

Abstract
This study characterizes the mouse β3a-adrenoceptor (AR) and the splice variant of the β3-AR (β3b-AR) expressed in Chinese hamster ovary cells (CHO-K1).
Stable clones with high (∼1200), medium (∼500) or low receptor expression (∼100 fmol mg protein−1) were determined by saturation binding with [125I]-(−)-cyanopindolol. Competition binding studies showed no significant differences in affinity of β-AR ligands for either receptor.
Several functional responses of each receptor were measured, namely extracellular acidification rate (EAR; cytosensor microphysiometer), cyclic AMP accumulation, and Erk1/2 phosphorylation. The β3-AR agonists BRL37344, CL316243, GR265162X, L755507, SB251023, the non-conventional partial β-AR agonist CGP12177 and the β-AR agonist (−)-isoprenaline caused concentration-dependent increases in EAR in cells expressing either splice variant. CL316243 caused concentration-dependent increases in cyclic AMP accumulation and Erk1/2 phosphorylation in cells expressing either receptor.
PTX treatment increased maximum EAR and cyclic AMP responses to CL316243 in cells expressing the β3b-AR but not in cells expressing the β3a-AR at all levels of receptor expression.
CL316243 increased Erk1/2 phosphorylation with pEC50 values and maximum responses that were not significantly different in cells expressing either splice variant. Erk1/2 phosphorylation was insensitive to PTX or H89 (PKA inhibitor) but was inhibited by LY294002 (PI3Kγ inhibitor), PP2 (c-Src inhibitor), genistein (tyrosine kinase inhibitor) and PD98059 (MEK inhibitor).
The adenylate cyclase activators forskolin or cholera toxin failed to increase Erk1/2 levels although both treatments markedly increased cyclic AMP accumulation in both β3a- or β3b-AR transfected cells.
These results suggest that in CHO-K1 cells, the β3b-AR, can couple to both Gs and Gi to stimulate and inhibit cyclic AMP production respectively, while the β3a-AR, couples solely to Gs to increase cyclic AMP levels. However, the increase in Erk1/2 phosphorylation following receptor activation is not dependent upon coupling of the receptors to Gi or the generation of cyclic AMP.
Keywords: β3-Adrenoceptor, splice variant, cytosensor microphysiometer, Erk1/2, cyclic AMP, mouse
Introduction
Although not universal, alternative splicing of transcripts encoding GPCRs has the potential to diversify the number of receptor subtypes beyond those encoded by distinct genes. Many GPCRs have isoforms with differing C-terminal tails. These include the prostaglandin EP3 receptor (Namba et al., 1993; Hasegawa et al., 1996; Jin et al., 1997; Irie et al., 1993), α1A-AR (Chang et al., 1998; Hirasawa et al., 1995), serotonin 5HT4 (Gerald et al., 1995) and 5HT7 receptors (Heidmann et al., 1997), and the somatostatin SSTR2 receptor (Vanetti et al., 1993; Schindler et al., 1998). Most splice variants share similar pharmacology, but some show marked differences in signalling properties. For instance, there are four splice variants of the EP3 receptor, and each variant couples to a different set of G-proteins (Namba et al., 1993; Irie et al., 1993).
The mouse β3-adrenoceptor (AR) gene contains two introns, both of which undergo alternate splicing (van Spronsen et al., 1993; Granneman & Lahners, 1995; Evans et al., 1999). Splicing of intron A at a novel acceptor site 100 bp upstream from the previously characterized start of exon 2 results in the production of mRNA encoding a β3-AR variant, termed the β3b-AR. This receptor has a unique C-terminal tail, with 17 amino acids (SSLLREPRHLYTCLGYP) that differ from the 13 in the known β3a-AR (RFDGYEGARPFPT). There is differential expression of the two isoforms in mouse tissues, with lowest proportion of β3b-AR transcripts in brown adipose tissue (BAT), and the highest in hypothalamus (Evans et al., 1999). Previous studies in mice have shown that alternate splice acceptor sites result in the generation of two β3-AR transcripts that differ in the 3′ untranslated regions (van Spronsen et al., 1993: Granneman & Lahners, 1995). These transcripts have differing tissue expression in WAT and BAT, with the shorter transcript predominant in WAT, and the longer transcript in BAT. The use of primers in the 3′ untranslated region demonstrated a total of five transcripts of the mouse β3-AR (Evans et al., 1999).
β3-ARs couple to Gs to increase cyclic AMP levels but also couple to Gi proteins and c-Src, resulting in activation of the MAP kinase pathway and phosphorylation of Erk1/2. Recent studies have been performed in several cell systems including adipocyte-like cell lines that express endogenous β3-ARs (3T3-F442A; Soeder et al., 1999; C3H10T1/2; Cao et al., 2000) and in cells transfected with the human (Gerhardt et al., 1999; Soeder et al., 1999) or mouse (Cao et al., 2000) β3-AR. These studies all demonstrated β3-AR mediated Erk1/2 phosphorylation that occurred via a pertussis toxin (PTX) sensitive Gi/o protein but was independent of adenylate cyclase/cyclic AMP-dependent protein kinase A (PKA). However another study (Lindquist et al., 2000) using mouse brown adipocytes in primary culture showed that this Gi/o pathway is not always necessary for β3-AR activation of Erk1/2 phosphorylation, as in this cell type activation was mediated via the cyclic AMP/PKA pathway and was not affected by PTX treatment.
In this study we have characterized the pharmacological properties of the mouse β3a-AR and β3b-AR expressed in CHO-K1 cells. Both receptors share similar pharmacological properties with regard to affinities and potencies of β-AR ligands in radioligand binding and functional assays. Stimulation of either receptor with CL316243 increases cyclic AMP production, extracellular acidification rate (EAR) and Erk1/2 phosphorylation. Whereas the β3a-AR increases cyclic AMP and extracellular acidification by signalling through Gs, the β3b-AR exerts its effects by signalling through both Gs and Gi/o. Both receptors increase Erk1/2 phosphorylation via a mechanism independent of their effect on Gi or on cyclic AMP levels.
Methods
Expression of the mouse β3a- and β3b-AR inCHO-K1 cells
We generated inserts carrying the coding region of the β3a- and β3b-AR by RT – PCR on brown adipose tissue RNA, using high-fidelity polymerase (Expand High Fidelity PCR System, Roche). The primers used were, forward (common to β3a- and β3b-AR): 5′-GGAAGCTTCCCACCCCAGGC-3′, reverse (β3a-AR): 5′-GAATCTAGATTCCTTGCTGGATCTTCACGG-3′, and reverse (β3b-AR): 5′-CCTTCTAGAGAGAGCGGGACTGAGGC-3′, and included HindIII or XbaI sites for subcloning fragments into the mammalian expression vector pcDNA3.1+ (Invitrogen). The complete inserts and junctions with pcDNA3.1+ were checked by DNA sequencing on both strands (Micromon, Monash University, Australia). Plasmids were linearized with ScaI prior to transfection. Fifteen μg of each plasmid was transfected into 5×106 CHO-K1 cells by electroporation (270 V, 960 μF) in a Bio-Rad Gene Pulser II. The cells were grown for 48 h, then stable transformants were selected in medium containing 800 μg ml−1 G418. Clonal cell lines were obtained by limiting dilution of mixed cell populations, and were expanded and analysed by a single point [125I]-(−)cyanopindolol (ICYP, 800 pM) binding screen. Suitable clones were grown further for a full saturation binding analysis.
