

Abstract
This study examined whether NO is involved in the in-vivo coronary vasodilator effects of amlodipine (a calcium channel blocker) and whether heart failure (HF) alters the coronary responses to amlodipine.
Nine conscious dogs were chronically instrumented to measure circumflex coronary blood flow (CBF) and coronary diameter (CD). Drugs were administered directly into the circumflex artery through an indwelling catheter to avoid systemic changes. HF was induced by right ventricular pacing (240 b.p.m., 3 weeks).
Compared with control (C), in HF, coronary responses to acetylcholine (1–10 ng kg−1) were reduced while responses to nitroglycerin (0.1–0.5 μg kg−1) were unchanged. In C, amlodipine (30–150 μg kg−1), increased dose-dependently CBF and CD. After LNA (a NO synthase inhibitor, 2 mg kg−1), amlodipine produced less increases in CBF and CD (+121±26 ml min−1 and +76±35 μm versus +196±40 ml min−1 and +153±39 μm respectively for 150 μg kg−1 amlodipine alone, both P<0.05). In HF, the coronary responses to amlodipine were reduced (150 μg kg−1 of amlodipine increased CBF and CD +121±23 ml min−1 and +77±21 μm respectively, both P<0.05). After LNA, the CBF responses to amlodipine tended to be reduced (+94±19 ml min−1 at 150 μg kg−1) but CD responses were significantly reduced (+41±16 μm, P<0.05). The supplementation with L-arginine did not enhance the coronary responses to amlodipine.
These results indicate that, in conscious dogs, NO participates in the coronary responses to amlodipine and in HF, the coronary responses to amlodipine are reduced, which is related to a reduced NO production.
Keywords: Amlodipine, nitric oxide, heart failure, coronary circulation
Introduction
Calcium channel blockers are widely used for the treatment of hypertension and angina pectoris because of their capability to induce potent coronary vasodilation. Calcium channel blockers are believed to increase coronary blood flow through inhibition of Ca2+ entry in smooth muscle cells. However, it has been shown that, in addition to this mechanism, amlodipine, a second generation dihydropyridine compound, can release nitric oxide (NO) in isolated coronary microvessels (Zhang & Hintze, 1998; Zhang et al., 1999). However, as vasomotor abnormalities related to impaired endothelial release of NO or decreased responsiveness of vascular smooth muscle cells occur during heart failure (HF) (Kuro et al., 1991; Katz et al., 1993, Wang et al., 1994; Zhao et al., 1995, Su et al., 1998), it might be hypothesized that, in HF, the role of NO in the coronary vasodilator action of amlodipine is decreased. Therefore, this study examined in conscious dogs whether NO is involved, in vivo, in the coronary responses to amlodipine and whether the development of HF alters the coronary in vivo responses to amlodipine. To accomplish these goals and to avoid the influence of systemic haemodynamic variations and neurohormonal activation usually observed when calcium channel blockers are given intravenously, experiments were conducted in conscious dogs chronically instrumented with an indwelling coronary catheter allowing direct intracoronary drug administrations before and after induction of HF induced by rapid ventricular pacing.
Methods
Surgical procedure and instrumentation
Nine mongrel dogs weighing 25–35 kg were used in this study. After injection of thiopenthal sodium (16 mg kg−1) and incubation, the dogs were ventilated and anaesthetized with 1 vol% halothane mixed with air. As previously described (Houel et al., 1997; Su et al., 2000), a left thoracotomy was performed under sterile conditions for implantation of tygon catheters in the descending aorta and left atrium, a micromanometer in the left ventricular (LV) cavity and two pacing leads on the right ventricle. A silastic indwelling catheter (0.3 mm i.d., 0.63 mm o.d.) was inserted into the circumflex coronary artery 1–1.5 cm after the bifurcation of the left main coronary artery. An ultrasonic Doppler flow probe (10 MHz) was implanted around the circumflex artery distal to the tip of the intracoronary catheter to measure coronary blood flow velocity (CBFv). A pair of 5 MHz piezoelectric crystal was implanted downstream from the Doppler flow probe on opposite side of the circumflex artery to measure coronary diameter (CD). Analgesia was obtained with subcutaneous hydrochlorate morphine (10 mg) after dogs had restored autonomic respiration. Ampicillin (2 g day−1), gentamycin (40 mg day−1) were administrated after intervention daily for 2 weeks. The animals were maintained in accordance with the official regulations of the French Ministry of Agriculture.
Experimental protocol
Experiments in control state (C) were performed in conscious dogs that had fully recovered from surgery. Systemic and coronary parameters including heart rate, LV pressure, LV dP/dtmax, aortic pressure, left atrial pressure, CBFv and CD were monitored. Throughout the experiments, the coronary catheter was infused with warm saline solution (37°C, 1 ml min−1) which did not modify systemic or coronary parameters.
