

Abstract
Nociceptin/orphanin FQ (N/OFQ) modulates several biological functions by activating a specific G-protein coupled receptor (NOP). Few molecules are available that selectively activate or block the NOP receptor. Here we describe the in vitro and in vivo pharmacological profile of a novel NOP receptor ligand, [Nphe1,Arg14,Lys15]N/OFQ-NH2 (UFP-101).
UFP-101 binds to the human recombinant NOP receptor expressed in Chinese hamster ovary (CHO) cells with high affinity (pKi 10.2) and shows more than 3000 fold selectivity over classical opioid receptors. UFP-101 competitively antagonizes the effects of N/OFQ on GTPγ35S binding in CHOhNOP cell membranes (pA2 9.1) and on cyclic AMP accumulation in CHOhNOP cells (pA2 7.1), being per se inactive at concentrations up to 10 μM.
In isolated peripheral tissues of mice, rats and guinea-pigs, and in rat cerebral cortex synaptosomes preloaded with [3H]-5-HT, UFP-101 competitively antagonized the effects of N/OFQ with pA2 values in the range of 7.3–7.7. In the same preparations, the peptide was inactive alone and did not modify the effects of classical opioid receptor agonists.
UFP-101 is also active in vivo where it prevented the depressant action on locomotor activity and the pronociceptive effect induced by 1 nmol N/OFQ i.c.v. in the mouse. In the tail withdrawal assay, UFP-101 at 10 nmol produces per se a robust and long lasting antinociceptive effect.
UFP-101 is a novel, potent and selective NOP receptor antagonist which appears to be a useful tool for future investigations of the N/OFQ-NOP receptor system.
Keywords: Nociceptin/orphanin FQ; NOP receptors; UFP-101; receptor antagonist; native and recombinant receptors; isolated tissues; rat cerebral cortex synaptosomes; tail withdrawal assay; locomotor activity, mice
Introduction
Nociceptin/orphanin FQ (N/OFQ) (Meunier et al., 1995; Reinscheid et al., 1995) is a neuropeptide that selectively interacts with the NOP receptor, a novel member of the opioid receptor family (Cox et al., 2000). The N/OFQ-NOP system has been reported to modulate several biological functions including pain transmission, stress and anxiety, learning and memory, locomotor activity, food intake, and the motivational properties of drugs of abuse (morphine and alcohol). N/OFQ may also intervene in the regulation of the cardiovascular, gastrointestinal, renal, genitourinary and respiratory systems (for review articles on these topics see Massi et al. (2000)). The pharmacology of the N/OFQ-NOP receptor system is still in its infancy. However, some peptide ligands have been described in the literature which behave as full agonists (N/OFQ(1-13)-NH2, [Tyr1]N/OFQ(1-13)-NH2, [(pF)Phe4]N/OFQ(1-13)-NH2), partial agonists ([Phe1ψ(CH2-NH)Gly2]N/OFQ(1-13)-NH2, Ac-RYYRIK-NH2) or antagonists (peptide III-BTD) for the NOP receptor (Calo et al., 2000a). Several non peptide compounds have also been described: these are both agonists (Ro 65-6570, NCC 63-0532, Ro 64-6198) and antagonists (J-113397, JTC 801) (Ronzoni et al., 2001).
Two other peptide ligands, selective for the NOP receptor, have been described: an antagonist, [Nphe1]N/OFQ(1-13)-NH2 (Calo et al., 2000b; Guerrini et al., 2000), and an agonist, [Arg14,Lys15]N/OFQ (Okada et al., 2000). The antagonist properties of [Nphe1]N/OFQ(1-13)-NH2 were documented with different techniques (GTPγS binding and cyclic AMP accumulation, bioassay, neurotransmitter release and electrophysiology) in a variety of in vitro preparations including recombinant systems, isolated tissues, brain slices and synaptosomes; pA2 values in the range of 6.0–6.7 were calculated for [Nphe1]N/OFQ(1-13)-NH2 from these studies. The antagonist properties of [Nphe1]N/OFQ(1-13)-NH2 were confirmed in vivo in a variety of assays (analgesiometric tests, food intake, learning and memory, locomotor activity, neurotransmitter release, cardiovascular and gastrointestinal function studies) where the peptide behaved as a low-potency, short lasting but pure and selective NOP antagonist (Calo et al., 2000a). The other compound, [Arg14,Lys15]N/OFQ, was identified as being a highly potent (17 fold more potent than N/OFQ) and selective agonist at recombinant human NOP receptors expressed in HEK-293 cells (Okada et al., 2000). Similar findings were obtained in our laboratory; indeed, [Arg14,Lys15]N/OFQ displayed higher potencies (from 5 to 20 fold) than N/OFQ in isolated tissues from rats, guinea-pigs and mice. In vivo in mice the compound was found to be 30 fold more potent than the natural peptide N/OFQ and its effects were longer lasting (Rizzi et al., 2002).
Based on these data, we decided to combine, in the sequence of N/OFQ, the chemical modification (Arg14, Lys15) which increases the potency of the agonist with that which confers antagonist properties (Nphe1). Moreover, N/OFQ was amidated at the C-terminus since N/OFQ-NH2 has been reported to be less susceptible to enzymatic degradation (Calo et al., 2000a). Thus, we synthesized the peptide [Nphe1, Arg14, Lys15]N/OFQ-NH2 hereafter referred to as UFP-101. This compound was tested in vitro (i) in binding and functional assays performed in CHOhNOP cells, (ii) in N/OFQ-sensitive isolated tissues and (iii) in a rat cerebral cortex synaptosome tritiated 5-HT release assay. We also evaluated the actions of UFP-101 in vivo in the locomotor activity and tail withdrawal assays in the mouse.
Methods
CHOhNOP cells
CHOhNOP cells were maintained in DMEM:F12 (50 : 50) containing 5% foetal calf serum (FCS), 2 mM glutamine, 200 μg ml−1 hygromycin B and 200 μg ml−1 G418. Cultures were maintained at 37°C in 5% CO2 / humidified air. When confluent, cells were harvested, membranes were then prepared and used fresh each day as described previously (Okawa et al., 1999). All binding assays were performed in 1 ml volumes of Tris-HCl (50 mM), containing MgSO4 (5 mM), 10 μM of the peptidase inhibitors (captopril, amastatin, bestatin, phosphoramidon), and bovine serum albumin 0.5%, pH 7.4. Approximately 5 μg of membrane protein and 0.2 nM [3H]-N/OFQ were incubated for 30 min at room temperature. Non-specific binding was defined in the presence of 1 μM N/OFQ. Bound and free radioactivities were separated by rapid vacuum filtration using a Brandel cell harvester. Harvester papers (Whatman GF/B) were presoaked in polyethyleneimine (0.5%) to reduce non-specific binding and loaded onto the harvester wet.