Cell culture and treatments
CHO-K1 cells were grown as monolayers in 50 : 50 Dulbecco's modified Eagle Medium (DMEM): Ham's F-12 medium containing 10% (v v−1) foetal bovine serum (FBS), glutamine (2 mM), penicillin (100 units ml−1) and streptomycin (100 μg ml−1). Clonal CHO-K1 lines transfected with the β3a- or β3b-AR were grown in the above media, but with the addition of G418 (400 μg ml−1). All cells were maintained under 5% CO2 at 37°C and cells passaged every 3 – 4 days. In experiments where cells were pre-treated with specific agents, concentration and time of treatment is indicated with the data.
Radioligand binding assay
Cells were grown to 95% confluence as a monolayer before membranes were harvested for binding studies. Cells were washed twice with HBS (10 mM HEPES, 150 mM NaCl, pH 7.4 room temperature), and scraped from flasks with lysis buffer (25 mM Tris pH 7.5 room temperature, 1 mM EDTA, 10 mg ml−1 bacitracin, 10 mg ml−1 leupeptin, 10 mg ml−1 pepstatin A, 0.5 mg ml−1 aprotinin). Cells were homogenized with a Dounce homogeniser (approximately 10 strokes per pestle), and centrifuged at low speed (800×g, 10 min) to remove cell debris. The supernatant containing membranes were retained and the pellet re-homogenized and centrifuged again. Supernatants were pooled and centrifuged (39,000×g, 15 min, 4°C). The pellet was homogenized in binding buffer (50 mM Tris pH 7.4 room temperature, 5 mM MgCl2, 1 mM EDTA, 10 mg ml−1 bacitracin, 10 mg ml−1 leupeptin, 10 mg ml−1 pepstatin A, 0.5 mg ml−1 aprotinin) and placed on ice for use on the same day.
Experiments were performed at room temperature in a volume of 100 μl of binding buffer in 96 well microtiter plates. Homogenate (∼10 – 20 μg protein) was incubated with ICYP (100 – 2000 pM) for 60 min in the absence or presence of (−)-alprenolol (1 mM) to define non-specific binding for saturation experiments. Competition experiments were performed using a range of concentrations of unlabelled drug using 500 pM ICYP. All reactions were terminated by rapid filtration through GF/C filters pre-soaked for 30 min in 0.5% polyethyleneimine using a Packard Cell Harvester. Filters were washed four times with wash buffer (50 mM Tris pH 7.4, 4°C), dried, 30 μl Microscint-O (Packard) added and radioactivity measured using a Packard Top Count. Experiments were performed in duplicate with n referring to the number of different membrane homogenate samples used.
Cytosensor microphysiometer studies
The cytosensor microphysiometer (Molecular Devices Corp., CA, U.S.A.) measures the cellular metabolic activity of isolated cells in terms of their rate of production of hydrogen ions. The basis of measurement involves the interaction of ligands with their respective receptor leading to a functional response requiring energy. Energy production and intracellular metabolic processes lead to the production and excretion of protons. The EAR is detected as a change in potential across a silicon light-addressable sensor during periods of cessation of flow of medium (McConnell et al., 1992). Cells were seeded into 12 mm transwell inserts (3 μm pore size) (Costar) at 5×105 cells per cup in media lacking FBS, and left to adhere overnight. Capsules were placed in sensor chambers in the cytosensor and maintained by a flow (100 μl min−1) of modified RPMI 1640 (Molecular Devices). The flow was stopped for 40 s at the end of each 2 min pump cycle and the rate of acidification (μ volts s−1) measured for 30 s during that period. Cells were superfused with media for a period of 2 h to stabilize baseline EAR before cumulative concentration – response curves to agonists were produced. Cells were exposed to each concentration of agonist for 8 min (isoprenaline) or 14 min (all other agonists used) until a stable state was achieved. All drugs were diluted in modified RPMI 1640. Baseline acidification rates were normalized using Cytosoft (Molecular Devices) prior to production of cumulative concentration – response curves. This allows comparison of results from different cell chambers in which the baseline EAR may be different due to cell density (where experiments are not performed on the same day; baseline values in cell chambers were generally similar in any given experiment performed on the same day) or other variations. The subsequent increases in EAR following agonist application to the cells is then expressed as a percentage over the increase of the normalized baseline rate. The metabolic stimulation is then normalized and scaled for each curve produced so that 0% represents the metabolic rate before addition of agonists and 100% represents the highest rate observed at the β3a-AR (elicited by the highest concentration of agonist used). In the case where cells are pretreated with PTX, 100% is defined as the maximum response produced by the highest concentration of agonist in control treated cells. All n values represent cells grown in different flasks before plating in transwell inserts, apart from experiments where cells were treated with PTX, where paired control and treated cells were obtained from the same flask of cells.
Cyclic AMP accumulation studies
Cells (1×105 per well) were grown in 12-well plates in DMEM/Ham's F-12 medium containing 0.5% FBS for 2 days. On the day of the experiment, medium was replaced with one containing IBMX (1 mM) and ascorbic acid (1 mM) for 2 h before cells were exposed to drugs for 30 min unless otherwise specified. The culture medium was aspirated, 0.8 ml ice cold 75% ethanol (containing 1 mM EDTA) added, cells scraped off, and the suspension transferred to microcentrifuge tubes before being dried in a Speedvac centrifuge. The dried samples were dissolved in 50 mM Tris pH 7.4 4°C (containing 1 mM EDTA) and sonicated for a few seconds. Cyclic AMP was measured using a commercial kit (Amersham Pharmacia TRK 432). All experiments were performed in duplicate with n referring to number of independent experiments performed.
Erk1/2 phosphorylation
Cells (1×105 per well) were grown in 12-well plates in DMEM/Ham's F-12 medium containing 0.5% FBS for 2 days, and the medium replaced 2 h before experiments were commenced. Cells were exposed to agonist for 5 min unless otherwise specified. Extraction of cells and subsequent Erk1/2 phosphorylation studies were performed as previously described (Lindquist et al., 2000). Primary antibodies used were phospho-p44/42 MAP kinase (Thr-202/Tyr-204) and p44/42 MAP kinase (Thr-202/Tyr-204) diluted 1 : 1000, detected using a secondary antibody (HRP linked anti-rabbit IgG) diluted 1 : 2000 and detected with the ECL kit (Amersham). Results are expressed as the ratio between phosphorylated and total Erk protein (α-p-Erk/α-Erk), with the ratio normalized in each experiment to that of control samples. All experiments were performed in duplicate with n referring to the number of independent experiments performed.
Data analysis
All results are expressed as mean±s.e.mean of n. Data was analysed using non-linear curve fitting (GraphPad PRISM version 2.0) to obtain pEC50 values (cytosensor microphysiometer, cyclic AMP and Erk1/2 experiments) or pKi values (competition experiments), or using a one-site fit to obtain KD and Bmax values (saturation experiments). Statistical significance was determined using two-way ANOVA tests or Student's t-test. Probability values less than or equal to 0.05 were considered significant.