To determine coronary endothelial function and responsiveness of large and small coronary vessels, acetylcholine (1–10 ng kg−1, bolus) and nitroglycerin (0.1–0.5 μg kg−1, bolus) were injected directly into coronary vessels. To assess coronary reserve, adenosine was infused into the coronary artery at the dose of 2–10 μg kg−1 min−1 (2 min for each dose).
On a different day, after recordings of baseline parameters, amlodipine was infused into circumflex artery at doses of 1.5 μg kg−1 min−1 for 5 min, 5 μg kg−1 min−1 for 15 min, and finally 15 μg kg−1 min−1 for 4.5 min. Amlodipine was perfused at a constant rate of 1 ml min−1. This perfusion rate did not produce any changes in CBF or CD. The selected dose of amlodipine was based on our preliminary experiments showing minimal systemic changes.
In eight dogs, on a different day, to examine the implication of the NO in the coronary effect of amlodipine, 5 min after intracoronary infusion of a NO synthase inhibitor, Nω-nitro-L-arginine (LNA, 2 mg kg−1 over 10 min), the same dose of amlodipine was again infused.
To examine whether the supply of substrate of NO synthase would increase the coronary response to amlodipine, on another day, the same doses of amlodipine was infused 5 min after L-arginine infusion (2 mg kg−1 min−1 over 15 min) in the same dogs.
Between two experiments, a delay of at least 24 h was respected.
After completion of the experiments in C, a continuous right ventricular pacing was initiated at 240–250 beats min−1 with a miniature pacemaker placed in a pocket on the back of the animals. Dogs were examined daily to evaluate cardiac function. After the development of HF (3 weeks of ventricular pacing), the same protocol as in C was repeated. Experiments were conducted after a 15-min period of stabilization following the pacemaker interruption.
Plasma atrial natriuretic peptide measurement
To measure arterial plasma atrial natriuretic peptide (ANP) level, blood samples were withdrawn from the aortic catheter at baseline in C and 15 min after interruption of pacemaker in HF. Blood samples were placed in plastic tubes containing EDTA, aprotinin, PMSF and trypsine inhibitor. Plasma was obtained by centrifugation and stored at −80°C. Plasma ANP was measured by radioimmunoassay with an antiserum directed against C-terminal region of α-rANP (rat, Novabiochem) and obtained after immunization of rabbits. The lowest limit for detection was 1.25 pg tube−1.
Data collection and statistical analysis
All signals were recorded on a microcomputer and analysed with HEM (v1.5, Notocord Systems, Croissy-sur-Seine, France). Statham P23ID transducers were used to measure aortic and left atrial pressures. LV pressure and dp/dt were measured by the micromanometer. CBFv was measured using a pulsed Doppler flowmeter and CD measurement was obtained by connecting the piezoelectric ultrasonic crystals to a sonomicrometer. In two dogs, for technical reasons, coronary diameter measurements were not available. Mean coronary blood flow (CBF, ml min−1) was calculated using the formula CBF=π r2·CBFv where r was the internal coronary radius calculated as half of external coronary diameter (CD) minus the wall thickness (which was considered as 20% CD).
Results are expressed as means±s.e.mean. ANOVA was performed with SuperANOVA software (v1.1, Abacus concept, Berkeley, CA, U.S.A.). A 1-way analysis of variance for repeated measures was used for intragroup interactions. A 2-way ANOVA of repeated measurements was used to compare the effects of one drug in two different states. When a significant trend was found by ANOVA, comparisons between means were performed by contrast analysis. When only two means were compared, a paired t-test was performed. A value of P<0.05 considered statistically significant.
Results
Baseline data and changes after induction of HF
After 3 weeks of rapid right ventricular pacing, dogs developed HF that was characterized by clinical symptoms of congestion (exertional dyspnea, ascites) and by haemodynamic changes (Table 1). In HF, at baseline (15 min after pacemaker interruption), heart rate and LV end-diastolic pressure were significantly higher than in C whereas mean aortic pressure, LV systolic pressure and dP/dtmax were significantly lower than in C. CBF was unchanged but calculated CBF and CD tended to decrease (P=0.07 and P=0.089 respectively). Plasma ANP was significantly increased from 56±13 pg ml−1 in C to 272±26 pg ml−1 in HF (P<0.01).
Table 1.