The binding of [3H]-diprenorphine (approximately 0.5 nM) to CHODOP, CHOKOP, and CHOMOP membranes was performed essentially as described previously (Calo et al., 2000b). As reference compounds, DPDPE was included for CHODOP, U-69593 for CHOKOP and fentanyl for CHOMOP cells.
GTPγS assay
Assay was performed essentially as described by Berger et al. (2000). Twenty μg of freshly prepared membranes were incubated for 1 h at 30°C in 0.5 ml volumes of Tris (50 mM) buffer supplemented with EGTA (0.2 mM), GDP (100 μM), bacitracin (0.15 mM), bovine serum albumin (1 mg ml−1), peptidase inhibitors (amastatin, bestatin, captopril, phosphoramidon; 10 μM), GTPγ35S (∼150 pM), N/OFQ (0.1 nM–10 μM), and UFP-101 (0, 10, 100, 1000 nM). Non-specific binding was measured in the presence of 10 μM unlabelled GTPγS. Bound and free radiolabel were separated by vacuum filtration onto Whatman GF/B filters.
cyclic AMP accumulation experiments
For measurement of cyclic AMP, whole CHOhNOP cells were incubated in 0.3 ml volumes of Krebs-HEPES buffer and bovine serum albumin, as described above. In addition 1-isobutyl-4-methylxanthine (1 mM) and forskolin (FSK, 1 μM) were also included. Concentration response curves to N/OFQ were constructed in the absence and presence of 1 μM of UFP-101. Cells were incubated at 37°C for 15 min. Reactions were terminated and cyclic AMP assayed using a protein binding assay as described by Okawa et al. (1999).
Electrically stimulated isolated organs
Tissues, taken from male Swiss mice (25–30 g), guinea-pigs (300–350 g), Sprague Dawley rats (300–350 g), and New Zealand albino rabbits (1.5–1.8 kg), were prepared as previously described (Bigoni et al., 1999; Calo et al., 2000b) and suspended in 5 ml organ baths containing oxygenated (95% O2 and 5% CO2) Krebs solution. The isolated organs were continuously stimulated through two platinum ring electrodes with supramaximal voltage rectangular pulses of 1 ms duration and 0.05 Hz frequency. Electrically evoked contractions were measured isotonically with a strain gauge transducer (Basile 7006) and recorded with the PC based acquisition system Autotrace 2.2 (RCS, Florence, Italy). Following an equilibration period of about 60 min, cumulative concentration response curves to N/OFQ were constructed (0.5 log unit steps) in the presence and absence of UFP-101. In the guinea-pig ileum, a Schild analysis was performed by testing UFP-101 in the concentration range 0.1–10 μM, while, in the other tissues, the antagonist was tested at the single concentration of 1 μM. In some experiments, UFP-101 10 μM was tested against the effects of classical opioid receptor agonists.
Rat cerebral cortex synaptosomes
Male Sprague-Dawley rats (180–240 g) were used. Synaptosomes were prepared as previously described (Morari et al., 1998; Sbrenna et al., 2000) and pre-loaded with [3H]-5HT by incubation in medium containing 50 nM [3H]-5HT for 20 min. One ml aliquots of the suspension (approximately 0.5 mg of protein) were slowly injected into nylon syringe filters connected to a peristaltic pump. Three minute sample collection was initiated after a 25 min washout period. Stimulation with 10 mM K+ (1 min pulse) was applied at 43 min. N/OFQ was added to the superfusion medium 12 min before the K+ pulse and maintained until the end of the experiment. The antagonist UFP-101 (0.3 μM) was added 3 min before N/OFQ. At the end of the experiment, radioactivity contained in the samples and retained in filters was determined using a Beckman LS 1800 β-spectrometer and Ultima Gold XR scintillation fluid (Packard Instruments, B.V., Groningen, The Netherlands).
In vivo studies
All experimental procedures adopted for in vivo studies were as humane as possible and complied with the standards of the European Communities Council directives (86/609/EEC) and national regulations (D.L. 116/92). Male Swiss albino mice weighing 20–25 g were used. They were housed under standard conditions (22°C, 12-h light–dark cycle) with food and water ad libitum for at least 2 days before experiments began. Each animal was used once. Intracerebroventricular (i.c.v.) injections (2 μl per mouse) were given into the left ventricle, according to the procedure described by Laursen & Belknap (1986).
Tail withdrawal assay
All experiments began at 10.00 a.m. and were performed according to the procedure described by Calo et al. (1998). Briefly, the animals were placed in a holder and the distal half of the tail was immersed in water at 48°C. Withdrawal latency time was measured by an experienced observer blind to drug treatment. A cut off time of 20 s was chosen to avoid tissue damage. Tail withdrawal time was determined immediately before and 5, 15, 30 and 60 min after i.c.v. injection of 2 μl of saline (control), N/OFQ (1 nmol), UFP-101 (3 or 10 nmol), or the co-application of N/OFQ and UFP-101.
Locomotor activity assay
Experiments were performed between 1400 h and 1800 h, following the procedure described by Rizzi et al. (2001). Briefly, the animals were routinely tested 3 min after i.c.v. injection of saline, N/OFQ (1 nmol), UFP-101 (3 or 10 nmol), or N/OFQ plus UFP-101. Locomotor activity was assessed using Basile activity cages. Animals were not accustomed to the cages before drug treatments and the experiment was performed in a quiet and dimly illuminated room. The total number of impulses were recorded every 5 min for 30 min.
Drugs
Peptides used in this study were prepared and purified as previously described (Guerrini et al., 1997). DPDPE, fentanyl, captopril, amastatin, bestatin, naloxone, cyclic AMP, 3-isobutyl-1-methylxanthine, HEPES and Tris were from Sigma Chemical Co. (Poole, U.K.); phosphoramidon from Peptide Institute (Osaka, Japan). U-69593 was from RBI (Milan, Italy). Naloxone was from Tocris Cookson (Bristol, U.K.). All tissue culture media and supplements were from Gibco (Paisley, U.K.). [2,8-3H]-cyclic AMP (28.4 Ci mmol−1), GTPγ35S (1250 Ci mmol−1) and (Leucyl-3,4,5-3H)-N/OFQ ([3H]-N/OFQ, 150 Ci mmol−1) were from NEN DuPont (Boston, MA, U.S.A.). [3H]-5HT and [15-16-3H]-diprenorphine (58 Ci mmol−1) were from NEN (Nen Life Science Products, Boston, U.S.A.). GDP was obtained from Sigma (Deisenhofen, Germany). Bacitracin, obtained from MERCK (Darmstadt, Germany), was heated for 1 h at 70°C in water to inactivate any enzymatic activity before use.