Drugs and reagents
The following drugs were gifts: CL316243 (Dr T. Nash, Wyeth-Ayerst), SR59230A (Dr L. Manara, Sanofi-Midi), GR265162X (Dr Conrad Cowan, Glaxo-Wellcome), SB251023 (Dr Jon Arch, SmithKline Beecham), L755507 (Dr Mike Fisher, Merck), labetalol (Prof B. Jarrott, Monash University, Australia). Drugs and reagents were purchased as follows: BRL37344 (Tocris Cookson Ltd, Bristol, U.K.); G418, H89, PP2 (CalBiochem Corp, La Jolla, CA, U.S.A.); (−)-[125I]-cyanopindolol (2200 Ci mmol−1, NEN Life Science Products, Boston, MA, U.S.A.); ICI118551 (Imperial Chemical Industries, Wilmslow, Cheshire, UK); CGP12177A (Research Biochemicals Inc., MA, U.S.A.); CGP20712A (Ciba-Geigy AG, Australia); PTX (Gibco – BRL, Life Technologies, Gaithersburg, MD, U.S.A.); (−)-alprenolol, bacitracin, genistein, IBMX, (−)-isoprenaline, LY294002, (−)-norepinephrine, PD98059, polyethyleneimine, (−)-propranolol, (Sigma Chemical Company, St. Louis, MO, U.S.A.); aprotinin, leupeptin, pepstatin A (ICN, Costa Mesa, CA, U.S.A.); tertatolol (Servier Laboratories, Neuilly Sur Seine, France); bupranolol (Schwartz Pharma AG, Manheim, Germany). All cell culture medium and supplements were obtained from Trace Biosciences (Castle Hill, NSW, Australia). Antibodies were obtained from Cell Signalling Technology (Beverly, MA, U.S.A.). All other drugs and reagents were of analytical grade.
Results
Radioligand binding studies
Stably transfected cells were chosen with three levels of expression as determined with saturation radioligand binding studies: high (β3a-AR Bmax 1118±48 fmol mg protein−1, KD 921±82 pM, n=3; β3b-AR Bmax 1243±53 fmol mg protein−1, KD 1321±103 pM, n=3 (two-way ANOVA n.s.)), medium (β3a-AR Bmax 587±97 fmol mg protein−1, KD 898±38 pM, n=3; β3b-AR Bmax 457±67 fmol mg protein−1, KD 853±24 pM, n=3 (two-way ANOVA ***P<0.001)),and low (β3a-AR Bmax 115±6 fmol mg protein−1, KD 344±48 pM, n=3; β3b-AR Bmax 101±5 fmol mg protein−1, KD 372±47 pM, n=3 (two-way ANOVA n.s.)) levels of expression. Non-specific binding represented ∼8%, ∼10% or ∼40% of the total binding measured in high, medium and low expressing clones respectively. The binding affinities (pKi) of β-AR ligands measured are shown in Table 1. The affinity values for all ligands examined were not statistically different between receptors (Student's t-test).
Table 1.
Binding affinities for β-AR ligands at the β3a- or β3b-AR. Values are mean pKi values±s.e.mean with n values in brackets

Cytosensor microphysiometer studies
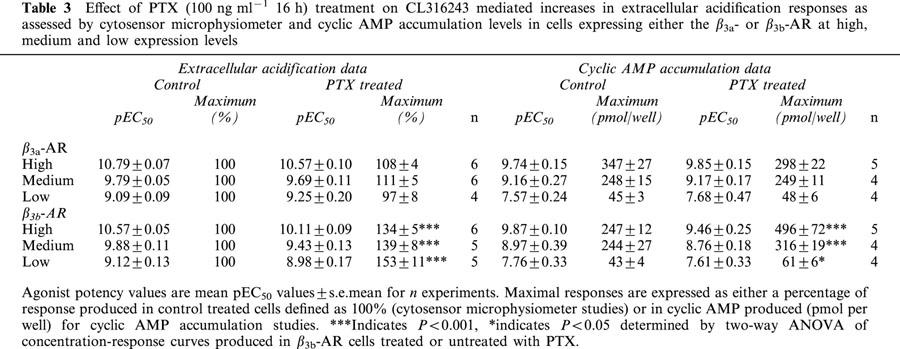
Exposure of CHO-K1 cells expressing high levels of the β3a- or β3b-AR to all β-AR ligands examined resulted in a concentration-dependent increase in the EAR relative to baseline acidification rates. Concentration-response curves to the non-selective β-AR agonist (−)-isoprenaline, β3-AR agonists (including the rodent (CL316243, BRL37344) and human (L755507) selective agonists), the β1-/β2-AR antagonist/β3-AR agonist CGP12177A, and to two new β3-AR agonists with reputedly high selectivity for the human β3-AR (GR265162X, SB251023) were measured (Figure 1). In all experiments performed, pEC50 values were similar for all agonists at both receptors when expressed at high levels, however maximal responses produced at the β3b-AR were significantly less than those at the β3a-AR as shown in Table 2 (two-way ANOVA ***P<0.001). The rank order of potency at both receptors was: BRL37344, CL316243, GR265162X>CGP12177A, L755507>SB251023, (−)-isoprenaline. Exposure of untransfected CHO-K1 cells to CL316243 (up to 10 μM) did not increase EAR (% increase of baseline acidification rate (defined as 100%): 10 μM CL316243 CHO-K1 cells, 102%; 10 nM CL316243 β3a-AR (high expressing cells), 218%). Additionally, when CL316243 concentration-response curves were produced in cells with all three expression levels in the same experiment, potency and efficacy of CL316243 was reduced when expression levels were reduced (data not shown). PTX treatment (100 ng ml−1 16 h) increased maximal responses to CL316243 in cells expressing the β3b-AR (two-way ANOVA ***P<0.001) but had no effect on responses in cells expressing the β3a-AR (two-way ANOVA n.s.) at all levels of cell expression examined (Figure 2; Table 3).
Figure 1.
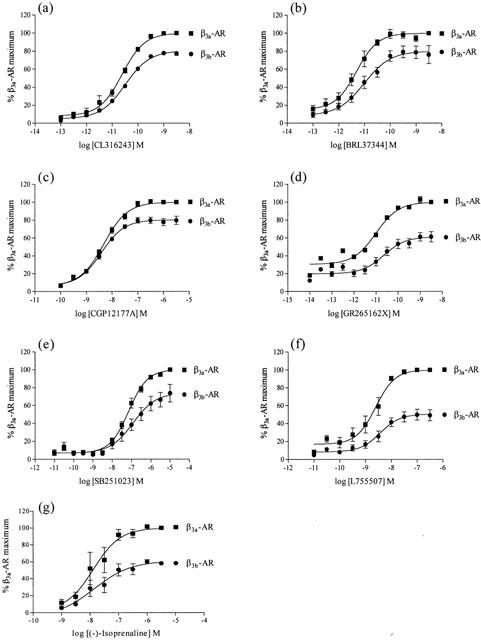
Concentration-response curves for CL316243 (a), BRL37344 (b), CGP12177A (c), GR265162X (d), SB251023 (e), L755507 (f) and (−)-isoprenaline (g) increasing EARs in the cytosensor microphysiometer in high expressing cells. Values represent means±s.e.mean obtained from 3 – 6 experiments. Responses from CHO-K1 cells expressing β3b-AR are presented as a percentage of the maximal response to the same agonist in cells expressing the β3a-AR. Note the difference in x-axis for graphs.
Table 2.
