

Abstract
A deficiency of constitutive nitric oxide synthase (cNOS)-derived nitric oxide (NO), due to reduced availability of L-arginine, importantly contributes to allergen-induced airway hyperresponsiveness (AHR) after the early asthmatic reaction (EAR). Since cNOS and arginase use L-arginine as a common substrate, we hypothesized that increased arginase activity is involved in the allergen-induced NO deficiency and AHR.
Using a guinea-pig model of allergic asthma, we addressed this hypothesis by examining the effects of the specific arginase inhibitor Nω-hydroxy-nor-L-arginine (nor-NOHA) on the responsiveness to methacholine of isolated perfused tracheae from unchallenged control animals and from animals 6 h after ovalbumin challenge. Arginase activity in these preparations was investigated by measuring the conversion of L-[14C]arginine to [14C]urea.
Airways from allergen-challenged animals showed a 2 fold (P<0.001) increase in responsiveness to intraluminal (IL) administration of methacholine compared to controls. A similar hyperresponsiveness (1.8 fold, P<0.01) was observed in control airways incubated with the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME, 0.1 mM, IL), while L-NAME had no further effect on the airways from challenged animals.
Remarkably, 5 μM nor-NOHA (IL) normalized the hyperresponsiveness of challenged airways to basal control (P<0.001), and this effect was fully reversed again by 0.1 mM L-NAME (P<0.05). Moreover, arginase activity in homogenates of the hyperresponsive airways was 3.5 fold (P<0.001) enhanced compared to controls.
The results indicate that enhanced arginase activity contributes to allergen-induced deficiency of cNOS-derived NO and AHR after the EAR, presumably by competition with cNOS for the common substrate, L-arginine. This is the first demonstration that arginase is involved in the pathophysiology of asthma.
Keywords: Arginase, constitutive nitric oxide synthase, nitric oxide, methacholine, Nω-hydroxy-nor-L-arginine, allergic asthma, early asthmatic reaction, airway hyperresponsiveness, tracheal perfusion, guinea-pig
Introduction
Characteristic features of allergic asthma are allergen-induced early (EAR) and late (LAR) asthmatic reactions (Booij-Noord et al., 1971), infiltration and activation of inflammatory cells in the airways (De monchy et al., 1985) and airway hyperresponsiveness (AHR), both after the EAR and LAR (Durham et al., 1988). The mechanisms behind these pathological features are complex and incompletely understood, involving a variety of neurotransmitters, mediators and other signalling molecules, including nitric oxide (NO).
NO is synthesized from the semi-essential amino acid L-arginine both by constitutive (cNOS) and inducible (iNOS) NO synthase isoforms (Barnes & Belvisi, 1993; Barnes, 1998). In the airways, cNOS is mainly expressed in inhibitory nonadrenergic noncholinergic (iNANC) nerves (neuronal NOS or nNOS), endothelial cells (endothelial NOS or eNOS), and airway epithelium (nNOS and eNOS) (Tucker et al., 1990; Fischer et al., 1993; Kobzik et al., 1993; Asano et al., 1994), and is primarily involved in the regulation of airway and vascular tone by the local production of small amounts of NO in response to neurogenic and non-neurogenic stimuli (Barnes & Belvisi, 1993). The involvement of endogenous cNOS-derived NO in the regulation of airway tone is indicated by observations that nonselective NOS inhibitors such as Nω-nitro-L-arginine methyl ester (L-NAME) enhanced contractile agonist-induced airway constriction in vitro (Nijkamp et al., 1993; De boer et al., 1996) and in vivo (Nijkamp et al., 1993; Ricciardolo et al., 1996; Schuiling et al., 1998b; Taylor et al., 1998).
iNOS, producing much larger amounts of NO than cNOS, may be induced by pro-inflammatory cytokines during airway inflammation, particularly in inflammatory and epithelial cells (Hamid et al., 1993; Asano et al., 1994; Barnes, 1998), and may be involved in mucosal swelling (Kuo et al., 1992), infiltration of inflammatory cells (Schuiling et al., 1998a) and epithelial damage (Flak & Goldman, 1996; Schuiling et al., 1998a). However, iNOS-derived NO may also have a beneficial bronchodilating action (de gouw et al., 1998; Schuiling et al., 1998a), indicating its dualistic action in the airways.
Several studies in animal models (de boer et al., 1996; 1999; Mehta et al., 1997; Miura et al., 1997; Schuiling et al., 1998a, 1998b; Samb et al., 2001) and in asthmatic patients (Ricciardolo et al., 1997; 2001; Silkoff et al., 2000) have indicated that a deficiency of cNOS-derived NO may be importantly involved in the pathophysiology of allergic asthma. Using a guinea-pig model of allergic asthma, characterized by allergen-induced EAR and LAR, we have established both ex vivo (de boer et al., 1996) and in vivo (Schuiling et al., 1998a, 1998b) that a deficiency of bronchodilating cNOS-derived NO contributes to the AHR observed after the EAR, and that this deficiency is due to limitation of L-arginine as substrate for cNOS (de boer et al., 1999).
Different mechanisms could account for this limitation. One mechanism involves inhibition of cellular uptake of L-arginine by specific cationic amino acid transporters induced by polycationic proteins, including eosinophil-derived major basic protein (Hammermann et al., 1999; Meurs et al., 1999). A second mechanism could be a reduced bioavailability of L-arginine due to enhanced activity of arginase, which hydrolyzes L-arginine to L-ornithine and urea. Arginase, classically an enzyme of the urea cycle in the liver, is also found in many other cells that do not express a complete urea cycle, including the lung (Que et al., 1998). Two distinct isoforms of mammalian arginase – arginase I and II – have been identified (Wu & Morris, 1998), which are constitutively expressed in the airways, particularly in the bronchial epithelium and in peribronchial connective tissue fibroblasts (Que et al., 1998).
The biological function of extrahepatic arginase is largely unclear, but has been implicated in the regulation of NO synthesis and cell growth in inflammatory conditions. In activated macrophages various studies have indicated that arginase activity may limit the utilization of L-arginine by iNOS and suppress the cytotoxic response by these cells (Wang et al., 1995; Modolell et al., 1995; Hey et al., 1997), while concomitant polyamine and proline synthesis from L-ornithine may cause cell proliferation and repair of inflammatory lesions (Shearer et al., 1997; Wu & Morris, 1998). Recent immunohistochemical studies in rat lung indicated that the expression of both arginase I and arginase II in the airways was upregulated during hyperoxic lung injury and recovery. This was associated with reduced NO production and increased expression of ornithine decarboxylase, indicating a role for pulmonary arginase in the repair of hyperoxic lung injury (Que et al., 1998). The role of arginase in asthmatic airway inflammation, however, is presently unknown.
