

Abstract
Intestinal xenobiotic transporters are a significant barrier to the absorption of many orally administered drugs. P-glycoprotein (PGP) is the best known, but several others, including members of the multidrug resistance-associated protein (MRP) family, are also expressed. Definitive information on their precise effect on intestinal drug permeability is scarce due to a lack of specific inhibitors and the difficulty of studying non-PGP activity in the presence of high PGP expression.
We have investigated the in vitro use of intestinal tissues from PGP knockout (mdr1a (−/−)) mice as a tool for dissecting the mechanisms of intestinal drug efflux. The permeability characteristics of digoxin (DIG), paclitaxel (TAX) and etoposide (ETOP) were measured in ileum from mdr1a (−/−) and wild-type (FVB) mice mounted in Ussing chambers.
DIG and TAX exhibited marked efflux across FVB tissues (B-A : A-B apparent permeability (Papp) ratio 10 and 17 respectively) which was absent in mdr1a (−/−) tissues, confirming that PGP is the sole route of intestinal efflux for these compounds. The A-B Papp of both compounds was 3 – 5 fold higher in mdr1a (−/−) than in FVB.
Polarized transport of ETOP in FVB tissues was reduced but not abolished in mdr1a (−/−) tissues. Residual ETOP efflux in mdr1a (−/−) tissues was abolished by the MRP inhibitor MK571, indicating involvement of both PGP and MRP.
MK571 abolished calcein efflux in mdr1a (−/−) tissues, while quinidine had no parallel effect in FVB tissues, suggesting involvement of MRP but not PGP.
Tissues from mdr1a (−/−) mice provide a novel approach for investigating the influence of PGP ablation on intestinal permeability and for resolving PGP and non-PGP mechanisms that modulate drug permeability.
Keywords: P-glycoprotein, intestinal permeability, mdr1a (−/−) mouse, paclitaxel, etoposide, digoxin, multidrug resistance-associated protein
Introduction
The intestinal epithelium presents a significant barrier to the absorption of some orally administered drugs. One of the key components of this barrier is P-glycoprotein (PGP), a member of the ABC transporter family (Hunter & Hirst, 1997), that is coded for by the MDR1 gene in humans and by the mdr1a and mdr1b genes in rodents, only mdr1a being found in intestine (Croop et al., 1989). PGP is a polyspecific transporter that can pump a very broad range of substrates, including vinca alkaloids, anthracyclines, digoxin, epipodophyllotoxins and β-adrenergic agonists, into the gut lumen (Hunter & Hirst, 1997). In vivo studies confirm that PGP significantly limits the oral bio-availability of several drugs (Fromm, 2000). As a result, screening of drug candidates for PGP interaction is becoming a routine part of the drug development process.
There is, however, a growing awareness that other non-PGP transporters with the potential for drug efflux may also be expressed by the intestinal epithelium (Suzuki & Sugiyama, 2000). These include members of the multidrug resistance-associated protein (MRP) family and others, such as lung resistance protein (LRP) and the breast cancer resistance protein (BCRP) (Gotoh et al., 2000; Makhey et al., 1998; Jonker et al., 2000). Relatively little is known about the transport characteristics of LRP and BCRP. MRP type transporters, studied primarily in over-expressing cell models, have the capacity to transport many substances including common drugs, often following enzymatic conjugation with glutathione, glucuronide or sulphate, although some MRPs may also act as unconjugated drug/glutathione co-transporters (Rappa et al., 1997; Borst et al., 2000). Several members of the MRP family have been identified at the molecular level in intestinal tissues (Mottino et al., 2000; Peng et al., 1999; Gotoh et al., 2000) and there is evidence that at least one of these, MRP2, is functionally active in this tissue (Gotoh et al., 2000). However, their contribution to intestinal drug efflux remains poorly defined.
Identifying the relative contribution of PGP and non-PGP processes to intestinal drug efflux has proved problematic. This is due mainly to the considerable overlap in substrate specificity between different transporters and a lack of reliable and selective inhibitors. This makes it difficult to differentiate between transport activities in cells or tissues such as the intestinal epithelium where multiple transporters are co-expressed. In addition, the high ‘background' level of efflux by PGP may swamp contributions from less highly expressed transporters.
Transgenic animal models, in which specific transporter genes have been disrupted (Schinkel et al., 1994; Lorico et al., 1997), represent important tools with which to address these questions. The mdr1a (−/−) mouse strain developed by Schinkel and co-workers has thus far, been used primarily to demonstrate PGP-mediated effects on in vivo toxicity and pharmacokinetics (Schinkel et al., 1994, 1995; Fromm et al., 1999; Yokogawa et al., 1999). Given that several factors in addition to intestinal absorption will determine drug bio-availability, such studies cannot provide direct information on the influence of PGP ablation on drug efflux and permeability at the level of intestinal tissue. To date, there appears to have been no attempt to use tissues isolated from PGP knockout animals in vitro to assess the contributions of PGP and other transporters to drug efflux at specific sites such as the intestine.
The present study has investigated the in vitro permeability characteristics of ileal tissues from mdr1a (−/−) mice compared with tissues from wild-type (FVB) control animals using Ussing chambers. Data are presented on the basic paracellular and transcellular permeability properties of these tissues and on their handling of four compounds, digoxin, paclitaxel, etoposide and calcein. In addition to definitive information on the extent to which PGP modulates intestinal permeability of these compounds, this technique also provides data on the extent to which non-PGP efflux transporters modulate drug permeability in intestinal tissues.
Methods
(G-[3H])-digoxin and L-(4-[3H])-propranolol were purchased from NEN Life Science Products (Hounslow, U.K.), (G-[3H])-etoposide and (G-[3H])-paclitaxel were obtained from Moravek Biochemicals, Inc. (Brea, CA, U.S.A.), and D-(1-[14C]-mannitol was obtained from Amersham Life Science Ltd (Little Chalfont, U.K.). MK571 was purchased from Affiniti Research Products Ltd (Exeter, U.K.). Calcein was obtained from Molecular Probes (Eugene, OR, U.S.A.). All other compounds were obtained from Sigma-Aldrich Chemical Co. Ltd (Poole, U.K.). Wild-type FVB (mdr1a (+/+)) mice (the background strain used to produce the mdr1a (−/−) and mdr1a/1b (−/−) strains) were obtained from local barrier maintained stock. Mdr1a (−/−) and mdr1a/1b (−/−) mice were obtained from M&B A/S (Bomholtgärd, Denmark).
Animals and tissues
Intestinal tissues were removed from non-fasting male mice (10 – 16 weeks, 20 – 36 g) killed by cervical dislocation. The ileum (the segment stretching from 1 to 13 cm proximal to the ileo – cecal junction) was immediately removed and flushed with ice-cold, bicarbonate-buffered Ringer solution containing (in mmol.l−1) Na+ 146, K+ 4.2, Ca2+ 1.2, Mg2+ 1.2, Cl− 126, HCO3− 27, HPO4− 1.4, D-glucose 10 mM, which had been equilibrated to pH 7.4 by bubbling with 5% CO2/ 95% O2. Tissues were mounted intact in modified Ussing chambers (0.52 cm2 cross-sectional area) without removal of the serosal muscle layer. Mounting was completed within 20 min of removal from the animal. All procedures involving animals conformed to current U.K. Home Office regulations.
