

Abstract
Interactions between A2A-adenosine receptors and α2-, A1- and P2- release-inhibitory receptors, on the modulation of noradrenaline release were studied in isolated rat tail artery. Preparations were labelled with [3H]-noradrenaline, superfused with desipramine-containing medium, and stimulated electrically (100 pulses at 5 Hz or 20 pulses at 50 Hz).
Blockade of α2-autoreceptors with yohimbine (1 μM) increased tritium overflow elicited by 100 pulses at 5 Hz but not by 20 pulses at 50 Hz.
The selective A2A-receptor agonist 2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine (CGS 21680; 1 – 100 nM) enhanced tritium overflow elicited by 100 pulses at 5 Hz. Yohimbine prevented the effect of CGS 21680, which was restored by the A1-receptor agonist N6-cyclopentyladenosine (CPA; 100 nM) or by the P2-receptor agonist 2-methylthioadenosine triphosphate (2-MeSATP; 80 μM).
CGS 21680 (100 nM) failed to increase tritium overflow elicited by 20 pulses at 50 Hz. The α2-adrenoceptor agonist 5-bromo-6-(2-imidazolin-2-ylamino)-quinoxaline (UK 14304; 30 nM), the A1-receptor agonist CPA (100 nM) or the P2-receptor agonist 2-MeSATP (80 μM) reduced tritium overflow. In the presence of these agonists CGS 21680 elicited a facilitation of tritium overflow.
Blockade of potassium channels with tetraethylammonium (TEA; 5 mM) increased tritium overflow elicited by 100 pulses at 5 Hz to values similar to those obtained in the presence of yohimbine but did not prevent the effect of CGS 21680 (100 nM) on tritium overflow.
It is concluded that, in isolated rat tail artery, the facilitation of noradrenaline release mediated by A2A-adenosine receptors is favoured by activation of release inhibitory receptors.
Keywords: A2A-adenosine receptors, noradrenaline release, A1-adenosine receptors, α2-autoreceptors, P2-receptors, rat tail artery, facilitation of neurotransmitter release, receptor interactions
Introduction
Noradrenaline release from postganglionic sympathetic nerve terminals is modulated by inhibitory α2-autoreceptors and also by receptors for other endogenous modulators (see Starke, 1977). Adenosine is one of these modulators (Fredholm & Hedqvist, 1980) and exerts its effects through activation of membrane A1-, A2A-, A2B- and A3-adenosine receptors (see Ribeiro, 1999). A1- and A2A-adenosine receptors may coexist in the same nerve terminal (rat neuromuscular junction: Correia-De-sá et al., 1991; rat vas deferens: Gonçalves & Queiroz, 1993; rat tail artery: Gonçalves & Queiroz, 1996) and mediate opposite effects on neurotransmitter release: an inhibition, mediated by A1-receptors, and a facilitation, mediated by A2A-receptors.
Several studies have provided evidence that the effects mediated by adenosine receptors influence or are influenced by the activation of other adenosine receptor subtypes or by receptors for other modulators (see Schlicker & Göthert, 1998; Ribeiro, 1999; Sebastião & Ribeiro, 2000). For instance, in rat hippocampal slices, activation of A1-receptors attenuates the inhibition of noradrenaline release mediated by α2-autoreceptors and by κ-opioid receptors (Limberger et al., 1988) whereas activation of α2-autoreceptors attenuates the inhibition of noradrenaline release mediated by A1-receptors (Allgaier et al., 1987; Limberger et al., 1988; Bucher et al., 1992). The facilitatory A2-receptors also seem to be involved in receptor-interactions. At motor nerve terminals, activation of A2-receptors potentiates the CGRP elicited facilitation of acetylcholine release, an effect opposite to that caused by activation of A1-receptors (Correia-De-sá & Ribeiro, 1994). Because the rat tail artery is also endowed with prejunctional A2A-adenosine receptors (Gonçalves & Queiroz, 1996), the aim of the present study was to investigate whether, as described for the A1-receptor-mediated inhibition, the A2A-adenosine receptor-mediated facilitation of noradrenaline release can be influenced by activation of release inhibitory receptors, namely α2-autoreceptors.
A preliminary account of this work has been presented previously (Diniz et al., 1999).
Methods
Adult male Wistar rats (290–400 g; Instituto Gulbenkian de Ciência, Oeiras, Portugal and CRIFFA, Barcelona, Spain) were sacrificed by cervical dislocation and exsanguination. The ventral tail artery was dissected out and cleaned of connective tissue. Eight tissue preparations (5–8 mg) were obtained from each artery. These were incubated in 2 ml medium containing 0.1 μM [3H]-noradrenaline, for 40 min at 37°C. Individual preparations were placed in superfusion chambers, between platinum electrodes, and superfused with [3H]-noradrenaline-free medium at a rate of 1 ml min−1. Successive 5-min samples of the superfusate were collected from t=55 min onwards (t=0 being the onset of superfusion). At the end of the experiments, tritium was determined in superfusate samples and in tissues by scintillation spectrometry (Beckman LS 6500, Beckman Instruments, Fullerton, U.S.A.).
The medium contained (mM): NaCl 118.6, KCl 4.70, CaCl2 2.52, MgSO4 1.23, NaHCO3 25.0, glucose 10.0, ascorbic acid 0.3 and dissodium EDTA 0.031; it was saturated with 95% O2–5% CO2 and kept at 37°C. Desipramine (400 nM; to inhibit neuronal uptake of noradrenaline) and, in some experiments, yohimbine (1 μM; to block α2-autoreceptors) were added throughout superfusion.
Up to five periods of electrical stimulation were applied (Stimulator II, Hugo Sachs Elektronik, March-Hugstetten, Germany; constant current mode; rectangular pulses; 1 ms width; current strength 50 mA; voltage drop between electrodes 18 V cm−1). The first, starting at t=30 min (S0) and consisting of 100 pulses at 5 Hz, was not used for determination of tritium outflow. The subsequent periods (S1 up to S4), consisting of either 100 pulses at 5 Hz or 20 pulses at 50 Hz, started at t=60 min with 30 min intervals, unless otherwise stated.
