

Abstract
Tramadol has been used clinically as an analgesic; however, the mechanism of its analgesic effects is still unknown.
We used bovine adrenal chromaffin cells to investigate effects of tramadol on catecholamine secretion, nicotine-induced cytosolic Ca2+ concentration ([Ca2+]i) increases and membrane current changes. We also investigated effects of tramadol on α7 nicotinic acetylcholine receptors (AChRs) expressed in Xenopus oocytes.
Tramadol concentration-dependently suppressed carbachol-induced catecholamine secretion to 60% and 27% of the control at the concentration of 10 and 100 μM, respectively, whereas it had little effect on veratridine- or high K+-induced catecholamine secretion.
Tramadol also suppressed nicotine-induced ([Ca2+]i) increases in a concentration-dependent manner. Tramadol inhibited nicotine-induced inward currents, and the inhibition was unaffected by the opioid receptor antagonist naloxone.
Tramadol inhibited nicotinic currents carried by α7 receptors expressed in Xenopus oocytes.
Tramadol inhibited both α-bungarotoxin-sensitive and -insensitive nicotinic currents in bovine adrenal chromaffin cells.
In conclusion, tramadol inhibits catecholamine secretion partly by inhibiting nicotinic AChR functions in a naloxone-insensitive manner and α7 receptors are one of those inhibited by tramadol.
Keywords: Tramadol, catecholamine secretion, nicotinic acetylcholine receptors, bovine adrenal chromaffin cells, Xenopus oocytes
Introduction
Tramadol, (1RS; 2RS)-2-[(dimethylamino) methyl]-1-(3-methoxyphenyl)-cyclohexanol hydrochloride, is used clinically as an analgesic. There have been some reports on the mechanism of the analgesic effects of tramadol: (1) actions via μ-opioid receptors, although its binding affinity is relatively low (Hennies et al., 1988) and only 30% of its analgesic effect could be antagonized by naloxone (Collart et al., 1993), and (2) inhibition of the reuptake of monoamines at nerve endings (Reimann & Hennies, 1994). However, the precise mechanism of its antinociception is still unknown.
Several investigators have identified that peripheral administration of acetylcholine (ACh), which is the classical neurotransmitter activating nicotinic as well as muscarinic receptors, activates nociceptors (Grunfeld et al., 1991; Steen & Reeh, 1993; Heyer & Hornstein, 1999). Recently, we found that tramadol has inhibitory effects on muscarinic M1 receptor function (Shiraishi et al., 2001); suggesting cholinergic receptors could be an acting site of tramadol. In capsaicin sensitive neurons including primary afferents and dorsal root ganglia, nicotinic as well as muscarinic AChRs are present (Roberts et al., 1995) and these nociceptive pathways express some subtypes of nicotinic AChRs (Zoli et al., 1995). On the other hand, nicotinic AChR-targeted compounds, such as ABT-594, are recently shown to be potential analgesic agents (Bannon et al., 1998). To date, there has been no report regarding the effect of tramadol on nicotinic AChRs. Thus, it seems to be interesting to examine if tramadol inhibits or enhances nicotinic AChRs.
The sympathetic nervous system has an important role in the generation and maintenance of chronic pain state (Mcmahon, 1991). Following nerve injury, sympathetic nerve stimulation can excite primary sensory afferent fibres via an action at α-adrenoceptors. There are innervations by sympathetic terminals in the dorsal root ganglion (Mclachlan et al., 1993) and sympathetic nerve blocks have been used for management of the pain. However, effects of tramadol on the sympathetic nervous system have not been studied to date.
Adrenal chromaffin cells are derived from the embryonic neural crest and are homologous with sympathetic postganglionic neurons. Cultured bovine adrenal chromaffin cells contain at least three distinct ion channels that are involved in catecholamine secretion; (1) nicotinic AChR-associated cation channels responsible for ACh-induced Na+ influx, (2) voltage-dependent Na+ channels responsible for veratridine-induced Na+ influx and (3) voltage-dependent Ca2+ channels which are activated by membrane depolarization (Wada et al., 1985). Moreover, GABAA receptor-associated Cl− channels also exist in adrenal chromaffin cells (Bormann & Clapham, 1985). Since ion channel-mediated catecholamine secretion has been well studied, the adrenal chromaffin cells appear to be a good model for detailed analysis of the action of drugs on the sympathetic nervous system (Minami et al., 1994).
In the present study, we investigated effects of tramadol on catecholamine secretion using cultured bovine adrenal chromaffin cells. We also examined effects of tramadol on cytosolic Ca2+ concentration ([Ca2+]i) in order to delineate the mechanisms by which tramadol alter catecholamine secretion from adrenal chromaffin cells. Moreover, we investigated effects of tramadol on nicotinic AChRs using the whole-cell patch-clamp technique in chromaffin cells, and on α7 nicotinic AChRs (α7 receptors) expressed in Xenopus oocytes using the two-electrode voltage clamp.
Methods
Isolation of adrenal chromaffin cells and their primary culture
Adrenal chromaffin cells were isolated by collagenase digestion of slices of the bovine adrenal medulla as described elsewhere (Minami et al., 1994). The cells were plated at a density of 4×106 cells dish−1 (35 mm in diameter) in Eagle's minimum essential medium containing 10% calf serum and antibiotics (60 mg ml−1 aminobenzylpenicilin, 100 mg ml−1 streptomycin, 0.3 mg ml−1 amphotericin B and 3.0 mM cytosine arabinoside). The cells were cultured in 5% CO2–95% air in a culture chamber and used on 1st to 5th day of culture.
Secretion of catecholamines
The secretion of catecholamines was measured as reported previously (Minami et al., 1994). In brief, oxygenated Krebs-Ringer phosphate (KRP) buffer was used throughout. It was composed as follows (mM): NaCl 154, KCl 5.6, MgSO4 1.1, CaCl2 2.2, NaH2PO4 0.85, Na2HPO4 2.15 and glucose 10, adjusted to pH 7.4. The cells were washed four times with 1 ml of KRP and they were stimulated at 37°C for 5 min by carbachol (0.3 mM), veratridine (0.1 mM) and K+ (56 mM) in the absence or presence of several concentrations of tramadol (100 nM–1 mM). Catecholamines (norepinephrine plus epinephrine) secreted into the medium were adsorbed to aluminum hydroxide and estimated by the ethylenediamine condensation method (Weil-Malherebe & Bone, 1952), using a fluorescence spectrophotometer (Hitachi Model 650-10S, Tokyo, Japan) with an excitation wavelength of 420 nm and an emission of 540 nm. Tramadol had no direct effect on the assay of catecholamine. The secreted catecholamines were expressed as percentage of total catecholamines (norepinephrine plus epinephrine) in the cells.
The whole cell patch clamp with chromaffin cells
The method used for electrophysiological experiments has been described elsewhere (Shibuya et al., 1999a). Adrenal chromaffin cells are plated on a glass cover slip (11 mm in diameter) and used after being maintained in culture for 2–4 days. Standard perfusion medium (HEPES-buffered solution; HBS) contained (mM) NaCl 140, KCl 5, CaCl2 2, MgCl2 1, HEPES 10 and glucose 10 (pH 7.4 adjusted with NaOH). The pipette solution used in the recording electrodes contained (in mM): KCl 140, MgCl2 1, CaCl2 1, EGTA 10, HEPES 10 and Mg-ATP 2 (pH 7.2 adjusted with Tris base). The electrodes were made with a pullar (P-97; Sutter Instrument Co., Novato, CA, U.S.A.) from thick-walled borosilicate glass (GD-1.5; Narishige, Tokyo, Japan) and had a final resistance of 3–6 MΩ when filled with the electrode solution. The volume of recording chamber was approximately 0.5 ml and the flow rate of the perfusion medium was 1.5 ml min−1 in most experiments. In experiments using α-bungarotoxin, the flow rate was increased up to 3.0 ml min−1 to records currents with faster kinetics. The solution level was kept constant by a low-pressure aspiration system. All electrophysiological recordings were carried out at a room temperature of around 23°C. Membrane currents were recorded with a patch-clamp amplifier (AxoPatch 200A; Axon Instrument Inc., Foster City, CA, U.S.A.) and were digitized using AxoGraph software (version 4.6; Axon Instrument Inc.) for subsequent off-line analysis. Data were analysed also using the AxoGraph software. The sampling rate was 1–10 KHz. Nicotine, GABA, naloxone and tramadol were added by changing the bath solution with a peristatic pump.
