

Abstract
The present study was aimed to determine whether propranolol improves contractile function of the ischaemic/reperfused heart through protection of the mitochondrial function during ischaemia.
Isolated perfused rat hearts were subjected to 35-min ischaemia followed by 60-min reperfusion. Pre-treatment with propranolol at the concentrations of 10 to 100 μM for the final 3 min of pre-ischaemia resulted in the improvement of ischaemia/reperfusion-induced contractile dysfunction, release of creatine kinase (CK) into perfusate, and decrease in myocardial high-energy phosphates. Propranolol also attenuated ischaemia-induced accumulation in Na+, suggesting that cytosolic sodium overload during ischaemia was prevented by propranolol.
The mitochondrial oxygen consumption rate of skinned bundles from the perfused heart decreased at the end of ischaemia and it further decreased at the end of reperfusion. These decreases were cancelled by treatment with propranolol. A release of cytochrome c from the perfused heart was observed during ischaemia, and this release was suppressed by treatment with propranolol.
To elucidate the direct effect of propranolol on mitochondria, the mitochondria were isolated from normal hearts and their activities were determined in the presence of various concentrations of Na+ and propranolol. The addition of sodium lactate, which mimicked sodium overload in the ischaemic heart, reduced the state 3 respiration, whereas this reduction was not attenuated by the presence of propranolol.
These results suggest that cardioprotection of propranolol may be exerted via attenuating Na+ influx into cardiac cells followed by prevention of the mitochondrial dysfunction in the ischaemic heart, leading to improvement of energy production of the heart during reperfusion.
Keywords: Contractile function, heart, ischaemia/reperfusion, mitochondria, sodium overload
Introduction
Propranolol has not only β-adrenoceptor blocking action (Nordenfelt, 1965; Luria et al., 1966) but also has other pharmacological effects such as a Na+ channel blocking action (Doggrell, 1988) and an antioxidant effect (Freedman et al., 1991; Kramer et al., 1991). This agent is known to protect the myocardium against ischaemia/reperfusion- or hypoxia/reoxygenation-induced injury in in vivo and in vitro experimental animals (Ichihara & Abiko, 1983; Fujioka et al., 1991; Kramer et al., 1991; Kirshnan & Antzelevitch, 1991; Liu et al., 1991; Okayama et al., 1993). Several studies suggested that the mechanisms underlying the cardioprotective effect against ischaemia/reperfusion and hypoxia/reoxygenation injuries may be exerted via an energy-sparing effect (Ichihara & Abiko, 1983), antioxidant effect (Freedman et al., 1991), preservation of membrane phospholipids (Liu et al., 1991), and/or attenuation of an increase in non-esterized fatty acids (Nakamura et al., 1989). However, the exact mechanism for the cardioprotective effects of propranolol against ischaemia/reperfusion injury has not been fully established.
Recently, we suggested that an attenuation of Na+ accumulation in the ischaemic myocardium may lead to improvement of post-ischaemic recovery of contractile function of perfused hearts (Takeo et al., 1995; Tanonaka et al., 1999). Furthermore, we also reported that Na+ channel blockade by tetrodotoxin and inhibition of the Na+/H+ exchanger by dimethyl amiloride attenuated sodium overload in the ischaemic myocardium and thereby enhanced post-ischaemic recovery of contractile function of perfused rat hearts (Tanonaka et al., 2000).
In addition, we showed in a previous study the close relationship between post-ischaemic contractile recovery and mitochondrial activity in the perfused heart (Yabe et al., 2000). Mitochondria are primary intracellular organelles to produce energy that is necessary for cardiac contraction. Therefore, protection of the mitochondria against ischaemia/reperfusion-induced cellular deterioration may be, at least, one of the critical mechanisms responsible for cardioprotection in ischaemic/reperfused hearts. In the present study, we examined the effect of treatment with propranolol on the mitochondrial function in the ischaemic and ischaemic/reperfused hearts.
Methods
Animals
Male Wistar rats (Japan Laboratory Animals Inc, Tokyo, Japan), weighing 230 – 270 g, were used in the present study. The animals were conditioned at 23±1°C with a constant humidity of 55±5%, a cycle of 12-h light and 12-h darkness, and were given free access to food and tap water according to the Guide for Care and Use of Laboratory Animals as promulgated by the National Research Council. The protocol of this study was approved by the Committee of Animal Use and Welfare of Tokyo University of Pharmacy and Life Science.
Perfusion of hearts
Perfusion of rat hearts was performed by the method described previously (Takeo et al., 1995). In brief, the rats were anaesthetized with diethyl ether. After thoracotomy, the hearts were rapidly isolated and perfused at 37°C in a Langendorff manner with a constant flow rate at 9 ml min−1 of Krebs-Henseleit bicarbonate buffer of the following composition (mM): NaCl 120; KCl 4.8; KH2PO4 1.2; MgSO4 1.2; CaCl2 1.25; NaHCO3 25; and glucose 11. The perfusion buffer was equilibrated with a gas mixture of 95% O2 and 5% CO2 to pH 7.4. A latex balloon with an uninflated diameter of 3.7 mm, connected to a pressure transducer (TP-200, Nihonkohden, Tokyo, Japan), was inserted into the left ventricular cavity through the mitral opening. A 5-mmHg of the initial ventricular end-diastolic pressure (LVEDP) was loaded onto the perfused heart. Left ventricular developed pressure (LVDP), a convenient maker of cardiac contractile function, was monitored by a pressure transducer (TP-200T, Nihonkohden) throughout the experiment. Heart rate (HR) was measured by means of a heart rate counter (AP-601G, Nihonkohden). These haemodynamic parameters were recorded on a thermal pen recorder (WT-645G, Nihonkohden).
Ischaemia/reperfusion
After 15-min equilibration, the heart was paced at 300 beats min−1 with an electrical stimulator (SEN-7023, Nihonkohden) via two silver electrodes attached to the right atrium and an additional 15 min of perfusion was carried out. Then the perfusion was stopped (ischaemia). The heart was immediately submerged in an organ bath filled with the Krebs-Henseleit bicarbonate buffer in which the 11 mM glucose had been replaced with 11 mM Tris/HCl. This buffer had been previously equilibrated with a gas mixture of 95% N2 and 5% CO2 (pH 7.4) and maintained at 37°C during the experiment to avoid hypothermia-induced cardioprotection. After 35 min of ischaemia, the buffer in the organ bath was drained, and the hearts were reperfused for 60 min at 37°C with the Krebs-Henseleit bicarbonate buffer equilibrated with a gas mixture 95% O2 and 5% CO2. The hearts were paced throughout the experiment except for the first 15 min of reperfusion, to prevent contractile irregularities, which might sometimes occur during this period. For the purpose of comparison, hearts were perfused for 95 min under normoxic conditions (normoxic group).
