman GF/C filters and subjected to three subsequent 4 ml rinses. Filters were then incubated with sodium dodecyl sulphate to dissolve membranes and release any trapped tritium. Radioactivity was quantified by liquid scintillation counting (l.s.c.) using a Beckman LS 3801 scintillation counter. In a given experiment, individual treatments were performed at least in duplicate and non-specific binding determined using 100 μM GABA, was 4% of total binding.
[3H]-EBOB binding assay
The isolation of brain membranes and the [3H]-EBOB binding assay was carried out according to methods published by Cole & Casida (1992). Brain membranes (circa 0.4 mg protein) were incubated with [3H]-EBOB (750 pM final concentration) together with study compounds or control solvent (DMSO) as necessary, for 90 min at 37°C. The binding reaction was stopped by rapid filtration through Whatman GF/C filters and membranes were given three washes with 4 ml ice-cold phosphate buffer prior to determination of radioactivity using l.s.c. Assays were performed at least in duplicate and non-specific binding was determined in the presence of lindane at a saturating concentration (5 μM).
Isolation of synaptoneurosomes and assay of [3H]-GABA uptake
Synaptoneurosomes were isolated from mouse brain essentially as described by Harris & Allen (1985), but with certain modifications (Bloomquist et al., 1986). Whole brains were removed from two animals and rapidly cooled in ice-cold isolation buffer (mM): NaCl 137, KCl 5, CaCl2 2.5, MgSO4 1.2, glucose 54, 1 mg/ml BSA and HEPES 20, adjusted to pH 7.4 with Tris base). Brain tissue was chopped thoroughly using a razor blade and then gently homogenized (eight excursions; by hand) in 2.5 ml isolation buffer. A further 12.5 ml of isolation buffer was added and the suspension was then passed through two layers of cheese cloth. The filtrate was centrifuged at 1000 g for 15 min and the resulting pellet resuspended in a further 10 ml of isolation buffer and centrifuged again. The final pellet (synaptoneurosomal fraction) was resuspended in isolation buffer containing BSA (1 mg ml−1) and held on ice. This fraction was diluted 1 in 16 for assay.
The transport of GABA was measured isotopically based on a method described by Martin & Smith (1972). Synaptoneurosomes (0.412 mg protein) were preincubated with test compounds for 10 min in saline (mM): NaCl 137, KCl 5, CaCl2 0.8, MgSO4 1.2, Na2HPO4 1, HEPES 20, glucose 14, buffered to pH 7.4 with Tris base), prior to being transferred to saline containing [3H]-GABA (final GABA concentration 0.95 μM; 0.2 μCi) and the same concentration of test compound. GABA uptake was continued for 3 min, whereupon 2 ml ice-cold saline was added followed by rapid mixing. Synaptoneurosomes were quickly filtered (Whatman GF/C; vacuum filtration) and then filters were washed with a further 5 ml ice-cold saline. Radioactivity was determined as described in preceding sections. In experiments examining the sodium dependence of synaptoneurosomal uptake of GABA, NaCl was replaced with an equivalent concentration of choline chloride. DMSO was used to introduce oleamide into the assay, and the concentration of this solvent did not exceed 0.33% v v−1.
Assay of protein
The assay of protein was carried out using the procedure of Peterson (1977), with BSA as the protein standard.
Analysis
Data are presented as mean±s.e.mean throughout. Repeated measures one-way ANOVA and Student's t-tests were used as appropriate using Prism (Graphpad U.S.A.). P values of <0.05 were considered significant. Composite figures were produced using Smart Draw 5 software (San Diego, CA, U.S.A.).
Results
Stereoselective modulation of both GABA and glycine currents by oleamide
Saturating dose – response profiles were obtained for glycine receptors, and GABAA α1β2γ2L receptors in the absence of drugs or hormones (not shown). The modulatory capacity of oleamide was studied at circa the EC20 for each neurotransmitter (1 μM for GABA and 100 μM for glycine). Recombinant GABAA receptors (α1β2γ2L) were modulated by oleamide in a dose dependent manner (Max=216%±35, EC50=28.94±1.4 μM, hillslope=2.4±1.59, n=4: see Figure 1). Glycine currents were also significantly increased by oleamide (Max=171%±30, EC50=22.12 μM±1.4, hillslope=1.9±0.89, n=11: Figure 1b). The EC50 for modulation of GABAA receptors by oleamide was significantly larger than that for glycine receptors (P<0.05, n=4 – 11), see Figure 1c. The maximum modulation and the hillslopes for modulation by oleamide were not significantly different between GABAA receptors and glycine receptors (P>0.05 for both parameters, n=4 – 11).
Figure 1.
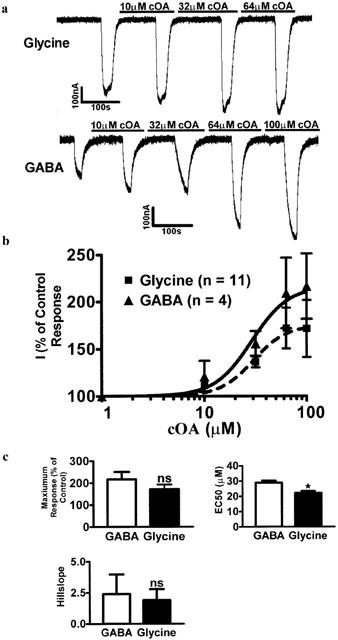
Oleamide enhanced the effects of both glycine and GABA at their respective receptors. (a) Perfusion of increasing concentrations of oleamide to oocytes responding to the EC20 of Glycine and GABA. Control responses are shown on the far left of each trace. (b) Concentration-response curves for oleamide modulation of glycine and GABAA receptors (Glycine: Max=1.72±0.34, EC50=22.12±1.4, Hillslope=1.9) (GABA: Max=2.16±0.35, EC50=28.94±1.4, Hillslope=2.4, n=4 – 11). (c) Compounded data from replicated experiments. Unpaired, 2-tailed Student's t-tests were used to compare the maximum responses, EC50s and hillslopes for the two curves. Only the EC50 was significantly different (P=0.305, 0.017, 0.781 respectively, n=4 – 11).