Baseline haemodynamic and coronary parameters in nine dogs in the control state and after induction of heart failure
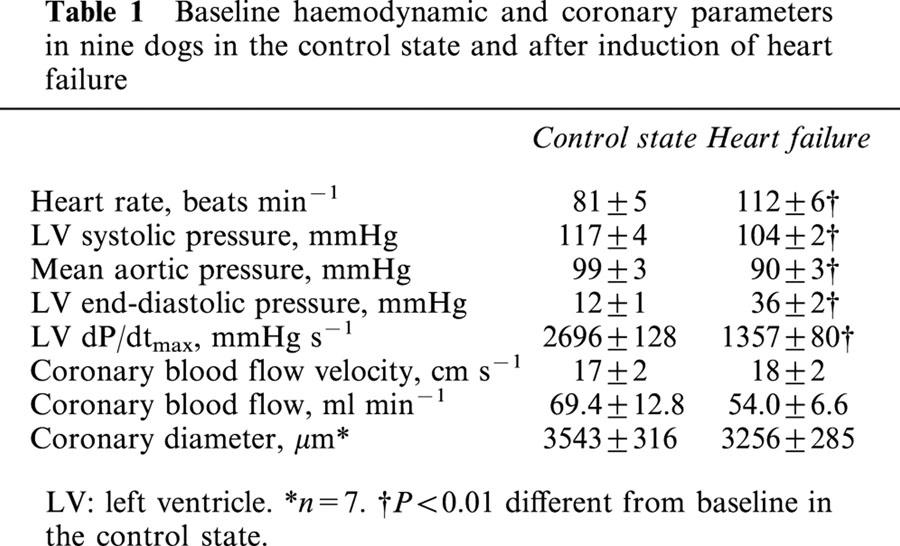
Systemic and coronary responses to acetylcholine, nitroglycerin and adenosine in C and in HF
Intracoronary injection of acetylcholine and nitroglycerin had no effects on systemic haemodynamics in both C and HF. At the maximal utilized dose, adenosine increased significantly heart rate in C by 5±1 beats min−1, and decreased LV end-diastolic pressure in HF (by 5±1 mmHg, P<0.05).
In both C and HF, each drug increased significantly and dose-dependently CBF and CD, but in HF, the coronary responses to acetylcholine and adenosine were significantly reduced while the coronary responses to nitroglycerin were not modified (Figure 1).
Figure 1.
Changes of mean coronary blood flow (CBF) and coronary diameter (CD) responses to intracoronary acetylcholine (Ach), nitroglycerin (NTG) and adenosine (Ade) before and after induction of heart failure. In heart failure, the coronary responses to acetylcholine and adenosine were impaired while the responses to nitroglycerin were unchanged. *P<0.05 versus corresponding baseline values. P value in each graph is the P value obtained by ANOVA for comparison between the control state and heart failure.
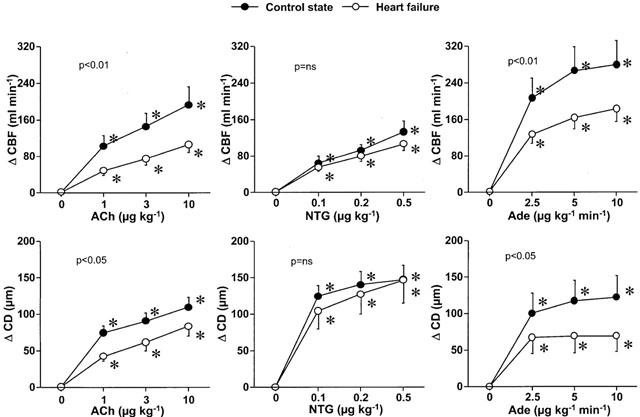
Systemic and coronary effects of amlodipine in C and in HF
In both C and HF, at doses of 100 and 150 μg kg−1, intracoronary amlodipine increased slightly but significantly heart rate. Amlodipine did not change significantly mean aortic pressure and LV systolic and end-diastolic pressures. Amlodipine decreased significantly LV dP/dtmax in C but not in HF (Table 2).
Table 2.
Haemodynamic responses to intracoronary infusion of amlodipine in nine dogs before and after induction of heart failure

Amlodipine increased significantly and dose-dependently CBF and CD in both C and HF, but the magnitudes of the response for both parameters were markedly reduced in HF (Figure 2).
Figure 2.
Changes of mean coronary blood flow (CBF) and coronary diameter (CD) in response to intracoronary amlodipine infusion in the control state and heart failure. Amlodipine increased CBF and CD in both states, but these responses were significantly reduced in heart failure. *P<0.05 versus corresponding baseline values. P value in each graph is the P value obtained by ANOVA for comparison between the control and heart failure.
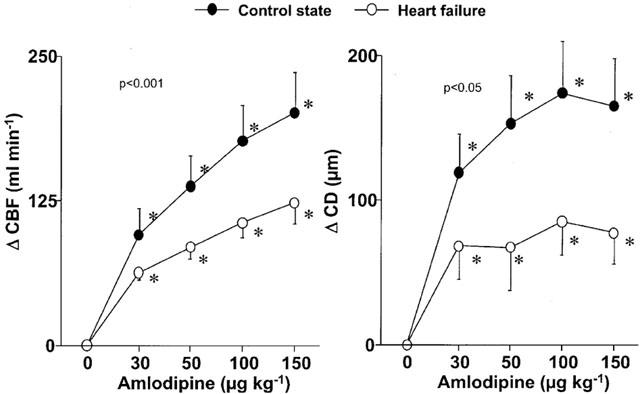
Systemic and coronary effects of amlodipine in the presence of LNA in C and in HF
Intracoronary infusion of 2 mg kg−1 LNA decreased heart rate from 77±6 to 68±4 and from 106±5 to 98±5 beats min−1 respectively in C and in HF (P<0.05) and decreased LV dP/dtmax from 2474±121 to 2369±129 mmHg s−1 (P<0.05) in C. LNA did not change significantly CBFv (from 17±2 to 15±2 cm s−1 in C and from 19±3 to 18±3 cm s−1 in HF) and calculated CBF (from 69.4±128 to 68.7±13.5 ml min−1 in C and from 54.0±6.6 to 57.1±16.4 ml min−1 in HF) but decreased CD from 3510±259 to 3479±258 μm (P<0.001) in C and from 3217±267 to 3193±263 μm (P<0.05) in HF.