Permeability studies
Drug transport across intestinal tissues was measured by methods similar to those described previously (Collett et al., 1999). Intestinal mucosa was bathed on the mucosal (apical) and serosal (basolateral) surfaces with 5 ml of bicarbonate-buffered Ringer, pH 7.4 at 37°C. Spontaneous tissue open-circuit potential difference (P.D.), short-circuit current (ISC) and transepithelial electrical resistance (RT) were monitored periodically throughout the experiment, otherwise tissues were maintained under open circuit conditions. A 30 min equilibration period was allowed prior to beginning permeability measurements to allow stabilization of electrical parameters. Tissues were excluded in cases where RT values fell by more than 15% from the value measured at the end of the equilibration period. Asymmetric permeability of 40 μM digoxin, 20 μM paclitaxel or 20 μM etoposide was measured following addition of unlabelled drug with the corresponding radiolabelled drug (0.2 μCi.ml−1, 7.4 kBq.ml−1) to apical (A) or basolateral (B) chambers. Asymmetric permeability of 80 μM calcein was measured fluorimetrically (see below). Drugs were added as stock solutions in DMSO, giving a final solvent concentration of 0.02 – 0.3%. In the case of digoxin, unlabelled drug was added to an aliquot of transport buffer from the ‘donor' chamber, which was vortexed for 1 min and returned to the chamber to ensure thorough mixing. For all radiolabelled compounds, 1 ml samples were removed from the ‘receiver' chamber at t=0 and after each of six 40 min flux periods and replaced with fresh transport buffer. Inhibitors were added to the chamber (where appropriate), after the third flux period (i.e. at t=120 min). Samples (100 μl) were also taken from the ‘donor' chamber at the beginning of the first period and at the end of the experiment to monitor any changes in ‘donor' drug concentrations during the experiment and to safeguard mass balance. In calcein experiments, samples taken from the receiver chamber were 100 μl rather than 1 ml.
As a further test of the viability of the tissues, the cyclic AMP agonist, forskolin (10 μM) was added basolaterally at the conclusion of the experiment. This elicited a sharp and sustained rise in ISC in viable tissues caused by the stimulation of electrogenic Cl− secretion (Warhurst et al., 1996).
Samples were analysed by liquid scintillation counting, except in calcein experiments, where tracer concentration was determined by fluorimetry (λexcitation: 497 nm, λemission: 517 nm) using a Perkin-Elmer LS50B fluorescence spectrometer.
Values of unidirectional transepithelial apparent permeability (Papp) in cm.s−1, for each chamber over each 40 min flux period, were calculated by:
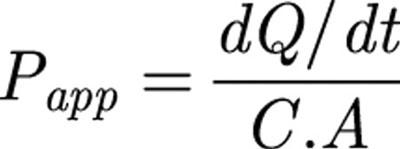 |
where dQ/dt is the rate of appearance of compound in the receiver chamber, C is the substrate concentration in the donor chamber and A is the cross-sectional area of the tissue (0.52 cm2). Values of Papp were then averaged over the first three 40 min flux periods (before the addition of the inhibitor) and over the second three 40 min flux periods (after addition of the inhibitor) to yield baseline and post-inhibition values. For all drugs (digoxin, paclitaxel, etoposide and calcein), dQ/dt was shown to be linear over the time course of the experiment. For analysis, the resulting Papp data from several experiments were then pooled (see ‘statistical methods' below).
Papp values shown are either unidirectional: apical to basolateral (A-B) or basolateral to apical (B-A) or net Papp (Papp B-A – Papp A-B). Where values for net Papp are shown, positive values represent net secretion in the B-A direction (i.e. B-A>A-B) while negative values represent net absorption in the A-B direction (i.e. B-A<A-B).
The effect of PGP ablation on the paracellular route was assessed using the paracellular marker mannitol. Following tissue equilibration, 100 μM mannitol, containing 0.2 μCi.ml−1 [14C]-mannitol, was added to the donor chamber and samples taken from the receiver chamber at 60 min intervals. Papp was calculated as described above.
Propranolol, which has been shown to be highly permeable across intestine (Ungell et al., 1998) was used to assess the inherent passive transcellular permeability of mouse tissues. Propranolol was added to the donor chamber at a concentration of 100 μM with 0.2 μCi.ml−1 [3H]-propranolol and samples were taken from the receiver chamber every 40 min for up to 4 h. For analysis, Papp values were averaged over 4 h (six 40 min flux periods).
Elucidation of the particular efflux processes responsible for net secretion of compounds in mouse ileum was investigated by apical and basolateral addition of the PGP inhibitor quinidine (200 μM) or the MRP-selective inhibitor, MK571 (20 μM). In some experiments, the PGP inhibitor verapamil (200 μM) was substituted for quinidine.
RT – PCR screen for transporter mRNA expression
Total RNA was prepared from mouse intestine, snap frozen in liquid N2, using ULTRASPEC reagent according to the manufacturer's instructions (AMS Biotechnologies Ltd, U.K.). Two μg of total RNA were reverse transcribed into cDNA using random primers and MMLV reverse transcriptase (Life Technologies Ltd, U.K.), as per manufacturer's instructions in a 10 μl reaction volume. One μl of the cDNA was then PCR-amplified on a Hybaid PCRSPRINT PCR block for 35 cycles. Reaction volumes were 50 μl, comprising: 0.2 mM each of dATP, dCTP, dTTP and dGTP, 1× PCR buffer (mM) TRIS/HCl 10, pH 8.3, MgCl2 1.5, KCl 50, and 0.5 μM of each primer (see Table 1). PCR products were visualized on 1% agarose/TAE gels, containing 0.01% Ethidium bromide, using a suitable DNA size ladder (Figure 2A: 100 bp DNA Ladder, Life Technologies, Paisley, U.K.; Figure 2B: DNA Molecular Weight Markers XIV, Roche Molecular Biochemicals, Lewes, U.K.) for verification of product sizes. In all cases, the identity of PCR products was confirmed by double-restriction digest, followed by visualization of restriction fragment sizes by agarose gel electrophoresis and commercial sequencing (Lark Technologies Inc., Saffron Walden, U.K.). Sequencing data indicated that identity of amplified products with corresponding published sequences (GENBank) was >99% in all cases.
Table 1.
Forward and reverse olignonucleotide primer sequences used for analysis of expression of MDR and MRP RNA in mouse intestine
Figure 2.