Both glycine and GABAA receptors were sensitive to 32 μM oleamide, but only the cis geometric isomer was effective (Figure 2). The trans isomer (also 32 μM) produced no significant modulation at GABAA receptors (control, 1738±240.9 nA; cOA, 2213±154.7 nA, P<0.05; tOA, 1704±223.5 nA, P>0.05, n=4) as previously reported in cultured cells (Verdon et al., 2000), or at glycine receptors (control, 591.1±135.2 nA; cOA, 668.5±155.0 nA, P<0.05; tOA, 563.8±155.2 nA, P>0.05, n=6).
Figure 2.
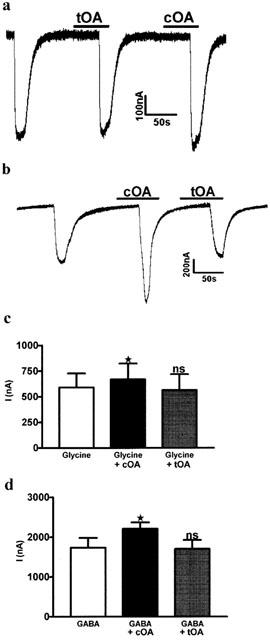
Modulatory effects of oleamide at chloride channels were only seen with the cis geometric isomer. The trans isomer was inactive at both Glycine and GABA receptors. (a) Representative trace showing the effects of 32 μM cOA and 32 μM tOA on glycine currents. (b) Trace showing the effects of 32 μM cOA and 32 μM tOA on GABAA receptors. (c) Replicated data shown as a bar graph. On Glycine receptors, 32 μM cOA produced a significant increase in response, whereas 32 μM tOA did not alter the peak current. (d) 32 μM cOA significantly enhanced GABA currents but 32 μM tOA did not. A repeated measures one-way ANOVA with a Dunnett's post test was used (see Results).
Glycine-evoked currents were outwardly rectifying, with a reversal potential of −17.8±0.6 mV (not shown) which is entirely consistent with the chloride equilibrium potential in mature oocytes. In the presence of 32 μM oleamide the reversal potential was not significantly different −19.6±0.5 (P=0.108, n=4). The per cent of modulation at each voltage (Vh ranging from 0 to −80 mV, in 20 mV increments) was not significantly different using a repeated measures one-way ANOVA (P=0.860, n=4). This suggests that modulation of inhibitory receptors by oleamide is not voltage dependent and that the enhanced currents do not reflect a change in ECl (not shown, but consistent with earlier results on GABAA receptors) (Lees et al., 1998). The sigmoid log concentration – response curve for glycine was shifted to the left (not shown) in the presence of 32 μM oleamide (pre-treatment: Max=1318 nA±573, EC50=322.2 μM±1.04, hillslope=3.6±1.5, oleamide: Max=1281 nA±573, EC50=239.4 μM±1.03, hillslope=3.3±0.24, n=4). The EC50 was significantly reduced in the presence of oleamide (P<0.001, n=4), whereas the maximum response and hillslopes were unchanged. This suggests that oleamide is increasing the affinity of glycine for its receptor without changing the maximum response, although other allosteric mechanisms might apply.
The β subunit alters the magnitude of the oleamide modulation of the GABAA receptor but is not an absolute determinant of sensitivity
Substitution of a β2 subunit for β1 did not change the sensitivity of the receptor to GABA. One mM GABA produced a saturating response, and 1 μM produced approximately 20% of the maximum response in receptors containing both types of subunit (β1, 1 μM, 21.46±2.71% of maximum response; β2 1 μM, 19.97±2.18% of maximum response, P=0.685, using an unpaired 2-tailed Student's t-test). To validate that the β1 had been successfully expressed, we examined the effects of the NSAID mefenamic acid (MFA) which has recently been characterized as a GABAA modulator and behavioural depressant in laboratory animals (Halliwell et al., 1999). We examined MFA at 10 μM (for β2 containing oocytes) and 100 μM (for β1 containing oocytes) according to protocols devised by Halliwell and co-workers (Halliwell et al., 1999). As noted in these earlier studies, mefenamic acid significantly enhanced the current produced by 1 μM GABA in oocytes containing α1β2γ2L (Control, 411.7±205.1 nA, 10 μM MFA, 1078.0±329.4 nA, P=0.02, n=5: Figure 3a). GABA currents in the β1 isoform were depressed by mefenamic acid (Control, 1270.0±520.4 nA, 100 μM MFA, 585.5±384.1 nA, wash, 901.9±448.2 nA, P=0.01, n=4: Figure 3b) which confirmed differential subunit expression. At GABA concentrations of 1 μM (EC20), in contrast to the NSAID, oleamide (32 μM) produced a significant positive modulatory effect on GABAA receptors (Figure 4) containing both the β1 (control; 410.4±143.3 nA, 32 μM cOA; 979.8±252.9 nA, P<0.01, wash; 552.8±146.2 nA, P>0.05; P=0.0003, n=8) and β2 subunits (control; 587.0±254 nA, 32 μM, cOA; 755.8±262.6 nA, P<0.01, wash; 660.3±240.5, P>0.05; P=0.0058, n=8). To determine which receptor subtype (β1 or β2 containing) was more sensitive to modulation by oleamide, the current in the presence of oleamide was also analysed as a percentage of the control response. The modulatory effect of oleamide on β1 containing receptors was significantly greater than β2 containing receptors (β1; 293.8±39.31%, β2; 157.4±16.94%, P=0.007, n=8: Figure 4c).
Figure 3.
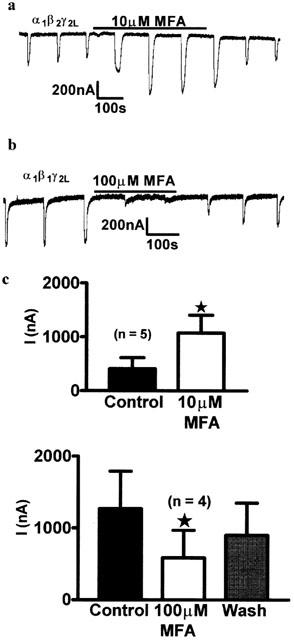
Mefenamic acid (MFA) enhanced GABA induced currents in β2 containing recombinant receptors, but depressed currents in β1 containing receptors. (a) Trace showing the effects of MFA on α1β2γ2L GABAA receptors. (b) Trace showing the effects of MFA (horizontal bar) on α1β1γ2L GABAA receptors. (c) Paired 2-tailed t-test and repeated measures one-way ANOVA show that MFA significantly enhanced responses in GABA receptors containing the β2 subunit (P=0.02, n=5), and significantly (and reversibly) blocked isoforms containing the β1 subunit (P=0.01, n=4).