Agonist potencies at the mouse β3a- and β3b-AR expressed at high levels in CHO-K1 cells assessed using the cytosensor microphysiometer
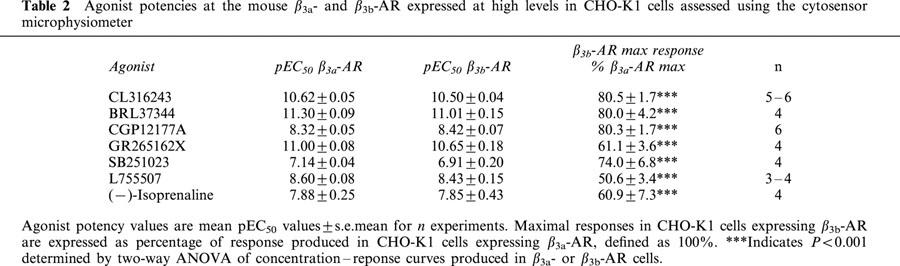
Figure 2.
Effect of PTX (100 ng ml−1 16 h) treatment on CL316243 extracellular acidification responses measured using the cytosensor microphysiometer in cells expressing either the β3a-AR (at high (a), medium (c) or low (e) receptor expression levels) or the β3b-AR (at high (b), medium (d) or low (f) receptor expression levels). Values represent means±s.e.mean from 4 – 6 experiments. Data is presented as a percentage of the maximal responses to the highest concentration of CL316243 in each experiment in control treated cells. Note that PTX treatment increased responses only in cells expressing β3b-AR.
Table 3.
Effect of PTX (100 ng ml−1 16 h) treatment on CL316243 mediated increases in extracellular acidification responses as assessed by cytosensor microphysiometer and cyclic AMP accumulation levels in cells expressing either the β3a- or β3b-AR at high, medium and low expression levels
Cyclic AMP accumulation studies
As seen with the cytosensor microphysiometer, cyclic AMP accumulation studies showed that in cells with high levels of receptor expression, responses produced by CL316243 at the β3b-AR were significantly decreased in comparison to those produced in cells expressing the β3a-AR (β3a-AR maximal response 100%; β3b-AR maximal response 75.16±4.95%; two-way ANOVA ***P<0.0001). PTX treatment (100 ng ml−1 16 h) increased responses in cells expressing the β3b-AR (two-way ANOVA ***P<0.001 high and medium expressing clones, *P<0.05 low expressing clones) while not affecting CL316243 responses in cells expressing the β3a-AR (two-way ANOVA n.s.) at all levels of receptor expression examined (Figure 3; Table 3). CL316243 (10 μM) did not increase cyclic AMP levels in untransfected CHO-K1 cells (Figure 4).
Figure 3.
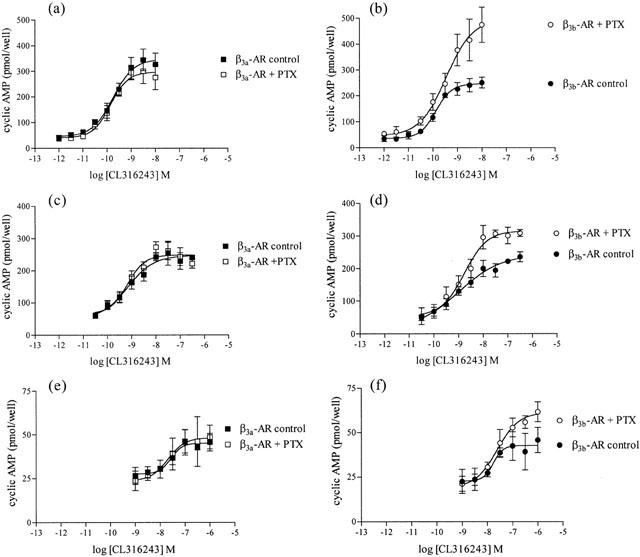
Effect of PTX (100 ng ml−1 16 h) treatment on CL316243 mediated increases in cyclic AMP accumulation levels in cells expressing either the β3a-AR (at high (a), medium (c) or low (e) levels of expression) or the β3b-AR (at high (b), medium (d) or low (f) levels of expression). Values represent means±s.e.mean obtained from 4 – 5 experiments performed in duplicate. Data is presented as the amount of cyclic AMP accumulated (pmol per well) following CL316243 treatment. Note that PTX treatment increased maximal cyclic AMP responses only in cells expressing β3b-AR. Note the difference in y-axis scale.
Figure 4.
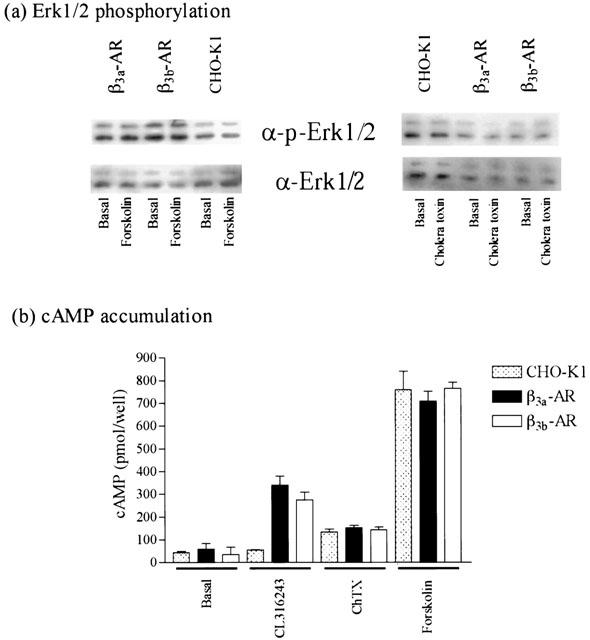
Effect of cholera toxin and forskolin treatment on (a) Erk1/2 phosphorylation and (b) cyclic AMP accumulation in transfected and untransfected CHO-K1 cells. Values represent means±s.e.mean obtained from three experiments performed in duplicate. Immunoblots are representative of three experiments performed in duplicate.
Signalling pathways contributing to Erk1/2 phosphorylation
All experiments examining CL316243 mediated increases in Erk1/2 phosphorylation levels were conducted in high expressing cells. CL316243 caused phosphorylation of Erk1/2 in a concentration-dependent manner (Figure 5) with pEC50 values of 7.28±0.26 and 7.36±0.24 in cells expressing the β3a- or β3b-AR respectively (n=3; two-way ANOVA n.s.), while having no effect in untransfected CHO-K1 cells (Figure 5). PTX (100 ng ml−1 16 h) treatment had no significant effect on CL316243 (10 μM) mediated increases in Erk1/2 phosphorylation in cells expressing either the β3a- or β3b-AR (Student's t-test n.s.). In cells expressing either receptor, the increase in Erk1/2 phosphorylation by CL316243 was not affected by treatment with H89 (PKA inhibitor, 10 μM, 1 h (Student's t-test n.s.), no effect also at 50 μM (data not shown)), but was inhibited by LY294002 (PI3Kγ inhibitor, 10 μM, 1 h; Student's t-test *P<0.05), PP2 (c-Src inhibitor, 10 μM, 1 h; Student's t-test *P<0.05), genistein (tyrosine kinase inhibitor, 50 μM, 1 h; Student's t-test *P<0.05) (Figure 6) and PD98059 (MEK inhibitor, 50 μM, 1 h, data not shown; Student's t-test *P<0.05). Treatment of untransfected or β3a- or β3b-AR transfected CHO-K1 cells with forskolin (10 μM, 5 min) or cholera toxin (2 μg ml−1, 90 min) produced no increase in Erk1/2 levels, although forskolin (10 mM, 30 min) and cholera toxin (2 μg ml−1, 90 min) markedly increased cyclic AMP accumulation in these cells (Figure 4). Cyclic AMP accumulation experiments were performed in parallel with Erk1/2 phosphorylation studies to assess the effects of these treatments on cyclic AMP levels. LY294002, H89, PP2, PD98059 and genistein treatment had no significant effect on the ability of CL316243 to increase cyclic AMP levels in cells expressing either receptor (data not shown), while PTX treatment increased cyclic AMP levels in cells expressing the β3b-AR but not the β3a-AR.