Peptidase inhibitors amastatin, bestatin, phosphoramidon and captopril were dissolved in water and frozen in 10 mM stocks. For in vitro experiments, the compounds were dissolved in physiological buffers and stock solutions (1 mM) were kept at −20°C until use. For in vivo studies, the substances were dissolved in physiological medium just before performing the experiment.
Data analysis and terminology
All data are expressed as means±s.e.mean of n experiments. For potency values the 95% confident limits are given. The pharmacological terminology adopted in this paper is consistent with IUPHAR recommendations (Jenkinson et al., 1995). Concentration of ligand producing 50% displacement of specific binding (IC50) was corrected for the competing mass of [3H]-N/OFQ or [3H]-diprenorphine to yield Ki. Curve fitting for binding data was performed using PRISM 2.0 (GraphPad Software In., San Diego, U.S.A.). Agonist potencies were measured as pEC50, which is the negative logarithm to base 10 of the agonist molar concentration that produces 50% of the maximal possible effect of that agonist. The Emax is the maximal effect that an agonist can elicit in a given preparation. Antagonist potencies are expressed in terms of pA2, which is the negative logarithm to base 10 of the antagonist molar concentration that makes it necessary to double the agonist concentration to elicit the original submaximal response. In the guinea-pig ileum and in GTPγ35S binding experiments, pA2 values were calculated by Schild analysis, while in the other preparations a single concentration of antagonist was used and the pA2 values were determined by applying the Gaddum Schild equation (pA2=−log((CR-1)/[Antagonist])), assuming a slope equal to unity.
In vivo data were analysed as follows: raw data from tail withdrawal experiments were converted to the area under the time×withdrawal latency curve and the data expressed as area under the curve were used for statistical analysis; locomotor activity data were analysed using the data expressed as cumulative impulses over the 30 min observation period. Data have been analysed statistically using Student's t-test or one-way ANOVA followed by the Dunnett's test, as specified in table and figure legends. P values less than 0.05 were considered to be significant.
Results
In vitro studies
Receptor binding, GTPγS stimulation and cyclic AMP accumulation assays in CHOhNOP
The ability of UFP-101 to bind to opioid receptors was evaluated using membranes of CHO cells expressing recombinant mouse DOP (δ), rat KOP (κ), rat MOP (μ), and human NOP receptors. As shown in Table 1, UFP-101 was essentially inactive at DOP and MOP sites, where about 30% inhibition of [3H]-diprenorphine binding was observed at 10 μM UFP-101. As internal positive assay controls DPDPE and fentanyl inhibited [3H]-diprenorphine binding with pKi values consistent with those previously reported (Hirst et al., 1998; Smart et al., 1997). In contrast to DOP and MOP, UFP-101 inhibited [3H]-diprenorphine binding to KOP receptors with a pKi of 6.69. In these cell membranes, U-69593 displayed a pKi value consistent with previous reports (Richardson et al., 1992).
Table 1.
Receptor binding profile of UFP-101 to recombinant DOP, KOP, MOP, and NOP receptors expressed in CHO cells

In CHOhNOP membranes, UFP-101 produced a concentration-dependent inhibition of [3H]-N/OFQ binding with a pKi value of 10.24. UFP-101 displayed an affinity for the NOP receptor very similar to that of N/OFQ (pKi=10.52) and selectivity over the classical opioid receptors of about 3000 fold.
In the same preparation, N/OFQ stimulated the GTPγ35S binding in a concentration-dependent manner with a pEC50 value of 8.73. The antagonistic properties of UFP-101 were evaluated over the 10–1000 nM range of concentrations, in order to obtain data for a Schild analysis. As shown in Figure 1A, UFP-101 displaced to the right the concentration response curve of N/OFQ in a concentration dependent manner, the curves remaining parallel to the control and reaching the same maximal effect. Figure 1B shows the corresponding Schild plot, which was linear (r=0.99) with a slope of 1.00±0.01. The extrapolated pA2 value is 9.12. These data suggest that the novel NOP receptor ligand UFP-101 behaves as a competitive antagonist at the recombinant human NOP receptor.
Figure 1.
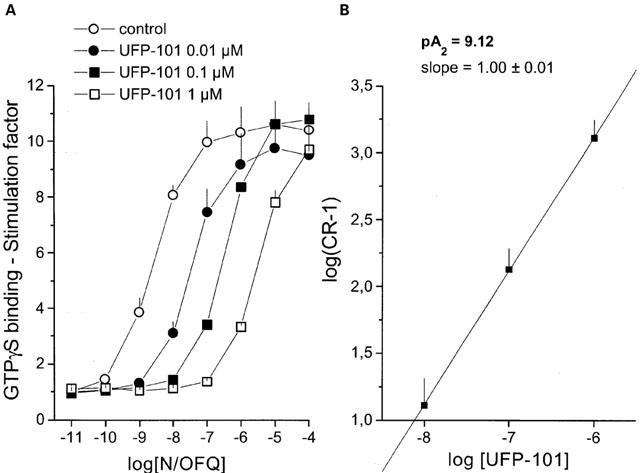
Stimulation of GTPγ35S binding to CHOhNOP cells membranes. (A) Concentration response curve to N/OFQ obtained in the absence (control) and presence of increasing concentrations of UFP-101. Corresponding Schild plot is shown in (B). Points indicate the means and vertical lines the s.e.mean of three experiments.
We have also examined the effects of this compound on FSK-stimulated cyclic AMP formation in whole CHOhNOP cells. UFP-101, applied at concentrations up to 10 μM, did not produce per se any significant inhibition of FSK-stimulated cyclic AMP formation (data not shown). When tested against N/OFQ, 1 μM UFP-101 displaced the concentration response curve of the natural peptide to the right, the curves being parallel and reaching the same maximal effects. The pA2 estimated from these experiments was 7.11 (Table 2).
Table 2.
Antagonist action of UFP-101 against N/OFQ in different preparations/assays

Electrically-stimulated isolated tissues
The rat and mouse vas deferens, and the guinea-pig ileum are N/OFQ-sensitive preparations, in which the peptide inhibits electrically-induced contractions (Bigoni et al., 1999). In the guinea-pig ileum, UFP-101, tested over the concentration range 0.1–10 μM, did not modify per se the electrically-induced twitches, but displaced to the right the concentration response curve to N/OFQ in a concentration dependent manner. Curves obtained in the presence of UFP-101 were parallel to the control, and reached the same maximal effects even in the presence of the highest concentration of antagonist (Figure 2A). The corresponding Schild plot was linear (r=0.99) with a slope not significantly different from unity, yielding a pA2 value of 7.18 (Figure 2B).