Using a perfused guinea-pig tracheal tube preparation, we recently demonstrated that the potent and highly specific arginase inhibitor Nω-hydroxy-nor-L-arginine (nor-NOHA) (Custot et al., 1997) caused a concentration-dependent inhibition of methacholine-induced airway constriction, which was reversed by L-NAME (Meurs et al., 2000). This indicates that arginase activity in the airways is also involved in the modulation of airway responsiveness, by limiting cNOS-derived bronchodilating NO production. For a number of cells, it has been reported that the expression of arginase may be enhanced by Th2 lymphocyte-derived cytokines such as IL-4, IL-10 and IL-13 and by cyclic AMP-elevating stimuli such as PGE2 (Modolell et al., 1995; Munder et al., 1999; Wei et al., 2000). Since these mediators are crucial in the development of asthmatic airway inflammation (Chung & Barnes, 1999), we hypothesized that induction of arginase activity after allergen challenge is involved in the reduction of cNOS activity and the initiation of AHR after the EAR. In the present study, we investigated this hypothesis by examining the effect of the specific arginase inhibitor nor-NOHA on cholinergic AHR in isolated perfused tracheae from sensitized guinea-pigs at 6 h after allergen challenge. In addition, we assessed the effect of allergen challenge on arginase activity in these preparations.
Methods
Animals
Outbred specified pathogen free guinea-pigs (Harlan, Heathfield, U.K.), weighing 600 – 800 g, were used in this study. Animals were actively IgE-sensitized to ovalbumin (OA) at 3 weeks of age as described by Van amsterdam et al. (1989). In short, 0.5 ml of an allergen solution containing 100 μg ml−1 ovalbumin and 100 mg ml−1 Al(OH)3 in saline was injected intraperitoneally, while another 0.5 ml was divided over seven intracutaneous injection sites in the proximity of lymphe-nodes in the paws, lumbar regions and the neck. The animals were used experimentally in weeks 4 to 8 after sensitization. The animals were group-housed in individual cages in climate controlled animal quarters and given water and food ad libitum, while a 12-h on/12-h off light cycle was maintained. All protocols described in this study were approved by the University of Groningen Committee for Animal Experimentation.
Allergen provocation
Ovalbumin provocations were performed by inhalation of aerosolized solutions. The provocations were performed in a specially designed animal cage, in which the guinea-pigs could move freely (Santing et al., 1992). The volume of the cage was 9 l, which ensured fast replacement of the air inside the cage with aerosol and vice versa. A DeVilbiss nebulizer (type 646, DeVilbiss, Somerset, PA, U.S.A.) driven by an airflow of 8 l min−1 provided the aerosol required, with an output of 0.33 ml min−1. Allergen provocations were performed by inhalation of an aerosol concentration of 0.05 mg ml−1 ovalbumin in saline. Allergen inhalations were discontinued when the first signs of respiratory distress were observed. Anti-histamines were not needed to prevent the development of anaphylactic shock. Previous studies measuring pleural pressure changes in ovalbumin sensitized, permanently instrumented, unrestrained guinea-pigs have indicated that the allergen-induced EAR induced by this procedure is maximal within 20 min and lasts for up to 5 h (Santing et al., 1992, 1994b). Six hours after ovalbumin challenge (between the EAR and LAR; Santing et al., 1992; 1994b), the guinea-pigs were sacrificed. The animals were killed by a sharp blow on the head and exsanguinated. Non-challenged animals were used as controls.
Tracheal perfusion
The tracheas were rapidly removed and placed in Krebs-Henseleit (KH) solution (37°C) of the following composition (mM): NaCl 117.50, KCl 5.60, MgSO4 1.18, CaCl2 2.50, NaH2PO4 1.28, NaHCO3 25.00, D-glucose 5.50; gassed with 5% CO2 and 95% O2; pH 7.4. The tracheas were prepared free of serosal connective tissue and cut into two halves of approximately 17 mm before mounting in a perfusion setup, as described previously (de boer et al., 1996).
To this aim, the tracheal preparations were attached at each end to stainless steel perfusion tubes fixed in a Delrin perfusion holder. The holder with the trachea was then placed in a water-jacketed organ bath (37°C) containing 20 ml of gassed KH (the serosal or extraluminal (EL) compartment). The lumen was perfused with recirculating KH from a separate 20 ml bath (mucosal or intraluminal (IL) compartment) at a constant flow rate of 12 ml min−1. Two axially centred side-hole catheters connected with pressure transducers (TC-XX,Viggo-Spectramed B.V., Bilthoven, The Netherlands) were situated at the distal and proximal ends of the trachealis to measure hydrostatic pressures (Poutlet and Pinlet, respectively). The signals were fed into a differential amplifier to obtain the difference between the two pressures (ΔP = Pinlet – Poutlet), which was plotted on a flatbed chart recorder (BD 41, Kipp en Zonen, Delft, The Netherlands). ΔP reflects the resistance of the tracheal segment to perfusion and is a function of the mean diameter of the trachea between the pressure taps. The transmural pressure in the trachea was set at 0 cm H2O. At the perfusion flow rate used, a baseline ΔP of 0.1 to 1.0 cm H2O was measured, depending on the diameter of the preparation. After a 45 min equilibration period with three washes with fresh KH (both IL and EL), 1 μM isoprenaline was added to the EL compartment for maximal smooth muscle relaxation to assess basal tone. After three washes during at least 30 min, the trachea was exposed to EL 40 mM KCl in KH to obtain a receptor-independent reference response. Subsequently, the preparation was washed four times with KH during 45 min until basal tone was reached and a cumulative concentration response curve (CCRC) was made with IL methacholine. When used, L-NAME (0.1, 0.5 or 1.0 mM) and nor-NOHA (5 or 10 μM) were applied to the IL reservoir, 40 min prior to agonist-addition.