Treatment with propranolol
Treatment of the perfused hearts with different concentrations of propranolol (Nacalai Tesque, Kyoto, Japan) ranging from 10 to 100 μM was carried out by perfusing the hearts with the agent in Krebs-Henseleit bicarbonate buffer for the final 3 min of pre-ischaemia. The agent was dissolved and diluted in the Krebs-Henseleit bicarbonate buffer.
Examination of perfusate
The perfusate eluted from the heart during reperfusion was collected. The creatine kinase (CK) activity of the perfusate was determined by the method of Bergmeyer et al. (1970). The perfusate was also used for determination of purine nucleosides and bases (ATP metabolites), such as adenosine, inosine, and hypoxanthine, by the HPLC method described previously (Takeo et al., 1988).
Myocardial Na+ and Ca2+ contents
The myocardial Na+ and Ca2+ contents of the ischaemic or reperfused heart were determined to assess ionic disturbances in the heart, as described previously (Tanonaka et al., 1999). After ischaemia, reperfusion, or normoxic perfusion, hearts were perfused with 8.0 ml of 320 mM sucrose-20 mM Tris/HCl, pH 7.4. Their wet weights were weighed and then the hearts were dried at 120°C for 48 h. The dried tissues were digested with 60% HNO3 and evaporated at 180°C. The residues were dissolved in 0.75N HNO3. Myocardial cation concentrations were determined by means of an atomic absorption spectrometer (AA-680, Shimazdu, Kyoto).
In a previous study, we extensively characterized this method (Tanonaka et al., 1999). We found that perfusion of the isolated heart with approximately 8 ml of sucrose buffer was necessary to eliminate various cations from the vascular and extracellular spaces. Values for the myocardial cations after the perfusion with 8 ml of the sucrose buffer were quite similar to those determined by the Co2+-EDTA method based on the theory that vascular and extracellular spaces are equilibrated with 1 mM Co2+-EDTA and the Krebs-Henseleit solution preperfused. Accordingly, it is suggested that cations determined by this method are originated from the intracellular milieu and various organelles in cardiac cells.
Determination of myocardial ATP and creatine phosphate
After appropriate sequences of perfusion, the hearts were freeze-clamped with aluminum tongs pre-cooled with liquid nitrogen to determine myocardial ATP and creatine phosphate contents as described previously (Iwai et al., 2000). The frozen ventricle was pulverized and mixed with 0.3 N HClO4 and 0.25 mM EDTA under liquid nitrogen cooling. The extract was centrifuged at 8000×g for 15 min at 4°C, and the resulting supernatant was sampled for determination of myocardial ATP and creatine phosphate (CP) by the HPLC method described previously (Iwai et al., 2000). Myocardial CP was converted to ATP by the enzymatic method of Lowry & Passonneau (1972).
Mitochondrial oxygen consumption rate of skinned bundles
The mitochondrial oxygen consumption rate was determined by the method of Sanbe et al. (1993). After appropriate sequences of perfusion, the untreated and propranolol-treated hearts were quickly removed from the perfusion apparatus. Myocardial bundles, 0.3 to 0.4 mm in diameter and 3 to 4 mm in length, were prepared from the left ventricular free wall by use of a McIlwain Tissue Chopper (Mickle Lab. Engineering Co., NY, U.S.A.) and transferred into relaxing medium A of the following composition (mM): EGTA, 10; MgSO4, 3; taurine, 20; dithiothreitol, 0.5; imidazole, 20; potassium 2-(N-morpholino)-ethanesulphonate, 160; ATP, 5; CP, 15 (pH 7.0). The bundles were incubated for 20 min in 1 ml of medium A containing 75 μg ml−1 saponin. After incubation, the bundles (skinned bundles) were washed for 10 min in fresh medium B (medium A without ATP and CP but supplemented with 0.5% bovine serum albumin; BSA) to remove the saponin. All procedures were carried out at 4°C. The oxygen consumption rate of skinned bundles was determined in 1.0 ml of medium B at 30°C by means of a Clark-type electrode connected to an Oxygraph (Central Kagaku, Tokyo). The basal oxygen consumption rate was measured following the addition of glutamate 5 mM, malate 3 mM and KH2PO4 3 mM. The maximal oxygen consumption rate was measured by addition of 1 mM ADP and 7.5 mM creatine. The velocity of ADP-stimulated oxygen consumption rate (OCR) of skinned bundles was taken as the difference between the maximal and basal glutamate/malate-stimulated oxygen consumption rates. After determination of the oxygen consumption rate, the skinned bundles were solubilized with 2 N NaOH for 30 min at 60°C, and then the protein concentration was determined according to the method of Lowry et al. (1951). The mitochondrial oxygen consumption rate was expressed as nano-atoms of oxygen consumed per min per mg protein.
Release of cytochrome c from mitochondria in ischaemic hearts
At the end of ischaemia, untreated and agent-treated hearts were quickly removed from the perfusion apparatus. Tissue was mildly homogenized in an ice-cold buffer containing (mM) mannitol 210, sucrose 70, EDTA 1, DTT 10, 2 μg ml−1 leupeptin, 2.5 μg ml−1 aprotonin, and 0.5 mM PMSF (pH 7.4). The homogenate was centrifuged at 900×g for 10 min at 2°C. The supernatant was centrifuged at 8000×g for 30 min at 2°C. The resulting supernatant solution was centrifuged at 100,000×g for 30 min at 2°C to remove any mitochondrial contamination. The cytosolic fraction was denatured in Laemmli buffer (Tris-HCl 250 mM, SDS 4%, glycerol 10%, bromophenol blue 0.006%, β-mercaptoethanol 2%, pH 6.8) at 100°C and fractionated by SDS electrophoresis on a 15% polyacrylamide gel (Laemmli, 1970). The fractionated proteins were then transferred onto a nitrocellulose filter, which was confirmed with a monoclonal anti-cytochrome c antibody (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A), followed by horseradish peroxidase-conjugated donkey anti-mouse IgG (Amersham Pharmacia Biotech, Buckinghamshire, U.K.). Bound antibody was detected by the chemiluminescence method using ECL reagents (Amersham Pharmacia Biotech). The concentration of cytochrome c in the sample was estimated by the densitometric method by comparing with the bands of the samples with the reference obtained with the standard solution of horse heart cytochrome c (1.25 ng).