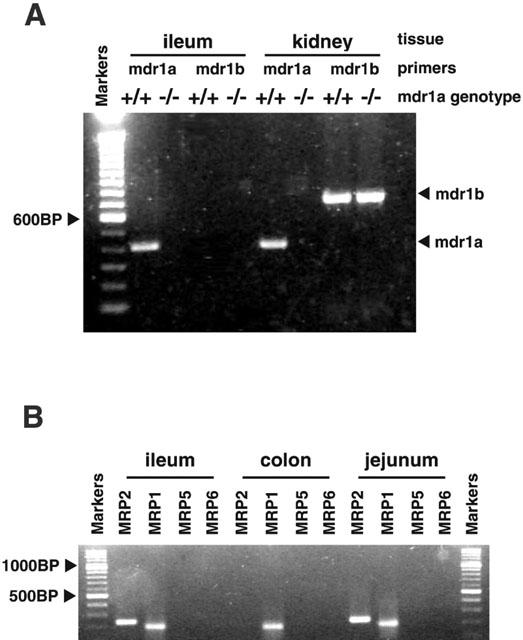
Molecular expression of efflux transporters in mouse intestine. (A) RT – PCR screen for mRNA expression of mdr1a and mdr1b genes in mdr1a (+/+) wild-type and mdr1a (−/−) knockout mouse tissues, visualized on a 1% agarose/TAE gel, containing 0.01% Ethidium bromide, using a 100 bp DNA Ladder (Life Technologies, Paisley, U.K.) for verification of product sizes. (B) RT – PCR screen for MRP1, MRP2, MRP5 and MRP6 genes in mdr1a (−/−) mouse intestine. Reaction products were visualized on a 1% agarose/TAE gel, containing 0.01% Ethidium bromide, using 100 base pair ladder (DNA Molecular Weight Markers XIV, Roche Molecular Biochemicals, Lewes, U.K.) for verification of product sizes.
Statistical methods
Values are expressed as mean±s.e.mean (n). Values of n represent the number of tissue segments used in each experimental group. The number of animals from which the tissues were collected is also shown in the figure legends. Statistical analyses were carried out using PRISM 2.01 (Graphpad Software Inc.). Single comparisons of unidirectional and net Papp values, electrical parameters and the effects of inhibitors on substrate fluxes were made using Student t-test with a significance level of 5%. Where multiple comparisons were made this was done using one-way analysis of variance (ANOVA) with a Bonferroni post test, using a significance level of 5%.
Results
Mannitol and propranolol permeability and electrical properties of normal and mdr1a (−/−) mouse ileum
Initial studies examined the baseline electrical parameters and stability of ileum isolated from FVB control and mdr1a (−/−) animals and mounted in Ussing chambers (Figure 1). A limited number of studies were also performed in mdr1a/1b (−/−) double knockout animals. Initial values for potential difference (PD) were similar in all strains (Figure 1A) and increased consistently by ≈20% over a 4-h period (P<0.05, paired t-test). Electrical resistance (RT) remained stable for at least 4 h following mounting in FVB and mdr1a (−/−) mice (Figure 1B), although the resistance of tissues from mdr1a/1b (−/−) animals was significantly lower (P<0.01, ANOVA) than the other two strains at all time points. In addition, tissues from mdr1a/1b animals exhibited a significantly higher Isc (49.2±2.5 compared to 42.9±2.9 and 40.5±3.0 μA.cm−2 in FVB and mdr1a (−/−), respectively; P<0.01, ANOVA, n=17 tissues in each group). Addition of 200 μM quinidine, which was used as a PGP inhibitor during the latter stages of permeability studies in tissues from control mice caused a significant decrease in PD (P<0.05) and concomitant significant rise in RT (P<0.001, Figure 1A and B, respectively) consistent with the blockade of membrane K+ channels (Richards & Dawson, 1986).
Figure 1.
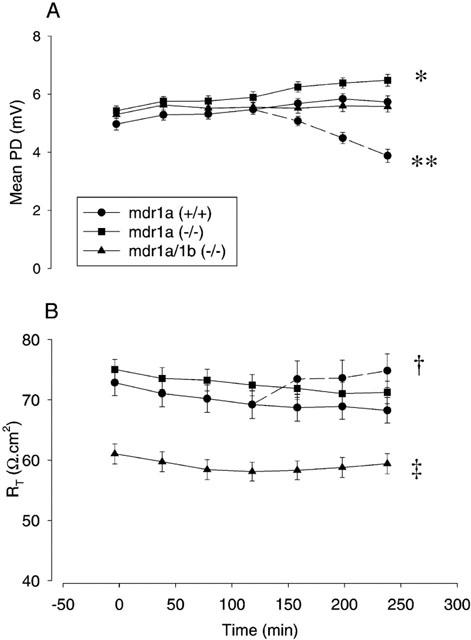
Electrical parameters of isolated mouse ileum. Tissue electrical parameters measured over a 4 h time course following mounting of unstripped intestine in Ussing chambers as indicated in Methods. Values at each time point are means±s.e.mean for 81, 38, and 19 tissues from 20 mdr1a (+/+), 11 mdr1a (−/−) and 5 mdr1a/1b (−/−) mice respectively. (A) Mean potential difference (PD, mV); *P<0.05 value at t=240 compared with value at t=0. In a subset of the mdr1a (+/+) group only, 200 μM quinidine was added from t=120 onwards as indicated by the dashed line, **P<0.05 value at t=240 compared with t=120; both comparisons made by paired t-test. (B) Mean transepithelial resistance (Rt, Ω.cm2); †P<0.001, value at t=240 compared with t=120 (paired t-test), ‡P<0.01 compared with mdr1a (−/−) and mdr1a (+/+) by one-way ANOVA.
The paracellular pathway in FVB and mdr1a (−/−) mice was also assessed by following mannitol permeability. Mannitol (Papp) across mdr1a (−/−) ileum was stable over 4 h (3.6±0.6 and 4.0±0.5×10−6 cm.s−1 at t=60 and t=240 respectively) and showed no evidence of asymmetry (A-B 3.6±0.6; B-A 4.4±0.8×10−6 cm.s−1 at t=60 ). Similar results were observed in ileum from FVB control mice with no significant differences in paracellular flux between the control and mdr1a (−/−) strains.
Permeability of the passive transcellular marker propranolol was approximately 10 fold greater than mannitol, and also symmetrical in both FVB (A-B 45.2±0.7×10−6 cm.s−1; B-A 40.4±0.8×10−6 cm.s−1, n=6) and mdr1a (−/−) tissues (A-B: 40.2±0.3×10−6 cm.s−1; B-A 37.9±0.5×10−6 cm.s−1, n=6). The values obtained for propranolol and mannitol permeability in unstripped mouse ileum are very similar to those reported for stripped rat intestine (Ungell et al., 1998) suggesting that the presence of an intact muscle layer in mouse ileum has no significant effect on passive permeability.
RT – PCR screen for multidrug resistance transporter expression
As expected, an RT – PCR screen for expression of mdr1a and mdr1b genes in mouse tissues (Figure 2A) revealed mdr1a expression in wild-type mouse ileum and kidney, but not in the same tissues taken from mdr1a (−/−) mice. Mdr1b was not detected in ileum from either mdr1a (+/+) or mdr1a (−/−) mice, but was detected in the kidneys of both strains. The expression of MRP isoforms in mdr1a (−/−) intestine was also investigated (Figure 2B). MRP1 mRNA could be detected in both small and large intestine of mdr1a (−/−) mice while MRP2 expression was seen only in jejunum and ileum. There was no detectable expression of MRP5 (MOAT-C) or MRP6 (MOAT-E) in mdr1a (−/−) intestine. MRP3 (MOAT-D) and MRP4 (MOAT-B) have yet to be cloned in the mouse and were not investigated.