Arginase assay
Tracheal tissue was snap-frozen in liquid nitrogen and stored at −80°C until homogenization. The tissue was ground using a pestle and mortar in the presence of liquid nitrogen prior to homogenization in approximately 6-vol ice-cold 20 mM Tris HCl, 2 μM phenylmethylsulphonyl fluoride, pH 7.4 using a polytron homogenizer (Kinematica GmbH, Luzern, Switzerland). The homogenate was centrifuged at 20,000×g for 30 min at 4°C and the supernatant was used for arginase assay. Arginase activity was determined by measuring the conversion of L-[guanidino-14C]arginine to [14C]urea essentially as described by Custot et al. (1996). In short, homogenate aliquots (50 μl) were incubated in a final volume of 150 μl, containing 25 mM Tris HCl, 0.67 mM MnCl2, 1 to 20 mM L-arginine (1.66 mM for standard conditions) and 200,000 d.p.m. of L-[guanidino-14C]arginine (51.5 mCi mmol−1), pH 7.4, for 20 min at 37°C. Specific inhibition of arginase activity was measured in the presence of 0.5 or 5.0 μM nor-NOHA. Reactions were terminated by adding 1 ml of ice-cold stop buffer, containing 20 mM sodium acetate, 100 mM urea, 10 mM EDTA and 1 mM L-citrulline, pH 3.0. Samples were applied to columns containing 2 ml Dowex AG 50W-X8 (H+ form), preequilibrated with 20 ml of stop buffer. Columns were eluted with stop buffer and aliquots (0.5 ml) were counted in 4 ml Ultima Gold scintillation fluid using a Beckman LS 1701 liquid scintillation counter. The protein concentrations of each sample were determined by the Bradford Coomassie brilliant blue method (Bradford, 1976), using bovine serum albumin as a standard. Arginase activity, expressed as pmol urea.mg protein−1.min−1, showed linear enzyme kinetics up to 30 min and was proportional to protein concentration.
Data analysis
To compensate for variations in baseline ΔP and in ΔP responses to contractile stimuli due to variation in resting internal diameter of the preparations used, IL responses of the tracheal tube preparations to methacholine were expressed as a percentage of the response induced by EL administration of 40 mM KCl. The contractile effect of 10 mM methacholine (highest concentration) was defined as Emax (De Boer et al., 1996; 1999; Meurs et al., 1999; 2000). Using this Emax, the sensitivity to methacholine was evaluated as pEC50 (−log10 EC50) value. Kinetic parameters of tracheal arginase activity and its inhibition by nor-NOHA were determined by Lineweaver-Burk analysis, by plotting arginase activity−1 versus arginine concentration−1 for different L-arginine and nor-NOHA concentrations. Results are expressed as means±s.e.mean. Statistical analysis was performed using the Student's t-test for paired or unpaired observations as appropriate. Differences were considered statistically significant at P<0.05.
Chemicals
Histamine hydrochloride, ovalbumin (grade III), aluminium hydroxide, (−)-isoprenaline hydrochloride, L-arginine hydrochloride, L-citrulline, Nω-nitro-L-arginine methyl ester and phenylmethylsulphonyl fluoride were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.) and methacholine chloride from Aldrich (Milwaukee, WI, U.S.A.). Dowex AG 50W-X8 resin was from Bio-Rad Laboratories (Hercules, CA, U.S.A.). L-[guanidino-14C]arginine (specific activity 51.5 mCi mmol−1) was purchased from New England Nuclear Life Science Products, Inc. (Boston, MA, U.S.A.). Ultima Gold scintillation fluid was obtained from Packard Bioscience (Groningen, The Netherlands). Nω-hydroxy-nor-L-arginine was kindly provided by Dr J.-L. Boucher (Université Paris V, Paris).
Results
In perfused tracheal preparations from unchallenged control guinea-pigs, the non-selective NOS inhibitor L-NAME (0.1 mM, IL) caused a significant 1.8 fold increase in the Emax of IL methacholine (P<0.01), without an effect on the sensitivity (pEC50) to this agonist (Figure 1a, Table 1). In the ovalbumin-challenged group of animals, the ΔP response to EL KCl was unchanged compared to the unchallenged control group (not shown); however, a 2 fold increase in the Emax to IL methacholine was observed (P<0.001), without a change in pEC50 (Figure 1b, Table 1). This increase was not further enhanced in the presence of L-NAME (Figure 1b, Table 1).
Figure 1.
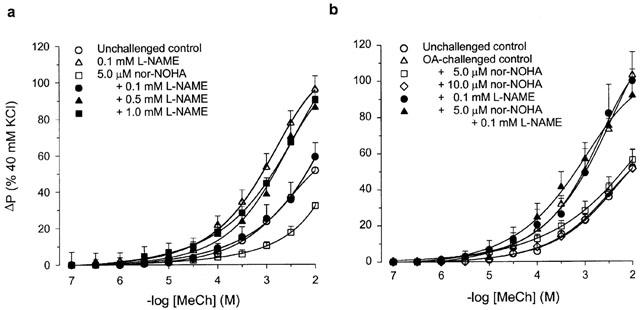
Methacholine (MeCh; IL)-induced constriction of intact perfused tracheal preparations obtained from (a) unchallenged guinea-pigs, in the absence (control) and presence of 0.1 mM L-NAME, 5.0 μM nor-NOHA, and 5.0 μM nor-NOHA plus 0.1, 0.5 or 1.0 mM L-NAME, and (b) ovalbumin (OA)-challenged guinea-pigs, in the absence (control) and presence of 5.0 and 10.0 μM nor-NOHA, 0.1 mM L-NAME, and 5.0 μM nor-NOHA plus 0.1 mM L-NAME. For comparison, methacholine-induced constriction of control preparations from unchallenged guinea-pigs is also shown in b. Methacholine, nor-NOHA and L-NAME were all applied to the IL compartment. Results are means±s.e.mean of 3 – 14 experiments.
Table 1.