In the presence of LNA, intracoronary amlodipine did not change systemic haemodynamics except an increase in heart rate in HF and a decrease in LV dP/dtmax in C (Table 3).
Table 3.
Haemodynamic responses to intracoronary infusion of amlodipine after LNA (0.2 mg kg−1 min−1, 10 min) in eight dogs before and after induction of heart failure

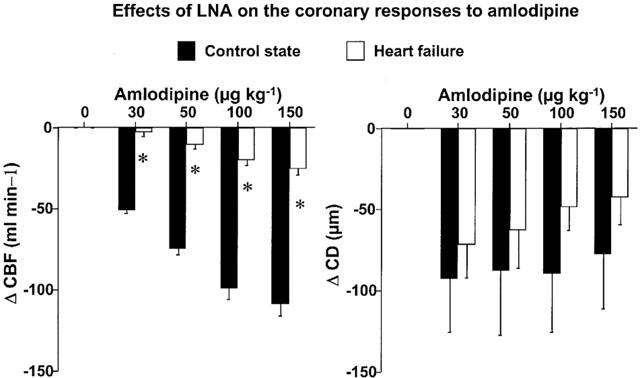
In C, in the presence of LNA, amlodipine increased dose-dependently CBF and CD but the magnitudes of the coronary effects were markedly reduced compared with amlodipine alone (Figure 3). In HF, in the presence of LNA, the CBF response to amlodipine tended to be reduced compared with that in the absence of LNA and CD response to amlodipine was significantly reduced (Figure 3). However, the effect of LNA on the coronary response to amlodipine was less in HF than in C, especially for CBF responses (Figure 4) indicating a reduced role of NO in the coronary responses to amlodipine in HF.
Figure 3.
Changes of mean coronary blood flow (CBF) and coronary diameter (CD) in response to intracoronary amlodipine infusion in the absence or presence of LNA in the control state and heart failure. In the presence of LNA, the coronary effects of amlodipine were markedly reduced in the control state. In heart failure, the CD response was also altered but the CBF response was minimally affected. *P<0.05 versus corresponding baseline values. P value in each graph is the P value obtained by ANOVA for comparison between the control and heart failure.
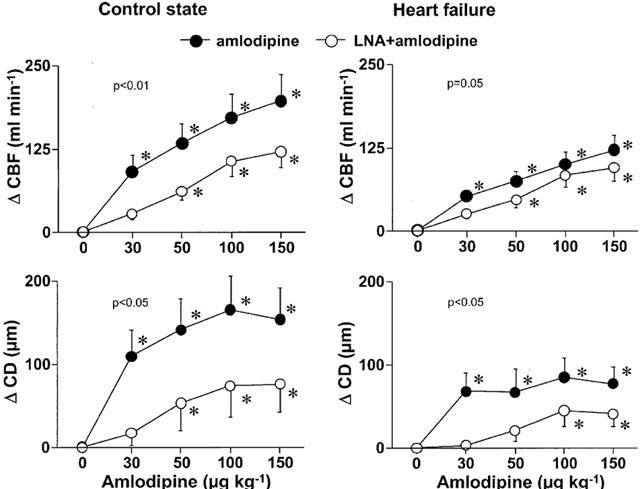
Figure 4.
The implication of nitric oxide in the coronary responses to amlodipine before and after development of heart failure was assessed by the difference between the effects of amlodipine in the presence and absence of LNA. The difference was obtained by subtracting the effects of amlodipine in the presence of LNA from the effects of amlodipine alone in each state. In heart failure, the difference was significantly smaller for the CBF and there was a trend to a reduction for the CD. *P<0.01 versus corresponding values at same dose.
In addition, CBF responses to amlodipine in the presence of LNA were similar in both C and HF, suggesting a preserved coronary microvessel responses related to the blockade of L-type Ca2+ channels in HF.
Systemic and coronary effects of amlodipine in the presence of L-arginine in C and in HF
Intracoronary infusion of L-arginine did not produce significant changes in systemic and coronary parameters in both C and HF (data not shown).
L-arginine did not enhance systemic effects of intracoronary amlodipine. The CBF and CD responses to amlodipine were not enhanced by L-arginine (Figure 5).
Figure 5.