Polarized efflux of digoxin and paclitaxel is mediated solely by P-glycoprotein in mouse ileum
Both digoxin and paclitaxel have been reported to exhibit polarized efflux across epithelial cell monolayers consistent with interaction with xenobiotic transporters such as PGP (Fromm et al., 1999; Fromm, 2000; Sparreboom et al., 1997). Comparative studies across FVB control and mdr1a (−/−) mouse ileum were used to define the degree to which PGP influences the permeability of these compounds in intestinal tissues. In tissues from control animals, digoxin permeability showed marked efflux (Figure 3) with permeability in the B-A direction being 10 fold greater than in the A-B direction. This efflux was abolished in the presence of quinidine (200 μM), which caused a 4 fold increase in A-B permeability of digoxin (11.9±1.3 vs 3.1±0.6×10−6 cm.s−1 in the presence and absence of quinidine respectively (n=4); P<0.01). In contrast, in mdr1a (−/−) tissues, digoxin permeability was virtually identical in both directions with no evidence of efflux (Figure 3). A-B permeability of digoxin was also markedly higher in mdr1a (−/−) animals compared to FVB controls (8.4±0.7 vs 3.1±0.6×10−6 cm.s−1 (n=6 and 4, respectively); P<0.001), these values being similar to quinidine-treated control tissues. Addition of the MRP inhibitor, MK571 to mdr1a (−/−) tissues had no effect on digoxin permeability in either direction (P>0.1).
Figure 3.
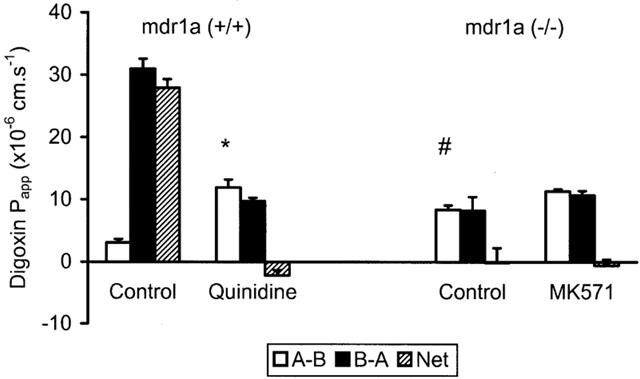
Digoxin permeability in control and mdr1a (−/−) mouse intestine. Unidirectional (A-B, B-A) and net permeability of digoxin (40 μM) across unstripped ileum from (A) FVB-control and (B) mdr1a (−/−) mice. Values shown are mean Papp±s.e.mean (cm.s−1) in the absence of inhibitor and after addition of 200 μM quinidine in the case of FVB-control or before and after addition of 20 μM MK571 in mdr1a (−/−) mice. n=4 – 6 tissues in each direction for both groups (3 – 5 animals in each group). *P<0.01 compared to A-B in the absence of quinidine, #P<0.005 compared to A-B in FVB-control in the absence of quinidine.
Paclitaxel exhibited similar permeability properties with a 17 fold greater permeability in the B-A than A-B direction in FVB-control ileum. (A-B: 2.3±0.2; B-A: 38.9±2.2×10−6 cm.s−1 (n=4); P<0.001) which was quinidine sensitive (Figure 4). Again, no asymmetry was observed for paclitaxel in mdr1a (−/−) ileum but the absorptive (A-B) permeability of the drug was 5 fold greater than in tissues from control animals expressing the mdr1a gene (Figure 4). The MRP inhibitor MK571 (20 μM) once again had no effect on Papp for paclitaxel (P<0.1). These data from mdr1a (−/−) tissues provide clear evidence that PGP is the sole mediator of intestinal efflux of both digoxin and paclitaxel and that the transporter has a significant inhibitory effect on the absorptive flux of these compounds in tissues.
Figure 4.
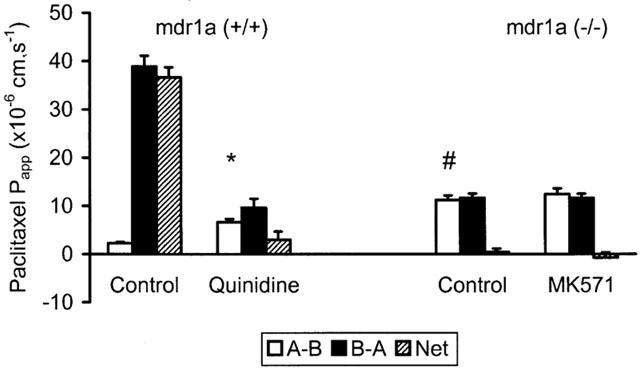
Paclitaxel permeability in control and mdr1a (−/−) mouse intestine: Unidirectional (A-B, B-A) and net permeability of paclitaxel (20 μM) across unstripped ileum from (A) FVB-control and (B) mdr1a (−/−) mice. Values shown are mean Papp±s.e.mean (cm.s−1) in the absence of inhibitor and after addition of 200 μM quinidine in the case of FVB-control or before and after addition of 20 μM MK571 in mdr1a (−/−) mice. n=4 tissues in each direction for both groups (three animals in each group). *P<0.01 compared to A-B in the absence of quinidine, #P<0.01 compared to A-B in FVB-control in the absence of quinidine.
Etoposide absorption across mouse ileum is influenced by P-glycoprotein and MRP transporters
In common with the other compounds used in this study, etoposide exhibited luminal secretion (efflux) in control mouse ileum characterized by a 9 fold asymmetry in the B-A direction (Figure 5). However, in this case, 200 μM quinidine only partially inhibited the asymmetry (77%) leaving a residual B-A/A-B ratio of ≈2 fold. The remaining component of etoposide efflux was abolished by addition of 20 μM MK571. The net effect of quinidine and MK571 addition was to increase the A-B permeability of etoposide by a factor of 4 (P<0.001, ANOVA).
Figure 5.
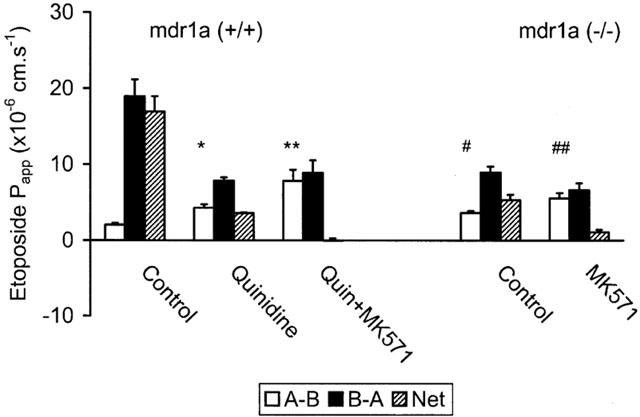
Influence of PGP and MRP transporters on etoposide permeability in mouse ileum. Unidirectional (A-B, B-A) and net permeability of etoposide (20 μM) across unstripped ileum from (A) FVB-control and (B) mdr1a (−/−) mice. Values are mean Papp±s.e.mean (cm.s−1) in the absence of inhibitor and following sequential addition of either 200 μM quinidine or a mixture of 200 μM quinidine and 20 μM MK571 in FVB control or following addition of 20 μM MK571 alone in mdr1a (−/−). n=4 – 6 tissues in each direction for both groups, 3 – 6 animals in each group. *P<0.02 compared to A-B in absence of quinidine, **P<0.01 compared to A-B in the presence of quinidine, #P<0.02 compared to A-B in FVB control in absence of quinidine, ##P<0.02 compared to A-B in mdr1a (−/−) in absence of MK571. All comparisons were made using one-way ANOVA.