Effects of L-NAME and nor-NOHA on the responsiveness to methacholine of intact perfused tracheae from unchallenged and ovalbumin-challenged guinea-pigs
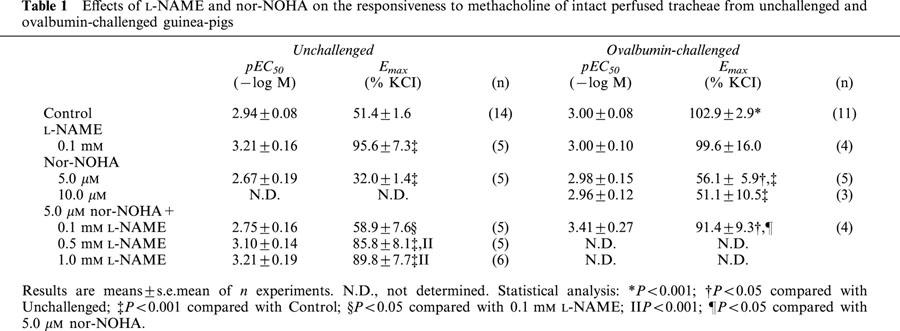
IL perfusion of tracheae from unchallenged control animals with the specific arginase inhibitor nor-NOHA (5 μM) caused a decrease in the Emax of methacholine-induced airway constriction by 38% (P<0.001), while no effect was observed on the pEC50 value for the agonist (Figure 1a, Table 1). As in a previous study (Meurs et al., 2000), we demonstrated that the reduced responsiveness in the presence of nor-NOHA can be concentration-dependently reversed by L-NAME, to the level of responsiveness of untreated airways in the presence of the NOS inhibitor (Figure 1a, Table 1). Remarkably, the enhanced Emax to IL methacholine at 6 h after allergen challenge was fully restored to the Emax value of unchallenged control animals by IL administration of 5 μM nor-NOHA, without an effect on the sensitivity to the agonist; no further effect on Emax was obtained in the presence of 10 μM nor-NOHA (Figure 1b, Table 1). The normalized responsiveness in the presence of 5 μM nor-NOHA was completely reversed in the additional presence of 0.1 mM L-NAME (Figure 1b, Table 1). Both in the unchallenged control preparations and in the preparations of allergen-challenged animals, L-NAME and nor-NOHA, alone or in combination, had no effect on basal airway tone (not shown).
Arginase activity in normal tracheal homogenates displayed Michaelis-Menten kinetics, and Lineweaver-Burk analysis indicated a Vmax of 2286±596 pmol urea.mg protein−1.min−1 and a Km for L-arginine of 2.74±0.33 mM at pH 7.4. Lineweaver-Burk plots of arginase inhibition experiments demonstrated that nor-NOHA acted as a competitive inhibitor with respect to L-arginine, with a Ki-value of 0.23±0.03 μM. The arginase activity in tracheae from allergen-challenged animals assessed in the presence of 1.66 mM L-arginine was 3.5 fold enhanced compared to unchallenged controls (2021±273 vs 577±47 pmol urea.mg protein−1.min−1, respectively; P<0.001). Both in control and in challenged airways 5 μM nor-NOHA caused approximately 95% inhibition of arginase activity, while the NOS inhibitor L-NAME (0.1 mM) had no effect (Figure 2).
Figure 2.
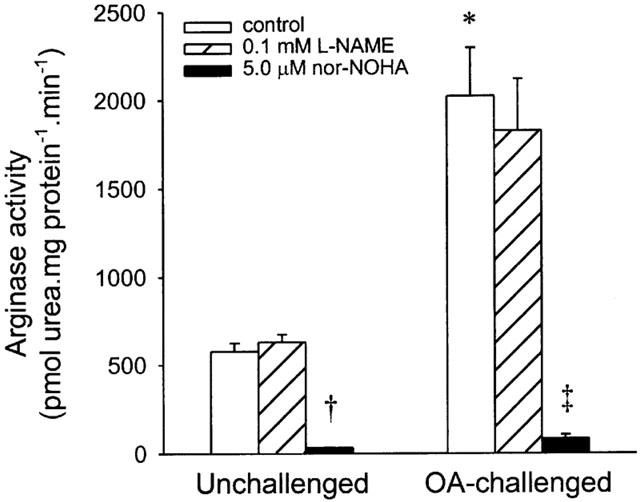
Arginase activity in tracheal homogenates from unchallenged and ovalbumin (OA)-challenged guinea-pigs, in the absence (control) and presence of 0.1 mM L-NAME and 5.0 μM nor-NOHA. Results are means±s.e.mean of 3 – 7 experiments. *P<0.001, †P<0.0001 compared to unchallenged control; ‡P<0.005 compared to OA-challenged control.
Discussion
The present study demonstrates for the first time the involvement of arginase in the pathophysiology of allergic asthma. Using a guinea-pig model of acute allergic asthma, we confirmed our previous finding that the responsiveness of isolated intact tracheal tube preparations to methacholine is considerably enhanced at 6 h after allergen challenge (de boer et al., 1996). The allergen-induced AHR was closely mimicked by the administration of L-NAME to unchallenged control preparations, while, in addition, the challenged preparations were unresponsive to the NOS inhibitor, indicating that a deficiency of agonist-induced airway dilating NO is a major determinant of the observed hyperresponsiveness. Remarkably, the enhanced responsiveness to methacholine at 6 h after allergen challenge was fully normalized in the presence of the specific arginase inhibitor nor-NOHA. This indicates that arginase activity, presumably by competition with cNOS for the common substrate, L-arginine, contributes to the deficiency of NO and subsequent AHR after allergen challenge. This hypothesis was confirmed by the observation that the decrease in AHR by nor-NOHA was reversed by administration of L-NAME.
As reported in a previous study (Meurs et al., 2000), endogenous arginase activity is also involved in the regulation of cholinergic airway tone under basal conditions. Thus, IL administration of nor-NOHA also reduced methacholine-induced bronchoconstriction in unchallenged control preparations, which was concentration-dependently reversed by L-NAME, indicating that the reduced airway responsiveness was caused by enhanced production of NO. In the nor-NOHA-treated preparations, a higher concentration of L-NAME (0.5 mM) was required for complete inhibition of the functional antagonism of cholinergic airway constriction by agonist-induced NO than in untreated controls (0.1 mM), indicating that L-NAME had to compete with a higher concentration of endogenous L-arginine in these preparations.
Since L-arginine is a limiting factor for bronchodilating NO production in the airways (de boer et al., 1999), it may be expected that increased arginase activity is involved in the allergen-induced AHR and its inhibition by nor-NOHA as discussed above. Indeed, biochemical measurement of arginase activity in tracheal homogenates indicated that allergen challenge caused a 3.5 fold increase in this activity, which was specifically reversed by 5 μM nor-NOHA, the same concentration as used in the pharmacological experiments. The Nω-hydroxy-L-aminoacid nor-NOHA is one of the most potent arginase inhibitors reported so far, and the observed Ki value for tracheal homogenates (0.23 μM) corresponds closely to the Ki of 0.5 μM found for purified rat liver arginase (Custot et al., 1997).