Figure 5.
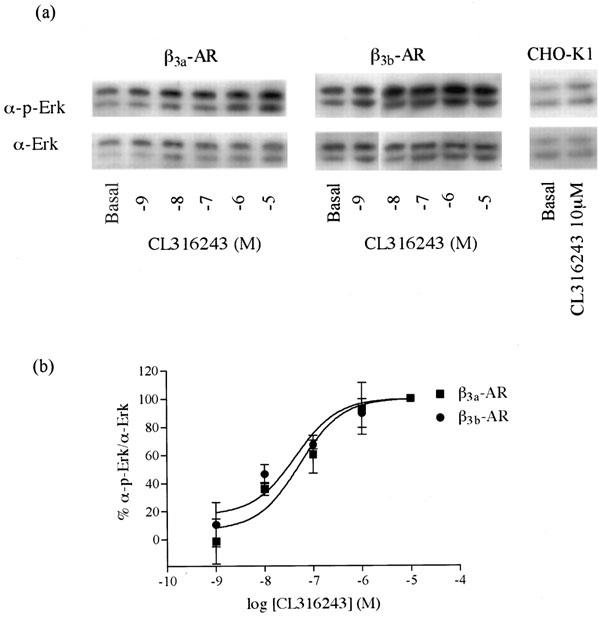
Concentration – response curve for Erk1/2 activation by CL316243 in high expression level cells expressing either the β3a-AR or β3b-AR. Values represent means±s.e.mzean obtained from three experiments performed in duplicate. Data is presented as percentage of the maximal responses to 10μM CL316243 at each receptor.
Figure 6.
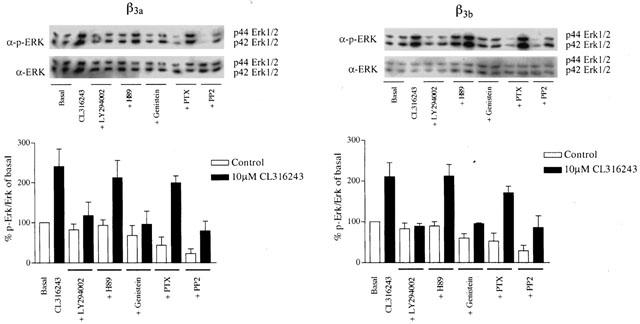
Effect of signal transduction inhibitors on Erk1/2 phosphorylation levels following CL316243 (10 μM, 5 min) stimulation in cells expressing either the β3a-AR (left panels) or the β3a-AR (right panels). Cells were treated either with PTX (100 ng ml−1 16 h), or genistein (50 μM), or H89 (10 μM), or PP2 (10 μM), or LY294002 (10 μM) for 1 h. Values represent means±s.e.mean obtained from four experiments performed in duplicate. Data is presented is a percentage of the response in control cells. ADP ribosylation of Gi by PTX or PKA inhibition by H89 failed to inhibit Erk1/2 phosphorylation following agonist stimulation of cells expressing either β3a- or β3b-AR.
Discussion
Alternate splicing of genes encoding GPCRs results in isoforms differing at their C-terminus that share similar pharmacological properties, but may show differences in distribution profiles and signalling properties. Here, we compare the pharmacology and signalling properties of mouse β3-AR splice variants (Evans et al., 1999) expressed in CHO-K1 cells. There was no difference in the affinity of β-AR ligands between the β3a- and β3b-AR, consistent with the isoforms differing at the C-terminus and not in the transmembrane regions that form the β3-AR ligand-binding pocket (Guan et al., 1995; Gros et al., 1998; Granneman et al., 1998). The rank order of potency of β-AR agonists assessed using the cytosensor microphysiometer was identical in cells expressing either receptor at high levels. The potency and efficacy of the β3-AR selective agonist CL316243 was reduced with levels of receptor expression for both the β3a- and β3b-AR, as assessed by cyclic AMP accumulation and cytosensor microphysiometer studies. This observation is consistent with standard drug-receptor interaction models, both in theoretical (Kenakin, 1995a) and practical applications (Wilson et al., 1996; McDonnell et al., 1998; Cordeaux et al., 2000). In the cytosensor microphysiometer, all β-AR agonists caused a larger maximal response with β3a-AR compared to β3b-AR, but the pEC50 values were similar at each receptor. This indicated that differential coupling of each receptor to intracellular signalling pathways and not differences in expression levels is responsible for the observed difference in maximal response.
β3-ARs are known to couple to Gs and activate adenylate cyclase, increasing intracellular cyclic AMP levels. The β3-AR agonist CL316243 increased intracellular cyclic AMP in a concentration-dependent manner in β3a- or β3b-AR cells, suggesting coupling of both receptors to Gs. However, as with the cytosensor microphysiometer, maximal cyclic AMP responses in β3b-AR cells were lower (by 25 – 30%) than in β3a-AR cells. It is known that the β3-AR can couple to Gi (Chaudhry et al., 1994; Soeder et al., 1999) and in 3T3-F442A adipocytes endogenously expressing β3-ARs, PTX treatment enhances cyclic AMP accumulation in response to CL316243 (Soeder et al., 1999). Brown adipocytes in primary culture also show a 50% increase in BRL37344-stimulated cyclic AMP production in the presence of PTX (Lindquist et al., 2000). We find that CL316243-mediated cyclic AMP or cytosensor responses are increased following PTX treatment in β3b-AR but not β3a-AR cells. The protocol used for PTX treatment in the present studies has been used extensively in previous studies with β3-AR (Cao et al., 2000; Gerhardt et al., 1999; Soeder et al., 1999) and was clearly effective in demonstrating the difference between β3a- and β3b-AR. Facilitation of cyclic AMP responses in 3T3-F442A cells or brown adipocytes by PTX treatment may therefore indicate that a fraction of the response to agonist treatment is mediated by endogenous β3b-ARs. Although unphysiological promiscuous receptor-second messenger coupling can occur at high receptor expression levels (Eason et al., 1992; Kenakin, 1995a, 1995b; Cordeaux et al., 2000), it is unlikely to be a factor in the medium (∼500 fmol mg protein−1) and low (∼100 fmol mg protein−1) expressing cells used here since these receptor levels are similar to physiological levels reported in rat white adipocytes (∼400 – 600 fmol mg protein−1; Germack et al., 1996), rat brown adipocytes (∼430 fmol mg protein−1; Sillence et al., 1993), and mouse ileum (∼60 and ∼150 fmol mg protein−1; Hutchinson et al., 2000 and unpublished data respectively).