Figure 4.
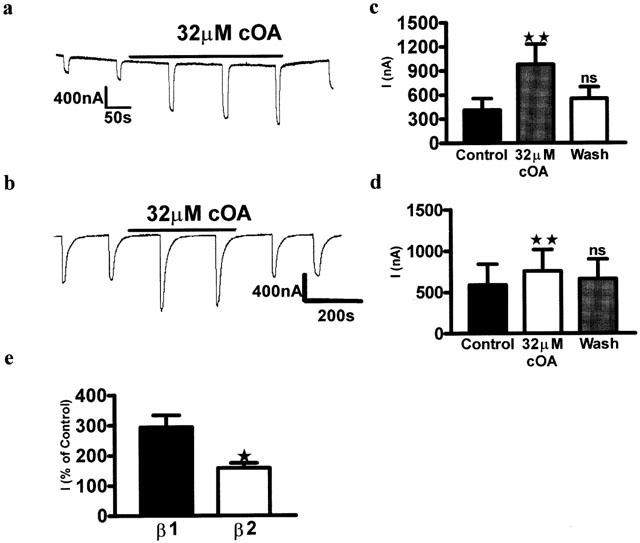
Oleamide differentially enhanced GABA induced currents in both β1 and β2 containing recombinant receptors. (a) Trace showing the effects of oleamide on α1β1γ2L GABAA receptors (b) Trace showing the effects of oleamide on α1β2γ2L GABAA receptors. (c) Repeated measures one-way ANOVA, with a Dunnett's post test shows that cOA significantly enhanced responses in GABA receptors containing the β1 subunit (P<0.01, n=8). (d) The β2 subunit (P<0.01, n=8). (e) Unpaired 2-tailed Student's t-test shows that β1 containing GABA receptors were modulated by cOA more than β2 containing receptors (P=0.011, n=8).
Influence of γ subunit
γ2L subunits bear a splice variant which confers sensitivity to protein kinase C and may be important in conferring sensitivity to the sedative effects of ethanol (Wafford et al., 1991). As in our pilot experiments (Lees et al., 1998b), oleamide (32 μM) was able to modulate both short and long forms of the receptor (α1β2γ2L; Control, 469.9±140.9 nA, 32 μM cOA, 694.2±169.0 nA, Wash, 537.1±153.8 nA, P=0.014, n=5: α1β2γ2S; Control, 807.1±245.7 nA, 32 μM cOA, 965.0±285.2 nA, Wash, 820.0±246.9 nA, P=0.007, n=4: Figure 5). When we compared the intrinsic efficacy of the response in short and long splice variants they were not significantly different (γ2L, 166.3±24.2%, n=5; γ2S, 128.3±9.668%, n=4, P=0.229).
Figure 5.
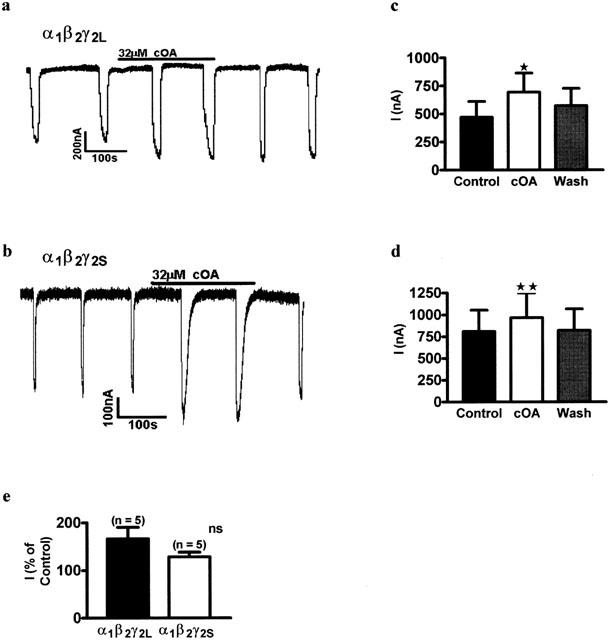
Oleamide enhanced GABA induced currents in both γ2L and γ2S containing receptors. (a) Representative trace showing the effects of oleamide on α1β2γ2L GABAA receptors. (b) Trace from a different oocyte showing the effects of oleamide on α1β2γ2S GABAA receptors. (c) Replicated data. Repeated measures, one-way ANOVA showed that cOA significantly enhanced responses in GABA receptors containing both the γ2L subunit (P=0.014, n=5), and (d) the γ2S subunit (P=0.007, n=4). (e) Unpaired 2-tailed Student's t-test showed that there was no significant difference in the per cent of modulation by oleamide in γ2L containing receptors and γ2S containing receptors (P=0.229, n=4 – 5).
Effects of oleamide on [3H]-muscimol and [3H]-EBOB binding
The basis for the positive modulation of GABAA receptor function by oleamide in mammalian brain was further examined using radioligand binding approaches. High micromolar concentrations of cOA failed to affect the specific binding of [3H]-muscimol to mouse brain preparations, under conditions where GABA and bicuculline produced characteristic inhibition (Figure 6). A very weak effect of cOA on [3H]-EBOB binding to brain membranes was detected (approximately 10 – 15% inhibition), however responses bore no relation to cOA concentration (Figure 7). The positive controls lindane and 12,14-dichlorodehydroabietic acid (Nicholson et al., 1999), produced full inhibition of [3H]-EBOB binding in these experiments.
Figure 6.
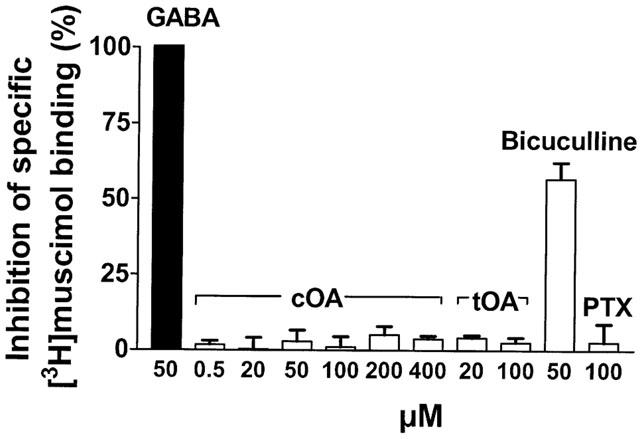
Lack of effect of cOA, tOA and picrotoxin on the binding of [3H]-muscimol to mouse brain membranes. GABA and bicuculline produced significant inhibition.