Figure 2.
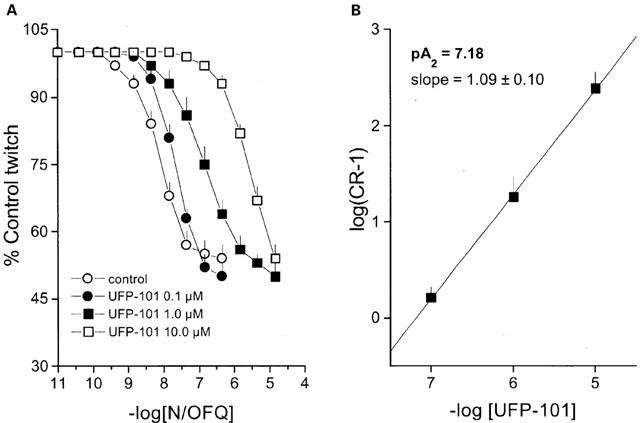
Electrically stimulated guinea-pig ileum. (A) Concentration response curve to N/OFQ obtained in the absence (control) and presence of increasing concentrations of UFP-101. Corresponding Schild plot is shown in (B). Points indicate the means and vertical lines the s.e.mean of at least four experiments.
In the mouse and rat vas deferens UFP-101 was tested at the single concentration of 1 μM against the effects of N/OFQ. In both preparations the concentration response curves to N/OFQ obtained in the absence and in presence of UFP-101 were parallel and reached similar maximal effects (Table 2). The estimated pA2 values were 7.30 and 7.29 in the rat and mouse vas deferens, respectively (Table 2). UFP-101 up to 10 μM did not show any residual agonist activity in these preparations.
To assess the selectivity of action of UFP-101, the peptide was tested at 10 μM against the inhibitory action of [DAla2]-deltorphin I in the mouse vas deferens, dermorphin in the guinea-pig ileum, etorphine in the rat vas deferens and U-69593 in the rabbit vas deferens. Emax (±s.e.mean) and pEC50(CL95%) of the agonists in the absence and presence of UFP-101 were: 93±4%, 10.28 (10.04–10.52) and 93±2%, 10.33 (10.01–10.65) in the mouse vas deferens, 84±4%, 8.91 (8.59–9.23) and 74±9%, 8.92 (8.67–9.17) in the guinea-pig ileum, 79±3%, 8.05 (7.70–8.40) and 73±9%, 7.99 (7.59–8.39) in the rat vas deferens, and 96±1%, 7.29 (7.01–7.57) and 96±3%, 7.27 (6.98–7.56) in the rabbit vas deferens. In summary, 10 μM UFP-101 did not modify the action of classical opioid receptor agonists in these preparations.
Rat cortical synaptosomes
N/OFQ inhibited 10 mM K+-evoked [3H]-5HT overflow in a concentration dependent manner, with a pEC50 value of 7.51 and an Emax value of −54±8% (Figure 3). These results are in line with previous findings (Sbrenna et al., 2000). UFP-101 0.3 μM did not modify per se the 10 mM K+-evoked [3H]-5HT overflow (data not shown), but produced a rightward shift in the concentration response curve to N/OFQ of about 1 log unit (pEC50=6.34), without any significant change in the maximal effect of the agonist (−59±14%) (Figure 3). The pA2 value, calculated for UFP-101 from this series of experiments, was 7.66.
Figure 3.
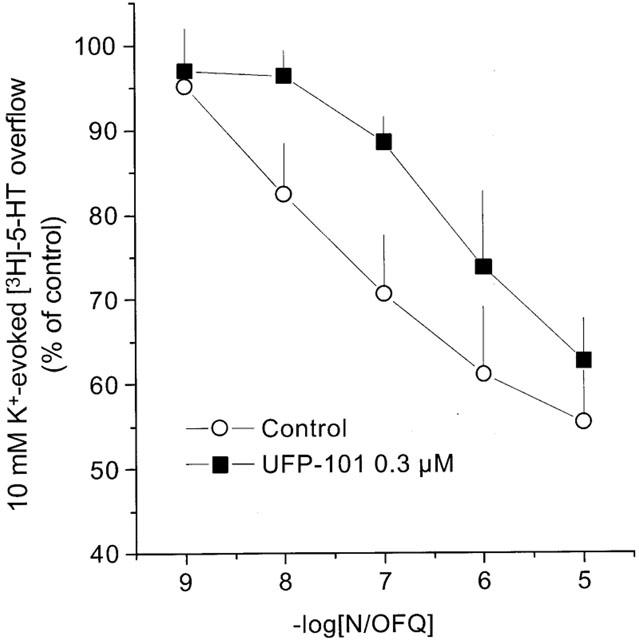
Rat cortical synaptosomes. Effects of UFP-101 on N/OFQ inhibition of 10 mM K+ evoked [3H]-5HT overflow. Points indicate the means and vertical lines the s.e.mean of at least four experiments.
In vivo studies
Tail withdrawal assay
Effects of UFP-101 at 3 and 10 nmol on the pronociceptive effects of 1 nmol N/OFQ in the mouse tail withdrawal assay are presented in Figure 4. Tail withdrawal latency in saline-injected mice was stable at around 4–5 s over the time course of the experiment. N/OFQ (1 nmol) applied i.c.v. significantly reduced tail withdrawal latency with a maximal effect (about 50% reduction in tail withdrawal latency) obtained at 5 min. Three nmol of UFP-101 applied via the same route, did not produce any significant effect per se, but prevented the pronociceptive effects of the natural peptide (Figure 4A). In contrast, at a dose of 10 nmol, UFP-101 produced a robust antinociceptive effect which peaked at 30 min and was still present 60 min following i.c.v. administration of the compound (Figure 4B). When N/OFQ (1 nmol) and UFP-101 (10 nmol) were coinjected, both the pronociceptive effect of the natural peptide and the antinociceptive effect of the pseudopeptide were abolished and tail withdrawal latencies were similar to those of saline injected animals (Figure 4B).
Figure 4.