Isolation of mitochondria
Cardiac mitochondria were prepared as described previously (Takeo et al., 1991). In brief, male rats were anaesthetized with diethyl ether, and their hearts were isolated. Heart tissue was homogenized in ice-cold buffer containing KCl 160 mM, EGTA (pH 7.4) 10 mM, and 0.5% fatty acid-free BSA. The homogenate was centrifuged at 1000×g for 10 min at 2°C, and the supernatant solution was centrifuged at 8000×g for 10 min at 2°C. The crude mitochondria were again suspended in buffer and centrifuged at 8000×g for 10 min at 2°C. Mitochondria were resuspended with suspension buffer (sucrose 320 mM, Tris/HCl 10 mM, pH 7.4) and used for measurement of mitochondrial activity. Protein concentrations were determined by the method of Lowry et al. (1951).
Mitochondrial respiration
Mitochondrial respiration activity was determined by the method described previously (Takeo et al., 1991). Mitochondria (approximately 1 mg of protein) were placed into 1 ml of incubation medium containing sucrose 250 mM, K2HPO4 10 mM, and glutamate 10 mM, pH 7.4. Mitochondrial oxidative phosphorylation was measured at 30°C in the chamber using a Clark-type oxygen electrode connected to an Oxygraph (Central Kagaku, Tokyo). The quality of the mitochondrial preparation was determined by using the respiratory control index calculated as the ratio of the rate of state 3 respiration to that of state 4 respiration. Mitochondria with a respiratory control index greater than 10 were used in the present study. The effects of sodium compounds, such as sodium chloride and sodium lactate, on state 3 respiration were assessed in the presence and absence of various concentrations (10 to 100 μM) of propranolol.
Statistics
Each value represents the mean±s.e.mean. Statistical analyses were performed with the aid of StatView® for Windows (SAS Institute Inc., Tokyo). Statistical significance was evaluated by one-way analysis of variance (ANOVA) followed by Bonferroni's or Dunnett's multiple comparisons if necessary. Differences with a probability of 5% or less were considered to be statistically significant.
Results
Contractile function of perfused hearts
The time course of changes in LVDP and LVEDP of ischaemic/reperfused hearts treated with 10, 30, or 100 μM propranolol is shown in Figure 1. Changes in LVDP were expressed as percentages of the initial values (the upper panel in Figure 1). The baseline (initial) values for LVDP of agent-untreated and treated groups ranged from 77.8±5.5 mmHg to 82.1±6.1 mmHg (n=5 for each group). Propranolol at 10, 30, or 100 μM decreased LVDP at the end of the agent-treatment in a concentration-dependent manner. After the onset of ischaemia, LVDP declined to zero within 2.5 min, and it remained at that value during ischaemia. The LVDP of the heart recovered to approximately 15 mmHg by the end of 60 min reperfusion. In contrast, LVDPs at the end of reperfusion of the hearts treated with 10, 30, and 100 μM propranolol were significantly recovered in a concentration-dependent manner.
Figure 1.
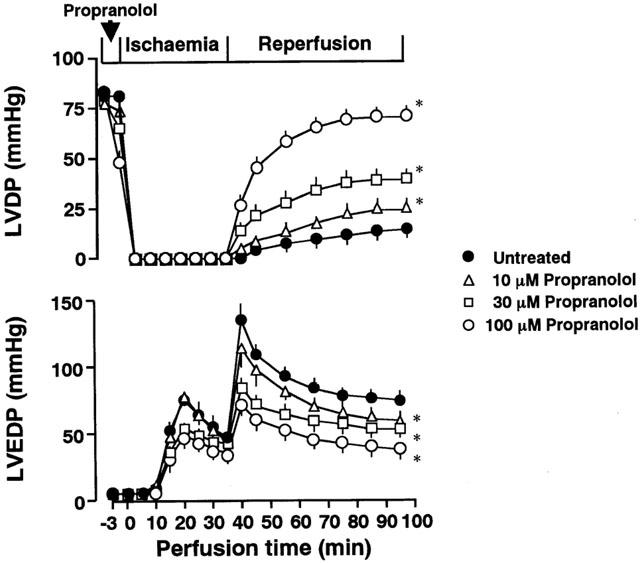
The time course of changes in left ventricular developed pressure (LVDP; the upper panel) and left ventricular end-diastolic pressure (LVEDP; the lower panel) of the ischaemic/reperfused untreated heart and the heart treated with 10, 30, or 100 μM propranolol. Each value represents the mean±s.e.mean of five experiments. Treatment with propranolol was conducted for the final 3 min of pre-ischaemia. *Significantly different from the corresponding untreated ischaemic/reperfused group (P<0.05).
The LVEDP of the untreated heart began to rise at 5 min after the onset of ischaemia and reached its peak level approximately 20 min after ischaemia (the lower panel in Figure 1). The LVEDP further increased upon reperfusion and reached the maximum level at 5 min after the onset of reperfusion. Although the LVEDP gradually declined during reperfusion, this high level of LVEDP was sustained throughout reperfusion. In contrast, treatment with various concentrations of propranolol attenuated the rise in LVEDP during reperfusion in a concentration-dependent manner, but not during ischaemia.
When perfused hearts were treated with 30 or 100 μM propranolol only during reperfusion, the recovery of LVDP was not enhanced at the end of the reperfusion (20.2±2.7 and 19.1±2.3 mmHg for 30 and 100 μM propranolol-treated heart, respectively, n=3 each).
Release of CK and ATP metabolites from reperfused hearts
To determine the amounts of CK and ATP metabolites released from perfused hearts, we collected the perfusate of the heart (Figure 2). During 30 min of pre-ischaemic perfusion or normoxia, CK activity in the perfusate was approximately 1 nmol NADPH min−1 g wet tissue−1 regardless of treatment with or without propranolol (n=5 each). CK activity in the perfusate from the untreated heart markedly increased during reperfusion (approximately 100 fold the value for the normoxic group). Treatment with propranolol attenuated the increase in CK release from reperfused hearts in a concentration-dependent manner (the left panel in Figure 2).
Figure 2.
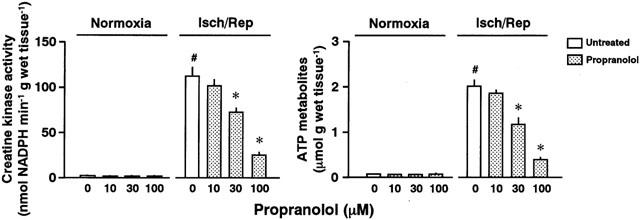
Creatine kinase activity (the left panel) and the amount of purines and bases (ATP metabolites; the right panel) in the perfusate eluted from normoxic (Normoxia) or ischaemic/reperfused hearts (Isch/Rep) untreated or treated with 10, 30, or 100 μM propranolol. Each value represents the mean±s.e.mean of five experiments. *Significantly different from the untreated group (0 μM propranolol; P<0.05). #Significantly different from the normoxic heart of the untreated ischaemic/reperfused group (P<0.05).