Figure 7.
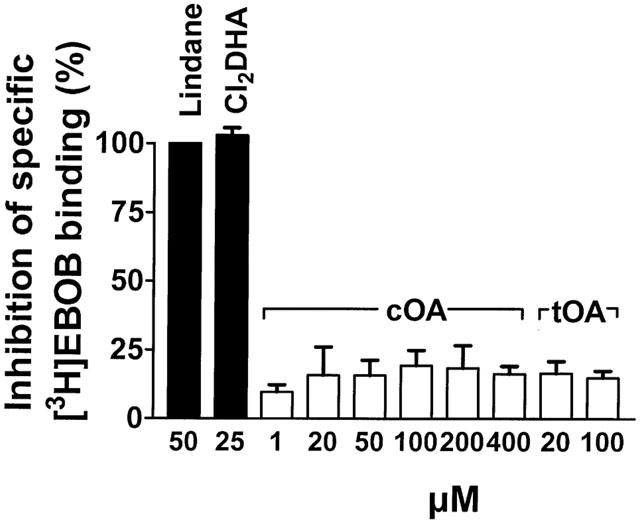
Failure of cOA to influence the binding of [3H]EBOB to brain membranes in a concentration-dependent fashion. tOA showed a similar effect, whereas lindane and 12,14-dichlorodehydroabietic acid produced full block.
GABA transport
In other experiments using synaptoneurosomes, cOA had no significant effect on [3H]-GABA uptake in situations where 10 mM nipecotic acid inhibited uptake by 98.5±0.5% (Figure 8). Using these assay conditions, replacement of Na+ by choline and conducting experiments at 0°C, reduced the association of [3H]-GABA with synaptoneurosomes by 97.3±0.3 and 89.2±0.8% respectively. The non-hypnotic isomer (tOA) showed a similar profile to cOA in both the binding and the uptake assays.
Figure 8.
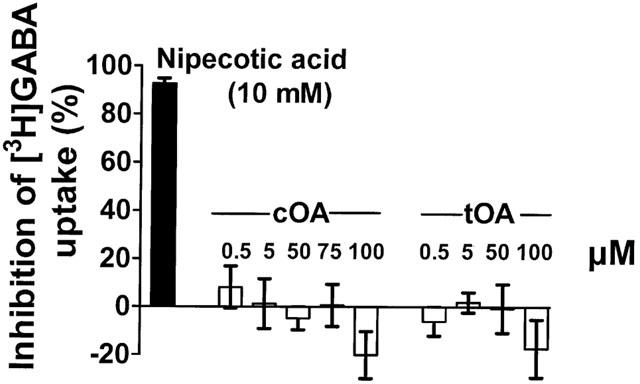
Inhibition of the transport of [3H]-GABA into synaptoneurosomes by nipecotic acid and inability of cOA or tOA to affect this process.
Discussion
Although GABA is the most prevalent inhibitory neurotransmitter in the brain, glycine also couples to a related ionotropic receptor. Glycinergic inhibition is important in the brain stem and spinal cord and has been associated with active sleep (Soja et al., 1991). Most anaesthetics and depressant drugs modulate glycine receptors, but to a lesser extent than GABA receptors (Belelli et al., 1999a, 1999b). Oleamide was also found to have a marginally less profound maximum modulatory effect on glycine receptors. However, oleamide had a significantly higher affinity for glycine receptors than GABA receptors. The glycine receptors expressed were human α1 homomers. Therefore, oleamide most likely binds to the α1 subunit to produce its effect, although indirect effects via 2nd messengers or kinases cannot be discounted. Oleamide is stereoselectively active on both glycine and GABAA receptors, i.e. the cis isoform positively modulates currents through these receptors, whereas the trans isoform produced no significant effect. This stereoselective effect suggests that the molecular structure of the molecule is important both in sleep induction and ion channel modulation. Both GABAA and glycine receptors produced outwardly rectifying currents in the presence and absence of oleamide (Lees et al., 1998): the modulatory effects were not voltage-dependent. Oleamide had no effect on the reversal potential, inferring that it does not regulate chloride pumping or transmembrane chloride gradient. As with GABAA receptors (Lees et al., 1998a) oleamide shifts the dose response curve for glycine to the left. This is evidence that oleamide increases the affinity of glycine for its receptor, without affecting the maximal current, although allosteric effects on gating can not be discounted.
Our GABAA data suggest that the positive modulatory effects of oleamide are not absolutely dependent upon the presence of β2 subunits in the receptor complex in contrast to the diverse depressant drugs mentioned earlier, suggesting that the sites of action differ. The work of Yost et al. (1998) suggests that oleamide cannot synergize inhalational anaesthetics, which is unusual for other depressant drugs which target the GABAA complex (e.g. benzodiazepines, barbiturates, steroids). On the other hand the in vivo effects of oleamide are highly dependent upon formulation strategies and routes/methods of administration used in different laboratories. Although isoforms of the GABAA receptor containing both the β1 and β2 subunits are sensitive to oleamide, this does not imply that the β subunit is not important in oleamide recognition. Clearly, β1 containing receptors are significantly more sensitive to oleamide that β2 containing receptors. This mirrors barbiturate (pentobarbital) effects on the GABAA receptor: β1 containing receptors are enhanced more than β2 containing subunits (Thompson et al., 1996). Comparing these data to those of Mihic et al. (1997) and Belelli et al. (1997): it becomes apparent that neither oleamide nor barbiturates bind to the site of action of volatile anaesthetics or loreclazole on the M2 domain of the β subunit. However, it is unlikely that oleamide produces its modulatory effect by binding to the same site of action as barbiturates. Evidence of this has been produced by recent work in our laboratories, which have shown that oleamide and pentobarbital have additive effects: see also binding data below (Laws & Lees, 2001). The concept of a shared receptor seems less likely when one considers that glycine receptors are insensitive to barbiturates (Kotchine et al., 1996). Furthermore, barbiturates but not oleamide can modulate α and β receptors in the absence of γ subunits (Lees et al., 1998).