Concentration-response curves for the A2A-adenosine receptor agonist 2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine (CGS 21680; Jarvis et al., 1989; Lupica et al., 1990) were obtained in experiments in which five stimulation periods (S0 to S4) were applied. The agonist was added, at increasing concentrations, 5 min before S2, S3 to S4 and kept until the end of the stimulation period. In all other experiments only three stimulation periods (S0 and S2) were applied. CGS 21680 and the other agonists tested were added 5 min before S2, as previously; antagonists were added 20 min before S2 (unless stated otherwise).
The outflow of tritium was expressed as the fraction of tissue content at the onset of the respective collection period (fractional rate of outflow; min−1). Effects of drugs on basal tritium outflow were estimated by the values bn/b1 and expressed as the percentage of the mean ratio obtained in the appropriate control; bn was the fractional rate of outflow in the 5-min period before S2, S3 and S4 (b2, b3 and b4, respectively) and b1 the fractional rate of outflow in the 5-min period before S1. The electrically evoked overflow of tritium was calculated as the difference between ‘total tritium outflow during and after stimulation' minus ‘basal outflow', and expressed as percentage of the tissue tritium content at the time of stimulation. Effects of drugs that were added after S1 on electrically evoked overflow were evaluated as ratios of the overflow elicited by S2, S3 and S4 and the overflow elicited by S1 (Sn/S1). Sn/S1 values obtained in individual experiments in which a test compound A was added after S1 were calculated as a percentage of the respective mean ratio in the appropriate control group (solvent instead of A). When the interaction of A, added after S1, and a drug B either added after S1 or at the beginning of superfusion, was studied, the ‘appropriate control' was a group in which B alone was used (von kügelgen et al., 1995).
Results are expressed as mean±s.e.mean; n denotes the number of tissue preparations. Differences between means were tested for significance using the unpaired Student's t-test or one-way ANOVA followed by Tukey's multiple comparison test. A P value lower than 0.05 was taken to indicate significant differences.
The following drugs were used: levo-[ring-2,5,6-3H]-noradrenaline, specific activity 46.8 Ci mmol−1 was from DuPont NEN (Garal, Lisboa, Portugal); 5-bromo-6-(2-imidazolin-2-ylamino)-quinoxaline tartrate (UK 14304), 2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride (CGS 21680), N6-cyclopentyladenosine (CPA), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), desipramine hydrochloride, tetraethylammonium chloride monohydrate (TEA), yohimbine hydrochloride were from Sigma and 2-methylthioadenosine triphosphate tetrasodium (2-MeSATP) was from RBI (Sigma Aldrich, Alcobendas, Spain); 4-(2[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino] ethyl)phenol (ZM 241385) was from Tocris (Bristol, U.K.).
Solutions of drugs were prepared with either distilled water or dimethylsulphoxide and diluted with medium immediately before use. Solvents were added to the superfusion medium in parallel control experiments.
Results
In the absence of drugs (except desipramine 400 nM, present in the superfusion medium of all experiments), the fractional rate of outflow immediately before S1 (b1) was 0.126±0.006% min−1 (n=154) and remained almost constant throughout the experiment with b2/b1, b3/b1 and b4/b1 of 0.86±0.02, 0.85±0.03 and 0.82±0.03, respectively (n=14–30). The fractional rate of outflow was not different when yohimbine (1 μM) was added throughout (0.129±0.004% min−1; n=78). None of the drugs used (except for TEA; see below) or their solvents altered the basal tritium outflow (not shown).
Effect of CGS 21680 on tritium overflow evoked by trains of 100 pulses at 5 Hz
Stimulation with trains of 100 pulses at 5 Hz caused an overflow of tritium. In the absence of other drugs, the S1 value was 0.520±0.029% (n=138) of the tritium content of the tissue and remained almost constant throughout the experiment with S2/S1, S3/S1 and S4/S1 values of 0.91±0.04, 0.86±0.04 and 0.88±0.04 (n=52), respectively. Yohimbine (1 μM), when introduced at the beginning of the superfusion and kept throughout, increased tritium overflow (S1 values of 1.799±0.059% of the tissue tritium content; n=77) which remained constant throughout the experiment (S2/S1, S3/S1 and S4/S1 values of 0.97±0.03, 0.97±0.03 and 0.93±0.04, respectively; n=31). The higher tritium overflow observed in the presence of 1 μM yohimbine indicated that a marked 2-autoinhibition of noradrenaline release was occurring. This was confirmed in experiments where yohimbine, introduced 20 min before S2, increased tritium overflow (S2/S1 value of 346±22%; n=6; P<0.05).
In the absence of yohimbine, the selective A2A-adenosine receptor agonist CGS 21680 enhanced the evoked overflow of tritium in a concentration dependent manner (Figure 1). The facilitatory effect of CGS 21680 was blocked not only by the A2A-adenosine-receptor antagonist ZM 241385 (20 nM; Poucher et al., 1995), but also by the α2-adrenoceptor antagonist yohimbine (Figure 1).
Figure 1.