Complementary studies in mdr1a (−/−) ileum were also performed (Figure 5). In this system devoid of PGP, etoposide exhibited a 2.7 fold asymmetry in the B-A direction, which was essentially abolished by MK571. The handling of etoposide in mdr1a (−/−) intestine was compared to that of calcein (Figure 6), which is known to interact with MRP (Fujita et al., 1996) but has a very low affinity for PGP. Although the unidirectional permeability of calcein was much lower than etoposide, it exhibited a similar level of asymmetry in the B-A direction (2.2 fold), which was also sensitive to inhibition by MK571 (P<0.05). Calcein also displayed asymmetry in FVB mice, which was similar in magnitude to that seen in mdr1a (−/−) intestine and which was insensitive to the PGP inhibitor quinidine (Figure 6). These data confirm that calcein permeability is influenced by MRP but not PGP in mouse small intestine, while etoposide permeability is influenced by a combination of both PGP and MRP transporters.
Figure 6.
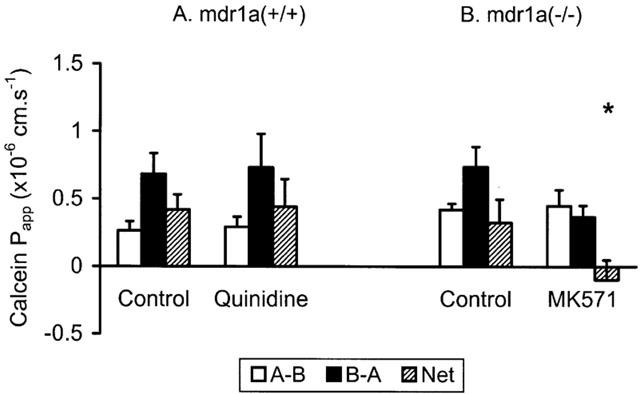
Calcein apparent permeability (Papp) across mouse intestine. Unidirectional (A-B, B-A) and net permeability of calcein (80 μM) across unstripped ileum from (A) FVB control and (B) mdr1a (−/−) mice (n=6 and 4 tissues in each direction, four and three animals in each group, respectively). Values shown are mean±s.e.mean in the absence of inhibitor and after addition of (A) 200 μM quinidine or (B) 20 μM MK571. *P<0.05 compared with net Papp in absence of inhibitor.
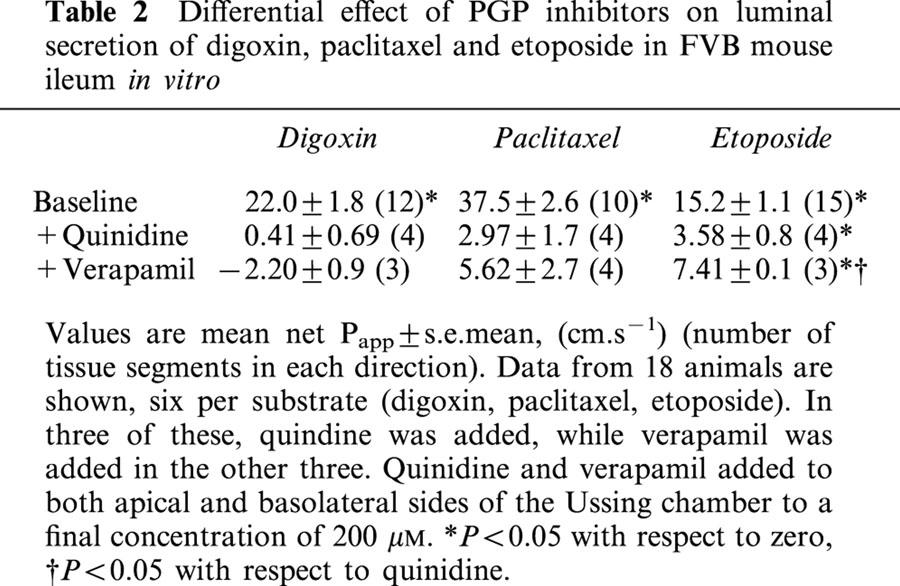
Studies were also performed in wild-type tissues comparing the inhibitory effects of verapamil and quinidine, both of which are used extensively as PGP inhibitors (Table 2). The data show significant differences in inhibitory potency, which were dependent on the compound being studied. For example, quinidine and verapamil at 200 μM abolished luminal secretion of digoxin and paclitaxel in FVB mouse ileum. However, quinidine was significantly more effective than verapamil in inhibiting etoposide secretion at the same concentration (% inhibition of etoposide secretion: 200 μM quinidine: 76.8±1.2% vs 200 μM verapamil: 44.5±2.5%, (n=four and three animals respectively); P<0.02).
Table 2.
Differential effect of PGP inhibitors on luminal secretion of digoxin, paclitaxel and etoposide in FVB mouse ileum in vitro
Lack of involvement of mdr1b in drug efflux in mouse intestine
Although mdr1b is absent from the intestine of normal mice (Croop et al., 1989), the expression of this gene is known to be upregulated in the kidney and liver of mdr1a (−/−) mice (Schinkel et al., 1994). To investigate potential compensatory effects in intestine the transport of paclitaxel and etoposide was measured in intestine from mice in which both mdr1a and mdr1b expression had been ablated (Figure 7). In common with its profile in mdr1a (−/−) ileum, paclitaxel permeability was symmetrical in mdr1a/1b (−/−) tissues. Similarly, etoposide showed a 2.2 fold asymmetry in the B-A direction, which was sensitive to MK571 (P<0.05). These data show that disruption of the mdr1b gene in addition to the mdr1a gene has no additional impact on drug permeability in small intestine suggesting that mdr1b is not involved in intestinal drug efflux.
Figure 7.
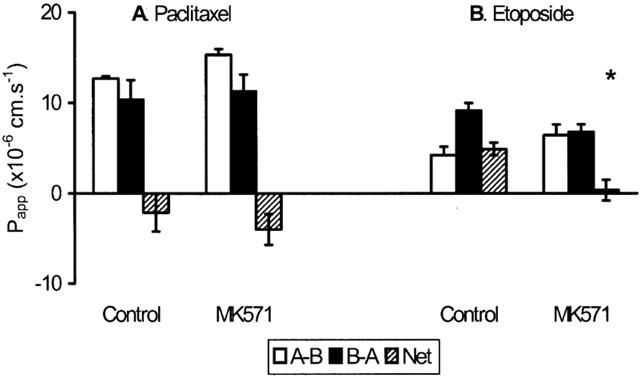
Drug transport in double knockout mice. Unidirectional (A-B, B-A) and net permeability of (A) paclitaxel (20 μM) or (B) etoposide (20 μM) across mdr1a/1b (−/−) double-knockout mouse ileum. Values shown are mean Papp±s.e.mean (cm.s−1) before and after addition of 20 μM MK571 (n=4 tissues in each direction, three animals in each group) *P<0.05 compared with net permeability in the absence of MK571.