We find that treatment of CHO-K1 cells expressing either β3a- or β3b-ARs with CL316243 increases Erk1/2 phosphorylation by 2 fold in a concentration-dependent manner, similar to other studies using CL316243 (Gerhardt et al., 1999; Soeder et al., 1999; Cao et al., 2000). The concentration-response curve for Erk1/2 activation by CL316243 in CHO-K1 cells expressing either receptor is shifted significantly to the right compared to cyclic AMP accumulation responses (over 100 fold shift), compared to 30 fold shift reported by Soeder et al. (1999). This suggests that activation of Gs/adenylate cyclase/cyclic AMP occurs at low concentrations of agonist, but that Erk1/2 phosphorylation becomes significant at high levels of receptor occupancy. The present experiments showed that there was no difference of Erk1/2 activation between the β3a- or β3b-AR. This would rule out an involvement of Gi in Erk1/2 activation, and in addition this was further supported by the observation that Erk1/2 activation was insensitive to treatment with PTX. Both forskolin and cholera toxin did not activate Erk1/2 in both untransfected and transfected CHO-K1 cells suggesting that Erk1/2 activation is not dependent upon cyclic AMP or Gs. The PKA inhibitor H89 was also ineffective in preventing the activation of Erk1/2 by the β3-AR agonist CL316243 in cells transfected with either β3a- or β3b-AR. On the other hand activation of Erk1/2 was sensitive to the inhibitors of Src (PP2), PI3K (LY294002) and the general tyrosine kinase inhibitor genistein. The potential involvement of Gβγ subunits cannot be ruled out and the use of the C-terminal fragment of β-ARK may be helpful for future studies investigating the role of Gβγ subunits on Erk1/2 activation in this system. Hence the differences in the concentration-response curves observed for cyclic AMP accumulation and Erk1/2 activation may reflect a lack of involvement of Gα-proteins in the stimulation of Erk1/2 phosphorylation in this system.
Phosphorylation of Erk1/2 occurs in cell systems expressing endogenous or transfected β2- or β3-ARs. The mechanism by which this occurs, however, differs according to the receptor and the cell type. β2-AR mediated activation of Erk1/2 is linked to receptor phosphorylation, binding of β-arrestin, and subsequently binding of c-Src to the β-arrestin (Luttrell et al., 1999; Maudsley et al., 2000). There is no phosphorylation of the β3-AR with agonist stimulation (Liggett et al., 1993) and this receptor is unable to bind β-arrestin (Cao et al., 2000). However, activated β3-ARs bind c-Src directly via four motifs (PXXP) in the third intracellular loop and the C-terminus (Cao et al., 2000). Mutation of proline residues in these motifs prevents both c-Src binding and Erk1/2 phosphorylation. Activation of the Erk1/2 pathway via β3-ARs in mouse adipocytes is inhibited by the selective c-Src inhibitor, PP2 (Lindquist et al., 2000). We also found that Erk1/2 phosphorylation was inhibited by PP2 in β3a- or β3b-AR CHO-K1 cells.
In most cell systems, Erk1/2 phosphorylation is inhibited by PTX or the C-terminus of β-ARK, which sequesters Gβγ subunits. The involvement of Gi or Gβγ subunits derived from Gi in Erk1/2 phosphorylation differs between brown and white adipocytes. In brown adipocytes a Gs/PKA-dependent rap1/B-raf pathway mediates β3-AR stimulation of Erk1/2 activation (Lindquist et al., 2000), while in white adipocytes H89 has no effect and there is clear inhibition by PTX, indicating a requirement for Gi (Soeder et al., 1999). Activation of Erk1/2 by human β3-ARs expressed in HEK293 (Soeder et al., 1999) or CHO-K1 (Gerhardt et al., 1999) cells is sensitive to PTX but not H89, indicating a role for Gi but not Gs. We found that H89 had no effect on Erk1/2 phosphorylation in β3a- or β3b-AR CHO-K1 cells. Also, treatment of untransfected or transfected CHO-K1 cells with forskolin or cholera toxin failed to increase Erk1/2 levels. This suggests that activation of Erk1/2 in these cells is not dependent upon increases in cyclic AMP, consistent with some studies (Gerhardt et al., 1999; Ai et al., 1999) but not others (Verheijen & Defize, 1997). We have demonstrated coupling of the β3b-AR to Gi, but unlike CHO-K1 cells expressing the human β3-AR, PTX had no effect on Erk1/2 phosphorylation in response to CL316243 in β3b-AR CHO-K1 cells. These differences in the PTX sensitivity of Erk1/2 phosphorylation may reflect sequence differences between the mouse and human β3-ARs. Whereas the mouse β3-AR has four PXXP motifs for the binding of c-Src, the human receptor has only two. Weaker association of c-Src with the human β3-AR may increase the dependence of the MAPK signalling pathway on Gi/Gβγ. Finally, in CHO-K1 cells expressing human (Gerhardt et al., 1999) or mouse β3a- or β3b-ARs, Erk1/2 phosphorylation is blocked by LY294002 (PI3K inhibitor). PI3Ks are stimulated by Gβγ subunits (Gerhardt et al., 1999), although this is inconsistent with our observation that Erk1/2 phosphorylation was unaffected by PTX. So while a PI3K pathway is present, the mechanism of activation remains to be elucidated. Recent reports show that Gβγ subunits from Gs can couple to other intracellular signalling events such as isoprenaline-mediated β2-AR inhibition of NADPH-dependent H2O2 generation in human adipocytes (Krieger-Brauer et al., 2000). Thus mechanisms involving Gs may not universally require elevation of cyclic AMP and activation of PKA.
The β3a- and β3b-AR isoforms differ at the C-terminus. Motifs present in the C-termini of GPCRs, including PDZ-binding and PSD-95-binding domains in the β2- and β1-AR C-terminus respectively (Cao et al., 1999; Hu et al., 2000) are important for interactions with a range of targeting and recycling proteins. The C-terminal motifs in both these receptors show complete conservation amongst nine mammalian species, indicative of their critical importance. In contrast, the final amino acid residues following the putative palmitoylation site (Cys361) in the C-terminus of β3-ARs are much less conserved amongst the nine mammalian species examined. There is only one conserved PXXP motif that is consistently present in the β3-AR C-terminus. At the moment it is not possible to tell whether the secondary structure of the β3-AR C-terminus facilitates protein – protein interactions. Our hypothesis is that the altered amino acid sequence of the β3b-AR affects the conformation of the proximal region of the C-terminus and the third intracellular loop due to hydrogen bonding or other interactions involving amino acids uniquely present in the β3b-AR, or because of an additional cysteine in the β3b-AR that may affect palmitoylation of Cys361 and hence formation of the ‘fourth intracellular loop'.
The present study demonstrates that the β3a- and β3b-AR share similar affinity and potency for all β-AR ligands examined. However, in cyclic AMP and cytosensor microphysiometer studies, the β3b-AR coupled to both Gs and Gi, while the β3a-AR coupled solely to Gs. Increases in Erk1/2 phosphorylation following β3a- or β3b-AR activation were independent of receptor coupling to Gi, and involved a mechanism that includes activation of PI3Kγ and c-Src. Activation of Erk1/2 signalling by β-ARs and other GPCRs is clearly a complex process modulated by multiple signalling proteins that differ between cell types.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia, the Tore Nilsons Stiftelse for Medicinsk Forskening (Sweden) and the Jeanssonska funds (Sweden). D.S. Hutchinson is a Monash University Postgraduate Scholar and is supported by the Monash University Postgraduate Publications Award. The authors wish to record their appreciation for technical assistance provided by Maria Papaioannou and Johanna Lindquist for parts of this study.