The sources of NO and constitutive and allergen-induced arginase activity, as well as the arginase isoforms in our perfused tracheal tube preparations were not assessed. Both in control and in challenged airways, L-NAME only increased the agonist-induced constriction and not basal tone, indicating that the NO was most likely produced by cNOS. Previous studies have indicated that agonist-induced bronchodilating NO is mainly derived from the airway epithelium (Nijkamp et al., 1993). Since these cells also constitutively express arginase (type I and type II) (Que et al., 1998), it can be hypothesized that the epithelium is an important site of competition between the two enzymes for their common substrate. This may not only apply to the basal condition, but also to the reduced cNOS activity after the allergen challenge. Thus, preliminary data have recently indicated that Th2 cytokines may cause enhanced expression of arginase I and arginase II in cultured A549 human lung epithelial cells (McCluskie et al., 2001). However, it cannot be excluded that other cell types expressing arginase and/or cNOS are involved.
Recently, a role for arginase has also been described in the regulation of inhibitory nonadrenergic noncholinergic (iNANC) nerve-stimulated, cNOS-derived NO-mediated relaxation of the penile corpus cavernosum smooth muscle (Cox et al., 1999) and the internal anal sfincter smooth muscle (Baggio et al., 1999). Although iNANC nerve activity was not measured in our tracheal preparations, upregulation of arginase in airway iNANC nerves could also be of importance in allergen-induced airway hyperresponsiveness, since it has been demonstrated that the bronchodilating iNANC activity is reduced after allergen challenge (Miura et al., 1997).
Different studies have indicated that the present ex vivo findings are relevant to the development of AHR in vivo. Using the same guinea-pig model of asthma, we have recently shown that inhalation of the nonselective NOS inhibitor L-NAME, but not a selective dose of the specific iNOS inhibitor aminoguanidine, caused AHR to histamine before allergen challenge, indicating that cNOS-derived NO counteracts histamine-induced bronchoconstriction under basal conditions (Schuiling et al., 1998a, 1998b). At 6 h after a single allergen challenge, both L-NAME and aminoguanidine had no effect on the allergen-induced AHR to histamine at this time point, indicating that a deficiency of (cNOS-derived) NO contributes to AHR after the EAR (Schuiling et al., 1998a, 1998b), confirming the earlier ex vivo data (de boer et al., 1996). A deficiency of cNOS activity and endogenous bronchodilating NO contributing to AHR was also demonstrated after repeated allergen challenge of sensitized guinea-pigs (Mehta et al., 1997; Samb et al., 2001). Very interestingly, evidence for a deficiency of cNOS-derived NO modulating airway reactivity has also been found in patients with severe asthma treated with corticosteroids, which suppresses the expression of iNOS (Ricciardolo et al., 1997). Thus, while the airway reactivity to bradykinin (and to methacholine) could be significantly increased by inhalation of NG-monomethyl-L-arginine (L-NMMA) in patients with mild asthma (Ricciardolo et al., 1996), a similarly enhanced AHR to bradykinin present in severe asthmatics was insensitive to the NOS inhibitor, indicating that a deficiency of NO contributed to the enhanced airway responsiveness in these patients. A recent study by the same authors suggested that this deficiency may indeed be induced by allergen challenge (Ricciardolo et al., 2001). Accordingly, from the assessment of physical diffusion parameters of NO by measuring the relationship between exhaled NO and expiratory flow, a recent study by Silkoff et al. (2000) suggested that residual airway obstruction and AHR in asthmatic patients after steroid therapy are associated with an apparent decrease in activity of cNOS-derived NO.
Our data correspond with a previous observation by Kochańsky et al. (1980), who found two decades ago that arginase activity is enhanced in expectorated sputum from asthmatic patients. The underlying mechanism as well as its functional relevance have thus far not been investigated, but could be related to the allergen-induced increase in arginase activity and AHR found in the present study. Interestingly, in the same study, arginase activity was also shown to be enhanced in patients with chronic bronchitis (Kochańsky et al., 1980).
Although speculative, the implications of increased arginase activity in the asthmatic lung could be more widespread. Thus, upregulation of arginase, emerging from Th2 cytokines and other mediators released during allergic airway inflammation, could possibly also limit the synthesis of iNOS-derived-NO known to be present in chronic asthma and to be induced during the LAR (Kharitonov et al., 1995; Schuiling et al., 1998a), and therefore protect the airways against exaggerated NO production and tissue injury. In addition, as a consequence of increased synthesis of arginase-induced ornithine as a precursor for polyamines and proline, arginase could be involved in the rapid repair process of epithelial damage as observed after allergen challenge in a guinea-pig model of acute asthma (Erjefält et al., 1997) and in inflammation-induced airway remodelling in chronic asthma (Bousquet et al., 2000), by promoting proliferation of structural subepithelial cells (fibroblasts and airway smooth muscle cells) and collagen deposition. However, the role of arginase in asthmatic airway inflammation and remodelling remains to be established.
The functional inhibition of AHR to methacholine by nor-NOHA in the challenged preparations appeared to be maximal in the presence of 5 μM nor-NOHA, since no additional effect was obtained with 10 μM of the arginase inhibitor. This maximal inhibitory effect did not fully reach the reduced level of responsiveness observed in the control preparations in the presence of nor-NOHA, indicating that additional mechanisms may be involved in the development of the allergen-induced NO deficiency and AHR. One of these mechanisms could be limitation of L-arginine by reduced cellular uptake of this amino acid, induced by inhibition of specific cationic amino acid transporters by polycations such as eosinophil-derived major basic protein (Hammermann et al., 1999; Meurs et al., 1999). As in humans, eosinophilic polycations have been demonstrated in the inflamed guinea-pig airways at 6 h after allergen challenge (Santing et al., 1994a).
In conclusion, using a guinea-pig model of allergic asthma we have established that enhanced arginase activity may be importantly involved in the allergen-induced NO deficiency and AHR after the EAR, presumbly by competition with cNOS for the common substrate, L-arginine. Since reduced cNOS-derived bronchodilating NO has also been recognized to contribute to AHR in asthmatics patients, the advent of new and selective arginase inhibitors could have therapeutic potential in allergic asthma. However, since enhanced bioavailability of L-arginine may also amplify the inflammatory effects of iNOS-derived NO present in most patients (Sapienza et al., 1998), their usefulness remains to be established.