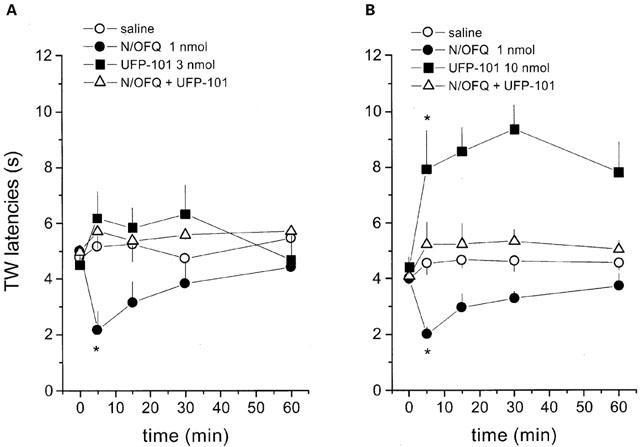
Tail withdrawal assay in mice. Effects of intracerebroventricular injection of N/OFQ (1 nmol) and UFP-101 (3 nmol, (A), 10 nmol, (B)) and their co-application on tail withdrawal latency in mice. Points indicate the means and vertical lines the s.e.mean of five (A) and six (B) experiments. Raw data were converted to the area under the time×withdrawal latency curve and the area under the curve data were used for statistical analysis. *P<0.05 vs saline, according to ANOVA followed by the Dunnett's test.
Locomotor activity
Effects of UFP-101 given at 3 and 10 nmol i.c.v. to counteract the depressant effects of 1 nmol N/OFQ on locomotor activity are shown in Figure 5. Saline-injected mice displayed a progressive reduction in spontaneous locomotor activity over the time course of the experiment (30 min) (Figure 5). In agreement with previously published data (Rizzi et al., 2001), 1 nmol N/OFQ produced a significant depression of locomotor activity in the first 10–15 min following i.c.v. administration (Figure 5). UFP-101 did not modify per se this animal behaviour, but partially prevented the effect of N/OFQ when given at 3 nmol (Figure 5A) and abolished this response when applied at 10 nmol (Figure 5B).
Figure 5.
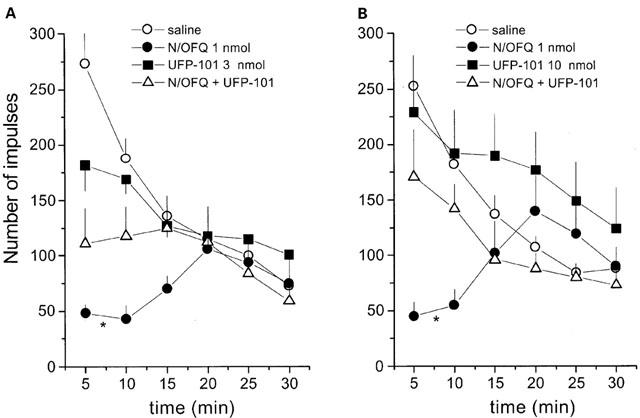
Locomotor activity assay in mice. Effects of N/OFQ (1 nmol) and UFP-101 (3 nmol, (A); 10 nmol, (B)) and their co-application on spontaneous locomotor activity in mice. Points indicate the means and vertical lines the s.e.mean of seven (A) and eight (B) experiments. Cumulative impulses over the 30 min observation period were used for statistical analysis. *P<0.05 vs saline, according to ANOVA followed by the Dunnett's test.
Discussion
The present study demonstrates that the peptide UFP-101 acts as a pure, competitive, and selective antagonist at the NOP receptor with greatly improved potency compared to [Nphe1]N/OFQ(1-13)NH2. UFP-101 is also active in vivo in the mouse where it antagonizes the effects of N/OFQ in the tail withdrawal and locomotor activity assays. UFP-101 competes with [3H]-N/OFQ for binding to the recombinant NOP receptor yielding an affinity similar to that of the natural peptide. In contrast, UFP-101 is unable to displace [3H]-diprenorphine from DOP and MOP sites and displays relatively low affinity for KOP sites (NOP/KOP ratio of selectivity about 3000). The selectivity of action of UFP-101 is confirmed by results obtained in isolated tissues where UFP-101 up to 10 μM did not modify the actions of opioid receptor agonists.
UFP-101 behaves as a pure antagonist devoid of agonist activity since it does not produce any effect per se but antagonizes N/OFQ actions in all the in vitro functional assays performed in the present study. These include cyclic AMP accumulation and GTPγS binding to recombinant receptors, bioassays in isolated tissues (native peripheral receptors) and neurochemical studies in synaptosomes (native central receptors).
The competitive nature of UFP-101 antagonism is suggested by the parallel displacement to the right of concentration response curves to N/OFQ in several preparations where maximal effects evoked by the natural peptide were maintained in the presence of the antagonist. More importantly, Schild analyses made on data obtained in the guinea-pig ileum and in CHOhNOP membranes (GTPγS binding) yielded regression lines which do not significantly differ from unity, thus confirming that the antagonist is indeed competitive.
The potency of UFP-101 (pA2 values are in the range 7.11–7.66) obtained in the present experiments are similar in the various in vitro preparations, with the only exception of the GTPγS binding experiments. This confirms previous findings obtained with NOP selective antagonists of peptide ([Nphe1]N/OFQ(1-13)-NH2 (Calo et al., 2000b; Rizzi et al., 1999; Sbrenna et al., 2000)) and non-peptide (J-113397 (Bigoni et al., 2000; Ozaki et al., 2000)) nature. The similarity of UFP-101 pA2 values suggests that NOP receptors, expressed by different species (mouse, rat, guinea-pig, human) and in the same species (the rat) by different tissues (central and peripheral), are of the same type.
In the GTPγS binding experiments UFP-101 (pA2 9.12) is significantly more potent than in other preparations (pA2≈#38;7.5). Similar findings were obtained with [Nphe1]N/OFQ(1-13)-NH2 on GTPγS binding stimulated by N/OFQ in rat cortex (pA2 7.7; (Berger et al., 2000) and CHOhNOP cell (pA2 7.0; (Hashiba et al., 2002)) membranes, where values of potency are higher than those obtained in other preparations (pA2 6.0–6.5, (Calo et al., 2000c)). It thus appears that potencies of antagonists evaluated in membrane preparations with the GTPγS binding assay are consistently higher than those obtained in other preparations/assays. The reader is referred to the paper by Berger et al. (2000) for a detailed discussion of this topic.
Worthy of mention is the fact that pA2 values of UFP-101 are about 10 fold higher than those of [Nphe1]N/OFQ(1-13)-NH2. This increase in antagonist potency is very similar to the increase in agonist potency obtained when the Arg14,Lys15 couple is used in the natural sequence N/OFQ. Indeed, [Arg14,Lys15]N/OFQ is a full agonist of the NOP receptor about 10 fold more potent than N/OFQ (Okada et al., 2000; Rizzi et al., 2002). These results suggest that the presence of Arg14,Lys15 increases the ligand affinity for the receptor without modifying its pharmacological activity (agonist or antagonist). In other words, the same result, i.e. a 10 fold increase in potency, has been obtained by using this modification in an agonist (N/OFQ) and in an antagonist ([Nphe1]N/OFQ) template. These findings support our hypothesis that the N/OFQ sequence can be separated into a N-terminal message domain (N/OFQ(1-4)) responsible for receptor activation, and a C-terminal address domain (N/OFQ(5-17)) responsible for receptor occupation (Guerrini et al., 1997; Salvadori et al., 1999). The Arg14, Lys15 couple is located in the address domain of the peptide and therefore it affects ligand affinity but not efficacy.