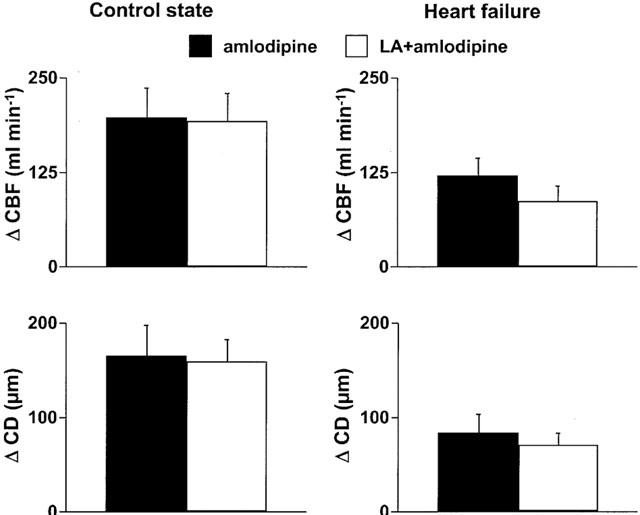
Changes of mean coronary blood flow (CBF) and coronary diameter (CD) in response to intracoronary amlodipine at the dose of 150 μg kg−1 in the absence or presence of L-arginine in the control state and in heart failure. There was no difference in coronary responses with or without L-arginine in both states.
Discussion
In isolated coronary microvessels, amlodipine promotes the release of NO (Zhang & Hintze, 1998; Zhang et al., 1999), the NO pathway could therefore contribute to the in vivo coronary vasodilator effects of amlodipine. Our results show that amlodipine strongly increase CBF and CD and NO is involved in the coronary vasodilator responses to amlodipine in control dogs since LNA partly inhibited these responses. The NO-dependent action has been reported with other calcium channel blockers such as nicardipine, benidipine, nifedipine (Ghaleh et al., 1995; Kitakaze et al., 1999; 2000), but the precise mechanism by which this action takes place remains unknown. It has been suggested that the biochemical pathway for the amlodipine induced NO production involved the kinin system since the NO production induced by amlodipine can be blocked by a bradykinin B2 receptor antagonist and by a serine protease inhibitor which blocks the action of local kallikrein (Zhang & Hintze, 1998; Zhang et al., 1999). This latter suggests that amlodipine may activate local kalikrein as benidipine in the kidney (Yoshida et al., 1996) and increase local formation of kinins to stimulate NO production through bradykinin B2 receptors.
Chronic ventricular pacing induced HF is a well-established HF model that shares many of the characteristics of human dilated cardiomyopathy. It is characterized by low cardiac output, ventricular dilation, ventricular diastolic and systolic dysfunction and neurohumoral activation (Armstrong et al., 1986; Shannon et al., 1991; Su et al., 1994; 1998). In the present study, most of those characteristics were observed. In this model, previous studies showed that coronary endothelium-dependent relaxation and NO production are impaired (Wang et al., 1994; Zhao et al., 1995). Using intracoronary catheter technique that allows direct assessment of coronary response to pharmacological agents with minimal systemic interaction, the present study shows a decreased vasodilator effect of acetylcholine on large and small coronary vessels which is usually used to assess vascular endothelial function (Kuro et al., 1991; Katz et al., 1993, Wang et al., 1994; Zhao et al., 1995; Su et al., 1998) and demonstrates for the first time that in conscious dogs with HF, the vasodilator effects of amlodipine on large and small coronary arteries are markedly reduced. This altered coronary response to amlodipine is related to a reduction or a loss of NO-dependent mechanism in this setting as indicated by reduced LNA effect on the coronary vasodilator response to amlodipine after induction of HF compared with C (Figure 4). This is particularly true for the CBF response that represents essentially coronary microvessels.
Another new finding of the present study is the different contribution of NO to amlodipine responses in large and small coronary vessels. In contrast with C, in HF, the CBF response to amlodipine became less sensitive to LNA while the CD response to amlodipine was still sensitive. This suggests that the response of large coronary vessels to amlodipine is more NO-dependent than that of small coronary vessels. It is known that vasodilation of large coronary vessels in responses to many vasodilator agents involves direct and indirect (e.g. flow mediated dilation) mechanisms (Holtz et al., 1983). In the present study, besides a direct effect, flow mediated dilation mechanism may have contributed to the vasodilator effect of amlodipine on large coronary vessels. Since the flow mediated dilation mechanism is also endothelial dependent in many cases, and since in-vitro studies showed a NO producing effect of amlodipine in large and small coronary vessel tissues where there is no flow mediated phenomena, the impaired vasodilator effects of amlodipine that we observed is likely the consequence of altered NO dependent mechanism secondary to endothelium dysfunction in HF although an altered flow mediated dilation may participate in this response.