Acknowledgments
The authors wish to thank Drs Fiona Westerhof for expert technical support and Dr Ad Nelemans for critically reading the manuscript. We thank Dr Jean-Luc Boucher and Dr Sandrine Vadon-Le Goff for providing nor-NOHA and helpfull discussion. We are indebted to Bartek Szilajtis-Nowicki for translating the paper by Kochańsky et al. (1980) from Polish. We thank the Netherlands Asthma Foundation for financial support (grant 00.23).
Abbreviations
- AHR
airway hyperreactivity
- cNOS
constitutive nitric oxide synthase
- EAR
early asthmatic reaction
- EL
extraluminal
- Emax
maximal effect
- eNOS
endothelial nitric oxide synthase
- IL
intraluminal
- iNANC
inhibitory nonadrenergic noncholinergic
- iNOS
inducible nitric oxide synthase
- KH
Krebs-Henseleit
- LAR
late asthmatic reaction
- L-NAME
Nω-nitro-L-arginine methyl ester
- L-NMMA
NG-monomethyl-L-arginine
- nNOS
neuronal nitric oxide synthase
- nor-NOHA
Nω-hydroxy-nor-L-arginine
- ΔP
differential (hydrostatic) pressure
- Pinlet
(hydrostatic) pressure at the inlet
- Poutlet
(hydrostatic) pressure at the outlet
- pEC50
−log10 of the concentration causing 50% of the effect
References
- ASANO K., CHEE C.B., GASTON B., LILLY C.M., GERARD C., DRAZEN J.M., STAMLER J.S. Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 1994;91:10089–10093. doi: 10.1073/pnas.91.21.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAGGIO R., EMIG F.A., CHRISTIANSON D.W., ASH D.E., CHAKDER S., RATTAN S. Biochemical and functional pr ofile of a newly developed potent and isozyme-selective arginase inhibitor. J. Pharmacol. Exp. Ther. 1999;290:1409–1416. [PubMed] [Google Scholar]
- BARNES P.J.Nitric Oxide Asthma: Basic Mechanisms and Clinical Management 1998London: Academic Press Limited; 369–388.ed. Barnes, P.J., Rodger, I.W. & Thomson, N.C. pp [Google Scholar]
- BARNES P.J., BELVISI M.G. Nitric oxide and lung disease. Thorax. 1993;48:1034–1043. doi: 10.1136/thx.48.10.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOOIJ-NOORD H., ORIE N.G., DE VRIES K. Immediate and late bronchial obstructive reactions to inhalation of house dust and protective effects of disodium cromoglycate and prednisolone. J. Allergy Clin. Immunol. 1971;48:344–354. doi: 10.1016/0091-6749(71)90080-7. [DOI] [PubMed] [Google Scholar]
- BOUSQUET J., JEFFERY P.K., BUSSE W.W., JOHNSON M., VIGNOLA A.M. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am. J. Respir. Crit. Care Med. 2000;161:1720–1745. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CHUNG K.F., BARNES P.J. Cytokines in asthma. Thorax. 1999;54:825–857. doi: 10.1136/thx.54.9.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COX J.D., KIM N.N., TRAISH A.M., CHRISTIANSON D.W. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat. Struct. Biol. 1999;6:1043–1047. doi: 10.1038/14929. [DOI] [PubMed] [Google Scholar]
- CUSTOT J., BOUCHER J.L., VADON S., GUEDES C., DIJOLS S., DELAFORGE M., MANSUY D. N-omega-hydroxy-alpha-amino acids as a new class of very strong inhibitors of arginases. JBIC. 1996;1:73–82. [Google Scholar]
- CUSTOT J., MOALI C., BROLLO M., BOUCHER J.L., DELAFORGE M., MANSUY D., TENU J.P., ZIMMERMANN J.L. The new alpha-amino acid N-omega-hydroxy-nor-L-arginine: a high affinity inhibitor of arginase well adapted to bind to its manganese cluster. J. Am. Chem. Soc. 1997;119:4086–4087. [Google Scholar]
- DE BOER J., DUYVENDAK M., SCHUURMAN F.E., POUW F.M., ZAAGSMA J., MEURS H. Role of L-arginine in the deficiency of nitric oxide and airway hyperreactivity after the allergen-induced early asthmatic reaction in guinea-pigs. Br. J. Pharmacol. 1999;128:1114–1120. doi: 10.1038/sj.bjp.0702882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE BOER J., MEURS H., COERS W., KOOPAL M., BOTTONE A.E., VISSER A.C., TIMENS W., ZAAGSMA J. Deficiency of nitric oxide in allergen-induced airway hyperreactivity to contractile agonists after the early asthmatic reaction: an ex vivo study. Br. J. Pharmacol. 1996;119:1109–1116. doi: 10.1111/j.1476-5381.1996.tb16011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE GOUW H.W., GRUNBERG K., SCHOT R., KROES A.C., DICK E.C., STERK P.J. Relationship between exhaled nitric oxide and airway hyperresponsiveness following experimental rhinovirus infection in asthmatic subjects. Eur. Respir. J. 1998;11:126–132. doi: 10.1183/09031936.98.11010126. [DOI] [PubMed] [Google Scholar]
- DE MONCHY J.G., KAUFFMAN H.F., VENGE P., KOËTER G.H., JANSEN H.M., SLUITER H.J., DE VRIES K. Bronchoalveolar eosinophilia during allergen-induced late asthmatic reactions. Am. Rev. Respir. Dis. 1985;131:373–376. doi: 10.1164/arrd.1985.131.3.373. [DOI] [PubMed] [Google Scholar]
- DURHAM S.R., CRADDOCK C.F., COOKSON W.O., BENSON M.K. Increases in airway responsiveness to histamine precede allergen-induced late asthmatic responses. J. Allergy Clin. Immunol. 1988;82:764–770. doi: 10.1016/0091-6749(88)90077-2. [DOI] [PubMed] [Google Scholar]