Sensitivity of the GABAA receptor to certain depressant drugs is dependent on the presence and/or type of γ subunit present. Benzodiazepines require the presence of a γ2 subunit in order to produce a modulatory response (Tecott, 2000). Sensitivity to the sedative effects of ethanol may be determined by the splice variant of the γ2 subunit present in the GABA receptor (Wafford et al., 1991). Oleamide, although requiring the presence of the γ subunit to produce its modulatory effect on GABA receptors (Lees et al., 1998) could not distinguish between the long and short form of the γ subunit, producing similar modulatory effects in receptors containing both γ2L and γ2S (Lees et al., 1998b).
We used mefenamic acid to confirm the expression of β1 (isoforms containing this receptor were insensitive to the positive modulatory effects of mefenamic acid). (Halliwell et al., 1999). Interestingly, we found that mefenamic acid sensitivity was altered (we saw a marked antagonist effect rather than no modulation at all) by using the γ2L subunit rather than the γ2S, which was used in earlier studies (Halliwell et al., 1999). This emphasises the complex manifestations of isomerism within the receptor superfamilies.
It is unclear at present whether cis-oleamide exerts its effect at the GABAA receptor by interacting with a novel receptor or by activating a second messenger system (e.g. a phosphorylation process). Moreover, we cannot rule out physical effects such as selective intercalation with lipid molecules in the receptor microenvironment, or by perturbing the lipid – protein interface of the receptor, which may lead to membrane fluidity changes. The latter proposal has been rejected by others (Gobbi et al., 1999).
The modulatory effects reported here have been characterized at 10 – 32 μM, although effects of oleamide at recombinant GABAA receptors have been reported at 0.1 μM (Yost et al., 1998). Analytical studies to date have revealed that the concentrations of oleamide in ‘bulk' CSF are relatively low (30 – 170 nM, increasing to 450 nM during sleep deprivation) (Hanus et al., 1999). These results did not take into account oleamide's high lipid solubility (log P=6.5), which causes it to be distributed locally in cell membranes nor do they acknowledge the short-range peri-synaptic roles of unidentified endocannabinoids at functional nerve terminals (Wilson & Nicoll, 2001). Furthermore, FAAH is present in bulk CSF, resulting in the degradation of oleamide as soon as it leaves the membrane. Pilot experiments suggest that FAAH does not limit the efficacy of oleamide in our cultures at room temperature. Our results and those of Gobbi et al. (1999), suggest that in functional terms oleamide is a low-affinity modulator of inhibitory receptor function.
In common with the anxiolytic benzodiazepines, synthetic anaesthetic steroids and barbiturates, cis-oleamide potentiates GABA-evoked currents in mammalian neuronal systems (Mehta & Ticku, 1999). The considerable body of evidence supporting allosteric modulation of GABAA receptors in the central nervous system by benzodiazepines neurosteroids, and barbiturates originates mostly from radioligand binding data. For example, neuroactive steroids act allosterically to enhance the equilibrium binding of [3H]-muscimol to the GABA recognition site and inhibit the binding of the bicyclic ligand [35S]-TBPS to the non-competitive antagonist binding site on this complex (Turner et al., 1989; Delory et al., 1993; Goodnough & Hawkinson, 1995; Ito & Ho, 1994). Barbiturates allosterically inhibit the binding of caged convulsants (e.g. [35S]-TBPS) to brain membranes (Squires et al., 1983; Ramanjaneyulu & Ticku, 1984). In contrast, the benzodiazepines, especially those of the anxiolytic type are generally considered poor inhibitors of the binding of both bicyclophosphorothionate (e.g. [35S]-TBPS) and bicyclo-orthobenzoate (e.g. [3H]-TBOB) radioligand probes (Squires et al., 1983; Lawrence et al., 1985), although, stimulatory and inhibitory effects on [3H]-TBOB binding with clonazepam and zolpidem has been reported (Sakurai et al., 1994). Allosteric enhancement of [3H]-muscimol binding by diazepam and the volatile anaesthetic isoflurane has also been demonstrated (Delory et al., 1993; Harris et al., 1994). [3H]-muscimol and [3H]-EBOB (an improved bicylo-orthobenzoate probe for the TBPS recognition site (Huang and Casida, 1997)) were therefore considered ideal radioligands to determine whether cis-oleamide facilitates GABAA receptor function through similar mechanisms. The results of the present investigation clearly show that high micromolar concentrations of cis-oleamide do not affect the equilibrium binding of either probe to neuronal membranes. Our findings therefore tend to exclude the possibility that cis-oleamide mimics GABA itself, or interacts directly with the neurosteroid, benzodiazepine or barbiturate recognition sites on GABAA-receptors. It is noteworthy that certain structurally similar unsaturated free fatty acids (e.g. arachidonic and oleic acids) have been reported to stimulate [3H]-muscimol binding and to inhibit the binding of [35S]-TBPS to brain membranes (Koenig & Martin, 1992), thus highlighting the selective pharmacology that the amide linkage of cOA imparts at this complex.
The rapid uptake of GABA from the synaptic cleft into the nerve ending by a high affinity, sodium-dependent transport system, serves as a critical mechanism for signal termination at GABA-ergic synapses (Iversen & Neal, 1968; Martin & Smith III, 1972). GAT1 provides the primary neuronal uptake mechanism for GABA (Itouji et al., 1996) and inhibition of GAT1 in vivo leads to substantial anticonvulsant effects (Suzdak et al., 1993). We reasoned that inhibition of GABA reuptake by GAT1 in synaptic regions may help explain the hypnotic and some of the in vitro effects of cis-oleamide (such as prolongation of IPSCs and potentiation of low concentrations of exogenous GABA). The results of the present investigation clearly exclude this possibility since the sleep inducer did not affect the uptake of GABA into synaptoneurosomes under conditions where nipecotic acid, a potent inhibitor of GAT1 and other GAT subtypes (Thomsen et al., 1997), produced substantial inhibition.