The ATP metabolites were minimally released from the normoxic heart as well as the propranolol-treated, normoxic heart (less than 0.1 μmol g wet tissue−1). Ischaemia/reperfusion induced a marked release of the ATP metabolites (the right panel in Figure 2). Treatment with propranolol attenuated the increase in the release of ATP metabolites (right panel in Figure 2).
Myocardial Na+ and Ca2+ contents
Myocardial Na+ and Ca2+ contents were determined at the end of pre-ischaemia, ischaemia, or reperfusion (Figure 3). Baseline values for myocardial Na+ and Ca2+ contents were 55.9±3.3 and 1.87±0.05 μmol g dry tissue−1, respectively (n=5). Changes in the Na+ or Ca2+ contents of the ischaemic hearts treated with and without different concentrations of propranolol are shown in Figure 3. The myocardial Na+ content increased to approximately double the baseline value at the end of ischaemia (the upper panel in Figure 3). In contrast, the myocardial Ca2+ content did not change under ischaemic conditions. There were no changes in Ca2+ content at the end of ischaemia regardless of the treatment with or without the agent (the lower panel in Figure 3).
Figure 3.
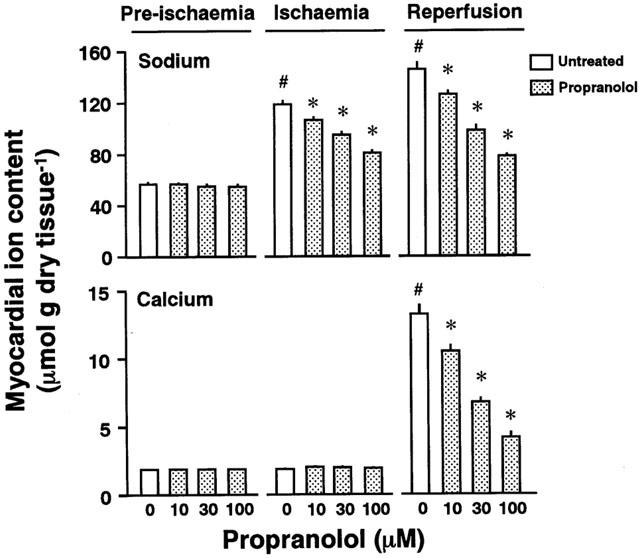
Na+ (the upper panel) and Ca2+ contents (the lower panel) of perfused hearts untreated or treated with 10, 30, or 100 μM propranolol at the ends of pre-ischaemia, ischaemia, and reperfusion, respectively. Each value represents the mean±s.e.mean of five experiments. *Significantly different from the untreated group (0 μM propranolol; P<0.05). #Significantly different from the normoxic heart of the untreated ischaemic/reperfused group (P<0.05).
Myocardial Na+ and Ca2+ contents at the end of 60 min reperfusion are shown in Figure 3. When the heart was subjected to 35 min ischaemia and then reperfused, a further increase in myocardial Na+ content was observed during reperfusion (the upper panel in Figure 3). This increase in myocardial Na+ content was attenuated by pre-treatment with propranolol in a concentration-dependent manner. The myocardial Ca2+ content of the ischaemic/reperfused heart also increased (the lower panel in Figure 3). The reperfusion-induced increase in myocardial Ca2+ content was attenuated by treatment with propranolol.
Myocardial ATP and CP contents
Figure 4 shows ATP and CP contents of the hearts treated with and without propranolol. The myocardial ATP and CP contents at the end of pre-ischaemia in the absence of propranolol were 24.74±0.73 and 34.61±1.53 μmol g dry tissue−1, respectively (n=5). The ATP and CP contents at the end of pre-ischaemia were not altered by treatment with various concentrations of propranolol. The myocardial ATP and CP contents at 95 min of normoxia were similar to those at the end of pre-ischaemia. The myocardial ATP and CP contents at the end of ischaemia were approximately 3 and 10% of the pre-ischaemic values, respectively. After reperfusion, the myocardial ATP and CP contents were restored to approximately 30 and 25% of the pre-ischaemic values, respectively (Reperfusion sections in Figure 4).
Figure 4.
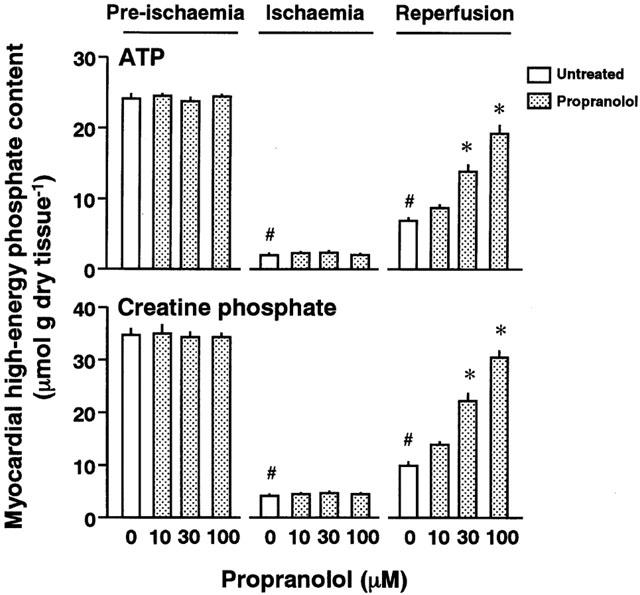
ATP (the upper panel) and creatine phosphate (CP) contents (the lower panel) of perfused hearts untreated or treated with 10, 30, or 100 μM propranolol at the ends of pre-ischaemia, ischaemia, and reperfusion, respectively. Each value represents the mean±s.e.mean of five experiments. *Significantly different from the untreated group (0 μM propranolol; P<0.05). #Significantly different from the normoxic heart of the untreated ischaemic/reperfused group (P<0.05).
When hearts were pre-treated with different concentrations of propranolol, the ATP and CP contents of the reperfused heart were restored in a concentration-dependent manner.