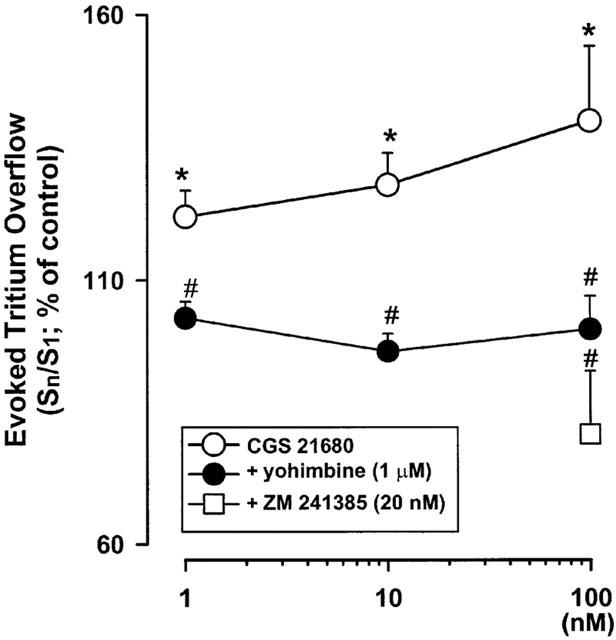
Effect of the A2A-adenosine receptor agonist CGS 21680 on the evoked tritium overflow from isolated rat tail artery in the absence of the α2-adrenoceptor antagonist yohimbine (α2-autoinhibition rich conditions: open circles, CGS 21680 alone; open squares, CGS 21680 in the presence of the A2A-receptor antagonist ZM 241385) and in the presence of 1 μM yohimbine (α2-autoinhibition poor conditions; filled circles). S1 to Sn (S2, S3 and S4) are the overflows elicited by trains of 100 pulses at 5 Hz (30 min intervals between trains). CGS 21680 was added, at increasing concentrations, 5 min before S2, S3 and S4; ZM 241385 was added 20 min before S4. When indicated, yohimbine was present throughout. Ordinate, Sn/S1 values obtained in individual tissue preparations and expressed as percentage of the corresponding average control Sn/S1 value. Values are means±s.e.mean from 7–20 tissue preparations. Significant differences from the appropriate control: *P<0.05 (unpaired Student's t-test); from CGS 21680 alone: #P<0.05 (ANOVA followed by Tukey's t-test).
In the presence of yohimbine (1 μM), either the A1-adenosine receptor agonist CPA (100 nM) or the P2-receptor agonist 2-MeSATP (80 μM), when added 5 min before S2, reduced the evoked overflow of tritium: S2/S1 values of 70±4% (n=9) and 73±12% (n=6), respectively (P<0.05). CGS 21680 (100 nM) that, under these experimental conditions, did not modify tritium overflow (S2/S1=100±9%; n=8), increased tritium overflow when tested in the presence of CPA (100 nM) or 2-MeSATP (80 μM): S2/S1 values of 151±24% (n=9) and 121±5% (n=10), respectively (P<0.05).
Effect of CGS 21680 on tritium overflow evoked by trains of 20 pulses at 50 Hz
Stimulation with trains of 20 pulses at 50 Hz caused an overflow of tritium. In the absence of other drugs, the S1 value was 0.362±0.025% (n=22) of the tritium content of the tissue and remained constant when a third period of stimulation was applied (S2/S1 values of 1.06±0.06; n=22). Stimulation with this brief train of pulses elicited a tritium overflow under α2-autoinhibition-poor conditions: yohimbine (1 μM), when introduced 20 min before S2, did not increase the evoked overflow of tritium (S2/S1=92±11%; n=7).
CGS 21680 (up to 100 nM), when added 5 min before S2, failed to increase the tritium overflow elicited by trains of 20 pulses at 50 Hz (S2/S1 value of 98±5%; n=17). However, the selective α2-adrenoceptor agonist UK 14304 (30 nM), which decreased the evoked overflow of tritium (S2/S1 value of 65±6%; n=12; P<0.05), restored the ability of CGS 21680 to enhance tritium overflow under these stimulation conditions (Figure 2). The A1-adenosine receptor agonist CPA (100 nM) or the P2-receptor agonist 2-MeSATP (80 μM), when added 5 min before S2 also reduced tritium overflow: S2/S1 values of 55±5% (n=8) and 32±11% (n=6), respectively (P<0.05) and restored the facilitatory effect of CGS 21680 (Figure 2).
Figure 2.
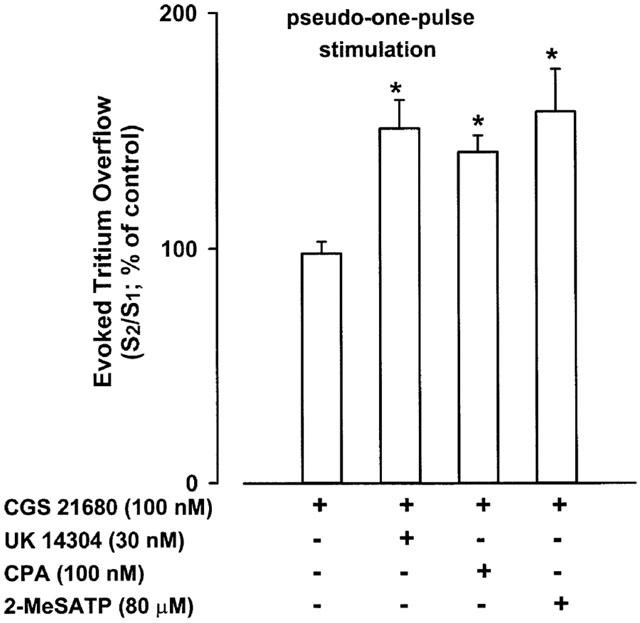
Effect of the A2A-adenosine receptor agonist CGS 21680 on the evoked tritium overflow from isolated rat tail artery in α2-autoinhibition poor conditions. S1 and S2 are the overflows elicited by trains of 20 pulses at 50 Hz (pseudo-one-pulse stimulation; 30 min intervals between trains). CGS 21680 and/or UK 14304 (an α2-adrenoceptor agonist), CPA (an A1-receptor agonist) or 2-MeSATP (P2-receptor agonist) were added 5 min before S2. When tested alone, UK 14304, CPA and 2-MeSATP reduced tritium overflow (S2/S1 values between 32 and 65%, see text). Ordinate, S2/S1 values obtained in individual tissue preparations and expressed as percentage of the corresponding average control S2/S1 value. Values are means±s.e.mean from 7–12 tissue preparations. Significant differences from the appropriate control: *P<0.05 (unpaired Student's t-test).