Discussion
The mdr1a (−/−) mouse developed by Schinkel and co-workers has proved an invaluable model for investigating the influence of PGP on drug pharmacokinetics in vivo (Schinkel et al., 1994, 1995). However, the potential usefulness of this model in understanding the precise role that PGP plays in controlling permeability in specific tissues such as the intestine has not been addressed. This study reports data on the influence of PGP ablation on the intestinal handling of four compounds, paclitaxel, digoxin, etoposide and calcein, using isolated ileum from the mdr1a (−/−) mouse compared to tissues from FVB control animals expressing the mdr1a gene. The first three of these compounds were chosen as prototypical PGP substrates for which there is clear evidence of both in vitro interaction with PGP and impaired bio-availability in in vivo systems (Schinkel et al., 1995; 1997; Cavet et al., 1996; Sparreboom et al., 1997; Makhey et al., 1998; Walle & Walle, 1998; Doppenschmitt et al., 1999; Fromm et al., 1999). The data presented here indicate that PGP plays an important role in defining the intestinal permeability of these compounds. For example, the permeability of digoxin and paclitaxel is an order of magnitude greater in the basolateral to apical direction in tissues from control animals. The complete abolition of this secretory flux in mdr1a (−/−) tissues indicates that it is wholly mediated by PGP. Perhaps more importantly, the observation that apical to basolateral permeability of digoxin and paclitaxel is increased 3 – 5 fold in mdr1a knockout tissues gives a key insight into the role of PGP in regulating the efficiency of intestinal absorption in general.
In contrast, the handling of etoposide by mouse intestine is far more complex than that of digoxin or paclitaxel, with evidence that multiple transporters are involved in limiting its permeability. While PGP is undoubtedly the major efflux protein present in mammalian intestine there is good evidence that other transporters are also expressed, including several members of the MRP family. Sugiyama and co-workers have shown MRP2 to be functionally expressed in Caco-2 and rodent intestine, transporting organic anions and glutathione conjugates in a blood-to-lumen direction (Gotoh et al., 2000; Hirohashi et al., 2000). MRP1 and MRP3 are also expressed in intestinal cells although the functional significance of these transporters remains to be determined (Peng et al., 1999; Ortiz et al., 1999; Hirohashi et al., 2000).
The present studies suggest that etoposide permeability is influenced by both PGP and MRP transporters in mouse intestine. Luminal secretion of etoposide was only partially reduced in mdr1a (−/−) tissues but could be completely abolished by the MRP-selective inhibitor, MK571. Etoposide is known to interact with PGP and studies in everted intestinal sacs show that the efficiency of etoposide absorption can be increased by incubation with a monoclonal antibody against PGP (Leu & Huang, 1995). However, there is also a close correlation between etoposide resistance or transport and MRP expression in several cell systems (Cole et al., 1994; Paul et al., 1996a, 1996b; Gaj et al., 1998), and Makhey et al. (1998) showed etoposide efflux in rat intestine to be reduced by leukotriene C4, an MRP substrate.
The efflux of calcein in mdr1a (−/−) ileum is similar to that described by Fujita et al. (1996) in rat jejunum and its abolition by MK571 (Figure 6) provides further evidence for the functional involvement of one or more MRP transporters in drug efflux in the mdr1a (−/−) mouse intestine. In contrast to etoposide, PGP inhibitors had no effect on calcein efflux in wild-type mouse intestine which is consistent with previous evidence that this compound is a substrate for MRP but not PGP transporters (Hollo et al., 1994).
The lack of selectivity of current MRP inhibitors does not allow the identification of the specific MRP protein(s) involved in etoposide and calcein efflux. Expression of MRP1 and MRP3 have both been shown to confer resistance to etoposide consistent with efflux (Gaj et al., 1998; Kool et al., 1999) but there is little direct evidence for interaction with MRP2 (Chen et al., 1999). Further studies are underway to examine how etoposide permeability varies regionally in the mdr1a (−/−) intestine, which may give us further insights into the identity of the transporter, given that MRP2, for example, is expressed at very low levels or not at all in rodent large intestine (Figure 2 and Gotoh et al., 2000; Stephens et al., 2001).
The profile of PGP expression in rodents is complicated by the presence of two genes mdr1a and mdr1b (Croop et al., 1989). However, both absorptive (A-B) permeability and the degree of efflux of paclitaxel and etoposide was essentially identical in tissues from mdr1a (−/−) and mdr1a/1b (−/−) animals, suggesting that mdr1b is not involved in drug efflux in the intestine. This is supported by molecular studies showing the absence of mdr1b expression in the intestine of both FVB and mdr1a (−/−) animals, although it is present in other tissues. The finding that mdr1a/1b (−/−) animals exhibit a ≈20% increase in Isc (accompanied by a similar reduction in RT) is interesting and has not previously been reported. Such a difference is consistent with changes in electrogenic ion transport although the physiological significance and mechanism of these changes will require further investigation.
Isolated tissues from mdr1a (−/−) animals offer several significant advantages over existing in vitro systems for studying drug efflux. Firstly, definitive information on the influence of PGP on a compound's permeability is produced without recourse to inhibitors. The efficacy of even common inhibitors such as verapamil and quinidine appears to vary considerably depending on the competing substrate and it can be difficult to achieve complete and selective inhibition. In the present study, quinidine and verapamil abolished luminal secretion of digoxin and paclitaxel but showed variable effectiveness in inhibiting etoposide secretion (Table 2). Similarly, a previous study reported significant regional variation in the interaction between verapamil and PGP along the rat intestine (Saitoh & Aungst, 1995). Variability in responsiveness to inhibitors makes it difficult to gather reliable, quantitative information on the true impact of PGP on intestinal permeability. It is also possible that inhibitors may influence other epithelial transport processes in addition to their effects on drug efflux so presenting a confused picture. For example, in this study the PGP inhibitor quinidine also caused a significant increase in tissue resistance and a decrease in potential difference which may be due to its known inhibitory action on epithelial K+ channels leading to membrane depolarization (Richards & Dawson, 1986).
Secondly, the mdr1a (−/−) model facilitates identification of non-PGP transporters and allows their relative contribution to intestinal efflux to be defined. The high level of PGP in intestine and the fact that PGP inhibitors exhibit significant cross-reactivity with other transporters confounds such studies in normal tissues. For example, both verapamil and quinidine have been shown to interact with MRP transporters (Tomonaga et al., 1996; Makhey et al., 1998; Vezmar & Georges, 2000). Recent studies have implicated non-PGP, non-MRP transporters as possible limiting factors for intestinal permeability (Soldner et al., 2000; Jonker et al., 2000) and there is a growing awareness that our understanding of the drug efflux systems present in the intestine is far from complete. The PGP knockout mouse may be an important in vivo tool in this respect, as demonstrated by a recent pharmacokinetic study pointing to breast cancer resistance protein (BCRP) as a limiting factor in intestinal uptake and bioavailability of topotecan (Jonker et al., 2000). Combining this with complementary studies using the ex vivo technique described here will provide a powerful approach for defining the influence of multiple efflux transporters on drug absorption.