Collectively, these in vitro data demonstrate that UFP-101 acts as a selective and competitive antagonist at NOP receptors from various species with a potency not far from that of the natural ligand N/OFQ.
N/OFQ has been shown by us and several other groups to depress locomotor activity (Noble and Roques, 1997; Reinscheid et al., 1995; Rizzi et al., 2001) and elicit pronociceptive effects (Calo et al., 1998; Meunier et al., 1995; Nishi et al., 1997; Reinscheid et al., 1995) following i.c.v. injection in the mouse. Results obtained in the present experiments using N/OFQ 1 nmol are therefore in line with data from the literature. These actions of N/OFQ were reduced or abolished by the coinjection of 3 or 10 nmol UFP-101, indicating that UFP-101 also acts as an effective NOP receptor antagonist in vivo. These results further corroborate the indications emerging from experiments with NOP receptor antagonists (Calo et al., 2000b; Ozaki et al., 2000; Rizzi et al., 2001) and NOP knockout mice (Nishi et al., 1997) that the supraspinal actions of N/OFQ on locomotor activity and pain threshold are exclusively due to NOP receptor activation.
It is worthy of note that statistically significant effects of UFP-101 against N/OFQ were obtained in both the locomotor activity and tail withdrawal assays using the dose of 3 nmol, while the same dose of [Nphe1]N/OFQ(1-13)-NH2 is inactive (A. Rizzi, personal communication). Therefore, in agreement with in vitro findings, UFP-101 was also found to be more potent than [Nphe1]N/OFQ(1-13)-NH2 in vivo.
In the tail withdrawal assay, UFP-101 at 10 nmol not only prevented the pronociceptive effects of N/OFQ but produced per se an antinociceptive effect similar to [Nphe1]N/OFQ(1-13)-NH2 (Calo et al., 2000b). However, the antinociceptive effect induced by UFP-101 reached a peak at 30 min and was still evident 60 min after i.c.v. injection, while that evoked (under the same experimental conditions) by [Nphe1]N/OFQ(1-13)-NH2 peaked at 5 min and lasted no more than 15 min (Calo et al., 2000b). These in vivo findings indicate that the presence of the Arg14, Lys15 couple not only contributes to an increase in potency of the antagonist but also its duration of action in vivo. This interpretation is supported by data obtained with the agonist [Arg14, Lys15]N/OFQ, whose effects in vivo are longer lasting than those of N/OFQ (Rizzi et al., 2002). The increase in duration of action of Arg14, Lys15 substituted ligands may be due to several factors including stronger binding to the NOP receptor and/or increased metabolic stability. Increased stability is suggested by in vitro experiments, performed in the rat vas deferens, where peptidase inhibitors potentiated N/OFQ but not [Arg14, Lys15]N/OFQ effects (Rizzi et al., 2002). Further studies are however required to validate this interpretation.
Irrespective of differences in kinetics for UFP-101 and [Nphe1]N/OFQ(1-13)-NH2, both these compounds are selective NOP antagonists and both produce per se antinociceptive effects. Analgesic actions of NOP antagonists have been reported using other molecules such as the peptide retro-nociceptin methylester (Jinsmaa et al., 2000) and the non-peptide JTC-801 (Shinkai et al., 2000). Moreover, the non-selective antagonist naloxone benzoylhydrazone produces antinociceptive effects in normal but not in NOP knockout mice (Noda et al., 1998). Recent findings from our laboratory also indicate that the antinociceptive properties of [Nphe1]N/OFQ(1-13)-NH2 are no longer evident in NOP receptor deficient mice (di giannuario et al., 2001). Taken together, these findings suggest that NOP receptor antagonists may be considered as analgesic agents. The antinociceptive properties of these molecules may be due to their ability to block endogenous N/OFQ signalling which may control pain threshold by exerting a tonic pronociceptive effect. However, other data in the literature contradict this suggestion. The non-peptide NOP receptor antagonist J-113397, which is probably the best NOP selective antagonist available to date, does not induce antinociception at doses that are able to prevent the pronociceptive effects of exogenously applied N/OFQ (Ozaki et al., 2000). In addition, NOP receptor knockout mice do not show any obvious change in pain threshold compared to their wild-type littermates (Nishi et al., 1997). Thus, as recently pointed out by Mogil & Pasternak (2001) ‘for now, at least, the mystery of the true endogenous role of N/OFQ in nociception has failed to yield to simple explanations and technological advances'.
Further studies and possibly the identification of other NOP selective antagonists are required for better defining the therapeutic potential of NOP antagonists as analgesics. Based on data obtained in vitro and in vivo, it is concluded that UFP-101 acts as a selective and competitive antagonist of the NOP receptor displaying long lasting effects in vivo. By comparing UFP-101 with the other available NOP antagonists of peptide ([Nphe1]N/OFQ(1–13)NH2, pA2 6.0–6.7, selectivity over classical opioid receptors ≈#38;250 fold (Calo et al., 2000b) and peptide III-BTD, pA2≈#38;7, selectivity ≈#38;10 fold (Becker et al., 1999)) and non peptide nature (J-113397 pA2≈#38;8, selectivity ≈#38;350 fold (Ozaki et al., 2000) and JTC-801 pA2 not determined, selectivity ≈#38;10 fold (Shinkai et al., 2000)) it can be said that UFP-101 is one of the most potent antagonists and, clearly, the most selective for NOP receptors. This comparison underlines the usefulness of UFP-101 as a pharmacological tool for future in vitro and in vivo studies aimed at evaluating the roles of the N/OFQ-NOP receptor system in physiology and pathology.
Acknowledgments
We would like to thank Dr F. Marshall and Mrs N. Bevan of Glaxo-Wellcome, Stevenage, Herts U.K. for providing CHO cells expressing the human NOP receptor, Dr D.K. Grandy, Vollum Institute for Advanced Biomedical Research, Portland, Oregon, U.S.A. for providing CHO cells expressing rat KOP and MOP receptors and Dr L.A. Devi, New York University Medical Center, New York, U.S.A. for providing CHO cells expressing the mouse DOP receptor. This work was supported by the University of Ferrara (60% Grants to S. Salvadori and G. Calo) and by Italian Ministry of the University (Cofin 1999–2000 grant to D. Regoli).