Effect of CGS 21680 under conditions leading to high tritium overflow levels
The overflow of tritium from preparations stimulated with 100 pulses at 5 Hz was markedly enhanced by yohimbine (see above). In order to test whether yohimbine prevents the effects of CGS 21680 by increasing tritium overflow so markedly that makes difficult to envisage further increases, the effect of CGS 21680 was investigated in the presence of the non-selective K+-channel blocker TEA (Cook, 1988).
TEA (5 mM) caused a marked increase on basal outflow which stabilized only after 25 min incubation: b2/b1 value of 139±3% (n=8). Therefore, stimulation periods (100 pulses at 5 Hz) were applied with 45 min intervals. TEA (5 mM), when introduced 10 min after S1 (35 min before S2), increased the evoked tritium overflow (S2/S1 value of 417±68%; n=8). Yohimbine (1 μM), when introduced 35 min before S2, increased tritium overflow (S2/S1 value of 328±30%; n=4; not different from that elicited by TEA alone). When yohimbine was combined with TEA, it potentiated the increase on tritium overflow caused by TEA alone (S2/S1 value of 774±164%; n=4; P<0.05, from the increase caused by TEA or yohimbine alone). Contrasting with the effect of CGS 21680 on tritium overflow in the presence of yohimbine, CGS 21680 significantly enhanced tritium overflow in the presence of TEA (Figure 3).
Figure 3.
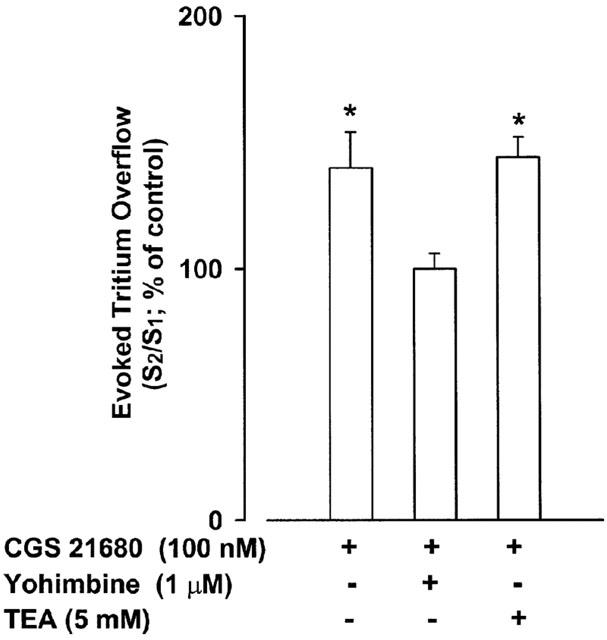
Effect of the A2A-adenosine receptor agonist CGS 21680 on the evoked tritium overflow from isolated rat tail artery in the absence or in the presence of yohimbine or tetraethylammonium (TEA). S1 and S2 are the overflows elicited by trains of 100 pulses at 5 Hz (45 min intervals between trains). Yohimbine (1 μM) or TEA (5 mM) were added 35 min and CGS 21680 (100 nM) 5 min before S2. S2 value (% of tritium content) in the absence of drugs was 0.480±0.077% (n=12), in the presence of yohimbine was 1.823±0.013% (n=4) and in the presence of TEA was 2.022±0.340% (n=8). Ordinate, S2/S1 values obtained in individual tissue preparations and expressed as percentage of the corresponding average control S2/S1 value. Values are means±s.e.mean from 6–8 tissue preparations. Significant differences from the appropriate control: *P<0.05 (unpaired Student's t-test).
Effect of α2-autoinhibition on the tonic effects mediated by adenosine receptors
The effects of selective A1- and A2A-adenosine receptor antagonists on tritium overflow elicited by trains of 100 pulses at 5 Hz were tested in the absence and in the presence of yohimbine in order to investigate if a putative modulation exerted by endogenous adenosine is influenced by the α2-autoinhibition.
The A1-adenosine receptor antagonist DPCPX (20 nM) did not change tritium overflow either in the absence or in the presence of 1 μM yohimbine, (Figure 4). However, yohimbine influenced the effects of the A1-adenosine receptor agonist N6-cyclopentyladenosine (CPA; 100 nM) which reduced tritium overflow less markedly in the absence (S2/S1=83±9%; n=9) than in the presence of 1 μM yohimbine (S2/S1=58±4%; n=12; P<0.05).
Figure 4.
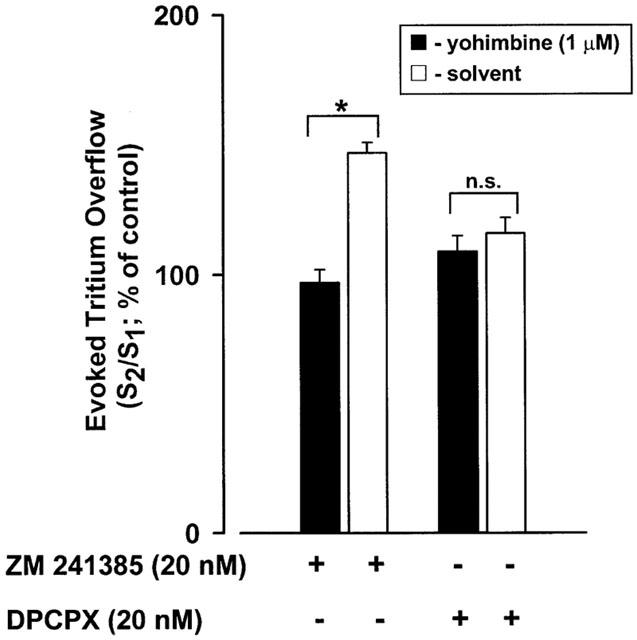
Effect of the A2A-adenosine receptor antagonist ZM 241385 and of the A1-adenosine receptor antagonist DPCPX on the evoked tritium overflows from isolated rat tail artery in the absence (solvent; open bars) or in the presence of the α2-adrenoceptor antagonist yohimbine (filled bars). S1 and S2 are the overflows elicited by trains of 100 pulses at 5 Hz (30 min intervals between trains). When indicated, yohimbine (1 μM) was added throughout the superfusion; ZM 241385 or DPCPX were added 20 min before S2. Ordinate, S2/S1 values obtained in individual tissue preparations and expressed as percentage of the corresponding average control S2/S1 value. Values are means±s.e.mean from 8–15 tissue preparations. Significant differences from the effect in the presence of yohimbine: *P<0.05; n.s., non significant (unpaired Student's t-test).