Conclusion
Oleamide can mimic the actions of many depressant drugs as modulators of both GABAA and glycine receptors which are crucial regulators of excitability throughout the brain and spinal cord. However, its site of action on the GABAA receptor chloride channel complex is not identical to those for benzodiazepines, barbiturates, etomidate, loreclazole or inhalational anaesthetics. Serotonergic metabotropic receptors and pre-synaptic voltage-gated Na+ channels respond to lower levels of the sleep lipid than these ligand-gated anion channels but a consensus mode of action for oleamide has not yet emerged. Brain permeant oleamide mimics clearly have the potential to induce sleep. Currently used hypnotics have several undesirable features including habituation, tolerance, hangovers and amnesia. Strategies which regulate the physiological levels of endogenous sleep hormones may yield safer long-term hypnotic drugs: inhibitors of FAAH and selective blockers of fatty-acid amide uptake transporters are particularly attractive pharmaceutical targets.
Acknowledgments
Thanks to the Wellcome trust (for equipment support to G. Lees), the European Social Fund (L. Coyne, studentship funding) and Adam Errington for technical assistance with cell cultures. This research was also supported by a Natural Sciences and Engineering Research Council of Canada Research grant to R.A. Nicholson. G. Lees and R.A. Nicholson contributed equally to this research.
Abbreviations
- BSA
bovine serum albumin
- cOA or ‘oleamide'
cis-9,10-octadecenoamide
- DMSO
dimethyl sulfoxide
- [3H]-EBOB
[propyl-2,3-3H]-ethynyl bicyclo-orthobenzoate
- FAAH
fatty acid amidohydrolase
- [3H]-GABA
γ-[2,3-3H]-aminobutyric acid
- NSAID
non-steroidal anti-inflammatory drug
- tOA
trans-9,10-octadecenoamide
References
- BASILE A.S., HANUS L., MENDELSON W.B. Characterisation of the Hypnotic Properties of Oleamide. Neuroreport. 1999;10:947–951. doi: 10.1097/00001756-199904060-00010. [DOI] [PubMed] [Google Scholar]
- BEAUMONT K. , CHILTON W.S., YAMAMURA H.I., ENNA S.J. Muscimol binding in rat brain:association with synaptic GABA receptors. Brain Res. 1978;148:153–162. doi: 10.1016/0006-8993(78)90385-2. [DOI] [PubMed] [Google Scholar]
- BELELLI D., LAMBERT J.J., PETERS J.A., WAFFORD K., WHITING P.J. The interaction of the general anaesthetic etomidate γ-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELELLI D., PISTIS M., PETERS J.A., LAMBERT J.J. General anaesthetic action at transmitter-gated inhibitory amino acid. Trends Pharmacol. Sci. 1999a;20:496–502. doi: 10.1016/s0165-6147(99)01405-4. [DOI] [PubMed] [Google Scholar]
- BELELLI D., PISTIS M., PETERS J.A., LAMBERT J.J. The interaction of general anaesthetics and neurosteroids with GABAA and Glycine Receptors. Neurochem. Int. 1999b;34:447–452. doi: 10.1016/s0197-0186(99)00037-6. [DOI] [PubMed] [Google Scholar]
- BLOOMQUIST J.R., ADAMS P.M., SODERLUND D.M. Inhibition of γ-aminobutyric acid-stimulated chloride flux in mouse brain vesicles by polychlorocycloalkane and pyrethroid insecticides. NeuroToxicol. 1986;7:11–20. [PubMed] [Google Scholar]
- BOGER D.L., PATTERSON J.E., GUAN X., CRAVATT B.F., LERNER R.A., GILULA N.B. Chemical requirements for inhibition of gap junction communication by the biologically active lipid oleamide. Proc. Natl. Acad. Sci. U.S.A. 1998b;95:4810–4815. doi: 10.1073/pnas.95.9.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOGER D.L., PATTERSON J.E., JIN Q. Structural requirements for 5-HT2A and 5-HT1A serotonin receptor potentiation by the biologically active lipid oleamide. Proc. Nat. Acad. Sci. U.S.A. 1998a;95:4102–4107. doi: 10.1073/pnas.95.8.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORING D.L., BERGLUND B.A., HOWLETT A.C. Cerebrodiene, arachidonyl-ethanolamide, and hybrid structures: potential for interaction with brain cannabinoid receptors. Prostaglandins Leukot. Essent. Fatty Acids. 1996;55:207–210. doi: 10.1016/s0952-3278(96)90100-3. [DOI] [PubMed] [Google Scholar]
- CHEER J.F., CADOGAN A.K., MARSDEN C.A., FONE K.C.F., KENDALL D.A. Modification of 5-HT2 receptor mediated behaviour in the rat by oleamide and the role of cannabinoid receptors. Neuropharmacology. 1999;38:533–541. doi: 10.1016/s0028-3908(98)00208-1. [DOI] [PubMed] [Google Scholar]
- COLE L.M., CASIDA J.E. GABA-gated chloride channel: Binding site for 4//-ethynyl-4-n-[2,3-3H2]propylbicycloorthobenzoate ([3H]-EBOB) in vertebrate brain and insect head. Pestic. Biochem. Physiol. 1992;44:1–8. [Google Scholar]
- CRAVATT B.F., PROSPEROGARCIA O., SIUZDAK G., GILULA N.B., HENRIKSEN S.J., BOGER D.L., LERNER R.A. Chemical characterization of a family of brain lipids that induce sleep. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- DELORY T.M., KISSIN I., BROWN P., BROWN G.B. Barbiturate-benzodiazepine interactions at the gamma-aminobutyric acid-A receptor in rat cerebrocortical synaptoneurosomes. Anesth. & Analg. 1993;77:598–605. doi: 10.1213/00000539-199309000-00030. [DOI] [PubMed] [Google Scholar]