Abbreviations
- CHO
Chinese hamster ovary
- DPDPE
[D-Pen2.5]enkephalin
- N/OFQ
nociceptin/orphanin FQ
- UFP-101
[Nphe1,Arg14,Lys15]N/OFQ-NH2
References
- BECKER J.A., WALLACE A., GARZON A., INGALLINELLA P., BIANCHI E., CORTESE R., SIMONIN F., KIEFFER B.L., PESSI A. Ligands for kappa-opioid and ORL1 receptors identified from a conformationally constrained peptide combinatorial library. J. Biol. Chem. 1999;274:27513–27522. doi: 10.1074/jbc.274.39.27513. [DOI] [PubMed] [Google Scholar]
- BERGER H., CALO G., ALBRECHT E., GUERRINI R., BIENERT M. [Nphe1]NC(1–13)NH2 selectively antagonizes nociceptin/orphanin FQ-stimulated G-protein activation in rat brain. J. Pharmacol. Exp. Ther. 2000;294:428–433. [PubMed] [Google Scholar]
- BIGONI R., CALO G., RIZZI A., GUERRINI R., DE RISI C., HASHIMOTO Y., HASHIBA E., LAMBERT D.G., REGOLI D. In vitro characterization of J-113397, a non-peptide nociceptin/orphanin FQ receptor antagonist. Naunyn Schmiedebergs Arch. Pharmacol. 2000;361:565–568. doi: 10.1007/s002100000220. [DOI] [PubMed] [Google Scholar]
- BIGONI R., GIULIANI S., CALO G., RIZZI A., GUERRINI R., SALVADORI S., REGOLI D., MAGGI C.A. Characterization of nociceptin receptors in the periphery: in vitro and in vivo studies. Naunyn Schmiedebergs Arch. Pharmacol. 1999;359:160–167. doi: 10.1007/pl00005338. [DOI] [PubMed] [Google Scholar]
- CALO G., BIGONI R., RIZZI A., GUERRINI R., SALVADORI S., REGOLI D. Nociceptin/orphanin FQ receptor ligands. Peptides. 2000a;21:935–947. doi: 10.1016/s0196-9781(00)00230-8. [DOI] [PubMed] [Google Scholar]
- CALO G., GUERRINI R., BIGONI R., RIZZI A., MARZOLA G., OKAWA H., BIANCHI C., LAMBERT D.G., SALVADORI S., REGOLI D. Characterization of [Nphe1]nociceptin(1-13)NH2, a new selective nociceptin receptor antagonist. Br. J. Pharmacol. 2000b;129:1183–1193. doi: 10.1038/sj.bjp.0703169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALO G., GUERRINI R., RIZZI A., SALVADORI S., REGOLI D. Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br. J. Pharmacol. 2000c;129:1261–1283. doi: 10.1038/sj.bjp.0703219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALO G., RIZZI A., MARZOLA G., GUERRINI R., SALVADORI S., BEANI L., REGOLI D., BIANCHI C. Pharmacological characterization of the nociceptin receptor mediating hyperalgesia in the mouse tail withdrawal assay. Br. J. Pharmacol. 1998;125:373–378. doi: 10.1038/sj.bjp.0702087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COX B.M., CHAVKIN C., CHRISTIE M.J., CIVELLI O., EVANS C., HAMON M.D., HOELLT V., KIEFFER B., KITCHEN I., MCKNIGHT A.T., MEUNIER J.C., PORTOGHESE P.S.Opioid receptors The IUPHAR Compendium of Receptor Characterization and Classification 2000London: IUPHAR Media Ltd; 321–333.ed. Girdlestone, D. pp [Google Scholar]
- DI GIANNUARIO A., RIZZI A., PIERETTI S., GUERRINI R., BERTORELLI R., SALVADORI S., REGOLI D., CALO' G. Studies on the antinociceptive effect of [Nphe1]nociceptin(1–13)NH2 in mice. Neurosci. Lett. 2001;316:25–28. doi: 10.1016/s0304-3940(01)02352-7. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO G., BIGONI R., RIZZI A., VARANI K., TOTH G., GESSI S., HASHIBA E., HASHIMOTO Y., LAMBERT D.G., BOREA P.A., TOMATIS R., SALVADORI S., REGOLI D. Further studies on nociceptin-related peptides: discovery of a new chemical template with antagonist activity on the nociceptin receptor. J. Med. Chem. 2000;43:2805–2813. doi: 10.1021/jm990075h. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO G., RIZZI A., BIANCHI C., LAZARUS L.H., SALVADORI S., TEMUSSI P.A., REGOLI D. Address and message sequences for the nociceptin receptor: a structure-activity study of nociceptin-(1-13)-peptide amide. J. Med. Chem. 1997;40:1789–1793. doi: 10.1021/jm970011b. [DOI] [PubMed] [Google Scholar]
- HASHIBA E., LAMBERT D.G., JENCK F., WICHMANN J., SMITH G. Characterization of the non-peptide nociceptin receptor agonist, Ro 64-6198 in chinese hamster ovary cells expressing recombinant human nociceptin receptors. Life Sci. 2002;70:1719–1725. doi: 10.1016/s0024-3205(02)01477-7. [DOI] [PubMed] [Google Scholar]
- HIRST R.A., SMART D., DEVI L.A., LAMBERT D.G. Effects of C-terminal truncation of the recombinant delta opioid receptor on phospholipase C and anenylyl cyclase coupling. J. Neurochem. 1998;70:2273–2278. doi: 10.1046/j.1471-4159.1998.70062273.x. [DOI] [PubMed] [Google Scholar]
- JENKINSON D.H., BARNARD E.A., HOYER D., HUMPHREY P.P.A., LEFF P., SHANKLEY N.P. International Union of Pharmacology Commitee on receptor nomenclature and drug classification XI Recommendations on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 1995;47:255–266. [PubMed] [Google Scholar]
- JINSMAA Y., TAKAHASHI M., FUKUNAGA H., YOSHIKAWA M. Retronociceptin methylester, a peptide with analgesic and memory-enhancing activity. Life Sci. 2000;67:3095–3101. doi: 10.1016/s0024-3205(00)00889-4. [DOI] [PubMed] [Google Scholar]
- LAURSEN S.E., BELKNAP J.K. Intracerebroventricular injections in mice. Some methodological refinements. J. Pharmacol. Methods. 1986;16:355–357. doi: 10.1016/0160-5402(86)90038-0. [DOI] [PubMed] [Google Scholar]
- MASSI M., POLIDORI C., CALO' G., REGOLI D. Nociceptin/orphanin FQ and its receptor. Peptides. 2000;21 [Google Scholar]