The A2A-adenosine receptor antagonist ZM 241385 (20 nM) caused, per se, an unexpected increase on tritium overflow (S2/S1 value of 148±6%; n=6) which did not occur when ZM 241385 was tested in the presence of 1 μM yohimbine (Figure 4).
Discussion
The electrically evoked tritium overflow from tissue preparations of rat tail artery preincubated with [3H]-noradrenaline was assumed to reflect action potential-evoked neuronal noradrenaline release and drug-induced changes in evoked tritium overflow assumed to reflect changes in neuronal release. This is consistent with the established release-inhibitory effects of α2-adrenoceptor, A1-adenosine and P2- receptor agonists observed in this preparation (Illes et al., 1989; Gonçalves & Queiroz, 1996) and with the release desinibiting effects of α2-adrenoceptor antagonists observed in the present experimental conditions.
The present study confirms previous observations (Gonçalves & Queiroz, 1996; Mota et al., 2000) indicating the presence of release-facilitatory A2A-adenosine receptors in the rat tail artery. However, in the present experimental conditions, the selective A2A-receptor agonist CGS 21680 enhanced noradrenaline release only when α2-autoinhibition was occurring: blockade of α2-autoreceptors with yohimbine prevented the A2A-mediated facilitation of noradrenaline release caused by CGS 21680. This observation is in agreement with results published recently (Mota et al., 2000). An A2A-receptor-mediated facilitation was previously observed even in the presence of yohimbine (Gonçalves & Queiroz, 1996). However, in this study noradrenaline release was estimated by measuring overflow of endogenous noradrenaline and, therefore, a long train of stimulation (2700 pulses) had to be used. The effects of CGS 21680 in the absence of yohimbine were not investigated and the possibility that the facilitatory effect of CGS 21680 would be greater in yohimbine-free medium cannot be excluded. The fact that CGS 21680 was still able to cause facilitation of noradrenaline in the presence of yohimbine may also be due to the occurrence of P2-autoinhibition under that experimental conditions: revealed by the P2-antagonist Reactive Blue 2 (Gonçalves & Queiroz, 1996): or due to a higher degree of α2-autoinhibition attained with the long train of pulses, not totally blocked with the concentration of yohimbine used. The possibility that an increase in the train length may strengthen the autoinhibition is compatible with the observation that tritium overflow elicited by a long train of pulses decreases along the train (Driessen et al., 1994). In the present study it was not feasible to estimate drug effects based on tritium overflow elicited by 2700 pulses because tritium overflow was markedly reduced along the experiments, with S2/S1 values lower than 0.2 (not shown).
A possible explanation for the failure of CGS 21680 to facilitate noradrenaline release in the presence of yohimbine could be that the levels of noradrenaline released under the present experimental conditions are so high that further increases would become difficult to envisage. However, the following observations argue against this hypothesis: (i) TEA enhances noradrenaline release by a mechanism that does not involve α2-adrenoceptor-mediated autoinhibition (Kirpekar et al., 1976) and, by opposition to what happened in the presence of yohimbine, the A2A-receptor-mediated facilitation of noradrenaline release was still observed when noradrenaline release was raised by TEA; (ii) by using a short train of high frequency stimulation it is possible to elicit noradrenaline release under α2-autoinhibition poor conditions (pseudo-one-pulse; brain slices: Singer, 1988; postganglionic sympathetic nerve terminals: Trendelenburg et al., 1997). Under this α2-autoinhibition poor conditions, the A2A-adenosine receptor agonist CGS 21680 failed to increase noradrenaline release although it occurred when α2-autoreceptors were activated by an exogenous agonist or when other release inhibitory receptors were activated.
Another tentative hypothesis could be that yohimbine is acting as an antagonist at A2A-adenosine receptors. The following observations argue against this hypothesis. In the rat vas deferens, CGS 21680 causes an A2A-adenosine-receptor-mediated inhibition of contractions to exogenous noradrenaline, and this effect of CGS 21680 was not changed by 1 μM yohimbine (Diniz & Gonçalves, unpublished observation). Furthermore, in other receptor systems, interactions of this kind have been revealed irrespective of the α2-antagonist used (phentolamine: Majewski & Rand, 1981; Johnston & Majewski, 1986; Costa & Majewski, 1988; Cox et al., 2000; rauwolscine: Bucher et al., 1992; yohimbine: Allgaier et al., 1987; Limberger et al., 1988).
In summary, the present study provides evidence that the occurrence of an A2A-adenosine receptor-mediated facilitation of noradrenaline release is neither dependent on the levels of the released noradrenaline nor on the presence of yohimbine per se: it occurs either with low (100 pulses, 5 Hz, yohimbine absent) or with high (100 pulses, 5 Hz, TEA present) noradrenaline released levels, but not in α2-autoinhibition poor conditions, irrespective of release noradrenaline levels being high (100 pulses, 5 Hz, yohimbine present) or low (20 pulses, 50 Hz, yohimbine absent).