Measurement of cytosolic Ca2+ concentration ([Ca2+]i)
The method used for [Ca2+]i measurements was described elsewhere in detail (Shibuya et al., 1999b). Cultured cells plated on a cover glass were incubated in HBS with the addition of acetoxymethyl esters of fura-2 (fura-2/AM; 3 μM) at room temperature for 1 h. The cells were then washed with dye-free HBS and kept at room temperature until used.
The arrangements for perfusing cells are essentially the same as those used for electrophysiological measurements described above. Fluorescence was measured from fura-2-loaded cells in the perfusion chamber, which had a glass coverslip bottom and was positioned on the stage of an inverted microscope (IX-70, Olympus, Tokyo, Japan) equipped with a Ca2+-imaging system (Quanticell/900, JOEL, Japan). Once cells were selected in the optical field under microscope, fluorescence intensities with excitation at 340 and 380 nm were alternatively recorded at an interval of 3–5 s with an intensified CCD camera. [Ca2+]i was calculated from the ratio (R) of the fluorescence measured with excitation at 340 nm to that at 380 nm using the following equation:
where Kd is the dissociation constant of the fura-2 (224 nM); Rmax and Rmin are the ratios for the unbound and bound forms of the fura-2/Ca2+ complex; and β is the ratio between the maximum and the minimum fluorescence intensities of fura-2 at 380 nm excitation. Rmax and Rmin were estimated with the fluorescence intensities of fura-2 solution (3 μM) containing CaCl2 5 mM and EGTA 5 mM, respectively. Autofluorescence in chromaffin cells was negligible compared with the fluorescence in the fura-2 loaded cells. The experiments were performed at room temperature of 23°C.
Two-electrode voltage clamp with Xenopus oocytes
Isolation and microinjection of Xenopus oocytes were performed as described by Sanna et al. (1994). The cDNA for the α7 was inserted into the pGEM-3Zf(−) vector and linearized with NaeI. The α7 cRNA was prepared using an mCAP mRNA Capping Kit, and transcribed with a SP6 RNA Polymerase in vitro Transcription Kit (Stratagene, La Jolla, CA, U.S.A.). Oocytes were injected with 50 ng of cRNA encoding the α7 subtype, and electrophysiological recording was performed 2–4 days after injection. Oocytes expressing α7 were placed in a rectangular chamber (100 μl volume) and perfused (2 ml min−1) with Ba2+ Ringer's solution ((mM) NaCl 115, KCl 2.5, BaCl2 1.8, and HEPES 10, pH 7.4) to minimize the effects of secondarily activated Ca2+-dependent Cl− currents. Recording electrodes (1–5 MΩ) filled with 3 M KCl were inserted into the animal pole. A Warner Oocyte-clamp OC 725-C (Warner, Hampden, CT, U.S.A.) was used to voltage clamp each oocyte at −100 mV. We measured the peak of the transient inward currents as the nicotine-induced currents, because this component is dependent on nicotine concentrations and is quite reproducible. Tramadol was pre-applied for 2 min to allow complete equilibration in the bath. Control responses to nicotine were measured before and after tramadol application to minimize possible shifts in the control currents as recording proceeded.
Analysis
The results are expressed as percentages of control responses due to variability in the cells. All values are presented as the mean±s.e.mean. The ‘n' values refer to the number of cells studied. Statistical analyses were performed using a one-way ANOVA (analysis of variance). Estimation of IC50 value for concentration-response curves was performed using GraphPad Prism software (San Diego, CA, U.S.A.).
Materials
Eagle's minimum essentials medium from Nissui Pharmaceuticals, (Tokyo, Japan); foetal calf serum, from Nacalai Tesque, (Kyoto, Japan); collagenase, Nitta Zerachine, (Osaka, Japan); tramadol hydrochloride was a kind gift from Nippon Shinyaku (Kyoto, Japan). Nicotine was from Nacalai Tesque (Kyoto, Japan); gamma aminobutyric acid (GABA) and naloxone were from Tocris Cookstone Ltd. (Bristol, U.K.); carbachol, veratridine and α-bungarotoxin were from Sigma (St. Louis, MO, U.S.A.); fura-2 AM was from Dojin Laboratory (Kumamoto, Japan). Adult Xenopus leavis female frogs were purchased from Seac Yoshitomi (Fukuoka, Japan); the Ultracomp E. coli Transformation Kit was from Invitrogen (San Diego, CA, U.S.A.). A Qiagen (Chatworth, CA, U.S.A.) Kit was used to purify plasmid cDNA. The chicken α7 nicotinic AChR cDNA was kindly provided by Dr Henry A. Lester (Division of Biology, California Institute of Technology).
Results
Effects of tramadol on carbachol, veratridine and high K+-evoked catecholamine secretion
Carbachol (0.3 mM), a non-selective AChR agonist, increased catecholamine secretion over the basal (0.3±0.01% of total catecholamines, n=4), by 12±0.6% of the total catecholamines in the cells. As shown in Figure 1A, tramadol significantly inhibited carbachol-induced catecholamine secretion in a concentration-dependent manner. The secretion was decreased to 60±4% of the control by 10 μM of tramadol and to 44±2% and 27±1% of the control by 30 and 100 μM of tramadol, respectively. The half-maximal inhibitory concentration (IC50) was 7.4 μM (Figure 1A). Veratridine (0.1 mM), an activator of voltage-dependent Na+ channels, increased catecholamine secretion, by 17±0.9% of the total catecholamines. Tramadol (0.1–100 μM) had little effect on veratridine-induced catecholamine secretion (Figure 1B). A high concentration of K+, which activates voltage-dependent Ca2+ channels, increased the secretion of catecholamines by 11±0.3% of the total catecholamines. Tramadol had little effect on 56 mM K+-induced catecholamine secretion (Figure 1C). We next investigated whether tramadol competes with carbachol for nicotinic AChRs. As shown in Figure 2A, the inhibitory effect of tramadol was not overcome when the concentration of carbachol was increased in the incubation medium. The double-reciprocal plot analysis revealed that tramadol inhibited the carbachol-induced catecholamine secretion non-competitively (Figure 2B).
Figure 1.
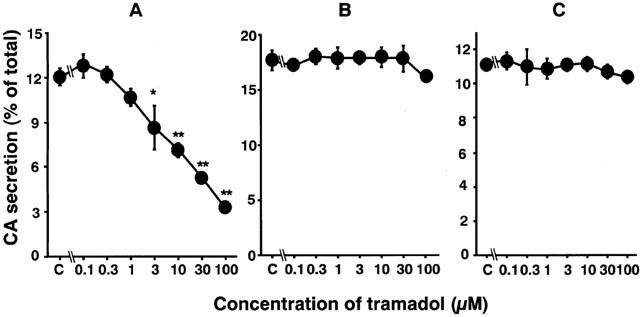
Effects of tramadol on catecholamine (CA) secretion induced by carbachol (A), veratridine (B) or high K+ (C) in bovine adrenal chromaffin cells. Cells were incubated with carbachol (300 μM), veratridine (0.1 mM) or a high K+ (56 mM) solution in the presence of various concentrations of tramadol for 5 min at 37°C. Catecholamine secreted into the medium were measured (see Methods), and expressed as percentage of total catecholamines. Data are means±s.e.mean of four experiments. *P<0.05, **P<0.01 compared with carbachol alone.
Figure 2.
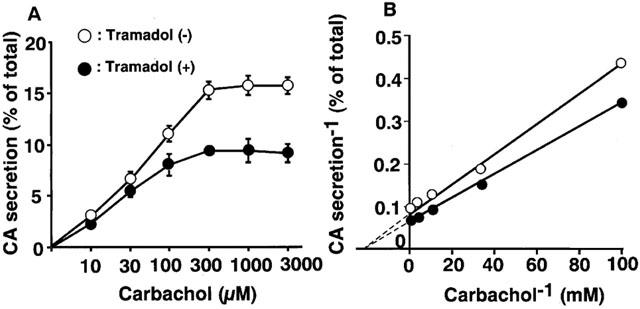
Characterization of inhibitory effects of tramadol on carbachol-evoked catecholamine (CA) secretion in bovine adrenal chromaffin cells. The inhibitory effects of tramadol on carbachol-evoked catecholamine secretion (A) and double-reciprocal plot analysis of the effects (B). Cells were incubated for 5 min with or without carbachol (10 μM to 3 mM) in the presence or absence of tramadol (10 μM). Data are means±s.e.mean of three experiments.