Mitochondrial oxygen consumption rate
Figure 5 shows the ADP-stimulated mitochondrial oxygen consumption rate (OCR) of the left ventricular skinned bundles of the propranolol-treated heart at the end of pre-ischaemia, ischaemia, or reperfusion. The OCR for pre-ischaemic hearts was 61.9±3.2 nano-atom O min−1 mg protein−1 (n=4). There were no significant differences in the OCR of perfused hearts under normoxic conditions regardless of treatment with or without propranolol. The OCR of the untreated heart under ischaemic conditions was significantly decreased to approximately 35% of the value for the normoxic heart (n=4) (Figure 5). The OCR of the reperfused heart was further decreased to approximately 25% of the value for the normoxic heart (n=5) (Figure 5). In contrast, treatment with 30 or 100 μM propranolol preserved the OCR at the ends of both ischaemia and reperfusion (n=4 each).
Figure 5.
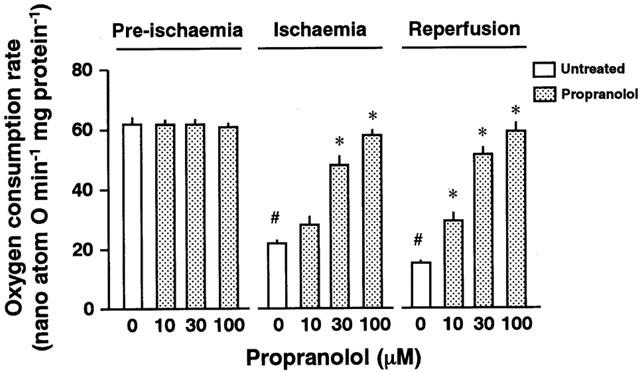
Mitochondrial oxygen consumption rate of the left ventricular skinned bundles prepared from perfused hearts untreated (0 μM) or pretreated with 10, 30, or 100 μM propranolol at the ends of pre-ischaemia, ischaemia, and reperfusion, respectively. Each value represents the mean±s.e.mean of five experiments. #Significantly different from the corresponding untreated ischaemic or ischaemic/reperfused group (P<0.05). *Significantly different from the untreated group (P<0.05).
Release of cytochrome c from mitochondria in the ischaemic heart
To test whether cytochrome c might have been released during ischaemia from the mitochondria in the perfused heart, we prepared the cytosolic fraction from the perfused heart after 35 min ischaemia and used it for Western blot analysis of cytochrome c (n=5 each). The amount of cytochrome c released from the mitochondria in the heart that was perfused under normoxic conditions was very low (the left panel in Figure 6). A marked increase in cytochrome c in the cytosolic fraction from mitochondria was seen at the end of ischaemia (the right panel in Figure 6). The increase in the release of cytochrome c was substantially inhibited by treatment with 100 μM propranolol.
Figure 6.
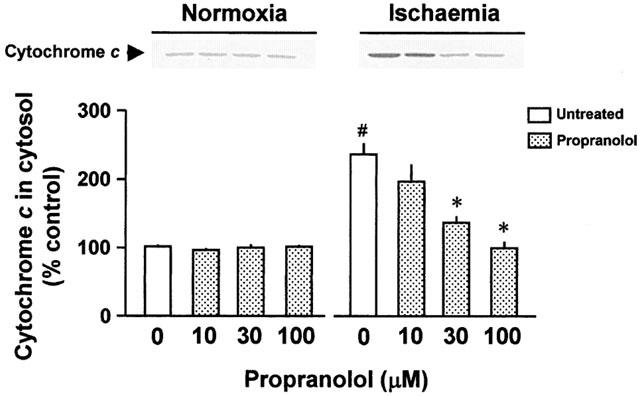
Representative Western blots of cytochrome c released from the mitochondria and effects of treatment without or with 10, 30, or 100 μM propranolol on the release of cytochrome c from the mitochondria into the cytosol in the normoxic (Normoxia) and ischaemic heart (Ischaemia), respectively. Values are expressed as the percentage of the heart without an exposure to ischaemia/reperfusion and propranolol treatment (the control heart). Each value represents the mean±s.e.mean of five experiments. #Significantly different from the normoxic, untreated group (P<0.05). *Significantly different from the corresponding untreated normoxic or ischaemic group (P<0.05).
Effect of propranolol on mitochondrial respiration in the presence of Na+
To examine the effects of Na+ on the mitochondrial ATP-generating ability, the mitochondrial state 3 respiration of normal hearts was determined in vitro in the presence and absence of different concentrations of sodium chloride or sodium lactate (the left panel in Figure 7). The state 3 respiration was significantly decreased along with the increased concentrations of sodium chloride in the incubation medium. When isolated mitochondria were incubated with sodium lactate, a possible metabolite in ischaemic hearts, the degree of decrease in state 3 respiration was greater than that of mitochondria incubated with sodium chloride. The increasing concentrations of sodium lactate deteriorated these parameters for the mitochondrial respiration.
Figure 7.
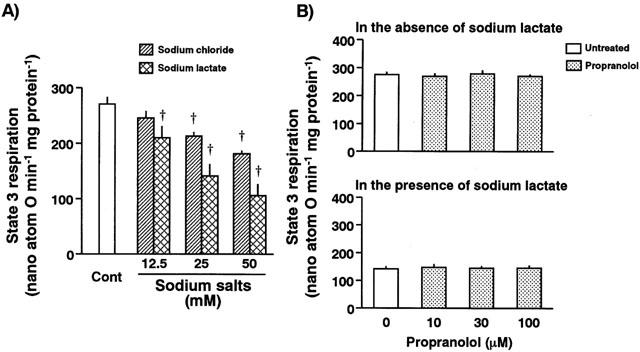
The left panel represents effects of sodium chloride and sodium lactate on the mitochondrial state 3 respiration. The mitochondrial respiration decreased in the presence of sodium chloride or sodium lactate in a concentration-dependent manner, respectively. Each value represents the mean±s.e.mean of five measurements. †Significantly different from the control group (Cont; P<0.05). The right panel represents the effect of propranolol on mitochondrial state 3 respiration in the absence (upper panel) and presence of 25 mM sodium lactate (lower panel). Treatment of isolated mitochondria with various concentrations (10 to 100 μM) of propranolol did not change the basal state 3 respiration. Sodium lactate-induced deterioration of mitochondrial activity was also unaffected in the presence of various concentrations of propranolol. Each value represents the mean±s.e.mean of five measurements.
To examine a direct effect of propranolol on the mitochondrial respiration, the mitochondrial respiration with or without Na+ was determined in the presence of propranolol. Propranolol at the concentration of 10, 30, or 100 μM altered neither the basal state 3 respiration nor the sodium lactate-induced decrease in the state 3 respiration (right panel in Figure 7).