In our view, the most likely hypothesis is that the inhibition by the α2-adrenoceptor antagonist of an effect mediated by A2A-adenosine receptors is due to the occurrence of an interaction between α2-autoreceptors and A2A-receptors in a way that activation of α2-autoreceptors would favour the A2A-receptor-mediated facilitation of noradrenaline release. Interactions between α2-autoreceptors and other release-inhibitory receptors have been previously described both at central and peripheral nervous systems (see Schlicker & Göthert, 1998). Interactions between α2-autoreceptors and release-facilitatory receptors had also been investigated but these led to conflicting results. The release-facilitatory effect of angiotensin II was reported to be increased (Costa & Majewski, 1988), decreased (Cox et al., 2000) or not influenced (Mota et al., 2000) by blockade of α2-autoreceptors. Furthermore, it was also reported that the release-facilitatory effect mediated by β-adrenoceptors was increased (Majewski & Rand, 1981; Johnston & Majewski, 1986) or not influenced (Cox et al., 2000; Mota et al., 2000) by blockade of α2-autoreceptors. The reasons for these differences are not understood but may be the use of different tissue preparations and/or different stimulation parameters.
Our experiments also provide evidences that activation of other release inhibitory receptors, such as the A1- or the P2-receptors, also favour the A2A-receptor-mediated facilitation of noradrenaline release, at least in conditions in which α2-autoinhibition is not operating. To our knowledge, the present is the first study to demonstrate that the A2A-adenosine receptor-mediated facilitation of noradrenaline release is favoured by activation of release inhibitory receptors. Nevertheless, previous observations can be interpreted according to the occurrence of such an interaction. One of the first studies on interactions between adenosine and other receptors in the brain showed that the α-adrenoceptor-mediated increase in cyclic AMP levels required the presence of endogenous adenosine (Sattin et al., 1975). Since none of the α1- and α2-receptor subtypes known is positively coupled to adenylate cyclase whereas the A2-adenosine receptors are, the explanation may be that it was the adenosine receptor-mediated increase in cyclic AMP that was potentiated by an α-adrenoceptor activation and not the opposite, as tentatively proposed.
Receptor interactions may be mutual (see Limberger et al., 1988). We did not study in detail the influence of A2A-receptor agonists on the α2-autoinhibition. However, the surprising observation that blockade of the A2A-adenosine receptor increases noradrenaline release, an effect not observed when α2-autoreceptors were blocked may indicate that the blockade of A2A-receptors may also disturb the α2-autoinhibition.
Interactions between adenosine A2-receptors and receptors for other transmitters have been recently reviewed (see Ribeiro, 1999; Sebastião & Ribeiro, 2000). The present study presents evidence that activation of α2-autoreceptors and other release inhibitory receptors favours the A2A-receptor-mediated facilitation of noradrenaline release. The molecular mechanism involved on this interaction is not known. In mouse atria, an interaction involving α2-autoreceptors and angiotensin or bradykinin receptors was explained by an interaction between Gq/11-protein, to which AT1- and B2-receptors are coupled, and Gi/o-protein, to which α2-autoreceptors are coupled (Cox et al., 2000). A2A-adenosine receptors are considered to be coupled to Gs (Marala & Mustafa, 1993) and the mechanism described above was shown not to be applicable to Gs coupled receptors such as the β-adrenoceptors (Cox et al., 2000). However, A2A-receptors may be promiscuous in their interaction with G proteins and be coupled to transducing mechanisms other than Gs (Gi/o; Cunha et al., 1999; or the Gs-like protein, Golf; Kull et al., 2000). Furthermore, A2A-receptors not only activate adenylate cyclase but also protein kinase C (Gubitz et al., 1996; Sheldon et al., 1996), phospholipase C/inositol triphosphate/calmodulin and calmodulin kinase II pathway (Wirkner et al., 2000), a serine/threonine protein phosphatase (Revan et al., 1996) or the mitogen-activated protein kinase cascade (Sexl et al., 1997). Facing the multiple coupling possibilities of A2A-adenosine receptors we cannot discard the possibility that, in rat tail artery, A2A-receptors may be coupled to transducing systems other than Gs and the occurrence of an interaction by a mechanism similar to that proposed for Gq/11 coupled receptors (Cox et al., 2000; see also Selbie & Hill, 1998) cannot be excluded.
In conclusion, the present study confirms the presence of adenosine A2A-receptors in the isolated rat tail artery mediating a facilitation of noradrenaline release and describes an interaction between A2A-adenosine receptors and release inhibitory receptors in a way that an inhibition mediated by α2-, A1- or P2-receptors favours the A2A-receptor-mediated facilitation of noradrenaline release.