Systemic haemodynamics, especially systolic pressure and LV end-diastolic pressure were changed after induction of HF. This may cause a reduction in perfusion gradient of coronary vessels and may have contributed to the reduced coronary response to amlodipine. If it was the case, this might also affect coronary response to other vasodilator agents. However, in the present study, the coronary responses to nitroglycerin were not modified in HF suggesting little role of systemic haemodynamic changes on nitroglycerin effects and probably on other vasodilator agents if there was no other changes in vascular reactivity. Similarly, after HF, there was a decrease in CD at baseline. This decreased coronary diameter may be related to the reduced systemic pressure and increased vasoconstriction secondary to the activation of vasoconstrictor systems. However, it is unlikely that this baseline change in CD have affected CD response to vasodilator agents as suggested by an unchanged vasodilator response of CD to nitroglycerin.
Although it has been recently shown that in contrast with an impaired vasodilator effect of acetylcholine, the vasodilator effect of exogenous bradykinin is preserved in this HF model (Su et al., 1998), it is not known whether or not coronary kinin system is changed and whether or not bradykinin contributed to the impaired coronary response to amlodipine. However, cellular mechanism involved in the impaired coronary vasodilator response to amlodipine is likely related to an altered endothelial L-arginine-NO synthase (eNOS) pathway. In the present study, in contrast with coronary responses to acetylcholine, the coronary blood flow and diameter responses to nitroglycerin were similar in both C and HF, suggesting an unchanged sensitivity of coronary smooth muscle cell to NO. In addition, our results suggest that the altered coronary responses to amlodipine are not due to reduced availability of L-arginine, since the supplementation with L-arginine did not enhance coronary response to amlodipine in HF as well as in C. This is in accordance with a clinical study showing a significant lower urinary excretion of [15N]-nitrate after infusion of L-[15N]-arginine in patients with HF compared with normal subjects (Katz et al., 1999). Although it has been shown that plasma nitrate and nitrite increased in pacing induced HF in dogs (Osorio et al., 2001) and in patients with HF (Winlaw et al., 1994), the increase in plasma NO metabolites does not mean an increased NO production. Recchia et al. (1998) showed a reduced cardiac NO production as indicated by a reduced NO metabolites in coronary sinus blood. In the other hand, it has been reported that the increased plasma NO metabolites are the consequence of renal impairment which results in a reduced excretion of NO metabolites and an increased reabsorption of NO metabolites (Osorio et al., 2001). The decreased activity of the L-arginine-NO metabolic pathway in HF is probably due to eNOS. Indeed, previous studies performed in the same model demonstrated a reduced level of mRNA for eNOS in aortic endothelial cells (Smith et al., 1996) and a decreased synthesis of NO in response to various vasodilator stimuli from isolated coronary vessels obtained from dogs with HF compared with that of control animals (Wang et al., 1994).
In contrast with the myocardium in which calcium channel abundance has been shown to be decreased in failing heart (Takahashi et al., 1992), there are no data concerning the possible modification of calcium channels in coronary vessels in HF. However, our data seem to indicate a preserved coronary vasodilator effect of amlodipine through L-calcium channel blockade at the microvessel level since the coronary blood flow responses to amlodipine after LNA were similar in both C and HF (Figure 3).
Finally, in the pacing induced HF, decreased coronary response to adenosine has been reported (Spinale et al., 1992; Saito et al., 2002). In our study, an impaired coronary response to adenosine was also observed. This means that the coronary reserve may be decreased. However, it is unlikely that this alteration has contributed to the reduced coronary responses to amlodipine because the CBF response to adenosine in HF was largely higher than that to amlodipine. Since the present study did not assess the role of NO in the effects of adenosine, we cannot determine what the contribution of impaired NO pathway is to the impaired coronary response to adenosine. Two previous studies showed that NO is not involved in the coronary response to adenosine in normal dogs as well as in humans since NO synthase inhibitors do not modify the coronary responses to adenosine (Canty & Schwartz, 1994; Shiode et al., 1996). However, opposed results have also been reported (Zanzinger et al., 1994). Therefore, the mechanisms involved in the reduced coronary response to adenosine in HF remain to be determined.
In conclusion, our study demonstrates that the vasodilator action of amlodipine acts partially through the NO pathway in both large and small coronary arteries in conscious dogs. However, in HF, coronary vasodilation induced by amlodipine is markedly impaired, which is related to an impaired endothelial NO synthesis rather than to a L-arginine shortage. The reduction or loss of NO-dependent mechanism of coronary vasodilation in response to amlodipine in coronary vessels may reduce the beneficial effect of amlodipine in HF secondary to dilated or ischaemic cardiomyopathy. This may be the case for other calcium channel blockers involving NO-dependent mechanism (direct or flow mediated NO release) in their vasodilator effects.