Discussion
Benefit of propranolol to ischaemia/reperfusion injury
In the present study, we showed that treatment with propranolol enhanced the post-ischaemic recovery of the contractile function and the suppression of LVEDP of the ischaemic/reperfused heart in a concentration-dependent manner. Furthermore, the release of CK, a marker of cell necrosis, was concentration-dependently blocked by treatment with propranolol. These findings suggest that propranolol has a cardioprotective action on the ischaemic/reperfused rat heart, which is consistent with the previous findings by other investigators with respect to functional and morphological improvement (Edoute et al., 1982; Nakamura et al., 1989; Freedman et al., 1991; Okayama et al., 1993).
Although ischaemia/reperfusion injury was suggested to occur during an early phase of reperfusion, we observed in previous studies that amelioration of post-ischaemic contractile function by agents always required treatment prior to ischaemia (Takeo et al., 1995; Tanonaka et al., 1999) and also observed that post-ischaemic recovery of cardiac contractile function was always associated with restoration of myocardial energy levels during reperfusion in perfused rat (Iwai et al., 2000; Yabe et al., 2000) and rabbit hearts (Takeo et al., 1988). Thus, we focused, in the present study, on myocardial energy-producing events that may occur during ischaemia.
Energy-sparing effect
Several studies have shown the possibility that propranolol may protect the heart from ischaemia/reperfusion injury through the mechanism underlying negative inotropic effects during and after pre-treatment with the agent and the following energy-sparing effect during ischaemia: HEPs preserved during ischaemia, even if any, might be beneficial for the recovery of cardiac contraction during reperfusion (Ichihara & Abiko, 1983; Nayler et al., 1985; Okayama et al., 1993). To elucidate this possibility under the present experimental conditions, we examined the negative inotropic effects on the perfused heart during administration of propranolol and myocardial energy levels prior to reperfusion. Indeed, a negative inotropic effect was seen in the propranolol-treated heart in a dose-dependent manner during the administration of this agent. However, no higher levels of myocardial HEPs were seen in the propranolol-treated hearts prior to ischaemia (at the end of the administration). In addition, there were no significant differences in myocardial HEPs at the end of ischaemia between the heart treated with propranolol and the untreated heart. These findings on the myocardial energy levels suggest that the energy-sparing effect would unlikely occur in the ischaemic/reperfused heart under the present experimental conditions.
Suppression of sodium overload during ischaemia
Propranolol has a membrane stabilizing action (Doggrell, 1988) or Na+ channel blocking action (Pollen et al., 1969). The exact role of this action in the protection of the heart against ischaemia/reperfusion injury remains elusive. Fujioka et al. (1991) reported that dl-propranolol and d-propranolol exerted cardioprotective effects on the hypoxic/reoxygenated heart. Since d-propranolol does not have a β-adrenoceptor blocking action but has a membrane stabilizing action or Na+ channel blocking action, this finding suggests that the membrane stabilizing action or Na+ channel blockade may play an important role in the cardioprotection against ischaemia/reperfusion or hypoxia/reoxygenation injury. It is well established that calcium overload in cardiac cells may play a key role in the ischaemia/reperfusion injury. Recently, accumulating evidence suggests that sodium overload may play a significant role in the cardioprotection of the ischaemic/reperfused heart (Karmazyn, 1991; Meng & Pierce, 1991; Takeo et al., 1995; Tanonaka et al., 1999; 2000). We examined changes in tissue ion content of the ischaemic or ischaemic/reperfused heart in the present study to elucidate ionic disturbance of the ischaemic/reperfused heart. The method for determination of tissue cations was based on detection of the absolute amount of tissue cations that were not present in the extracellular and vascular spaces, that is, cations that were present in the intracellular space and cellular organelles in cardiac cells. We observed a marked increase in Na+ content during ischaemia and during reperfusion, whereas Ca2+ content did not alter during ischaemia but increased during reperfusion. This suggests that ionic disturbance during ischaemia/reperfusion is initiated by cytosolic sodium overload during ischaemia and that at least massive accumulation of Ca2+ may not occur in cardiac cells during ischaemia. Propranolol prevented the ischaemia-induced increase in the Na+ content and reperfusion-induced increases in the Na+ and Ca2+ contents, but the agent did not affect the Ca2+ content during ischaemia. This suggests that prevention of cytosolic sodium overload during ischaemia may play a critical role in the cardioprotection of the perfused rat heart and that propranolol elicits this prevention probably through suppression of Na+ influx.
Recently, we examined the cardioprotective effect of TTX against ischaemia/reperfusion injury and found that TTX improved post-ischaemic contractile function, prevented Na+ accumulation in the ischaemic heart, preserved the mitochondrial function during ischaemia, attenuated the release of cytochrome c in the perfused heart and suppressed Na+-induced damage to isolated mitochondria in vitro as similar to the results in the preset study (Iwai et al., 2002). This finding also suggested the above idea concerning the role of the prevention of sodium overload during ischaemia.
Route of Na+ influx during ischaemia
There are several pathways for Na+ entry into cardiac cells, including fast Na+ channels, Na+/H+ exchanger, and Na+/Ca2+ exchanger in ischaemic/reperfused hearts. Recently, several studies have shown the presence of a persistent and inactivation-resistant Na+ current that is evoked by depolarization in cardiac cells (Silverman & Stern, 1994; Ju et al., 1996). This current is blocked by TTX or lidocaine and may be entirely different from the physiological fast Na+ current. Since the inactivation-resistant Na+ current opens when the cell is hypoxic, the contribution of this Na+ current to the observed Na+ accumulation during ischaemia is likely. The question arises as to which Na+ influx routes are affected by treatment with propranolol. So far as we know, there is no report that propranolol affects Na+/H+ exchanger, whereas several reports have shown that propranolol affected Na+ channels under normal conditions (Doggrell, 1988). Thus, propranolol-induced inhibition of Na+ accumulation appears to be attributable to the blockade of Na+ channels during ischaemia. A study on the effect of propranolol on the inactivation-activated Na+ current in hypoxia or ischaemia would be required to elucidate the exact route in the mechanism.