- EDWARDS M.D., LEES G. Modulation of a recombinant invertebrate gamma-aminobutyric acid receptor-chloride channel complex by isoflurane: effects of a point mutation in the M2 domain. Br. J. Pharmacol. 1997;122:726–732. doi: 10.1038/sj.bjp.0701417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANKS N.P., LIEB W.R. Molecular and Cellular Mechanisms of Anaesthesia. Nature. 1994;367:607–613. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- GOBBI M., MENNINI T., VALLE F., CERVO L., SALMONA M., DIOMEDE L. Oleamide-mediated sleep induction does not depend on pertubation of membane homeoviscosity. Febs. Letters. 1999;463:281–284. doi: 10.1016/s0014-5793(99)01630-0. [DOI] [PubMed] [Google Scholar]
- GOODNOUGH D.B., HAWKINSON J.E. Neuroactive steroid modulation of [3H]muscimol binding to the GABAA receptor complex in rat cortex. Eur J. Pharmacol. Mol. Pharmacol. Sect. 1995;288:157–162. doi: 10.1016/0922-4106(95)90190-6. [DOI] [PubMed] [Google Scholar]
- HALLIWELL R.F., THOMAS P., PATTEN D., JAMES C.H., MARTINEZTORRES A., MILEDI R., SMART T.G. Subunit-selective modulation of GABA(A) receptors by the non- steroidal anti-inflammatory agent, mefenamic acid. Eur. J. Neurosci. 1999;11:2897–2905. doi: 10.1046/j.1460-9568.1999.00709.x. [DOI] [PubMed] [Google Scholar]
- HARRIS R.A., ALLEN A.M. Functional coupling of γ-aminobutyric acid receptors to chloride channels in brain membranes. Science. 1985;228:1108–1100. doi: 10.1126/science.2581319. [DOI] [PubMed] [Google Scholar]
- HARRIS B.D., MOODY E.J., BASILE A.S., SKOLNICK P. Volatile anesthetics bidirectionally and stereospecifically modulate ligand binding to GABA receptors. Eur. J. Pharmacol. Mol. Pharm. Sect. 1994;15:269–274. doi: 10.1016/0922-4106(94)90150-3. [DOI] [PubMed] [Google Scholar]
- HANUS L.O., FALES H.M., SPANDE T.F., BASILE A.S. A gas chromatographic-mass spectral assay for the quantitative determination of oleamide in biological fluids. Anal. Biochem. 1999;270:159–166. doi: 10.1006/abio.1999.4083. [DOI] [PubMed] [Google Scholar]
- HUANG J., CASIDA J.E. Characterization of [3H]ethynylbicycloorthobenzoate ([3H]EBOB) binding and the action of insecticides on the γ-aminobutyric acid-gated chloride channel in cultured cerebellar granule neurons. J. Pharm. Exp. Ther. 1997;279:1191–1196. [PubMed] [Google Scholar]
- HUIDOBRO TORO J.P., HARRIS R.A. Brain lipids that induce sleep are novel modulators of 5-hydroxytryptamine receptors. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8078–8082. doi: 10.1073/pnas.93.15.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO Y., HO I.K. Studies on picrotoxin binding sites of GABA-A receptors in rat cortical synaptoneurosomes. Brain Res. Bull. 1994;33:373–378. doi: 10.1016/0361-9230(94)90279-8. [DOI] [PubMed] [Google Scholar]
- ITOUJI A., SAKAI N., TANAKA C., SAITO N. Neuronal and glial localization of two GABA transporters (GAT1 and GAT3) in the rat cerebellum. Mol. Brain Res. 1996;37:309–316. doi: 10.1016/0169-328x(95)00342-p. [DOI] [PubMed] [Google Scholar]
- IVERSEN L.L., NEAL M.J. The uptake of [3H]GABA by slices of rat cerebral cortex. J. Neurochem. 1968;15:1141–1149. doi: 10.1111/j.1471-4159.1968.tb06831.x. [DOI] [PubMed] [Google Scholar]
- KOENIG J.A., MARTIN I.L. Effect of free fatty acids on GABAA receptor ligand binding. Biochem. Pharmacol. 1992;44:11–15. doi: 10.1016/0006-2952(92)90031-d. [DOI] [PubMed] [Google Scholar]
- KOTCHINE V.V., QING Y.E., FINN S.E., HARRISON N.L. Chimeric GABAa/Glycine Receptors: Expression and Barbiturate Pharmacology. Neuropharmacology. 1996;35:1445–1456. doi: 10.1016/s0028-3908(96)00088-3. [DOI] [PubMed] [Google Scholar]
- KRASOWSKI M.D., HARRISON N.L. The actions of ether, alcohol and alkane general anaesthetics on GABA(A) and glycine receptors and the effects of TM2 and TM3 mutations. Br. J. Pharmacol. 2000;129:731–743. doi: 10.1038/sj.bjp.0703087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWRENCE L.J., PALMER C.J., GEE K.W., WANG X., YAMAMURA H.I. and , CASIDA J.E. t-[3H]Butylbicycloorthobenzoate: new radioligand probe for the γ-aminobutyric acid-regulated chloride ionophore. J. Neurochem. 1985;45:798–804. doi: 10.1111/j.1471-4159.1985.tb04063.x. [DOI] [PubMed] [Google Scholar]
- LAWS D., LEES G. Oleamide: a putative endogenous ligand for anaesthetic receptor sites. Br. J. Anaesth. 2001;87:172P. doi: 10.1093/bja/87.3.380. [DOI] [PubMed] [Google Scholar]
- LAWS D., VERDON B., COYNE L., LEES G. Fatty acid amides are putative endogenous ligands for anaesthetic recognition sites in mammalian CNS. Br. J. Anaesth. 2001;87:380–384. doi: 10.1093/bja/87.3.380. [DOI] [PubMed] [Google Scholar]
- LEES G., EDWARDS M.D., HASSONI A.A., GANELLIN C.R., GALANAKIS D. Modulation of GABA(A) receptors and inhibitory synaptic currents by the endogenous CNS sleep regulator cis-9,10-octadecenoamide (cOA) Br. J. Pharmacol. 1998a;124:873–882. doi: 10.1038/sj.bjp.0701918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEES G., WILLOTT R., EDWARDS M.D. Effects of isoflurane and cis-9,10-octadecenoamide (cOA) on the rho(1) GABA receptor. J. Physiol. (London) 1998b;509:P190–P191. [Google Scholar]
- LERNER R.A. A Hypothesis About the Endogenous Analogue of General Anaesthesia. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13375–13377. doi: 10.1073/pnas.94.25.13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN D.L., SMITH A.A. , III Ions and the transport of gamma-aminobutyric acid by synaptosomes. J. Neurochem. 1972;19:841–855. doi: 10.1111/j.1471-4159.1972.tb01398.x. [DOI] [PubMed] [Google Scholar]
- MEHTA A.K., TICKU M.K. An update on GABAA receptors. Brain Research Reviews. 1999;29:196–217. doi: 10.1016/s0165-0173(98)00052-6. [DOI] [PubMed] [Google Scholar]