Abbreviations
- C
control
- CBFv
coronary blood flow velocity
- CD
coronary diameter
- HF
heart failure
- LNA
Nω-nitro-L-arginine
- NO
nitric oxide
References
- ARMSTRONG P.W., STOPPS T.P., FORD S.E., DE BOLD A.J. Rapid ventricular pacing in the dog: pathophysiologic studies of heart failure. Circulation. 1986;74:1075–1084. doi: 10.1161/01.cir.74.5.1075. [DOI] [PubMed] [Google Scholar]
- CANTY J.M., Jr, SCHWARTZ J.S. Nitric oxide mediates flow-dependent epicardial coronary vasodilation to changes in pulse frequency but not mean flow in conscious dogs. Circulation. 1994;89:375–384. doi: 10.1161/01.cir.89.1.375. [DOI] [PubMed] [Google Scholar]
- GHALEH B., DUBOIS-RANDÉ J.L., HITTINGER L., GIUDICELLI J.F., BERDEAUX A. Comparisons of the effects of nicorandil, pinacidil, nicardipine and nitroglycerin on coronary vessels in the conscious dog: role of the endothelium. Br. J. Pharmacol. 1995;114:496–502. doi: 10.1111/j.1476-5381.1995.tb13254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLTZ J., GIESLER M., BASSENGE E. Two different mechanisms of anti-anginal drugs on epicardial coronary arteries in vivo: indirect, flow-dependent, endothelium mediated dilation and direct smooth mucle relaxation. Z. Kardiol. 1983;72 Suppl 3:98–106. [PubMed] [Google Scholar]
- HOUEL R., SU J.B., BARBE F., CHOUSSAT R., CROZATIER B., HITTINGER L. Regional myocardial effects of intracoronary bradykinin in conscious dogs. Am. J. Physiol. 1997;272:H1266–H1274. doi: 10.1152/ajpheart.1997.272.3.H1266. [DOI] [PubMed] [Google Scholar]
- KATZ S.D., KHAN T., ZEBALLOS G.A., MATHEW L., POTHARLANKA P., KNECHT M., WHELAN J. Decreased activity of the L-arginine-nitric oxide metabolic pathway in patients with congestive heart failure. Circulation. 1999;99:2113–2117. doi: 10.1161/01.cir.99.16.2113. [DOI] [PubMed] [Google Scholar]
- KATZ S.D., SCHWARZ M., YUEN J., LEJEMTEL T.H. Impaired acetylcholine-mediated vasodilation in patients with congestive heart failure: role of endothelium-derived vasodilating and vasoconstricting factors. Circulation. 1993;88:55–61. doi: 10.1161/01.cir.88.1.55. [DOI] [PubMed] [Google Scholar]
- KITAKAZE M., ASANUMA H., TAKASHIMA S., MINAMINO T., UEDA Y., SAKATA Y., ASAKURA M., SANADA S., KUZUYA T., HORI M. Nifedipine-induced vasodilation in ischemic heart is attributable to bradykinin- and NO-dependent mechanisms in dogs. Circulation. 2000;101:311–317. doi: 10.1161/01.cir.101.3.311. [DOI] [PubMed] [Google Scholar]
- KITAKAZE M., NODE K., MINAMIDO T., ASANUMA H., KUZUYA T., HORI M. A Ca channel blocker, benidipine, increases coronary blood flow and attenuates the severity of myocardial ischemia via NO-dependent mechanism in dogs. J. Am. Coll. Cardiol. 1999;33:242–249. doi: 10.1016/s0735-1097(98)00556-7. [DOI] [PubMed] [Google Scholar]
- KURO S.H., RECTOR T.S., BANK A.J., WILLIAMS R.E., HEIFETZ S.M. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation. 1991;84:1589–1596. doi: 10.1161/01.cir.84.4.1589. [DOI] [PubMed] [Google Scholar]
- OSORIO J.C., XU X., VOGEL T., OCHOA M., LAYCOCK S., HINTZE T.H. Plasma nitrate accumulation during the development of pacing-induced dilated cardiac myopathy in conscious dogs is due to renal impairment. Nitric Oxide. 2001;5:7–17. doi: 10.1006/niox.2000.0326. [DOI] [PubMed] [Google Scholar]
- RECCHIA F.A., MCCONNELL P.I., VOGEL T.R., XU X., HINTZE T.H. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure. Circ. Res. 1998;83:969–979. doi: 10.1161/01.res.83.10.969. [DOI] [PubMed] [Google Scholar]
- SAITO T., MAEHARA K., TAMAGAWA K., OIKAWA Y., NIITSUMA T., SAITOH S.-I., MARUYAMA Y. Alterations of endothelium-dependent and independent regulation of coronary blood flow during heart failure. Am. J. Physiol. 2002;282:H80–H86. doi: 10.1152/ajpheart.2002.282.1.H80. [DOI] [PubMed] [Google Scholar]
- SHANNON R.P., KOMAMURA K., STAMBLER B.S., BIGAUD M., MANDERS W., VATNER S.F. Alterations in myocardial contractility in conscious dogs with dilated cardiomyopathy. Am. J. Physiol. 1991;260:H1903–H1911. doi: 10.1152/ajpheart.1991.260.6.H1903. [DOI] [PubMed] [Google Scholar]