Preservation of mitochondrial function during ischaemia
Questions arise concerning the role of cytosolic sodium overload in the cardioprotection against ischaemia/reperfusion injury. In this context, we focused on three mitochondrial events, mitochondrial respiration, myocardial OCR, and release of cytochrome c from the heart. At first, we observed that excess Na+, in particular a possible metabolite in ischaemia sodium lactate, inhibited the mitochondrial state 3 respiration in vitro in a concentration-dependent manner. This depression in the activity was not prevented by the presence of propranolol at any concentrations ranging from 10 to 100 μM, suggesting that excessive Na+ may reduce the ATP-generating activity and that direct effects of propranolol on the mitochondria are unlikely. Secondly, propranolol prevented ischaemia- or reperfusion-induced decrease in the mitochondrial oxygen consumption rate of the skinned bundles prepared from the perfused heart, but no change in oxygen consumption rate was seen by the presence of propranolol in the skinned bundles prepared from pre-ischaemic hearts. These findings suggest that propranolol preserved the ability of the mitochondria to produce ATP during ischaemia and further confirmed that the direct effect of propranolol on the mitochondria is unlikely. Thirdly, in perfused rat hearts, the ischaemia induced a release of cytochrome c. A release of cytochrome c is considered to represent alterations in the mitochondrial membrane integrity, and in some cases this release is considered to be a sign of activation of apoptic cell death (Klohn et al., 1998; Ouyang et al., 1999). In either case, this release was concentration-dependently attenuated by treatment with propranolol. However, there was no significant release of cytochrome c from the normoxic hearts when treated with propranolol. This suggests that propranolol prevented ischaemia-induced mitochondrial membrane perturbation and also eliminated the possibility that propranolol directly affected the mitochondria. These mitochondrial events in vitro or in perfused hearts strongly suggest that the mitochondrial activity in perfused hearts is greatly influenced by the excessive cytosolic Na+ concentration and that the pharmacological benefit of propranolol to ischaemia/reperfusion injury may be attributed to suppression of ischaemia-induced Na+ influx during ischaemia.
Clinical implications
In the present study, we used propranolol at the concentrations of 10 – 100 μM to demonstrate the benefit of the agent in the prevention of cytosolic sodium overload during ischaemia. The concentration of propranolol may be relatively high as compared with its clinical dose (1 – 3 μM). Since the conditions that can induce the ischaemia/reperfusion injury in the present study is very severe, relatively high concentrations of the agent would be required to reverse the injury. A further examination in the therapeutic implications of the agent is awaited.
Conclusion
In conclusion, ischaemia induces cytosolic sodium overload and this may initiate ionic disturbances of the heart during ischaemia. Sodium overload during ischaemia may deteriorate the mitochondrial function to produce ATP. This deterioration is supported by the findings of the release of mitochondrial component and the reduction of mitochondrial respiration in the presence of Na+. Such deterioration of mitochondria may not produce HEPs appropriately during reperfusion. In addition to reperfusion-induced deleterious sequences such as calcium overload and free radical attack, this mitochondrial damage may play a significant role in the genesis of ischaemia/reperfusion injury in perfused rat hearts. The cardioprotective effect of propranolol may be attributed to prevention of cytosolic sodium overload during ischaemia.
Abbreviations
- BSA
bovine serum albumin
- CK
creatine kinase
- CP
creatine phosphate
- HR
heart rate
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end-diastolic pressure
- OCR
oxygen consumption rate
References
- BERGMEYER H.U., RICH W., BUTHER H., SCHMIDT E., HILLMAN G., KREU F.H., STAMM D., LANG H., SZASZ G., LAUE D. Standardization of methods for estimation of enzyme activity in biological fluids. Z. Klin. Chem. Biochem. 1970;8:658–669. [Google Scholar]
- DOGGRELL S.A. Simultaneous assessment of membrane-stabilizing and β-adrenoceptor blocking activity of drugs with the rat isolated left atria. J. Pharmacol. Methods. 1988;19:93–107. doi: 10.1016/0160-5402(88)90030-7. [DOI] [PubMed] [Google Scholar]
- EDOUTE Y., GRANEY D., SANAN D., KOTZE J.C., LOCHNER A. Normothermic ischaemic cardiac arrest of isolated working rat heart: effects of reserpine and propranolol on functional, metabolic and morphological recovery. Cardiovasc. Res. 1982;16:428–438. doi: 10.1093/cvr/16.8.428. [DOI] [PubMed] [Google Scholar]
- FREEDMAN A.M., KRAMER J.H., CASSIDY M.M., WEGLICKI W.B. Propranolol preserves ultrastructure in adult cardiomyocytes exposed to anoxia/reoxygenation: a morphometric analysis. Free Radic. Biol. Med. 1991;11:197–206. doi: 10.1016/0891-5849(91)90172-y. [DOI] [PubMed] [Google Scholar]
- FUJIOKA H., YOSHIHARA S., TANAKA T., FUKUMOTO K., KUROIWA A., TANONAKA K., HAYASHI M., TAKEO S. Enhancement of post-hypoxic contractile and metabolic recovery of perfused rat hearts by dl-propranolol: Possible involvement of non-beta-receptor mediated activity. J. Mol. Cell. Cardiol. 1991;23:949–962. doi: 10.1016/0022-2828(91)90137-b. [DOI] [PubMed] [Google Scholar]
- ICHIHARA K., ABIKO Y. Effects of diltiazem and propranolol on irreversibility of ischemic cardiac function and metabolism in the isolated perfused rat heart. J. Cardiovasc. Pharmacol. 1983;5:745–751. doi: 10.1097/00005344-198309000-00007. [DOI] [PubMed] [Google Scholar]
- IWAI T., TANONAKA K., KOSHIMIZU M., TAKEO S. Presevation of mitochondrial function by diazoxide during sustained ischaemia in the rat heart. Br. J. Pharmacol. 2000;129:1219–1227. doi: 10.1038/sj.bjp.0703148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IWAI T., TANONAKA K., INOUE R., KASAHARA S., KAMO N., TAKEO S.Mitochondrial damage during ischemia determines post-ischemic contractile dysfunction in perfused rat hearts J. Mol Cell. Cardiol. 2002. in press [DOI] [PubMed]