- MENDELSON W.B., BASILE A.S. The hypnotic actions of oleamide are blocked by a cannabinoid receptor antagonist. Neuroreport. 1999;10:3237–3239. doi: 10.1097/00001756-199910190-00021. [DOI] [PubMed] [Google Scholar]
- MIHIC S.J., YE Q., WICK M.J., KOLTCHINE V.V., KRASOWSKI M.D., FINN S.E., MASCIA M.P., VALENZUELA C.F., HANSON K.K., GREENBLATT E.P., HARRIS R.A., HARRISON N.L. Sites of alcohol and volatile anaesthetic action on GABA and glycine receptors. Nature. 1997;389:385–388. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- NEGRO M., CHINCHETRU M.A., FERNANDEZ A., CALVO P. Effect of ethanol treatment on rate and equilibrium constants for [3H]muscimol binding to rat brain membranes: alteration of two affinity states of the GABAA receptor. J. Neurochem. 1995;64:1379–1389. doi: 10.1046/j.1471-4159.1995.64031379.x. [DOI] [PubMed] [Google Scholar]
- NICHOLSON R.A., LEES G., ZHENG J., VERDON B. Inhibition of GABA-gated chloride channels by 12,14-dichlorodehydroabietic acid in mammalian brain. Brit. J. Pharmacol. 1999;126:1123–1132. doi: 10.1038/sj.bjp.0702419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLSON R.A., ZHENG J., GANELLIN C.R., VERDON B., LEES G. Anaesthetic-like interaction of the sleep inducing lipid oleamide with voltage-gated sodium channels in mammalian brain. Anesthesiology. 2001;94:120–128. doi: 10.1097/00000542-200101000-00022. [DOI] [PubMed] [Google Scholar]
- PASCOE P.A. Drugs and the sleep-wakefulness continuum. Pharmacol. Ther. 1994;61:227–236. doi: 10.1016/0163-7258(94)90064-7. [DOI] [PubMed] [Google Scholar]
- PETERSON G.L. A simplification of the protein assay of Lowry et al. which is more generally applicable. Anal. Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- RAMANJANEYULU R., TICKU M.K. Binding characteristics and interactions of depressant drugs with [35S]t-butylbicyclophosphorothionate, a ligand that binds to the picrotoxin in site. J. Neurochem. 1984;42:221–229. doi: 10.1111/j.1471-4159.1984.tb09721.x. [DOI] [PubMed] [Google Scholar]
- SAKURAI S.Y., KUME A., BURDETTE D.E., ABIN R.L. Quantitative autoradiography of [3H]t-butylbicycloorthobenzoate binding to the gamma-aminobutyric acid receptor-A complex. J. Pharmacol. Exp. Ther. 1994;270:362–370. [PubMed] [Google Scholar]
- SOJA P.J., LOPEZRODRIGUEZ F., MORALES F.R., CHASE M.H. The postsynaptic inhibitory control of lumbar motoneurons during the atonia of active sleep–effect of strychnine on motoneuron properties. J. Neurosci. 1991;11:2804–2811. doi: 10.1523/JNEUROSCI.11-09-02804.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SQUIRES R.F., CASIDA J.E., RICHARDSON M., SAEDERUP E. [35S]t-Butylbicyclophosphorothionate binds with high affinity to brain-specific sites coupled to γ-aminobutyric acid-A and ion recognition sites. Mol. Pharmacol. 1983;23:326–336. [PubMed] [Google Scholar]
- SUZDAK P. Lipophilic GABA uptake inhibitors: biochemistry, pharmacology and therapeutic potential. Drugs of the Future. 1993;18:1129–1136. [Google Scholar]
- TECOTT L.H. Designer genes and anti-anxiety drugs. Nature Neuroscience. 2000;3:529–530. doi: 10.1038/75692. [DOI] [PubMed] [Google Scholar]
- THOMSEN C., SORENSEN P.O., EJEBJERG J. 1-(3-(9H-Carbazol-9-yl)-1-propyl)-4-(2-methoxyphenyl)-4-piperinol, a novel subtype selective inhibitor of the mouse type II GABA-transporter. Brit. J. Pharmacol. 1997;120:983–985. doi: 10.1038/sj.bjp.0700957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON S.A., WHITING P.J., WAFFORD K. Barbiturate Interactions at the Human GABAA Receptor: Dependence on Receptor Subunit Combination. Bri. J. Pharmacol. 1996;117:521–527. doi: 10.1111/j.1476-5381.1996.tb15221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURNER D.M., RANSOM R.W., YANG J. S.-J., OLSEN R.W. Steroid anesthetics and naturally occurring analogs modulate the γ-aminobutyric acid receptor complex at a site distinct from barbiturates. J. Pharmacol. Exp. Ther. 1989;248:960–966. [PubMed] [Google Scholar]
- VERDON B., ZHENG J., NICHOLSON R.A., GANELLIN C.R., LEES G. Stereoselective modulatory actions of oleamide on GABA(A) receptors and voltage-gated Na+ channels in vitro: a putative endogenous ligand for depressant drug sites in CNS. Br. J. Pharmacol. 2000;129:283–290. doi: 10.1038/sj.bjp.0703051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAFFORD K.A., BAIN C.J., QUIRK K., MCKERNAN R.M., WINGROVE P.B., WHITING P.J., KEMP J.A. A novel allosteric modulatory site on the GABAA receptor β subunit. Neuron. 1994;12:775–782. doi: 10.1016/0896-6273(94)90330-1. [DOI] [PubMed] [Google Scholar]
- WAFFORD K.A., BURNETT D.M., LEIDENHEIMER N.J., BURT D.R., WANG J.B., KOFUJI P., DUNWIDDIE T.V., HARRIS R.A., SIKELA J.M. Ethanol sensitivity of the GABAA receptor expressed in xenopus oocytes requires 8 amino acids contained in the γ2L subunit. Neuron. 1991;7:27–33. doi: 10.1016/0896-6273(91)90071-7. [DOI] [PubMed] [Google Scholar]
- WILSON R.I., NICOLL R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- YOST C.S., HAMPSON A.J., LEONOUDAKAS D., KOBLIN D.D., BORNHEIM L.M., GRAY A.T. Oleamide potentiates benzodiazepine-sensitive gamma-aminobutyric acid receptor activity but does not alter minimum alveolar anaesthetic concentration. Anesth. Analg. 1998;86:1294–1300. doi: 10.1097/00000539-199806000-00031. [DOI] [PubMed] [Google Scholar]