Abbreviations
- β-AR
β-adrenoceptor
- BAT
brown adipose tissue
- CHO-K1
Chinese hamster ovary
- EAR
extracellular acidification rate: GPCR, G-protein coupled receptor
- Erk1/2
extracellular-signal regulated kinase 1/2
- ICYP
[125I]-(−)-cyanopindolol
- MAP
mitogen-activated protein
- PI3K
phosphatidylinositol 3-kinase
- PKA
cyclic AMP dependent protein kinase A
- PTX
pertussis toxin
- WAT
white adipose tissue
References
- AI W., GONG J., YU L. MAP kinase activation by mu opioid receptor involves phosphatidylinositol 3-kinase but not the cAMP/PKA pathway. FEBS Lett. 1999;456:196–200. doi: 10.1016/s0014-5793(99)00949-7. [DOI] [PubMed] [Google Scholar]
- CAO T.T., DEACON H.W., RECZEK D., BRETSCHER A., VON ZASTROW M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the β2-adrenergic receptor. Nature. 1999;401:286–290. doi: 10.1038/45816. [DOI] [PubMed] [Google Scholar]
- CAO W., LUTTRELL L.M., MEDVEDEV A.V., PIERCE K.L., DANIEL K.W., DIXON T.M., LEFKOWITZ R.J., COLLINS S. Direct binding of activated c-Src to the β3-adrenergic receptor is required for MAP kinase activation. J. Biol. Chem. 2000;275:38131–38134. doi: 10.1074/jbc.C000592200. [DOI] [PubMed] [Google Scholar]
- CHANG D.J., CHANG T.K., YAMANISHI S.S., SALAZAR F.H., KOSAKA A.H., KHARE R., BHAKTA S., JASPER J.R., SHIEH I.S., LESNICK J.D., FORD A.P., DANIELS D.V., EGLEN R.M., CLARKE D.E., BACH C., CHAN H.W. Molecular cloning, genomic characterization and expression of novel human α1A-adrenoceptor isoforms. FEBS Lett. 1998;422:279–283. doi: 10.1016/s0014-5793(98)00024-6. [DOI] [PubMed] [Google Scholar]
- CHAUDHRY A., MACKENZIE R.G., GEORGIC L.M., GRANNEMAN J.G. Differential interaction of β1- and β3-adrenergic receptors with Gi in rat adipocytes. Cell. Signall. 1994;6:457–465. doi: 10.1016/0898-6568(94)90093-0. [DOI] [PubMed] [Google Scholar]
- CORDEAUX Y., BRIDDON S.J., MEGSON A.E., MCDONNELL J., DICKENSON J.M., HILL S.J. Influence of receptor number on functional responses elicited by agonists acting at the human adenosine A(1) receptor: evidence for signalling pathway-dependent changes in agonist potency and relative intrinsic activity. Mol. Pharmacol. 2000;58:1075–1084. doi: 10.1124/mol.58.5.1075. [DOI] [PubMed] [Google Scholar]
- EASON M.G., KUROSE H., HOLT B.D., RAYMOND J.R., LIGGETT S.B. Simultaneous coupling of α2-adrenergic receptors to two G-proteins with opposing effects. Subtype-selective coupling of α2C10, α2C4, and α2C2 adrenergic receptors to Gi and Gs. J. Biol. Chem. 1992;267:15795–15801. [PubMed] [Google Scholar]
- EVANS B.A., PAPAIOANNOU M., HAMILTON S., SUMMERS R.J. Alternative splicing generates two isoforms of the β3-adrenoceptor which are differentially expressed in mouse tissues. Br. J. Pharmacol. 1999;127:1525–1531. doi: 10.1038/sj.bjp.0702688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERALD C., ADHAM N., KAO H.T., OLSEN M.A., LAZ T.M., SCHECHTER L.E., BARD J.A., VAYSSE P.J., HARTIG P.R., BRANCHEK T.A., WEINSHANK R.L. The 5-HT4 receptor: molecular cloning and pharmacological characterization of two splice variants. EMBO J. 1995;14:2806–2815. doi: 10.1002/j.1460-2075.1995.tb07280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERHARDT C.C., GROS J., STROSBERG A.D., ISSAD T. Stimulation of the extracellular signal-regulated kinase 1/2 pathway by human β3 adrenergic receptor: new pharmacological profile and mechanism of activation. Mol. Pharmacol. 1999;55:255–262. doi: 10.1124/mol.55.2.255. [DOI] [PubMed] [Google Scholar]
- GERMACK R., ADLI H., VASSY R., PERRET G.Y. Triiodothyronine and amiodarone effects on β3-adrenoceptor density and lipolytic response to the β3-adrenergic agonist BRL 37344 in rat white adipocytes. Fundam. Clin. Pharmacol. 1996;10:289–297. doi: 10.1111/j.1472-8206.1996.tb00308.x. [DOI] [PubMed] [Google Scholar]
- GRANNEMAN J.G., LAHNERS K.N. Regulation of mouse β3-adrenergic receptor gene expression and mRNA splice variants in adipocytes. Am. J. Physiol. 1995;268:C1040–C1044. doi: 10.1152/ajpcell.1995.268.4.C1040. [DOI] [PubMed] [Google Scholar]
- GRANNEMAN J.G., LAHNERS K.N., ZHAI Y. Agonist interactions with chimeric and mutant β1- and β3-adrenergic receptors: involvement of the seventh transmembrane region in conferring subtype specificity. Mol. Pharmacol. 1998;53:856–861. [PubMed] [Google Scholar]
- GROS J., MANNING B.S., PIETRI-ROUXEL F., GUILLAUME J.L., DRUMARE M.F., STROSBERG A.D. Site-directed mutagenesis of the human β3-adrenoceptor–transmembrane residues involved in ligand binding and signal transduction. Eur. J. Biochem. 1998;251:590–596. doi: 10.1046/j.1432-1327.1998.2510590.x. [DOI] [PubMed] [Google Scholar]
- GUAN X.M., AMEND A., STRADER C.D. Determination of structural domains for G protein coupling and ligand binding in β3-adrenergic receptor. Mol. Pharmacol. 1995;48:492–498. [PubMed] [Google Scholar]
- HASEGAWA H., NEGISHI M., ICHIKAWA A. Two isoforms of the prostaglandin E receptor EP3 subtype different in agonist-independent constitutive activity. J. Biol. Chem. 1996;271:1857–1860. doi: 10.1074/jbc.271.4.1857. [DOI] [PubMed] [Google Scholar]
- HEIDMANN D.E., METCALF M.A., KOHEN R., HAMBLIN M.W. Four 5-hydroxytryptamine7 (5-HT7) receptor isoforms in human and rat produced by alternative splicing: species differences due to altered intron-exon organization. J. Neurochem. 1997;68:1372–1381. doi: 10.1046/j.1471-4159.1997.68041372.x. [DOI] [PubMed] [Google Scholar]
- HIRASAWA A., SHIBATA K., HORIE K., TAKEI Y., OBIKA K., TANAKA T., MURAMOTO N., TAKAGAKI K., YANO J., TSUJIMOTO G. Cloning, functional expression and tissue distribution of human α1c-adrenoceptor splice variants. FEBS Lett. 1995;363:256–260. doi: 10.1016/0014-5793(95)00330-c. [DOI] [PubMed] [Google Scholar]