Effects of tramadol on [Ca2+]i rises induced by carbachol, nicotine and high K+
Effects of tramadol on carbachol-induced [Ca2+]i rises were studied. Carbachol induced a rapid [Ca2+]i increase in single bovine adrenal chromaffin cells. Bath application of tramadol (1 μM), which was added 100 s before carbachol-stimulation, inhibited the [Ca2+]i increase (Figure 3). The inhibitory effects were concentration-dependent at tramadol concentrations ranging from 1–100 μM. Taramadol inhibited carbachol-induced [Ca2+]i increase to 22±2% and 74±3% of control at 100 μM and 10 μM, respectively (n=16). Likewise, the effect of tramadol on [Ca2+]i rises in response to nicotine, a selective nicotinic AChR agonist, was examined. Nicotine (10 μM) induced a rapid [Ca2+]i increase and the inhibitory effects of tramadol on the nicotine-induced [Ca2+]i increase were also concentration-dependent (Figure 4, n=16). By contrast, muscarine at 100 μM produced a small rise in [Ca2+]i (157.2±16.5 nM from basal; n=24) as a result of IP3-dependent Ca2+ release from intracellular Ca2+ stores (Tanaka et al., 1998). Tramadol at 10 μM did not significantly affect the response to muscarine (92.4±5.2% of control, n=24; data not shown). Moreover, tramadol did not inhibit high K+-induced [Ca2+]i increases (Figure 5). The [Ca2+]i increases by high K+-solution obtained with 1 μM tramadol were 103±2% of the control values (n=16).
Figure 3.
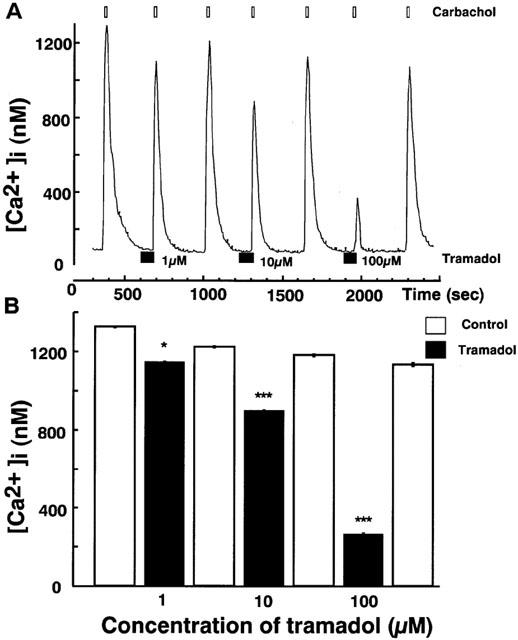
Effects of tramadol on [Ca2+]i rises induced by carbachol in bovine adrenal chromaffin cells. These figures show average changes of the cytosolic Ca2+ concentration induced by carbachol (300 μM) with or without various concentrations of tramadol (1,10 and 100 μM) (A and B). Carbachol was added for 20 s after pretreatment with tramadol (10 μM). Tramadol alone did not induce any change in basal [Ca2+]i rises. Open bars mean the period for carbachol applications and closed bars indicate the period for tramadol applications. *P<0.05, ***P<0.001 (n=16).
Figure 4.
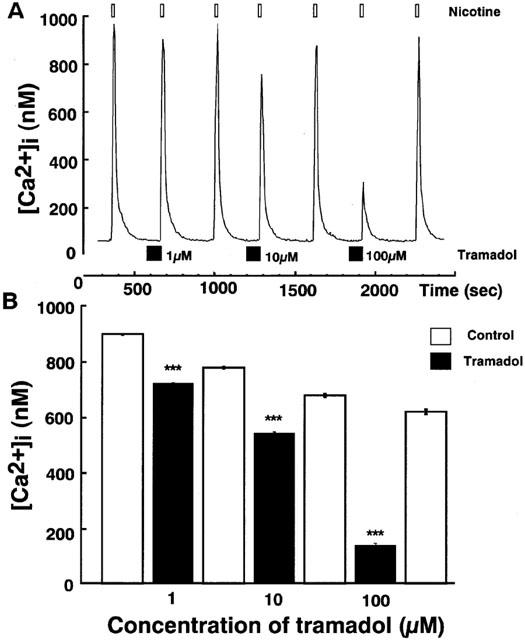
Effects of tramadol on [Ca2+]i rises induced by nicotine in bovine adrenal chromaffin cells. These figures show average changes of the cytosolic Ca2+ concentration induced by nicotine (10 μM) with or without various concentrations of tramadol (1,10 and 100 μM) A and B. Nicotine was added for 20 s after pretreatment with tramadol (10 μM). Tramadol alone did not induce any change in basal [Ca2+]i rises. Open bars mean the period for nicotine applications and closed bars indicate the period for tramadol applications. ***P<0.001 (n=16).
Figure 5.
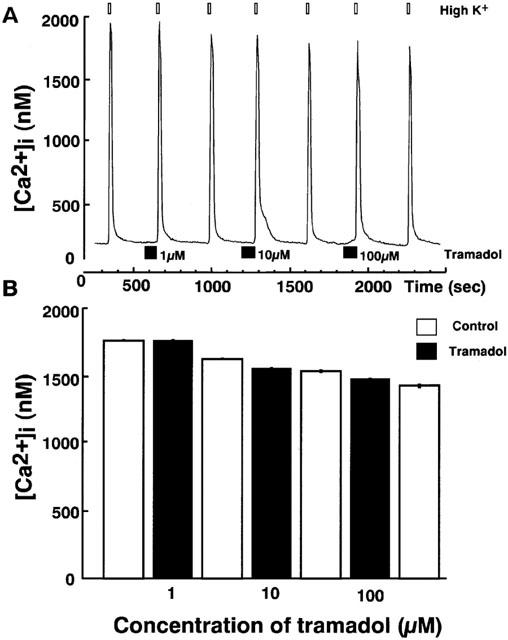
Effects of tramadol on [Ca2+]i rises induced by high K+ in bovine adrenal chromaffin cells. These figures show average changes of the cytosolic Ca2+ concentration induced by high K+ (50 mM) with or without various concentrations of tramadol (1,10 and 100 μM) (A and B). High K+ was added for 20 s after pretreatment with tramadol (10 μM). Tramadol alone did not induce any change in basal [Ca2+]i rises. Open bars mean the period for high K+ applications and closed bars indicate the period for tramadol applications (n=16).
Effects of tramadol on membrane currents mediated by ligand-gated ion-channels, nicotinic AChRs and GABAA receptors
Bovine adrenal chromaffin cells were voltage-clamped at −60 mV by the whole-cell patch-clamp technique. Bath application of nicotine (10 μM) induced inward currents. Bath application of tramadol, which was added 100 s before nicotinic stimulation, potently suppressed the nicotine-induced currents (Figure 6A). Tramadol itself did not induce any ionic currents. Tramadol inhibited nicotine-induced inward currents in a concentration-dependent manner (10 nM–100 μM) and the inhibition was reversible and repeatable. The magnitude of the inhibition was expressed as percentage inhibition of the control currents, which were calculated by averaging two control currents recorded before the test and after the recovery. Tramadol inhibited the nicotine-induced inward currents to 53±6% and 29±3% of control at 1 and 10 μM tramadol, respectively (Figure 6B). The half maximal inhibitory concentration (IC50) of tramadol for the nicotine-induced currents was 1.2 μM.
Figure 6.