Acknowledgments
The authors thank Associação Nacional das Farmácias for the scintillation spectrometry equipment and M.C. Pereira for the technical assistance. Supported by FCT (I&D n. 226/94, POCTI - QCAIII and FEDER)
Abbreviations
- CGS 21680
2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine
- CPA
N6-cyclopentyladenosine
- DPCPX
8-cyclopentyl-1,3-dipropylxanthine
- 2-MeSATP
2-methylthioadenosine triphosphate
- TEA
tetraethylammonium
- UK 14304
5-bromo-6-(2-imidazolin-2-ylamino)-quinoxaline
- ZM 241385
4-(2[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol
References
- ALLGAIER C., HERTING G., KÜGELGEN O.V. The adenosine receptor-mediated inhibition of noradrenaline release possibly involves a N-protein and is increased by α2-autoreceptor blockade. Br. J. Pharmacol. 1987;90:403–412. doi: 10.1111/j.1476-5381.1987.tb08970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUCHER B., CORRIU C., STOCLET J-C. Prejunctional opiod μ-receptors and adenosine A1-receptors on the sympathetic nerve endings of the rat tail artery interact with the α2-adrenoceptors. Naunyn-Schmiedeberg's. Arch. Pharmacol. 1992;345:37–43. doi: 10.1007/BF00175467. [DOI] [PubMed] [Google Scholar]
- COOK N.S. The pharmacology of potassium channels and their therapeutic potential. Trends Pharmacol. Sci. 1988;9:21–28. doi: 10.1016/0165-6147(88)90238-6. [DOI] [PubMed] [Google Scholar]
- CORREIA-DE-SÁ P., RIBEIRO J.A. Potentiation by tonic A2a-adenosine receptor activation of CGRP-facilitated [3H]-ACh release from rat motor nerve endings. Br. J. Pharmacol. 1994;111:582–588. doi: 10.1111/j.1476-5381.1994.tb14777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORREIA-DE-SÁ P., SEBASTIÃO A.M., RIBEIRO J.A. Inhibitory and excitatory effects of adenosine receptor agonists on evoked transmitter release from phrenic nerve endings of the rat. Br. J. Pharmacol. 1991;103:1614–1620. doi: 10.1111/j.1476-5381.1991.tb09836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSTA M., MAJEWSKI H. Facilitation of noradrenaline release from sympathetic nerves through activation of ACTH receptors, β-adrenoceptors and angiotensin II receptors. Br. J. Pharmacol. 1988;95:993–1001. doi: 10.1111/j.1476-5381.1988.tb11730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COX S.L., SCHELB V., TRENDELENBURG A.U., STARKE K. Enhancement of noradrenaline release by angiotensin II and bradykinin in mouse atria: evidence for cross-talk between Gq/11 protein- and Gi/o protein-coupled receptors. Br. J. Pharmacol. 2000;129:1095–1102. doi: 10.1038/sj.bjp.0703167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNHA R.A., CONSTANTINO M.D., RIBEIRO J.A. G Protein coupling of CGS 21680 binding sites in the rat hippocampus and cortex is different from that of adenosine A1 and striatal A2A receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;359:295–302. doi: 10.1007/pl00005355. [DOI] [PubMed] [Google Scholar]
- DINIZ C., FRESCO P., QUEIROZ G., GONÇALVES J. Influence of α2-autoreceptor blockade on the A2A-mediated facilitation of noradrenaline release in rat isolated tail artery. Br. J. Pharmacol. 1999;127:114P. doi: 10.1038/sj.bjp.0704686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRIESSEN B., VON KÜGELGEN I., BÜLTMANN R., ELRICK D.B., CUNNANE T.C., STARKE K. The fade of the purinergic neurogenic contraction of the guinea-pig vas deferens: analysis of possible mechanisms. Naunyn-Schmiedeberg's Arch. Pharmacol. 1994;350:482–490. doi: 10.1007/BF00173017. [DOI] [PubMed] [Google Scholar]
- FREDHOLM B.B., HEDQVIST P. Modulation of neurotransmission by purine nucleotides and nucleosides. Biochem. Pharmacol. 1980;29:1635–1643. doi: 10.1016/0006-2952(80)90117-3. [DOI] [PubMed] [Google Scholar]
- GONÇALVES J., QUEIROZ G. Facilitatory and inhibitory modulation by endogenous adenosine of noradrenaline release in the epididymal portion of rat vas deferens. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;348:367–371. doi: 10.1007/BF00171335. [DOI] [PubMed] [Google Scholar]
- GONÇALVES J., QUEIROZ G. Purinoceptor modulation of noradrenaline release in rat tail artery: tonic modulation mediated by inhibitory P2Y- and facilitatory A2A-receptors. Br. J. Pharmacol. 1996;117:156–160. doi: 10.1111/j.1476-5381.1996.tb15168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUBITZ A.K., WIDDOWSON L., KUROKAWA M., KIRKPATRICK K.A., RICHARDSON P.J. Dual signalling by the adenosine A2a receptor involves activation of both N- and P-type calcium channels by different G proteins and protein kinases in the same striatal nerve terminals. J. Neurochem. 1996;67:374–381. doi: 10.1046/j.1471-4159.1996.67010374.x. [DOI] [PubMed] [Google Scholar]
- ILLES P., RICKMANN H., BROD I., BUCHER B., STOCLET J.C. Subsensitivity of presynaptic adenosine A1-receptors in caudal arteries of spontaneously hypertensive rats. Eur. J. Pharmacol. 1989;174:237–251. doi: 10.1016/0014-2999(89)90316-6. [DOI] [PubMed] [Google Scholar]
- JARVIS M.F., SCHULZ R., HUTCHISON A.J., DO U.H., SILLS M.A., WILLIAMS M. [3H]CGS 21680, a selective A2 adenosine receptor agonist directly labels A2 receptors in rat brain. J. Pharmacol. Exp. Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- JOHNSTON H., MAJEWSKI H. Prejunctional β-adrenoceptors in rabbit pulmonary artery and mouse atria: effect of α-adrenoceptor blockade and phosphodiesterase inhibition. Br. J. Pharmacol. 1986;87:553–562. doi: 10.1111/j.1476-5381.1986.tb10197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIRPEKAR S.M., WAKADE A.R., PRAT J.C. Effect of tetraethylammonium and barium on the release of noradrenaline from the perfused cat spleen by nerve stimulation and potassium. Naunyn-Schmiedeberg's Arch. Pharmacol. 1976;294:23–29. doi: 10.1007/BF00692781. [DOI] [PubMed] [Google Scholar]