- SHIODE N., MORISHIMA N., NAKAYAMA K., YAMAGATA T., MATSUURA H., KAJIYAMA G. Flow-mediated vasodilation of human epicardial coronary arteries: effect of inhibition of nitric oxide synthesis. J. Am. Coll. Cardiol. 1996;27:304–310. doi: 10.1016/0735-1097(95)00465-3. [DOI] [PubMed] [Google Scholar]
- SMITH C.J., SUN D., HOEGLER C., ROTH B.S., ZHANG X., ZHAO G., XU X.B., KOBARI Y., PRITCHARD K., SESSA W.C., HINTZE T.H. Reduced gene expression of vascular endothelial NO synthase and cyclooxygenase-1 in heart failure. Circ. Res. 1996;78:58–64. doi: 10.1161/01.res.78.1.58. [DOI] [PubMed] [Google Scholar]
- SPINALE F.G., TANAKA R., GRAWFORD F.A., ZILE M.R. Changes in myocardial blood flow during development of and recovery from tachycardia-induced cardiomyopathy. Circulation. 1992;85:717–729. doi: 10.1161/01.cir.85.2.717. [DOI] [PubMed] [Google Scholar]
- SU J.B., BARBE F., HOUEL R., GUYENE T.T., CROZATIER B., HITTINGER L. Preserved vasodilator effects of bradykinin in dogs with heart failure. Circulation. 1998;98:2911–2918. doi: 10.1161/01.cir.98.25.2911. [DOI] [PubMed] [Google Scholar]
- SU J.B., HOUEL R., HELOIRE F., BEVERELLI F., SAMBIN L., CASTAIGNE A., BERDEAUX A., CROZATIER B., HITTINGER L. Stimulation of bradykinin B1 receptors induces vasodilation in both conductance and resistance coronary vessels in conscious dogs. Comparison with B2 receptor stimulation. Circulation. 2000;101:1848–1853. doi: 10.1161/01.cir.101.15.1848. [DOI] [PubMed] [Google Scholar]
- SU J.B., RENAUD N., CARAYON A., CROZATIER B., HITTINGER L. Effects of the calcium channel blockers, diltiazem and Ro 40-5967, on systemic haemodynamics and plasma noradrenaline levels in conscious dogs with pacing-induced heart failure. Br. J. Pharmacol. 1994;113:395–402. doi: 10.1111/j.1476-5381.1994.tb17002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKAHASHI T., ALLEN P.D., LACRO R.V., MARKS A.R., DENNIS A.R., SCHOEN F.J., GROSSMAN W., MARSH J.D., IZUMO S. Expression of dihydropyridine receptor (Ca2+ channel) and calsequestrin genes in the myocardium of patients with end-stage heart failure. J. Clin. Invest. 1992;90:927–935. doi: 10.1172/JCI115969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J., SEYEDI N., XU X.-B., WOLIN M.S., HINTZE T.H. Defective endothelium-mediated control of coronary circulation in conscious dogs after heart failure. Am. J. Physiol. 1994;266:H670–680. doi: 10.1152/ajpheart.1994.266.2.H670. [DOI] [PubMed] [Google Scholar]
- WINLAW D.S., SMYTHE G.A., KEOGH A.M., SCHYVENS C.G., SPRATT P.M., MACDONALD P.S. Increased nitric oxide production in heart failure. Lancet. 1994;344:373–374. doi: 10.1016/s0140-6736(94)91403-6. [DOI] [PubMed] [Google Scholar]
- YOSHIDA K., KOHZUKI M., YASUJIMA M., KANAZAWA M., ABE K. Effects of benidipine, a calcium antagonist, on urinary kalikrein excretion and renal impairment in experimental diabetes. J. Hypertens. 1996;14:215–222. doi: 10.1097/00004872-199602000-00010. [DOI] [PubMed] [Google Scholar]
- ZANZINGER J., ZHENG X., BASSENGE E. Endothelium dependent vasomotor responses to endogenous agonists are potentiated following ACE inhibition by a bradykinin dependent mechanism. Cardiovasc. Res. 1994;28:209–214. doi: 10.1093/cvr/28.2.209. [DOI] [PubMed] [Google Scholar]
- ZHANG X., HINTZE T.H. Amlodipine releases nitric oxide from canine coronary microvessels. An unexpected mechanism of action of a calcium channel-blocking agent. Circulation. 1998;97:576–580. doi: 10.1161/01.cir.97.6.576. [DOI] [PubMed] [Google Scholar]
- ZHANG X., KICHUK M.R., MITAL S., OZ M., MICHLER R., NASJLETTI A., KALEY G., HINTZE T.H. Amlodipine promotes kinin mediated nitric oxide production in coronary microvessels of failing human hearts. Am. J. Cardiol. 1999;84:27L–33L. doi: 10.1016/s0002-9149(99)00362-8. [DOI] [PubMed] [Google Scholar]
- ZHAO G., SHEN W., XU X., OCHOA M., BERNSTEIN R., HINTZE T.H. Selective impairment of vagally mediated nitric oxide-dependent coronary vasodilation in conscious dogs after pacing-induced heart failure. Circulation. 1995;91:2655–2663. doi: 10.1161/01.cir.91.10.2655. [DOI] [PubMed] [Google Scholar]