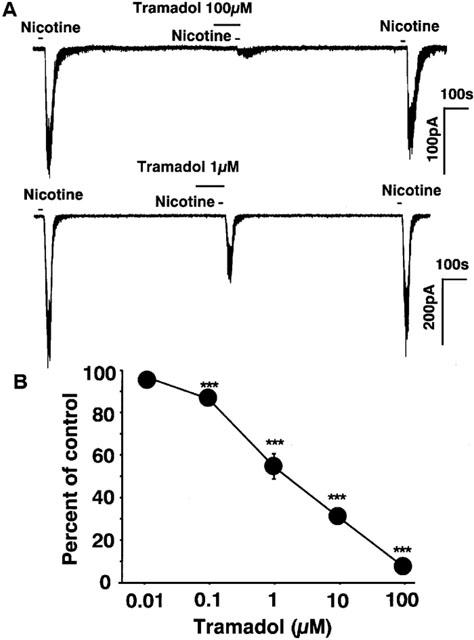
Effects of tramadol on nicotine-induced inward currents recorded by the whole-cell patch-clamp technique in bovine adrenal chromaffin cells. The membrane potential was voltage-clamped to −60 mV. (A) Tracings obtained from a single adrenal chromaffin cell show the effect of tramadol on 10 μM nicotine-induced currents. Nicotine was applied for 20 s with or without 2 min treatment with 100 and 1 μM tramadol. (B) Concentration-response relationship of tramadol on the nicotine-induced currents. Tramadol (10 nM–100 μM) was applied to the cell for 2 min and then 10 μM nicotine was applied for 20 s. Data represent the mean±s.e.mean (n=4–10). ***P<0.001 compared with the control response, using ANOVA.
As it is known that tramadol binds μ-opioid receptors (Hennies et al., 1988) and that μ-opioid receptors are present in adrenal chromaffin cells (Castanas et al., 1985), we investigated whether tramadol inhibits nicotine-induced currents via μ-opioid receptors. As shown in Figure 7, effects of naloxone on the inhibition of nicotinic currents by high concentrations of tramadol were examined. Naloxone (10 μM) had little effect on the inhibition by tramadol (100 and 10 μM).
Figure 7.
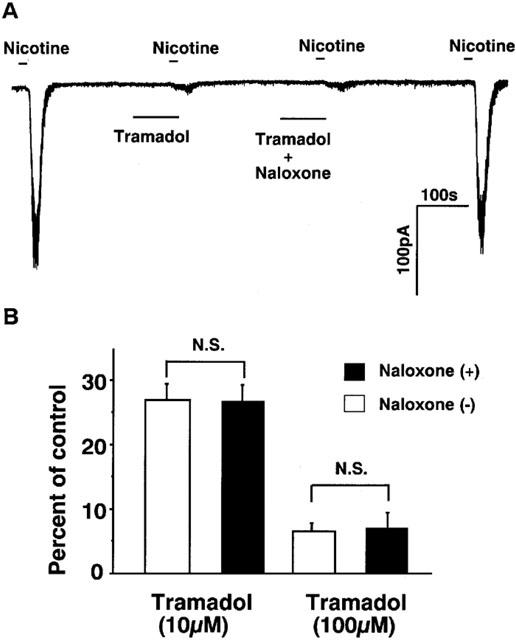
Effects of naloxone on the inhibition by tramadol of nicotine induced inward currents in bovine adrenal chromaffin cells. The effects of tramadol with or without co-application of naloxone were measured to know whether opioid effect of tramadol affect on nicotinic receptor ion channels or not. Membrane potential was voltage-clamped at −60 mV. (A) Tracings were obtained from a single cell showing the effect of tramadol (100 μM) on 10 μM nicotine-induced currents with or without co-application of naloxone (10 μM). Note that naloxone alone did not induce any ionic currents. (B) The effects of naloxone (10 μM) on the inhibition by 100 and 10 μM tramadol on 10 μM nicotine-induced currents. Open bars mean the tramadol applications without naloxone and closed bars indicate the tramadol applications with naloxone. Values are the mean±s.e.mean (n=4).
Next, effects of tramadol on GABAA receptor-mediated inward currents were studied to examine whether or not the inhibitory effects of tramadol are non-selective for ligand-gated ion-channels. GABA caused inward currents at concentrations ranging from 1 μM to 1 mM. Tramadol had little effect on GABA-induced inward currents as shown Figure 8 (n=6).
Figure 8.
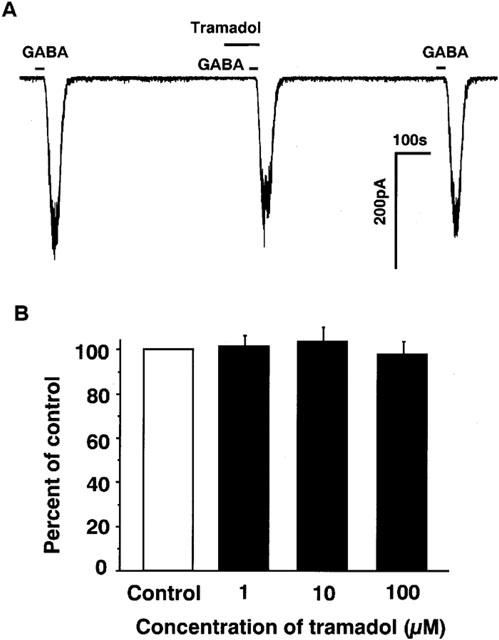
Effects various concentrations of tramadol on inward currents induced by GABA in bovine adrenal chromaffin cells. To know the effects of tramadol on the other ligand-gated ion channels, GABAA receptors instead of nicotinic receptors were examined in adrenal chromaffin cells. (A) Tracings obtained from a single adrenal chromaffin cell show the effect of tramadol (10 μM) on 1 μM GABA-induced currents. (B) Effects of tramadol (1–100 μM) on 1 μM GABA-induced inward currents. GABA was applied for 20 s with or without 2 min treatment with various concentrations of tramadol (1–100 μM). Membrane potential was voltage-clamped at −60 mV (n=6).
Effects of tramadol on nicotine-induced currents in Xenopus oocytes expressing α7 receptors
Effects of tramadol on nicotine-induced currents in the Xenopus oocytes expressing cloned α7 receptors were examined. Nicotine (1 mM) induced inward currents at a holding potential of −100 mV (80±30 nA, n=20) (Figure 9A). Tramadol inhibited nicotine-induced currents (Figure 9). Again, the magnitude of the tramadol-induced inhibition was normalized by the control currents, which were measured by averaging two control currents recorded before the test and after the recovery. Tramadol inhibited nicotine-induced inward currents to 63±7 and 43±7% of control at 1 and 10 μM tramadol, respectively (Figure 9B). The half maximal inhibitory concentration (IC50) of tramadol for α7 receptor-stimulated currents was 1.3 μM.
Figure 9.
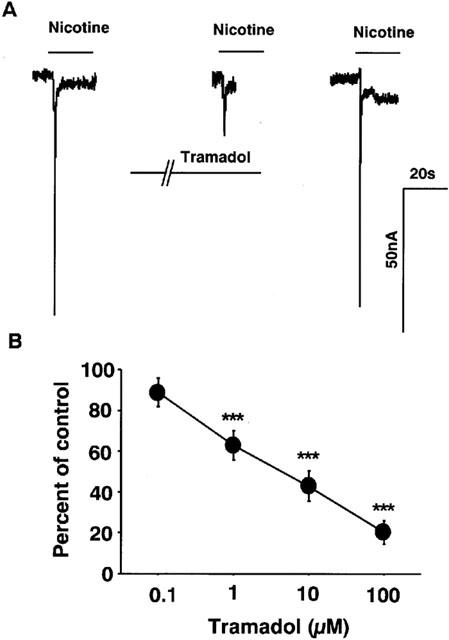
Effects of tramadol on nicotinic receptor-stimulated currents in Xenopus oocytes expressing α7 receptors. (A) Tracings obtained from a single oocyte expressing α7 nicotinic AChRs show the effect of tramadol on 1 mM nicotine-induced currents. Nicotine was applied for 20 s with or without 2 min treatment with 10 μM tramadol. (B) Concentration-response relationship of tramadol on nicotine-induced currents. Tramadol (100 nM–100 μM) was applied to the oocytes for 2 min and then 1 mM nicotine was applied for 20 s. Data represent the mean±s.e.mean (n=3–6). ***P<0.001 compared with the control response, using ANOVA.