- MEUNIER J.C., MOLLEREAU C., TOLL L., SUAUDEAU C., MOISAND C., ALVINERIE P., BUTOUR J.L., GUILLEMOT J.C., FERRARA P., MONSERRAT B., MAZARGUIL H., VASSART G., PARMENTIER M., COSTENTIN J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- MOGIL J.S., PASTERNAK G.W. The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol. Rev. 2001;53:381–415. [PubMed] [Google Scholar]
- MORARI M., SBRENNA S., MARTI M., CALIARI F., BIANCHI C., BEANI L. NMDA AND NON-NMDA ionotropic glutamate receptors modulate striatal acetylcholine release via pre- and postsynaptic mechanisms. J. Neurochem. 1998;71:2006–2017. doi: 10.1046/j.1471-4159.1998.71052006.x. [DOI] [PubMed] [Google Scholar]
- NISHI M., HOUTANI T., NODA Y., MAMIYA T., SATO K., DOI T., KUNO J., TAKESHIMA H., NUKADA T., NABESHIMA T., YAMASHITA T., NODA T., SUGIMOTO T. Unrestrained nociceptive response and disregulation of hearing ability in mice lacking the nociceptin/orphaninFQ receptor. EMBO J. 1997;16:1858–1864. doi: 10.1093/emboj/16.8.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOBLE F., ROQUES B.P. Association of aminopeptidase N and endopeptidase 24.15 inhibitors potentiate behavioral effects mediated by nociceptin/orphanin FQ in mice. FEBS Lett. 1997;401:227–229. doi: 10.1016/s0014-5793(96)01476-7. [DOI] [PubMed] [Google Scholar]
- NODA Y., MAMIYA T., NABESHIMA T., NISHI M., HIGASHIOKA M., TAKESHIMA H. Loss of antinociception induced by naloxone benzoylhydrazone in nociceptin receptor-knockout mice. J. Biol. Chem. 1998;273:18047–18051. doi: 10.1074/jbc.273.29.18047. [DOI] [PubMed] [Google Scholar]
- OKADA K., SUJAKU T., CHUMAN Y., NAKASHIMA R., NOSE T., COSTA T., YAMADA Y., YOKOYAMA M., NAGAHISA A., SHIMOHIGASHI Y. Highly potent nociceptin analog containing the Arg-Lys triple repeat. Biochem. Biophys. Res. Commun. 2000;278:493–498. doi: 10.1006/bbrc.2000.3822. [DOI] [PubMed] [Google Scholar]
- OKAWA H., NICOL B., BIGONI R., HIRST R.A., CALO G., GUERRINI R., ROWBOTHAM D.J., SMART D., MCKNIGHT A.T., LAMBERT D.G. Comparison of the effects of [Phe1psi(CH2-NH)Gly2]nociceptin(1-13)NH2 in rat brain, rat vas deferens and CHO cells expressing recombinant human nociceptin recetors. Br. J. Pharmacol. 1999;127:123–130. doi: 10.1038/sj.bjp.0702539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OZAKI S., KAWAMOTO H., ITOH Y., MIYAJI M., AZUMA T., ICHIKAWA D., NAMBU H., IGUCHI T., IWASAWA Y., OHTA H. In vitro and in vivo pharmacological characterization of J-113397, a potent and selective non-peptidyl ORL1 receptor antagonist. Eur. J. Pharmacol. 2000;402:45–53. doi: 10.1016/s0014-2999(00)00520-3. [DOI] [PubMed] [Google Scholar]
- REINSCHEID R.K., NOTHACKER H.P., BOURSON A., ARDATI A., HENNINGSEN R.A., BUNZOW J.R., GRANDY D.K., LANGEN H., MONSMA F.J., Jr, CIVELLI O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- RICHARDSON A., DEMOLIOU-MASON C., BARNARD E.A. Guanine nucleotide-binding protein-coupled and -uncoupled states of opioid receptors and their relevance to the determination of subtypes. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10198–10202. doi: 10.1073/pnas.89.21.10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIZZI A., BIGONI R., CALO' G., GUERRINI R., SALVADORI S., REGOLI D. [Nphe1]nociceptin(1-13)NH2 antagonizes nociceptin effects in the mouse colon. Eur. J. Pharmacol. 1999;385:R3–R5. doi: 10.1016/s0014-2999(99)00730-x. [DOI] [PubMed] [Google Scholar]
- RIZZI A., BIGONI R., MARZOLA G., GUERRINI R., SALVADORI S., REGOLI D., CALO G. Characterization of the locomotor activity-inhibiting effect of nociceptin/orphanin FQ in mice. Naunyn. Schmiedebergs. Arch. Pharmacol. 2001;363:161–165. doi: 10.1007/s002100000358. [DOI] [PubMed] [Google Scholar]
- RIZZI D., RIZZI A., BIGONI R., CAMARDA V., MARZOLA G., GUERRINI R., DE RISI C., REGOLI D., CALO G. [Arg14,Lys15]NC, a highly potent agonist of the nociceptin/orphanin FQ receptor: in vitro and in vivo studies. J. Pharmacol. Exp. Ther. 2002;300:57–63. doi: 10.1124/jpet.300.1.57. [DOI] [PubMed] [Google Scholar]
- RONZONI S., PERETTO I., GIARDINA G.A.M. Lead generation and lead optimization approaches in the discovery of selective, non-peptide ORL-1 receptor agonists and antagonists. Exp. Opin. Ther. Patents. 2001;11:525–546. [Google Scholar]
- SALVADORI S., GUERRINI R., CALO G., REGOLI D. Structure-activity studies on nociceptin/orphanin FQ: from full agonist, to partial agonist, to pure antagonist. Farmaco. 1999;54:810–825. doi: 10.1016/s0014-827x(99)00108-1. [DOI] [PubMed] [Google Scholar]
- SBRENNA S., MARTI M., MORARI M., CALO G., GUERRINI R., BEANI L., BIANCHI C. Modulation of 5-hydroxytryptamine efflux from rat cortical synaptosomes by opioids and nociceptin. Br. J. Pharmacol. 2000;130:425–433. doi: 10.1038/sj.bjp.0703321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHINKAI H., ITO T., IIDA T., KITAO Y., YAMADA H., UCHIDA I. 4-Aminoquinolines: novel nociceptin antagonists with analgesic activity. J. Med. Chem. 2000;43:4667–4677. doi: 10.1021/jm0002073. [DOI] [PubMed] [Google Scholar]
- SMART D., HIRST R.A., HIROTA K., GRANDY D.K., LAMBERT D.G. The effects of recombinant rat mu opioid receptor activation in CHO cells on phospholipase C, [Ca2+]i and adenylyl cyclase. Br. J. Pharmacol. 1997;120:1165–1171. doi: 10.1038/sj.bjp.0701012. [DOI] [PMC free article] [PubMed] [Google Scholar]