- KULL B., SVENNINGSSON P., FREDHOLM B.B. Adenosine A2A receptors are colocalized with and activate Golf in rat striatum. Mol. Pharmacol. 2000;58:771–777. doi: 10.1124/mol.58.4.771. [DOI] [PubMed] [Google Scholar]
- LIMBERGER N., SPÄTH L., STARKE K. Presynaptic α2-adrenoceptor, opioid κ-receptor and adenosine A1-receptor interactions on noradrenaline release in rabbit brain cortex. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;338:53–61. doi: 10.1007/BF00168812. [DOI] [PubMed] [Google Scholar]
- LUPICA C.R., CASS W.A., ZAHNISER N.R., DUNWIDDIE T.V. Effects of the selective adenosine A2 receptor agonist CGS 21680 on in vitro electrophysiology, cAMP formation and dopamine release in rat hippocampus and striatum. J. Pharmacol. Exp. Ther. 1990;252:1134–1141. [PubMed] [Google Scholar]
- MAJEWSKI H., RAND M.J. An interaction between prejunctional α-adrenoceptors and prejunctional β-adrenoceptors. Eur. J. Pharmacol. 1981;69:493–498. doi: 10.1016/0014-2999(81)90455-6. [DOI] [PubMed] [Google Scholar]
- MARALA R.B., MUSTAFA S.J. Direct evidence for the coupling of A2-adenosine receptor to stimulatory guanine nucleotide-binding-protein in bovine brain striatum. J. Pharmacol. Exp. Ther. 1993;266:294–300. [PubMed] [Google Scholar]
- MOTA A., PAIVA M.Q., MOURA D., GUIMARÃES S. Lack of interaction between α2-autoreceptors and prejunctional receptors mediating a facilitatory effect on noradrenaline release. Pharmacological Res. 2000;42:383–387. doi: 10.1006/phrs.2000.0707. [DOI] [PubMed] [Google Scholar]
- POUCHER S.M., KEDDIE J.R., SINGH P., STOGGALL S.M., CAULKETT P.W.R., JONES G., COLLIS M.G. The in vitro pharmacology of ZM 241385, a potent, non-xanthine, A2a selective adenosine receptor antagonist. Br. J. Pharmacol. 1995;115:1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REVAN S., MONTESINOS M.C., NAIME D., LANDAU S., CRONSTEIN B.N. Adenosine A2 receptor occupancy regulates stimulated neutrophil function via activation of a serine/threonine protein phosphatase. J. Biol. Chem. 1996;271:17114–17118. doi: 10.1074/jbc.271.29.17114. [DOI] [PubMed] [Google Scholar]
- RIBEIRO J.A. Adenosine A2A receptor interactions with receptors for other neurotransmitters and neuromodulators. Eur. J. Pharmacol. 1999;375:101–113. doi: 10.1016/s0014-2999(99)00230-7. [DOI] [PubMed] [Google Scholar]
- SATTIN A., RALL T.W., ZANELLA J. Regulation of cyclic adenosine 3′,5′-monophosphate levels in guinea-pig cerebral cortex by interaction of alpha adrenergic and adenosine receptor activity. J. Pharmacol. Exp. Ther. 1975;192:22–32. [PubMed] [Google Scholar]
- SCHLICKER E., GÖTHERT M. Interactions between the presynaptic α2-autoreceptor and presynaptic inhibitory heteroreceptors on noradrenergic neurones. Brain Res. Bull. 1998;47:129–132. doi: 10.1016/s0361-9230(98)00068-9. [DOI] [PubMed] [Google Scholar]
- SEBASTIÃO A.M., RIBEIRO J.A. Fine-tuning neuromodulation by adenosine. Trends Pharmacol. Sci. 2000;21:341–346. doi: 10.1016/s0165-6147(00)01517-0. [DOI] [PubMed] [Google Scholar]
- SELBIE L.A., HILL S.J. G protein-coupled-receptor cross-talk: the fine-tuning of multiple receptor-signalling pathways. Trends Pharmacol. Sci. 1998;19:87–93. doi: 10.1016/s0165-6147(97)01166-8. [DOI] [PubMed] [Google Scholar]
- SEXL V., MANCUSI G., HÖLLER C., GLORIA-MAERCKER E., SCHÜTZ W., FREISSMUTH M. Stimulation of the mitogen-activated protein kinase via the A2A-adenosine receptor in primary human endothelial cells. J. Biol. Chem. 1997;272:5792–5799. doi: 10.1074/jbc.272.9.5792. [DOI] [PubMed] [Google Scholar]
- SHELDON R.L., BONASERA L.K., EICHBERG J. Activation of adenosine A2 receptors stimulates phosphoinositide metabolism in rat peripheral nerve. J. Neurochem. 1996;66:613–619. doi: 10.1046/j.1471-4159.1996.66020613.x. [DOI] [PubMed] [Google Scholar]
- SINGER E.A. Transmitter release from brain slices elicited by single pulses: a powerful method to study presynaptic mechanisms. Trends Pharmacol. Sci. 1988;9:274–276. doi: 10.1016/0165-6147(88)90004-1. [DOI] [PubMed] [Google Scholar]
- STARKE K. Regulation of noradrenaline release by presynaptic receptor systems. Rev. Physiol. Biochem. Pharmacol. 1977;77:1–125. doi: 10.1007/BFb0050157. [DOI] [PubMed] [Google Scholar]
- TRENDELENBURG A-U., SUTEJ I., WAHL C.A., MOLDERINGS G.J., RUMP L.C., STARKE K. A re-investigation of questionable subclassifications of α2-autoreceptors: rat vena cava, rat atria, human kidney and guinea-pig urethra. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:721–737. doi: 10.1007/pl00005111. [DOI] [PubMed] [Google Scholar]
- VON KÜGELGEN I., STOFFEL D., STARKE K. P2-purinoceptor-mediated inhibition of noradrenaline release in rat atria. Br. J. Pharmacol. 1995;115:247–254. doi: 10.1111/j.1476-5381.1995.tb15870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIRKNER K., ASSMANN H., KÖLES L., GEREVICH Z., FRANKE H., NÖRENBERG W., BOEHM R., ILLES P. Inhibition by adenosine A2A receptors of NMDA but not AMPA currents in rat neostriatum neurons. Br. J. Pharmacol. 2000;130:259–269. doi: 10.1038/sj.bjp.0703234. [DOI] [PMC free article] [PubMed] [Google Scholar]