Effects of α-bungarotoxin on nicotine-induced currents in bovine adrenal chromaffin cells
To examine whether α7 mediated responses are involved in the tramadol-induced inhibition of the nicotinic AChR functions, we studied the effects of 1 μM of α-bungarotoxin, a selective blocker of α7 receptors, on nicotine-induced inward currents in bovine adrenal chromaffin cells. In this experiment, to apply drugs more rapidly the flow rate of the perfusion medium was doubled (application time of nicotine was 5 s) and a cell closest to the inlet was selected. Bath application of nicotine induced transient inward currents, which appeared faster and desensitized faster than those recorded in the experiments performed with slower perfusion (Figure 10A). α-Bungarotoxin (1 μM), which was administered 1 min before nicotine application, inhibited the currents to 60±5% of the control response (n=7) and the effect was reversible. Tramadol (10 μM) inhibited nicotinic currents recorded with the same fast perfusion to 32±2% of control and reduced nicotinic currents that had been suppressed by α-bungarotoxin (n=4) (Figure 10B). The difference between the nicotinic currents obtained with α-bungarotoxin and those with the combined application of α-bungarotoxin and tramadol was 30±12% of control currents. As 1 μM of α-bungarotoxin has been reported to abolish currents carried by α7 receptors expressed in Xenopus oocytes without affecting currents carried by α3β4 receptors (Lopez et al., 1998), this difference represents the α-bungarotoxin-insensitive component of tramadol inhibition. The α-bungarotoxin-sensitive component was calculated by subtracting this component from the total tramadol inhibition obtained in the absence of α-bungarotoxin (Figure 10C). The α-bungarotoxin-sensitive and -insensitive components were 56 and 44%, respectively, of the total tramadol inhibition. There was no significant difference between the currents recorded with tramadol and with tramadol and α-bungarotoxin.
Figure 10.
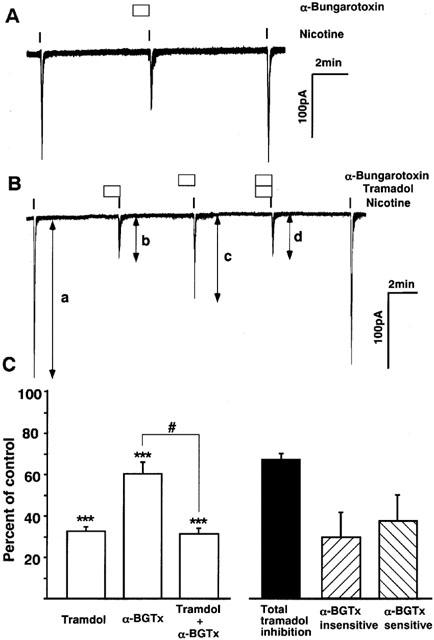
Effects of α-bungarotoxin on nicotine-induced inward currents and on the inhibition by tramadol in bovine adrenal chromaffin cells. The effects of α-bungarotoxin, a selective blocker of α7 nicotinic ACh receptors with or without co-application of tramadol were measured to know whether α7 receptors are involved in the tramadol inhibition on nicotinic currents. The rate of perfusion was doubled and the duration of nicotine application was set to 5 s in this series of experiments. Note that current kinetics is faster than that seen in the previous figures obtained with slower perfusion. Membrane potential was voltage-clamped at −60 mV. (A) a representative tracing obtained from a single cell showing reversible inhibition by α-bungarotoxin (1 μM). (B) The effects of tramadol (10 μM) on control nicotinic currents and currents that had been suppressed by α-bungarotoxin. Values are the mean±s.e.mean (n=4). (C) Summary of the results shown in B. The columns ‘Tramadol', ‘α-BGTx' and ‘Tramadol + α-BGTx' were calculated by dividing ‘b' by ‘a', ‘c' by ‘a' and ‘d' by ‘a' in B, respectively. The columns ‘Total tramadol inhibition', ‘α-BGTx-insensitive' and ‘α-BGTx-sensitive' were calculated by subtracting ‘Tramadol' from 100, ‘Tramadol + α-BGTx' from ‘α-BGTx' and ‘α-BGTx-insensitive' from ‘Total tramadol inhibition', respectively. ***P<0.001 compared with the control response, using ANOVA. #P<0.05 between the two columns.
Discussion
Effects of tramadol on catecholamine secretion from adrenal chromaffin cells
Our main conclusion from this study is that tramadol inhibits catecholamine secretion in response to cholinergic stimulation from adrenal chromaffin cells. Tramadol inhibited the catecholamine secretion to 60% of the control at 10 μM. According to clinical data presented by Lintz et al. (1986), the concentration of tramadol in human serum reaches about 2 μM after intravenous injection of 100 mg of tramadol. In the mouse tail-flick test, plasma concentrations of tramadol are reported to range from 0.8 μM (at the threshold dose) to 10.8 μM (at the maximum effective dose) (Friderichs & Becker, 1991). From our present findings and these previous reports, it seems likely that clinically relevant concentrations of tramadol inhibits the catecholamine secretion from the adrenal chromaffin cells. It is also possible that tramadol suppresses the sympathetic nervous activity through a similar inhibitory mechanism related with nicotinic AChRs.
It is well known that muscarinic stimulation does not evoke catecholamine secretion from bovine adrenal chromaffin cells (Yanagihara et al., 1979; Cheek & Burgoyne, 1985). Therefore, the inhibition of catecholamine secretion in response to carbachol appears to be a result of inhibition of nicotinic AChRs by tramadol. This is supported by the present result that the muscarine-evoked [Ca2+]i release response was virtually unaffected by tramadol. Na+ influx occurs via nicotinic AChRs; this stimulates Ca2+ influx via voltage-dependent Ca2+ channels and triggers secretion of catecholamines (Wada et al., 1985). In the present study, tramadol given at clinically relevant concentrations (below 10 μM) inhibited catecholamine secretion induced by carbachol and [Ca2+]i rises induced by nicotine or carbachol, but had little effect on catecholamine secretion induced by veratridine or high K+ and [Ca2+]i rises induced by high K+. Moreover, tramadol (1–100 μM) had little effect on the currents carried by the other ligand-gated ion-channels in adrenal chromaffin cells, GABAA receptors. These results indicate that clinically relevant concentrations of tramadol reduce catecholamine secretion by inhibiting the nicotinic AChRs without influencing voltage-dependent Ca2+ channels, voltage-dependent Na+ channels or GABAA receptors.
The IC50 value for the inhibition of nicotinic currents by tramadol was smaller than that for the inhibition of carbachol-induced catecholamine secretion by tramadol. One reason for this difference may be due to the fact that a maximally effective concentration of carbachol was used for the catecholamine secretion experiments but a submaximal concentration of nicotine was used for the nicotinic current measurement. However, tramadol was less potent in suppressing [Ca2+]i rises induced by nicotine than in inhibiting nicotinic currents even when the same concentration of nicotine was used for the two experiments. The difference in the efficacy of tramadol on the three parameters could be due to a non-linear relationship between nicotinic currents and a [Ca2+]i rise or exocytotic events. Ca2+ entry through voltage-gated Ca2+ channels (that is large enough to evoke catecholamine secretion) could still be triggered by nicotinic currents, even when the currents were reduced to a certain level by tramadol. This raises the possibility that tramadol may be more effective in suppressing nicotinic synaptic transmission in the central as well as the peripheral nervous system than in inhibiting nicotine-induced secretory events.
The inhibition by tramadol of nicotinic AChR-ion channels
To further investigate the mechanism of the inhibition by tramadol of nicotinic AChRs, we next studied the effects of tramadol on nicotinic AChR-mediated currents using the whole-cell patch-clamp technique. Tramadol inhibited nicotine-induced currents in a concentration-dependent manner and the inhibition by tramadol was reversible and repeatable. The inhibition was not affected by the maximally effective concentration of naloxone, indicating that the inhibitory effect on nicotinic AChRs is not mediated by opioid receptors. Although it has been shown that adrenal chromaffin cells possess μ-opioid receptors, activation of which lead to inhibition of voltage-dependent Ca2+ channels as well as inhibition of depolarization-dependent catecholamine secretion (Castanas et al., 1985), the present results provide evidence that the inhibitory effect of tramadol on nicotinic AChRs is independent of opioid receptors.