- JU Y-K, , SAINT D.A., GAGE P.W. Hypoxia increases persistent sodium current in rat ventricular myocytes. J. Physiol. (London) 1996;497.2:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARMAZYN M. Amiloride enhances postischemic ventricular recovery: possible role of Na+/H+ exchange. Am. J. Physiol. 1991;255:H608–H615. doi: 10.1152/ajpheart.1988.255.3.H608. [DOI] [PubMed] [Google Scholar]
- KIRSHNAN S.C., ANTZELEVITCH C. Sodium channel block produces opposite electrophysiological effects in canine ventricular epicardium and endocardium. Circ. Res. 1991;69:277–291. doi: 10.1161/01.res.69.2.277. [DOI] [PubMed] [Google Scholar]
- KLOHN P.C., BITSCH A., NEUMANN H.G. Mitochondrial permeability transition is altered in early stages of carcinogenesis of 2-acetylaminofluorene. Carcinogenesis. 1998;19:1185–1190. doi: 10.1093/carcin/19.7.1185. [DOI] [PubMed] [Google Scholar]
- KRAMER J.H., MAK I.T., FREEDMAN A.M., WEGLICKI W.B. Propranolol reduces anoxia/reoxygenation-mediated injury of adult myocytes through an anti-radical mechanism. J. Mol. Cell. Cardiol. 1991;23:1231–1244. doi: 10.1016/0022-2828(91)90081-v. [DOI] [PubMed] [Google Scholar]
- LAEMMLI U.K. Cleavage of structural proteins during the assembly of the head of bacteriopharge T4. Nature. 1970;277:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LIU X.K., ENGELMAN R.M., AGRAWAL H.R., DAS D.K. Preservation of membrane phospholipids by propranolol, pindolol, and metoprolol: a novel mechanism of action of beta-blockers. J. Mol. Cell. Cardiol. 1991;23:1091–1100. doi: 10.1016/0022-2828(91)90199-v. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;261:6300–6306. [PubMed] [Google Scholar]
- LOWRY O.H., PASSONNEAU J.R.A collection of metabolite assays A Flexible System of Enzymatic Analysis 1972New York: Academic Press; 20–152.ed. Lowry, O.H., Passonneau, J.R. pp [Google Scholar]
- LURIA M.H., ADELSON E.I., MILLER A.J. Acute and chronic effects of an adrenergic beta-receptor blocking agent (propranolol) in treatment of cardiac arrhythmias. Circulation. 1966;34:767–773. doi: 10.1161/01.cir.34.5.767. [DOI] [PubMed] [Google Scholar]
- MENG H-.P., PIERCE G.N. Involvement of sodium in the protective effect of 5-(N,N-dimethyl)-amiloride on ischemia-reperfusion injury in isolated rat ventricular wall. J. Pharmacol. Exp. Ther. 1991;256:772–777. [PubMed] [Google Scholar]
- NAKAMURA K., ICHIHARA K., ABIKO Y. Effect of propranolol on accumulation of NEFA in the ischemic perfused rat heart. Eur. J. Pharmacol. 1989;24:61–69. doi: 10.1016/0014-2999(89)90654-7. [DOI] [PubMed] [Google Scholar]
- NAYLER W.G., GORDON M., STEPHENS D.J., STURROCK W.J. The protective effect of prazosin on ischaemic and reperfused myocardium. J. Mol. Cell. Cardiol. 1985;17:685–699. doi: 10.1016/s0022-2828(85)80068-7. [DOI] [PubMed] [Google Scholar]
- NORDENFELT O. Orthostatic ECG changes and the adrenergic beta-receptor blocking agent, propranolol (Inderal) Acta Med. Scand. 1965;178:393–401. doi: 10.1111/j.0954-6820.1965.tb04284.x. [DOI] [PubMed] [Google Scholar]
- OKAYAMA Y., KOBAYASHI A., FUJISE Y., YAMAZAKI N. Effects of propranolol and diltiazem on the rate of high-energy phosphate metabolism in reperfused rat hearts. 31P-NMR saturation transfer study. Jpn. Circ. J. 1993;57:521–532. doi: 10.1253/jcj.57.521. [DOI] [PubMed] [Google Scholar]
- OUYANG Y.B., TAN Y., COMB M., LIU C.L., MARTONE M.E., SIESJO B.K., HU B.R. Survival- and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cyochrome c and activation of caspase-like protease. J. Cereb. Blood Flow. Metab. 1999;19:1126–1135. doi: 10.1097/00004647-199910000-00009. [DOI] [PubMed] [Google Scholar]
- POLLEN D.W., SCOTT A.C., WALLACE W.F. A comparison of the direct effects and adrenergic blocking activity of D/L- and D-propranolol on the electrical and mechanical behavior of isolated frog ventricle. Cardiovasc. Res. 1969;3:7–13. doi: 10.1093/cvr/3.1.7. [DOI] [PubMed] [Google Scholar]
- SANBE A., TANONAKA K., HANAOKA Y., KATOH T., TAKEO S. Regional energy metabolism of failing hearts following myocardial infarction. J. Mol. Cell. Cardiol. 1993;25:995–1013. doi: 10.1006/jmcc.1993.1113. [DOI] [PubMed] [Google Scholar]
- SILVERMAN HS, STERN MD. Ionic basis of ischemic cardiac injury: insights from cellular studies. Cardiovasc. Res. 1994;28:581–597. doi: 10.1093/cvr/28.5.581. [DOI] [PubMed] [Google Scholar]
- TAKEO S., TANONAKA K., MIYAKE K., FUKUMOTO T. Role of ATP metabolites in induction of incomplete recovery of cardiac contractile force after hypoxia. Can. J. Cardiol. 1988;4:193–200. [PubMed] [Google Scholar]
- TAKEO S., TANONAKA R., TANONAKA K., MIYAKE K., HISAYAMA H., UEDA N., KAWAKAMI K., TSUMURA H., KATSUSHIKA S., TANIGUCHI Y. Alterations in cardiac function and subcellular membrae activities after hypervitaminosis D3. Mol. Cell. Biochem. 1991;107:169–183. doi: 10.1007/BF00225520. [DOI] [PubMed] [Google Scholar]
- TAKEO S., TANONAKA K., HAYASHI M., YAMAMOTO K., LIU J.-X., KAMIYAMA T., YAMAGUCHI N., MIURA A., NATSUKAWA T. A possible involvement of sodium channel blockade of class-I-type antiarrhythmic agents in postischemic contractile recovery of isolated, perfused hearts. J. Pharmacol. Exp. Ther. 1995;273:1403–1409. [PubMed] [Google Scholar]
- TANONAKA K., KAJIWARA H., KAMEDA H., TAKASAKI A., TAKEO S. Relationship between myocardial cation content and injury in reperfused rat hearts treated with cation channel blockers. Eur. J. Pharmacol. 1999;372:37–48. doi: 10.1016/s0014-2999(99)00172-7. [DOI] [PubMed] [Google Scholar]
- TANONAKA K., TAKASAKI A., KAJIWARA H., TAKEO S. Contribution of sodium channel and sodium/hydrogen exchanger to sodium accumulation in the ischemic myocardium. Gen. Pharmacol. 2000;34:167–174. doi: 10.1016/s0306-3623(00)00057-4. [DOI] [PubMed] [Google Scholar]
- YABE K., TANONAKA K., KOSHIMIZU M., KATSUNO T., TAKEO S. A role of PKC in the improvement of energy metabolism in preconditioned heart. Basic Res. Cardiol. 2000;95:215–227. doi: 10.1007/s003950050184. [DOI] [PubMed] [Google Scholar]