- ERJEFÄLT J.S., KORSGREN M., NILSSON M.C., SUNDLER F., PERSSON C.G. Prompt epithelial damage and restitution processes in allergen challenged guinea-pig trachea in vivo. Clin. Exp. Allergy. 1997;27:1458–1470. doi: 10.1046/j.1365-2222.1997.1200932.x. [DOI] [PubMed] [Google Scholar]
- FISCHER A., MUNDEL P., MAYER B., PREISSLER U., PHILIPPIN B., KUMMER W. Nitric oxide synthase in guinea pig lower airway innervation. Neurosci. Lett. 1993;149:157–160. doi: 10.1016/0304-3940(93)90760-i. [DOI] [PubMed] [Google Scholar]
- FLAK T.A., GOLDMAN W.E. Autotoxicity of nitric oxide in airway disease. Am. J. Respir. Crit Care Med. 1996;154:S202–S206. doi: 10.1164/ajrccm/154.4_Pt_2.S202. [DOI] [PubMed] [Google Scholar]
- HAMID Q., SPRINGALL D.R., RIVEROS M., V, , CHANEZ P., HOWARTH P., REDINGTON A., BOUSQUET J., GODARD P., HOLGATE S., POLAK J.M. Induction of nitric oxide synthase in asthma. Lancet. 1993;342:1510–1513. doi: 10.1016/s0140-6736(05)80083-2. [DOI] [PubMed] [Google Scholar]
- HAMMERMANN R., HIRSCHMANN J., HEY C., MOSSNER J., FOLKERTS G., NIJKAMP F.P., WESSLER I., RACKÉ K. Cationic proteins inhibit L-arginine uptake in rat alveolar macrophages and tracheal epithelial cells. Implications for nitric oxide synthesis. Am. J. Respir. Cell Mol. Biol. 1999;21:155–162. doi: 10.1165/ajrcmb.21.2.3574. [DOI] [PubMed] [Google Scholar]
- HEY C., BOUCHER J.L., VADON L.G., KETTERER G., WESSLER I., RACKÉ K. Inhibition of arginase in rat and rabbit alveolar macrophages by N omega-hydroxy-D,L-indospicine, effects on L-arginine utilization by nitric oxide synthase. Br. J. Pharmacol. 1997;121:395–400. doi: 10.1038/sj.bjp.0701143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHARITONOV S.A., O'CONNOR B.J., EVANS D.J., BARNES P.J. Allergen-induced late asthmatic reactions are associated with elevation of exhaled nitric oxide. Am. J. Respir. Crit. Care Med. 1995;151:1894–1899. doi: 10.1164/ajrccm.151.6.7767537. [DOI] [PubMed] [Google Scholar]
- KOBZIK L., BREDT D.S., LOWENSTEIN C.J., DRAZEN J., GASTON B., SUGARBAKER D., STAMLER J.S. Nitric oxide synthase in human and rat lung: immunocytochemical and histochemical localization. Am. J. Respir. Cell Mol. Biol. 1993;9:371–377. doi: 10.1165/ajrcmb/9.4.371. [DOI] [PubMed] [Google Scholar]
- KOCHAŃSKY L., KOSSMAN S., ROGALA E., DWORNICKI J. Arginase activity in sputum of patients with bronchial asthma. Pneum. Pol. 1980;48:329–332. [PubMed] [Google Scholar]
- KUO H.P., LIU S., BARNES P.J. The effect of endogenous nitric oxide on neurogenic plasma exudation in guinea-pig airways. Eur. J. Pharmacol. 1992;221:385–388. doi: 10.1016/0014-2999(92)90728-m. [DOI] [PubMed] [Google Scholar]
- MCCLUSKIE M., MITCHELL J.A., YACOUB M.H., BELVISI M.G. Differential gene expression of arginase and NOS in a human lung epithelial cell line. Am. J. Respir. Crit Care Med. 2001;163:A946. [Google Scholar]
- MEHTA S., DRAZEN J.M., LILLY C.M. Endogenous nitric oxide and allergic bronchial hyperresponsiveness in guinea pigs. Am. J. Physiol. 1997;273:L656–L662. doi: 10.1152/ajplung.1997.273.3.L656. [DOI] [PubMed] [Google Scholar]
- MEURS H., HAMER M.A., PETHE S., VADON-LE GOFF S., BOUCHER J.L., ZAAGSMA J. Modulation of cholinergic airway reactivity and nitric oxide production by endogenous arginase activity. Br. J. Pharmacol. 2000;130:1793–1798. doi: 10.1038/sj.bjp.0703488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEURS H., SCHUURMAN F.E., DUYVENDAK M., ZAAGSMA J. Deficiency of nitric oxide in polycation-induced airway hyperreactivity. Br. J. Pharmacol. 1999;126:559–562. doi: 10.1038/sj.bjp.0702372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIURA M., YAMAUCHI H., ICHINOSE M., OHUCHI Y., KAGEYAMA N., TOMAKI M., ENDOH N., SHIRATO K. Impairment of neural nitric oxide-mediated relaxation after antigen exposure in guinea pig airways in vitro. Am. J. Respir. Crit. Care Med. 1997;156:217–222. doi: 10.1164/ajrccm.156.1.9606040. [DOI] [PubMed] [Google Scholar]
- MODOLELL M., CORRALIZA I.M., LINK F., SOLER G., EICHMANN K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur. J. Immunol. 1995;25:1101–1104. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
- MUNDER M., EICHMANN K., MORAN J.M., CENTENO F., SOLER G., MODOLELL M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J. Immunol. 1999;163:3771–3777. [PubMed] [Google Scholar]
- NIJKAMP F.P., VAN DER LINDE H.J., FOLKERTS G. Nitric oxide synthesis inhibitors induce airway hyperresponsiveness in the guinea pig in vivo and in vitro. Role of the epithelium. Am. Rev. Respir. Dis. 1993;148:727–734. doi: 10.1164/ajrccm/148.3.727. [DOI] [PubMed] [Google Scholar]
- QUE L.G., KANTROW S.P., JENKINSON C.P., PIANTADOSI C.A., HUANG Y.C. Induction of arginase isoforms in the lung during hyperoxia. Am. J. Physiol. 1998;275:L96–L102. doi: 10.1152/ajplung.1998.275.1.L96. [DOI] [PubMed] [Google Scholar]
- RICCIARDOLO F.L., DI MARIA G.U., MISTRETTA A., SAPIENZA M.A., GEPPETTI P. Impairment of bronchoprotection by nitric oxide in severe asthma. Lancet. 1997;350:1297–1298. doi: 10.1016/s0140-6736(05)62474-9. [DOI] [PubMed] [Google Scholar]