These results raised the question how tramadol inhibits nicotinic AChR-mediated responses. The inhibitory effect of tramadol was not overcome when the concentration of carbachol was increased in the incubation medium, suggesting that the inhibition by tramadol is a noncompetitive type of inhibition with the respect to carbachol. These results suggest that tramadol acts at a site different from that for carbachol or acetylcholine binding. The precise sites of actions of the non-competitive blockers of the acetylcholine receptors remain to be determined. A previous review by Lena & Changeux (1993) demonstrated that non-competitive nicotinic blockers inhibit nicotinic AChRs by acting at the different sites from those at which competitive blockers act. To provide further information concerning the inhibitory mechanism of tramadol on nicotinic AChRs, it is necessary to study the mechanism using site directed mutagenesis of AChRs.
The inhibition by tramadol of α7 receptors
Recently, several reports have pointed out that α7 may be one of the targets of intravenous anaesthetics. For example, ketamine inhibits responses to ACh in the human α7 nAChRs (Coates & Flood, 2001) and thiopental is a competitive inhibitor at the human α7 nicotinic acetylcholine receptors expressed in Xenopus oocytes (Coates et al., 2001). In the present study, tramadol inhibited the currents carried by α7 receptors expressed in Xenopus oocytes, suggesting that α7 receptors may be one of the targets for the tramadol action. In adrenal chromaffin cells, several distinct subclasses of nicotinic AChRs have been identified; α3 (Criado et al., 1992), α5 and β4, (Campos-Caro et al., 1997) and α7 (Garcia-Guzman et al., 1995) subunits of neuronal nicotinic AChRs were cloned and confirmed to be expressed in chromaffin cells. However, the nicotinic AChR subclass mediating catecholamine secretion in bovine chromaffin cells is still controversial. It was demonstrated that the functions controlling the electrical activity, the entry of Ca2+, and ensuring exocytotic response of chromaffin cells were attributable not only to α3β4 nicotinic AChRs but also to α7 nicotinic AChRs (Lopez et al., 1998). On the other hand, it has been reported that the subunit combinations of nicotinic AChRs associated with the catecholamine secretion from bovine adrenal chromaffin cells are α3β4 or α3β4α5 but not α7 (Tachikawa et al., 2001), and that α-bungarotoxin sensitive nicotinic AChRs had no effect on catecholamine secretion and functional response in bovine adrenal chromaffin cells (Quik & Trifaro, 1982; Broxton et al., 1999). The discrepancy seems to be explained by the different application methods used in the studies. Because α7 receptors show faster kinetics with quick desensitization than the others (Lopez et al., 1998), their contribution may have been masked in experiments using a slower drug application method. In our present experiments where faster perfusion was used, α-bungarotoxin suppressed the nicotine-induced currents in adrenal chromaffin cells. Moreover, the combined application of tramadol and α-bungarotoxin revealed that tramadol inhibited both α-bungarotoxin-sensitive and -insensitive components of nicotinic currents. These results indicate that tramadol inhibits both α7 and the other subclasses of nicotinic AChRs such as α3β4 in adrenal chromaffin cells. The result that there was no significant difference between the currents recorded with tramadol alone and with the combination of tramadol and α-bungarotoxin indicates that α7 receptor medicated currents were virtually abolished by the submaximal concentration (10 μM) of tramadol. The result that approximately one third of the total nicotinic currents persisted in the presence of this concentration of tramadol suggest that α7 receptors are more susceptible to the inhibition by tramadol than the other subclasses of nicotinic receptors. This needs to be confirmed in experiments using cloned nicotinic receptors other than α7 receptors.
Clinical implications of inhibitory effects of tramadol on nicotinic AChR functions
Tramadol had no effect on GABA-induced currents, whereas other anaesthetics, such as propofol (Concas et al., 1990) or barbiturates (Tomlin et al., 1999) potentiate GABA receptors. The result is consistent with the property of tramadol that this drug has little hypnotic effect for patients in a clinical situation (Lee et al., 1993), unlike propofol, benzodiazepines or barbiturate.
We have recently studied the effects of tramadol on blood pressure in anaesthetized rats. In the study, continuous infusion of tramadol had little effect on blood pressure (Nagaoka et al., 2002). We have also reported the inhibitory effects of tramadol on noradrenaline transporters (NAT) (Sagata et al., 2002). The plasma concentration of noradrenaline is the result of a balance between the rate of noradrenaline spillover into the circulation and the rate of noradrenaline clearance from the circulation. The spillover is determined by the balance between the release and uptake of noradrenaline in sympathetic nerve terminals. Thus, the lack of effects of tramadol on the blood pressure could be due to inhibition of NAT by tramadol. Another explanation is that in anaesthetized animals, the sympathetic nerve activity would be very low and synaptic transmission by ACh in the sympathetic ganglia might be so small that inhibition of nicotinic receptors by tramadol caused only a minimal effect.
Peripheral administration of nicotine is known to activate nociceptors (Grunfeld et al., 1991; Steen & Reeh, 1993). In our present results, tramadol inhibited the nicotinic AChR-mediated actions, and raised a possibility that the analgesic effects of tramadol could be due to its inhibitory effects on nicotinic AChRs in the peripheral nervous system. Moreover, since the sympathetic nervous system has an important role in the generation and maintenance of chronic pain state (Mcmahon, 1991), tramadol could decrease the sympathetic nervous activity as well as catecholamine secretion from the adrenal gland to suppress pain. Recent discoveries in the nicotinic AChR field have revealed that nicotinic AChR-targeted compounds such as ABT-594 are potential analgesic agents (Bannon et al., 1998). Furthermore, mice lacking α4 or β2 subtypes display a reduced antinociceptive effect of nicotine in the hot plate test and diminished sensitivity to nicotine in the tail-flick test. (Marubio et al., 1999; Cordero-Erausquin et al., 2000). The antinociceptive effects of nicotinic agonists might be due to receptor desensitization, rather than activation. In such case, antagonist inhibition of nicotinic receptors could still have an analgesic effect. Moreover, there have been several reports showing the inhibitory effects of analgesics on neuronal nicotinic AChR functions. At clinically relevant concentrations, inhalation anaesthetics suppressed nicotine-induced dopamine release in rat striatum (Salord et al., 1997) and carbachol-induced catecholamine release in chromaffin cells (Yashima et al., 1986; Minami et al., 1994). More recently, Mori et al. (2001) reported inhibition of acetylcholine receptors by halothane in rat cortical neurons. Because nicotinic AChRs located in presynaptic sites modulate the release of various neurotransmitters such as norepinephrine, dopamine, GABA, glutamate, serotonin, and ACh itself (Alkondon et al., 1997; Role & Berg, 1996; Wonnacott, 1997), tramadol could modulate the neurotransmitter release in the CNS through the inhibitory mechanism via nicotinic AChRs revealed in the present study. Taken together, the inhibition of tramadol on both muscarinic and nicotinic receptors could be one of the important mechanisms for its analgesic effects, however, further studies are needed to elucidate the interrelation between the inhibitory effects on AChRs by tramadol and its analgesic effects.
In conclusion, tramadol inhibits catecholamine secretion at least partly by inhibiting nicotinic AChR functions at clinically relevant concentrations in a manner independent of opioid receptors. The inhibitory effects of tramadol on nicotinic AChR functions might be one of the antinociceptive mechanisms exerted by tramadol.
Acknowledgments
We thank Dr. Y. Toyohira and Ms T. Tomihisa for assistance in the isolation of bovine adrenal medullary cells and technical supports. This research was supported as follows; Grants-in-Aid from the Ministry of Education, Science and Culture of Japan, (Nos. 11671532, 13671626, 12770851, 11770878, 12671515, 12671516, 12770849 and 12470013); UOEH (University of Occupational and Environmental Health) Research Grant for Promotion of Occupational Health; Grant-in Aid for the Promotion of Occupational Health for the Post Graduate Student of UOEH (University of Occupational and Environmental Health); The Japan Research Foundation for Clinical Pharmacology; The Uehara Memorial Foundation; The Kanehara-Ichiro Memorial Medical Foundation.