- HU L.A., TANG Y., MILLER W.E., CONG M., LAU A.G., LEFKOWITZ R.J., HALL R.A. β1-adrenergic receptor association with PSD-95. Inhibition of receptor internalization and facilitation of β1-adrenergic receptor interaction with N-methyl-D-aspartate receptors. J. Biol. Chem. 2000;275:38659–38666. doi: 10.1074/jbc.M005938200. [DOI] [PubMed] [Google Scholar]
- HUTCHINSON D.S., EVANS B.A., SUMMERS R.J. β3-Adrenoceptor regulation and relaxation responses in mouse ileum. Br. J. Pharmacol. 2000;129:1251–1259. doi: 10.1038/sj.bjp.0703160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IRIE A., SUGIMOTO Y., NAMBA T., HARAZONO A., HONDA A., WATABE A., NEGISHI M., NARUMIYA S., ICHIKAWA A. Third isoform of the prostaglandin-E-receptor EP3 subtype with different C-terminal tail coupling to both stimulation and inhibition of adenylate cyclase. Eur. J. Biochem. 1993;217:313–318. doi: 10.1111/j.1432-1033.1993.tb18248.x. [DOI] [PubMed] [Google Scholar]
- JIN J., MAO G.F., ASHBY B. Constitutive activity of human prostaglandin E receptor EP3 isoforms. Br. J. Pharmacol. 1997;121:317–323. doi: 10.1038/sj.bjp.0701121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENAKIN T. Agonist-receptor efficacy I: mechanisms of efficacy and receptor promiscuity. Trends Pharmacol. Sci. 1995a;16:188–192. doi: 10.1016/s0165-6147(00)89020-3. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Agonist-receptor efficacy II: agonist trafficking of receptor signals. Trends Pharmacol. Sci. 1995b;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- KRIEGER-BRAUER H.I., MEDDA P.K., SATTEL B., KATHER H. Inhibitory effect of isoprenaline on NADPH-dependent H2O2 generation in human adipocyte plasma membranes is mediated by βγ-subunits derived from Gs. J. Biol. Chem. 2000;275:2486–2490. doi: 10.1074/jbc.275.4.2486. [DOI] [PubMed] [Google Scholar]
- LIGGETT S.B., FREEDMAN N.J., SCHWINN D.A., LEFKOWITZ R.J. Structural basis for receptor subtype-specific regulation revealed by a chimeric β3/β2-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3665–3669. doi: 10.1073/pnas.90.8.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINDQUIST J.M., FREDRIKSSON J.M., REHNMARK S., CANNON B., NEDERGAARD J. β3- and α1-adrenergic Erk1/2 activation is Src but not Gi-mediated in brown adipocytes. J. Biol. Chem. 2000;275:22670–22677. doi: 10.1074/jbc.M909093199. [DOI] [PubMed] [Google Scholar]
- LUTTRELL L.M., DAAKA Y., LEFKOWITZ R.J. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr. Opin. Cell. Biol. 1999;11:177–183. doi: 10.1016/s0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- MAUDSLEY S., PIERCE K.L., ZAMAH A.M., MILLER W.E., AHN S., DAAKA Y., LEFKOWITZ R.J., LUTTRELL L.M. The β2-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J. Biol. Chem. 2000;275:9572–9580. doi: 10.1074/jbc.275.13.9572. [DOI] [PubMed] [Google Scholar]
- MCCONNELL H.M., OWICKI J.C., PARCE J.W., MILLER D.L., BAXTER G.T., WADA H.G., PITCHFORD S. The cytosensor microphysiometer: biological applications of silicon technology. Science. 1992;257:1906–1912. doi: 10.1126/science.1329199. [DOI] [PubMed] [Google Scholar]
- MCDONNELL J., LATIF M.L., REES E.S., BEVAN N.J., HILL S.J. Influence of receptor number on the stimulation by salmeterol of gene transcription in CHO-K1 cells transfected with the human β2-adrenoceptor. Br. J. Pharmacol. 1998;125:717–726. doi: 10.1038/sj.bjp.0702139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAMBA T., SUGIMOTO Y., NEGISHI M., IRIE A., USHIKUBI F., KAKIZUKA A., ITO S., ICHIKAWA A., NARUMIYA S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature. 1993;365:166–170. doi: 10.1038/365166a0. [DOI] [PubMed] [Google Scholar]
- SCHINDLER M., KIDD E.J., CARRUTHERS A.M., WYATT M.A., JARVIE E.M., SELLERS L.A., FENIUK W., HUMPHREY P.P. Molecular cloning and functional characterization of a rat somatostatin sst2(b) receptor splice variant. Br. J. Pharmacol. 1998;125:209–217. doi: 10.1038/sj.bjp.0702064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SILLENCE M.N., MOORE N.G., PEGG G.G., LINDSAY D.B. Ligand binding properties of putative β3-adrenoceptors compared in brown adipose tissue and in skeletal muscle membranes. Br. J. Pharmacol. 1993;109:1157–1163. doi: 10.1111/j.1476-5381.1993.tb13743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOEDER K.J., SNEDDEN S.K., CAO W., DELLA ROCCA G.J., DANIEL K.W., LUTTRELL L.M., COLLINS S. The β3-adrenergic receptor activates mitogen-activated protein kinase in adipocytes through a Gi-dependent mechanism. J. Biol. Chem. 1999;274:12017–12022. doi: 10.1074/jbc.274.17.12017. [DOI] [PubMed] [Google Scholar]
- VANETTI M., VOGT G., HOLLT V. The two isoforms of the mouse somatostatin receptor (mSSTR2A and mSSTR2B) differ in coupling efficiency to adenylate cyclase and in agonist-induced receptor desensitization. FEBS Lett. 1993;331:260–266. doi: 10.1016/0014-5793(93)80349-y. [DOI] [PubMed] [Google Scholar]
- VAN SPRONSEN A., NAHMIAS C., KRIEF S., BRIEND-SUTREN M.M., STROSBERG A.D., EMORINE L.J. The promoter and intron/exon structure of the human and mouse β3-adrenergic-receptor genes. Eur. J. Biochem. 1993;213:1117–1124. doi: 10.1111/j.1432-1033.1993.tb17861.x. [DOI] [PubMed] [Google Scholar]
- VERHEIJEN M.H.G., DEFIZE L.H.K. Parathyroid hormone activates mitogen-activated protein kinase via a cAMP-mediated pathway independent of Ras. J. Biol. Chem. 1997;272:3423–3429. doi: 10.1074/jbc.272.6.3423. [DOI] [PubMed] [Google Scholar]
- WILSON S., CHAMBERS J.K., PARK J.E., LADURNER A., CRONK D.W., CHAPMAN C.G., KALLENDER H., BROWNE M.J., MURPHY G.J., YOUNG P.W. Agonist potency at the cloned human β3-adrenoceptor depends on receptor expression level and nature of assay. J. Pharmacol. Exp. Ther. 1996;279:214–221. [PubMed] [Google Scholar]