- RICCIARDOLO F.L., GEPPETTI P., MISTRETTA A., NADEL J.A., SAPIENZA M.A., BELLOFIORE S., DI MARIA G.U. Randomised double-blind placebo-controlled study of the effect of inhibition of nitric oxide synthesis in bradykinin-induced asthma. Lancet. 1996;348:374–377. doi: 10.1016/s0140-6736(96)04450-9. [DOI] [PubMed] [Google Scholar]
- RICCIARDOLO F.L., TIMMERS M.C., GEPPETTI P., VAN SCHADEWIJK A., BRAHIM J.J., SONT J.K., DE GOUW H.W.F.M., HIEMSTRA P.S., VAN KRIEKEN H.J.M., STERK P.J. Allergen-induced impairment of bronchoprotective nitric oxide synthesis in asthma. J. Allergy Clin. Immunol. 2001;108:198–204. doi: 10.1067/mai.2001.116572. [DOI] [PubMed] [Google Scholar]
- SAMB A., PRETOLANI M., DINH-XUAN A.T., OUKSEL H., CALLEBERT J., LISDERO C., AUBIER M., BOCZKOWSKI J. Decreased pulmonary and tracheal smooth muscle expression and activity of type 1 nitric oxide synthase (nNOS) after ovalbumin immunization and multiple aerosol challenge in guinea pigs. Am. J. Respir. Crit. Care Med. 2001;164:149–154. doi: 10.1164/ajrccm.164.1.2004030. [DOI] [PubMed] [Google Scholar]
- SANTING R.E., HOEKSTRA Y., PASMAN Y., ZAAGSMA J., MEURS H. The importance of eosinophil activation for the development of allergen-induced bronchial hyperreactivity in conscious, unrestrained guinea-pigs. Clin. Exp. Allergy. 1994a;24:1157–1163. doi: 10.1111/j.1365-2222.1994.tb03322.x. [DOI] [PubMed] [Google Scholar]
- SANTING R.E., MEURS H., VAN DER MARK T.W., REMIE R., OOSTEROM W.C., BROUWER F., ZAAGSMA J. A novel method to assess airway function parameters in chronically instrumented, unrestrained guinea-pigs. Pulm. Pharmacol. 1992;5:265–272. doi: 10.1016/0952-0600(92)90069-s. [DOI] [PubMed] [Google Scholar]
- SANTING R.E., OLYMULDER C.G., ZAAGSMA J., MEURS H. Relationships among allergen-induced early and late phase airway obstructions, bronchial hyperreactivity, and inflammation in conscious, unrestrained guinea pigs. J. Allergy Clin. Immunol. 1994b;93:1021–1030. doi: 10.1016/s0091-6749(94)70051-6. [DOI] [PubMed] [Google Scholar]
- SAPIENZA M.A., KHARITONOV S.A., HORVATH I., CHUNG K.F., BARNES P.J. Effect of inhaled L-arginine on exhaled nitric oxide in normal and asthmatic subjects. Thorax. 1998;53:172–175. doi: 10.1136/thx.53.3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHUILING M., MEURS H., ZUIDHOF A.B., VENEMA N., ZAAGSMA J. Dual action of iNOS-derived nitric oxide in allergen-induced airway hyperreactivity in conscious, unrestrained guinea pigs. Am. J. Respir. Crit. Care Med. 1998a;158:1442–1449. doi: 10.1164/ajrccm.158.5.9803027. [DOI] [PubMed] [Google Scholar]
- SCHUILING M., ZUIDHOF A.B., BONOUVRIE M.A., VENEMA N., ZAAGSMA J., MEURS H. Role of nitric oxide in the development and partial reversal of allergen-induced airway hyperreactivity in conscious, unrestrained guinea-pigs. Br. J. Pharmacol. 1998b;123:1450–1456. doi: 10.1038/sj.bjp.0701738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEARER J.D., RICHARDS J.R., MILLS C.D., CALDWELL M.D. Differential regulation of macrophage arginine metabolism: a proposed role in wound healing. Am. J. Physiol. 1997;272:E181–E190. doi: 10.1152/ajpendo.1997.272.2.E181. [DOI] [PubMed] [Google Scholar]
- SILKOFF P.E., SYLVESTER J.T., ZAMEL N., PERMUTT S. Airway nitric oxide diffusion in asthma: Role in pulmonary function and bronchial responsiveness. Am. J. Respir. Crit. Care Med. 2000;161:1218–1228. doi: 10.1164/ajrccm.161.4.9903111. [DOI] [PubMed] [Google Scholar]
- TAYLOR D.A., MCGRATH J.L., ORR L.M., BARNES P.J., O'CONNOR B.J. Effect of endogenous nitric oxide inhibition on airway responsiveness to histamine and adenosine-5′-monophosphate in asthma. Thorax. 1998;53:483–489. doi: 10.1136/thx.53.6.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUCKER J.F., BRAVE S.R., CHARALAMBOUS L., HOBBS A.J., GIBSOM A. L-NG-nitro arginine inhibits non-adrenergic, non-cholinergic relaxations of guinea-pig isolated tracheal smooth muscle. Br. J. Pharmacol. 1990;100:663–664. doi: 10.1111/j.1476-5381.1990.tb14072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN AMSTERDAM R.G., BROUWER F., ZAAGSMA J. Analysis of the beta-adrenoceptor mediated inhibition of IgG1 and IgE dependent guinea-pig anaphylactic tracheal smooth muscle contraction. Agents Actions. 1989;26:48–51. doi: 10.1007/BF02126559. [DOI] [PubMed] [Google Scholar]
- WANG W.W., JENKINSON C.P., GRISCAVAGE J.M., KERN R.M., ARABOLOS N.S., BYRNS R.E., CEDERBAUM S.D., IGNARRO L.J. Co-induction of arginase and nitric oxide synthase in murine macrophages activated by lipopolysaccharide. Biochem. Biophys. Res. Commun. 1995;210:1009–1016. doi: 10.1006/bbrc.1995.1757. [DOI] [PubMed] [Google Scholar]
- WEI L.H., JACOBS A.T., MORRIS S.M., IGNARRO L.J. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2000;279:C248–C256. doi: 10.1152/ajpcell.2000.279.1.C248. [DOI] [PubMed] [Google Scholar]
- WU G., MORRIS S.M. Arginine metabolism: nitric oxide and beyond. Biochem. J. 1998;336:1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]