Abbreviations
- AChR
acetylcholine receptor
- CA
catecholamine
- GABA
gamma aminobutyric acid
- α-BGTx
α-bungarotoxin
References
- ALKONDON M., PEREIRA E.F., BARBOSA C.T., ALBUQUERQUE E.X. Neuronal nicotinic acetylcholine receptor activation modulates gamma-aminobutyric acid release from CA1 neurons of rat hippocampal slices. J. Pharmacol. Exp. Ther. 1997;283:1396–1411. [PubMed] [Google Scholar]
- BANNON A.W., DECKER M.W., HOLLADAY M.W., CURZON P., DONNELLY-ROBERTS D., PUTTFARCKEN P.S., BITNER R.S., DIAZ A., DICKENSON A.H., PORSOLT R.D., WILLIAMS M., ARNERIC S.P. Broad-spectrum, non-opioid analgesic activity by selective modulation of neuronal nicotinic acetylcholine receptors. Science. 1998;279:77–81. doi: 10.1126/science.279.5347.77. [DOI] [PubMed] [Google Scholar]
- BORMANN J., CLAPHAM D.E. gamma-Aminobutyric acid receptor channels in adrenal chromaffin cells: a patch-clamp study. Proc. Natl. Acad. Sci. U.S.A. 1985;82:2168–2172. doi: 10.1073/pnas.82.7.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROXTON N.M., DOWN J.G., GEHRMANN J., ALEWOOD P.F., SATCHELL D.G., LIVETT B.G. Alpha-conotoxin ImI inhibits the alpha-bungarotoxin-resistant nicotinic response in bovine adrenal chromaffin cells. J. Neurochem. 1999;72:1656–1662. doi: 10.1046/j.1471-4159.1999.721656.x. [DOI] [PubMed] [Google Scholar]
- CAMPOS-CARO A., SMILLIE F.I., DOMINGUEZ DEL TORO E., ROVIRA J.C., VICENTE-AGULLO F., CHAPULI J., JUIZ J.M., SALA S., SALA F., BALLESTA J.J., CRIADO M. Neuronal nicotinic acetylcholine receptors on bovine chromaffin cells: cloning, expression, and genomic organization of receptor subunits. J. Neurochem. 1997;68:488–497. doi: 10.1046/j.1471-4159.1997.68020488.x. [DOI] [PubMed] [Google Scholar]
- CASTANAS E., BOURHIM N., GIRAUD P., BOUDOURESQUE F., CANTAU P., OLIVER C. Interaction of opiates with opioid binding sites in the bovine adrenal medulla: I. Interaction with delta and mu sites. J. Neurochem. 1985;45:677–687. doi: 10.1111/j.1471-4159.1985.tb04046.x. [DOI] [PubMed] [Google Scholar]
- CHEEK T.R., BURGOYNE R.D. Effect of activation of muscarinic receptors on intracellular free calcium and secretion in bovine adrenal chromaffin cells. Biochim. Biophys. Acta. 1985;846:167–173. doi: 10.1016/0167-4889(85)90122-3. [DOI] [PubMed] [Google Scholar]
- COATES K.M., FLOOD P. Ketamine and its preservative, benzethonium chloride, both inhibit human recombinant alpha7 and alpha4beta2 neuronal nicotinic acetylcholine receptors in Xenopus oocytes. Br. J. Pharmacol. 2001;134:871–879. doi: 10.1038/sj.bjp.0704315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COATES K.M., MATHER L.E., JOHNSON R., FLOOD P. Thiopental is a competitive inhibitor at the human alpha7 nicotinic acetylcholine receptor. Anesth. Analg. 2001;92:930–933. doi: 10.1097/00000539-200104000-00026. [DOI] [PubMed] [Google Scholar]
- COLLART L., LUTHY C., FAVARIO-CONSTANTIN C., DAYER P. [Duality of the analgesic effect of tramadol in humans] Schweiz. Med. Wochenschr. 1993;123:2241–2243. [PubMed] [Google Scholar]
- CONCAS A., SANTORO G., MASCIA M.P., SERRA M., SANNA E., BIGGIO G. The general anesthetic propofol enhances the function of gamma-aminobutyric acid-coupled chloride channel in the rat cerebral cortex. J. Neurochem. 1990;55:2135–2138. doi: 10.1111/j.1471-4159.1990.tb05807.x. [DOI] [PubMed] [Google Scholar]
- CORDERO-ERAUSQUIN M., MARUBIO L.M., KLINK R., CHANGEUX J.P. Nicotinic receptor function: new perspectives from knockout mice. Trends. Pharmacol. Sci. 2000;21:211–217. doi: 10.1016/s0165-6147(00)01489-9. [DOI] [PubMed] [Google Scholar]
- CRIADO M., ALAMO L., NAVARRO A. Primary structure of an agonist binding subunit of the nicotinic acetylcholine receptor from bovine adrenal chromaffin cells. Neurochem. Res. 1992;17:281–287. doi: 10.1007/BF00966671. [DOI] [PubMed] [Google Scholar]
- FRIDERICHS E., BECKER R. Correlation of tramadol and M1 serum levels with antinociceptove activity in mice. Naunyn Schmiedebergs Arch. Pharmacol. 1991;343:R9. [Google Scholar]
- GARCIA-GUZMAN M., SALA F., SALA S., CAMPOS-CARO A., STUHMER W., GUTIERREZ L.M., CRIADO M. alpha-Bungarotoxin-sensitive nicotinic receptors on bovine chromaffin cells: molecular cloning, functional expression and alternative splicing of the alpha 7 subunit. Eur. J. Neurosci. 1995;7:647–655. doi: 10.1111/j.1460-9568.1995.tb00668.x. [DOI] [PubMed] [Google Scholar]
- GRUNFELD J.A., TIEDEMANN G.J., WESTERMAN R.A. Chronic nicotine exposure enhances cutaneous axon reflexes in the rat. Neuroreport. 1991;2:421–424. doi: 10.1097/00001756-199108000-00002. [DOI] [PubMed] [Google Scholar]
- HENNIES H.H., FRIDERICHS E., SCHNEIDER J. Receptor binding, analgesic and antitussive potency of tramadol and other selected opioids. Arzneimittelforschung. 1988;38:877–880. [PubMed] [Google Scholar]
- HEYER G.R., HORNSTEIN O.P. Recent studies of cutaneous nociception in atopic and non-atopic subjects. J. Dermatol. 1999;26:77–86. doi: 10.1111/j.1346-8138.1999.tb03516.x. [DOI] [PubMed] [Google Scholar]
- LEE C.R., MCTAVISH D., SORKIN E.M. Tramadol. A preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in acute and chronic pain states. Drugs. 1993;46:313–340. doi: 10.2165/00003495-199346020-00008. [DOI] [PubMed] [Google Scholar]
- LENA C., CHANGEUX J.P. Allosteric modulations of the nicotinic acetylcholine receptor. Trends. Neurosci. 1993;16:181–186. doi: 10.1016/0166-2236(93)90150-k. [DOI] [PubMed] [Google Scholar]
- LINTZ W., BARTH H., OSTERLOH G., SCHMIDT-BOTHELT E. Bioavailability of enteral tramadol formulations. 1st communication: capsules. Arzneimittelforschung. 1986;36:1278–1283. [PubMed] [Google Scholar]
- LOPEZ M.G., MONTIEL C., HERRERO C.J., GARCIA-PALOMERO E., MAYORGAS I., HERNANDEZ-GUIJO J.M., VILLARROYA M., OLIVARES R., GANDIA L., MCINTOSH J.M., OLIVERA B.M., GARCIA A.G. Unmasking the functions of the chromaffin cell alpha7 nicotinic receptor by using short pulses of acetylcholine and selective blockers. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14184–14189. doi: 10.1073/pnas.95.24.14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARUBIO L.M., DEL MAR ARROYO-JIMENEZ M., CORDERO-ERAUSQUIN M., LENA C., LE NOVERE N., DE KERCHOVE D'EXAERDE A., HUCHET M., DAMAJ M.I., CHANGEUX J.P. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- MCLACHLAN E.M., JANIG W., DEVOR M., MICHAELIS M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature. 1993;363:543–546. doi: 10.1038/363543a0. [DOI] [PubMed] [Google Scholar]