Thirdly, the ability to directly compare permeability in tissues in the presence and absence of PGP not only allows quantitative determination of PGP effects in a physiologically relevant system but also provides definitive measures of a compound's passive permeability, information that may be useful in developing mechanistic models of intestinal drug efflux (Doppenschmitt et al., 1999). Such information can be gathered from tissues isolated from different regions of the intestine and such studies are currently underway.
In conclusion, this study has examined the effects of PGP ablation on the intestinal permeability of paclitaxel, digoxin and etoposide using a novel approach based on the use of tissues from PGP-knockout mice. The data shows the profound effect of PGP on intestinal permeability of these compounds and also provides evidence that both PGP and MRP-type transporters in intestinal tissues influence absorption of etoposide.
Abbreviations
- BCRP
breast cancer resistance protein
- DIG
digoxin
- ETOP
etoposide
- ISC
short circuit current
- MRP
multidrug resistance-associated protein
- PD
open circuit potential difference
- PGP
P-glycoprotein
- RT
transepithelial electrical resistance
- TAX
paclitaxel
References
- BORST P., EVERS R., KOOL M., WIJNHOLDS J. A family of drug transporters: the multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000;92:1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- CAVET M.E., WEST M., SIMMONS N.L. Transport & epithelial secretion of the cardiac glycoside, digoxin, by human intestinal epithelial (Caco-2) cells. Br. J. Pharmacol. 1996;118:1389–1396. doi: 10.1111/j.1476-5381.1996.tb15550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Z., KAWABE T., ONO M., AOKI S., SUMIZAWA T., FURUKAWA T., UCHIUMI T., WADA M., KUWANO M., AKIYAMA S. Effect of multidrug resistance-reversing agents on transporting activity of human canalicular multispecific organic anion transporter. Mol. Pharmacol. 1999;56:1219–1228. doi: 10.1124/mol.56.6.1219. [DOI] [PubMed] [Google Scholar]
- COLE S.P.C., SPARKS K.E., FRASER K., LOE D.W., GRANT C.E., WILSON G.M. , DEELEY R.G. Pharmacological characterisation of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 1994;54:5902–5910. [PubMed] [Google Scholar]
- COLLETT A., HIGGS N.B., SIMS E., ROWLAND M., WARHURST G. Modulation of the permeability of H2 receptor antagonists cimetidine & ranitidine by P-glycoprotein in rat intestine and the human colonic cell line Caco-2. J. Pharmacol. Exp. Ther. 1999;288:171–178. [PubMed] [Google Scholar]
- CROOP J.M., RAYMOND M., HABER D., DEVAULT A., ARCECI R.J., GROS P., HOUSMAN D.E. The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol. Cell. Biol. 1989;9:1346–1350. doi: 10.1128/mcb.9.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOPPENSCHMITT S., SPAHN-LANGGUTH H., REGARDH C.G., LANGGUTH P. Role of P-glycoprotein-mediated secretion in absorptive drug permeability: an approach using passive membrane permeability and affinity to P-glycoprotein. J. Pharm. Sci. 1999;88:1067–1072. doi: 10.1021/js980378j. [DOI] [PubMed] [Google Scholar]
- FROMM M.F. P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs. Int. J. Clin. Pharmacol. Ther. 2000;38:69–74. doi: 10.5414/cpp38069. [DOI] [PubMed] [Google Scholar]
- FROMM M.F., KIM R.B., STEIN C.M., WILKINSON G.R., RODEN D.M. Inhibition of P-glycoprotein-mediated drug transport – A unifying mechanism to explain the interaction between digoxin & quinidine. Circulation. 1999;99:552–557. doi: 10.1161/01.cir.99.4.552. [DOI] [PubMed] [Google Scholar]
- FUJITA T., YAMADA H., FUKUZUMI M., NISHIMAKI A., YAMAMOTO A., MURANISHI S. Calcein is excreted from the intestinal mucosal cell membrane by the active transport system. Life Sci. 1996;60:307–313. doi: 10.1016/s0024-3205(96)00631-5. [DOI] [PubMed] [Google Scholar]
- GAJ C.L., ANYANWUTAKU I., CHANG Y.H., CHENG Y.C. Decreased drug accumulation without increased drug efflux in a novel MRP-overexpressing multidrug-resistant cell line. Biochem. Pharmacol. 1998;55:1199–1211. doi: 10.1016/s0006-2952(97)00614-x. [DOI] [PubMed] [Google Scholar]
- GOTOH Y., SUZUKI H., KINOSHITA S., HIROHASHI T., KATO Y., SUGIYAMA Y. Involvement of an organic anion transporter (canalicular multispecific organic anion transporter/multidrug resistance-associated protein 2) in gastrointestinal secretion of glutathione conjugates in rats. J. Pharmacol. Exp. Ther. 2000;292:433–439. [PubMed] [Google Scholar]
- HIROHASHI T., SUZUKI H., CHU X., TAMAI I., TSUJI A., SUGIYAMA Y. Function and expression of multidrug resistance-associated protein family in human colon adenocarcinoma cells (Caco-2) J. Pharmacol. Exp. Ther. 2000;292:265–270. [PubMed] [Google Scholar]
- HOLLO Z., HOMOLYA L., DAVIS C.W., SARKADI B. Calcein accumulation as a fluorimetric functional assay of the multidrug transporter. Biochim. Biophys. Acta. 1994;1191:384–388. doi: 10.1016/0005-2736(94)90190-2. [DOI] [PubMed] [Google Scholar]
- HUNTER J., HIRST B.H. Intestinal secretion of drugs. The role of P-glycoprotein and related drug efflux systems in limiting oral drug absorption. Adv. Drug. Del. Rev. 1997;25:129–157. [Google Scholar]
- JONKER J.W., SMIT J.W., BRINKHUIS R.F., MALIEPAARD M., BEIJNEN J.H., SCHELLENS J.H.M., SCHINKEL A.H. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J. Natl. Cancer. Inst. 2000;92:1651–1656. doi: 10.1093/jnci/92.20.1651. [DOI] [PubMed] [Google Scholar]
- KOOL M., VAN DER LINDEN M., DE HAAS M., SCHEFFER G.L., DE VREE J.M., SMITH A.J., JANSEN G., PETERS G.J., PONNE N., SCHEPER R.J., ELFERINK R.P., BAAS F., BORST P. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6914–6919. doi: 10.1073/pnas.96.12.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEU B.L., HUANG J. Inhibition of intestinal P-glycoprotein and effects of etoposide absorption. Cancer Chemother. Pharmacol. 1995;355:432–436. doi: 10.1007/s002800050258. [DOI] [PubMed] [Google Scholar]