- MCMAHON S.B. Mechanisms of sympathetic pain. Br. Med. Bull. 1991;47:584–600. doi: 10.1093/oxfordjournals.bmb.a072494. [DOI] [PubMed] [Google Scholar]
- MINAMI K., YANAGIHARA N., TOYOHIRA Y., TSUTSUI M., SHIGEMATSU A., WADA A., IZUMI F. Isoflurane inhibits nicotinic acetylcholine receptor-mediated 22Na+ influx and muscarinic receptor-evoked cyclic GMP production in cultured bovine adrenal medullary cells. Naunyn Schmiedebergs Arch. Pharmacol. 1994;349:223–229. doi: 10.1007/BF00169287. [DOI] [PubMed] [Google Scholar]
- MORI T., ZHAO X., ZUO Y., AISTRUP G.L., NISHIKAWA K., MARSZALEC W., YEH J.Z., NARAHASHI T. Modulation of neuronal nicotinic acetylcholine receptors by halothane in rat cortical neurons. Mol. Pharmacol. 2001;59:732–743. doi: 10.1124/mol.59.4.732. [DOI] [PubMed] [Google Scholar]
- NAGAOKA E., MINAMI K., SHIGA Y., UEZONO Y., SHIRAISHI M., AOYAMA K., SHIGEMATSU A. Tramadol has no effect on cortical renal blood flow-despite increased serum catecholamine levels-in anesthetized rats: implications for analgesia in renal insufficiency. Anesth. Analg. 2002;94:619–625. doi: 10.1097/00000539-200203000-00026. [DOI] [PubMed] [Google Scholar]
- QUIK M., TRIFARO J.M. The alpha-bungarotoxin site and its relation to the cholinergic and nerve growth factor mediated increases in tyrosine hydroxylase activity in cultures of sympathetic ganglia and chromaffin cells. Brain Res. 1982;244:331–336. doi: 10.1016/0006-8993(82)90092-0. [DOI] [PubMed] [Google Scholar]
- REIMANN W., HENNIES H.H. Inhibition of spinal noradrenaline uptake in rats by the centrally acting analgesic tramadol. Biochem. Pharmacol. 1994;47:2289–2293. doi: 10.1016/0006-2952(94)90267-4. [DOI] [PubMed] [Google Scholar]
- ROBERTS R.G., STEVENSON J.E., WESTERMAN R.A., PENNEFATHER J. Nicotinic acetylcholine receptors on capsaicin-sensitive nerves. Neuroreport. 1995;6:1578–1582. doi: 10.1097/00001756-199507310-00028. [DOI] [PubMed] [Google Scholar]
- ROLE L.W., BERG D.K. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- SAGATA K., MINAMI K., YANAGIHARA N., SHIRAISHI M., TOYOHIRA Y., UENO S., SHIGEMATSU A. Tramadol inhibits norepinephrine transporter function at desipramine-binding sites in cultured bovine adrenal medullary cells. Anesth. Analg. 2002;94:901–906. doi: 10.1097/00000539-200204000-00024. [DOI] [PubMed] [Google Scholar]
- SALORD F., KEITA H., LECHARNY J.B., HENZEL D., DESMONTS J.M., MANTZ J. Halothane and isoflurane differentially affect the regulation of dopamine and gamma-aminobutyric acid release mediated by presynaptic acetylcholine receptors in the rat striatum. Anesthesiology. 1997;86:632–641. doi: 10.1097/00000542-199703000-00016. [DOI] [PubMed] [Google Scholar]
- SANNA E., DILDY-MAYFIELD J.E., HARRIS R.A. Ethanol inhibits the function of 5-hydroxytryptamine type 1c and muscarinic M1 G protein-linked receptors in Xenopus oocytes expressing brain mRNA: role of protein kinase C. Mol. Pharmacol. 1994;45:1004–1012. [PubMed] [Google Scholar]
- SHIBUYA I., TANAKA K., HATTORI Y., UEZONO Y., HARAYAMA N., NOGUCHI J., UETA Y., IZUMI F., YAMASHITA H. Evidence that multiple P2X purinoceptors are functionally expressed in rat supraoptic neurones. J. Physiol. 1999a;514:351–367. doi: 10.1111/j.1469-7793.1999.351ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIBUYA I., TANAKA K., UEZONO Y., UETA Y., TOYOHIRA Y., YANAGIHARA N., IZUMI F., YAMASHITA H. Prostaglandin E2 induces Ca2+ release from ryanodine/caffeine-sensitive stores in bovine adrenal medullary cells via EP1-like receptors. J. Neurochem. 1999b;73:2167–2174. [PubMed] [Google Scholar]
- SHIRAISHI M., MINAMI K., UEZONO Y., YANAGIHARA N., SHIGEMATSU A. Inhibition by tramadol of muscarinic receptor-induced responses in cultured adrenal medullary cells and in Xenopus laevis oocytes expressing cloned M1 receptors. J. Pharmacol. Exp. Ther. 2001;299:255–260. [PubMed] [Google Scholar]
- STEEN K.H., REEH P.W. Actions of cholinergic agonists and antagonists on sensory nerve endings in rat skin, in vitro. J. Neurophysiol. 1993;70:397–405. doi: 10.1152/jn.1993.70.1.397. [DOI] [PubMed] [Google Scholar]
- TACHIKAWA E., MIZUMA K., KUDO K., KASHIMOTO T., YAMATO S., OHTA S. Characterization of the functional subunit combination of nicotinic acetylcholine receptors in bovine adrenal chromaffin cells. Neurosci. Lett. 2001;312:161–164. doi: 10.1016/s0304-3940(01)02211-x. [DOI] [PubMed] [Google Scholar]
- TANAKA K., SHIBUYA I., UEZONO Y., UETA Y., TOYOHIRA Y., YANAGIHARA N., IZUMI F., KANNO T., YAMASHITA H. PACAP causes Ca2+ release from ryanodine/caffeine stores through a novel pathway independent of both IP3 and cAMP in bovine adrenal medullary cells. J. Neurochem. 1998;70:1652–1661. doi: 10.1046/j.1471-4159.1998.70041652.x. [DOI] [PubMed] [Google Scholar]
- TOMLIN S.L., JENKINS A., LIEB W.R., FRANKS N.P. Preparation of barbiturate optical isomers and their effects on GABA(A) receptors. Anesthesiology. 1999;90:1714–1722. doi: 10.1097/00000542-199906000-00029. [DOI] [PubMed] [Google Scholar]
- WADA A., TAKARA H., IZUMI F., KOBAYASHI H., YANAGIHARA N. Influx of 22Na through acetylcholine receptor-associated Na channels: relationship between 22Na influx, 45Ca influx and secretion of catecholamines in cultured bovine adrenal medulla cells. Neuroscience. 1985;15:283–292. doi: 10.1016/0306-4522(85)90135-6. [DOI] [PubMed] [Google Scholar]
- WEIL-MALHEREBE H., BONE A.D. The chemical estimation of adrenaline-like substances in blood. Biochem. J. 1952;51:311–318. doi: 10.1042/bj0510311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WONNACOTT S. Presynaptic nicotinic ACh receptors. Trends. Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- YANAGIHARA N., ISOSAKI M., OHUCHI T., OKA M. Muscarinic receptor-mediated increase in cyclic GMP level in isolated bovine adrenal medullary cells. FEBS, Lett. 1979;105:296–298. doi: 10.1016/0014-5793(79)80633-x. [DOI] [PubMed] [Google Scholar]
- YASHIMA N., WADA A., IZUMI F. Halothane inhibits the cholinergic-receptor-mediated influx of calcium in primary culture of bovine adrenal medulla cells. Anesthesiology. 1986;64:466–472. doi: 10.1097/00000542-198604000-00009. [DOI] [PubMed] [Google Scholar]
- ZOLI M., LE NOVERE N., HILL J.A., , JR, CHANGEUX J.P. Developmental regulation of nicotinic ACh receptor subunit mRNAs in the rat central and peripheral nervous systems. J. Neurosci. 1995;15:1912–1939. doi: 10.1523/JNEUROSCI.15-03-01912.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]