- LORICO A., RAPPA G., FINCH R.A. , YANG D., FLAVELL R.A., SARTORELLI A.C. Disruption of the murine MRP (multidrug resistance protein) gene leads to increased sensitivity to etoposide (VP-16) and increased levels of glutathione. Cancer Res. 1997;57:5238–5242. [PubMed] [Google Scholar]
- MAKHEY V.D., GUA A., NORRIS D.A., HU P., YAN J., SINKO P. Characterization and regional intestinal kinetics of drug efflux in rat and human intestine & Caco-2 cells. Pharm. Res. 1998;15:1160–1167. doi: 10.1023/a:1011971303880. [DOI] [PubMed] [Google Scholar]
- MOTTINO A.D., HOFFMAN T., JENNES L., VORE M. Expression and localization of multidrug resistant protein mrp2 in rat small intestine. J. Pharmacol. Exp. Ther. 2000;293:717–723. [PubMed] [Google Scholar]
- ORTIZ D.F., LI S.H., IYER R., ZHANG X.M., NOVIKOFF P., ARIAS I.M. MRP3, a new ATP-binding cassette protein localized to the canalicular domain of the hepatocyte. Am. J. Physiol. 1999;276:G1493–G1500. doi: 10.1152/ajpgi.1999.276.6.G1493. [DOI] [PubMed] [Google Scholar]
- PAUL S., BREUNINGER L.M., KRUH G.D. ATP-dependent transport of lipophilic cytotoxic drugs by membrane vesicles prepared from MRP-overexpressing HL60/ADR cells. Biochemistry. 1996a;35:14003–14011. doi: 10.1021/bi9618528. [DOI] [PubMed] [Google Scholar]
- PAUL S., BREUNINGER L.M., TEW K.D., SHEN H., KRUH G.D. ATP-dependent uptake of natural product cytotoxic drugs by membrane vesicles establishes MRP as a broad specificity transporter. Proc. Natl. Acad. Sci. U.S.A. 1996b;93:6929–6934. doi: 10.1073/pnas.93.14.6929. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- PENG K.C., CLUZEAUD F., BENS M., VAN HUYEN J.P., WIOLAND M.A., LACAVE R., VANDEWALLE A. Tissue and cell distribution of the multidrug resistance-associated protein (MRP) in mouse intestine and kidney. J. Histochem. Cytochem. 1999;47:757–768. doi: 10.1177/002215549904700605. [DOI] [PubMed] [Google Scholar]
- RAPPA G., LORICO A., FLAVELL R.A., SARTORELLI A.C. Evidence that the multidrug resistance protein (MRP) functions as a co-transporter of glutathione and natural product toxins. Cancer Res. 1997;57:5232–5237. [PubMed] [Google Scholar]
- RICHARDS N.W., DAWSON D.C. Single potassium channels blocked by lidocaine and quinidine in isolated turtle colon epithelial cells. Am. J. Physiol. 1986;251:C85–C89. doi: 10.1152/ajpcell.1986.251.1.C85. [DOI] [PubMed] [Google Scholar]
- SAITOH H., AUNGST B.J. Possible involvement of multiple P-glycoprotein-mediated efflux systems in the transport of verapamil and other organic cations across rat intestine. Pharm. Res. 1995;12:1304–1310. doi: 10.1023/a:1016217505990. [DOI] [PubMed] [Google Scholar]
- SCHINKEL A.H., MAYER U., WAGENAAR E., MOL C.A.A.M., VAN DEEMTER L., SMIT J.J.M., VAN DER VALK M.A., VOORDOUW A.C., SPITS H., VAN TELLINGEN O., ZIJLMANS J.M.J.M., FIBBE W.E., BORST P. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHINKEL A.H., SMIT J.J.M., VAN TELLINGEN O., BEIJNEN J.H., WAGENAAR E., VAN DEETMER L., MOL C.A.A.M., VAN DER VALK M.A., ROBANUS-MAANDAG B.C., TE TIELE H.P.J., BERNS A.J.M., BORST P. Disruption of the mouse mdr1a P-Glycoprotein gene leads to a deficiency in the blood brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- SCHINKEL A.H., WAGENAAR E., VAN DEEMTER L., MOL C.A.A.M., BORST P. Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin and cyclosporin A. J. Clin. Invest. 1995;96:1698–1705. doi: 10.1172/JCI118214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOLDNER A., BENET L.Z., MUTSCHLER E., CHRISTIANS U.S. Active transport of the angiotensin-II antagonist losartan and its main metabolite EXP 3174 across MDCK-MDR1 and Caco-2 cell monolayers. Br. J. Pharmacol. 2000;129:1235–1243. doi: 10.1038/sj.bjp.0703150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPARREBOOM A., VAN ASPEREN J., MAYER U., SCHINKEL A.H., SMIT J.W., MEIJER D.W., BORST P., NOOJIEN W.J., BEIJNEN J.H., VAN TELLINGEN O. Limited oral bioavailability of an active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc. Natl. Acad. Sci. U.S.A. 1997;94:2031–2035. doi: 10.1073/pnas.94.5.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEPHENS R.H., O'NEILL C.A., WARHURST A., CARLSON G.L., ROWLAND M., WARHURST G. Kinetic profiling of P-glycoprotein mediated drug efflux in rat and human intestinal epithelia. J. Pharmacol. Exp. Ther. 2001;296:584–591. [PubMed] [Google Scholar]
- SUZUKI H., SUGIYAMA Y. Role of metabolic enzymes and efflux transporters in the absorption of drugs from the small intestine. Eur. J. Pharm. Sci. 2000;12:3–12. doi: 10.1016/s0928-0987(00)00178-0. [DOI] [PubMed] [Google Scholar]
- TOMONAGA M., OKA M., NARASAKI F., FUKUDA M., NAKANO R., TAKATANI H., IKEDA K., TERASHI K., MATSUO I., SODA H., COWAN K.H., KOHNO S. The multidrug resistance-associated protein gene confers drug resistance in human gastric and colon cancers. Japan. J. Cancer Res. 1996;87:1263–1270. doi: 10.1111/j.1349-7006.1996.tb03142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNGELL A.L., NYLANDER S., BERGSTRAND S., SJOBERG A., LENNERNAS H. Membrane transport of drugs in different regions of the intestinal tract of the rat. J. Pharm. Sci. 1998;87:360–366. doi: 10.1021/js970218s. [DOI] [PubMed] [Google Scholar]
- VEZMAR M., GEORGES E. Reversal of MRP-mediated doxorubicin resistance with quinoline-based drugs. Biochem. Pharmacol. 2000;59:1245–1252. doi: 10.1016/s0006-2952(00)00270-7. [DOI] [PubMed] [Google Scholar]
- WALLE U.K., WALLE T. Taxol transport by human intestinal epithelial Caco-2 cells. Drug Metab. Disp. 1998;26:343–346. [PubMed] [Google Scholar]
- WARHURST G., HIGGS N.B., FAKHOURY H., WARHURST A.C., GARDE J., COY D.H. Somatostatin receptor subtype 2 mediates somatostatin inhibition of ion secretion in rat distal colon. Gastroenterology. 1996;111:325–333. doi: 10.1053/gast.1996.v111.pm8690197. [DOI] [PubMed] [Google Scholar]
- YOKOGAWA K., TAKAHASHI M., TAMAI I., KONISHI H., NOMURA M., MORITANI S., MIYAMOTO K., TSUJI A. P-glycoprotein-dependent disposition kinetics of tacrolimus: studies in mdr1a knockout mice. Pharm. Res. 1999;16:1213–1218. doi: 10.1023/a:1018993312773. [DOI] [PubMed] [Google Scholar]