

Abstract
Challenge of COS1 cells with the adenylyl cyclase activator forskolin led to the activation of recombinant PDE4A8, PDE4B1, PDE4C2 and PDE4D5 cAMP-specific phosphodiesterase long isoforms.
Forskolin challenge did not activate mutant long PDE4 isoforms where the serine target residue (STR) within the protein kinase A (PKA) consensus phosphorylation site in Upstream Conserved Region 1 (UCR1) was mutated to alanine.
The PKA inhibitor, H89, ablated forskolin activation of wild-type long PDE4 isoforms.
Activated PKA caused the in vitro phosphorylation of recombinant wild-type long PDE4 isoforms, but not those where the STR was mutated to alanine.
An antiserum specific for the phosphorylated form of the STR detected a single immunoreactive band for recombinant long PDE4 isoforms expressed in COS1 cells challenged with forskolin. This was not evident in forskolin-challenged cells treated with H89. Neither was it evident in forskolin-challenged cells expressing long isoforms where the STR had been mutated to alanine.
In transfected COS cells challenged with forskolin, only the phosphorylated PDE4D3 long form showed a decrease in mobility in Western blotting analysis. This decreased mobility of PDE4D3 was ablated upon mutation of either of the two serine targets for PKA phosphorylation in this isoform, namely Ser54 in UCR1 and Ser13 in the isoform-specific N-terminal region.
Activation by forskolin challenge did not markedly alter the sensitivity of PDE4A8, PDE4B1, PDE4C2 and PDE4D5 to inhibition by rolipram.
Long PDE4 isoforms from all four sub-families can be phosphorylated by protein kinase A (PKA). This leads to an increase in their activity and may thus contribute to cellular desensitization processes in cells where these isoforms are selectively expressed.
Keywords: PDE4 cAMP phosphodiesterase, rolipram, phosphorylation, PKA, protein kinase A
Introduction
cAMP is a ubiquitous second messenger that is pivotal in controlling a wide variety of cellular functions (Houslay & Milligan, 1997). The only known means of degrading cAMP is through the action of cyclic nucleotide phosphodiesterases. It is now well appreciated that a large multigene family of enzymes exhibits the ability to hydrolyse cAMP (Beavo, 1995; Conti & Jin, 1999; Houslay, 2001; Manganiello et al., 1995a, 1995b; Soderling & Beavo, 2000; Thompson, 1991). Recently, however, the use of selective inhibitors has demonstrated the importance of the PDE4 cAMP-specific phosphodiesterase family in controlling inflammatory responses, depression and cognitive function (Barnette, 1999; Bolger, 1994; Cavalla & Frith, 1995; Giembycz, 2000; He et al., 1998; Houslay, 2001; Houslay et al., 1998; Rogers & Giembycz, 1998; Schudt et al., 1995; Souness & Rao, 1997; Spina et al., 1998; Torphy, 1998). Such discoveries have led to the development of PDE4-selective inhibitors as potential therapeutic agents in a variety of inflammatory disease states. Notwithstanding this, it is only recently that we are beginning to understand the complex regulatory mechanisms that control the functioning of the large number of PDE4 isoforms that have been recognized to date (Conti & Jin, 1999; Houslay, 2001; Houslay et al., 1998).
PDE4 enzymes are encoded by four genes (PDE4A, PDE4B, PDE4C and PDE4D), each capable of producing a number of isoforms through alternative mRNA splicing and the use of alternative promoters (Conti & Jin, 1999; Houslay, 2001; Houslay et al., 1998). Each PDE4 isoform within a particular PDE4 sub-family possesses a common core region, which consists of the catalytic unit and the C-terminal portion and is defined by its unique extreme N-terminal region. The various PDE4 isoforms are classified further as being either ‘long' or ‘short' isoforms. This classification relates to the presence or absence of two highly conserved sequences that are unique to the PDE4 enzyme family (Bolger, 1994; Bolger et al., 1993). These are the 55 amino acid Upstream Conserved Region 1 (UCR1) and the 76 amino acid Upstream Conserved Region 2 (UCR2) that are both located immediately N-terminal to the catalytic unit. Thus long isoforms exhibit both UCR1 and UCR2 whilst short isoforms lack UCR1 and ‘super-short' isoforms lack UCR1 and have a truncated UCR2 (Houslay, 2001; Houslay et al., 1998). All four PDE4 gene families are known to encode long-form enzymes. N-terminal truncation analyses have led to the suggestion that UCR1 and UCR2 may influence PDE4 catalytic activity (see e.g. Conti & Jin, 1999 for review). More recent studies on the PDE4D3 long isoform have led to an appreciation that UCR1 and UCR2 interact together to form a regulatory module (Beard et al., 2000; Lim et al., 1999) that integrates the regulatory effect of phosphorylation by both protein kinase A (PKA) and by ERK MAP kinase (MacKenzie et al., 2000).
Increased intracellular cAMP levels have been demonstrated to increase cellular PDE4 activity (see Houslay, 2001; Houslay et al., 1998 for reviews). This is believed to perform an adaptive role in desensitising cellular processes to increased levels of cAMP. The long-term elevation of intracellular cAMP levels has been shown to cause an increase in the levels of mRNA and protein expression for various PDE4 isoforms, in particular the PDE4D1 and PDE4D2 short forms (Erdogan & Houslay, 1997; Kovala et al., 1994; Sette et al., 1994b; Seybold et al., 1998; Swinnen et al., 1989; Verghese et al., 1995; Vicini & Conti, 1997) where a cAMP-controlled promoter has been identified (Vicini & Conti, 1997). However, a second cAMP-driven controlling mechanism that has been identified is the rapid activation of the PDE4D3 isoform that is achieved through direct PKA-mediated phosphorylation (Alvarez et al., 1995; Hoffmann et al., 1998; Sette & Conti, 1996; Sette et al., 1994a, 1994b). PKA appears to phosphorylate PDE4D3 at two sites, one being Ser13 in the isoform-specific N-terminal region and the other at Ser54 in UCR1. However, only modification of Ser54 in UCR1 is required to elicit activation (Hoffmann et al., 1998; Sette & Conti, 1996). This activation appears to contribute to short-term desensitization, at least in certain cell types (Oki et al., 2000). Such PKA-mediated phosphorylation of PDE4D3 has also been suggested to lead to an increase in the sensitivity of PDE4D3 to inhibition by the PDE4 selective inhibitor, rolipram (Alvarez et al., 1995). Additionally a decrease in mobility of PDE4D3, on SDS – PAGE, has been observed after its phosphorylation by PKA (Sette & Conti, 1996; Sette et al., 1994a). This has been suggested to be diagnostic for the presence of an activated PKA-phosphorylated enzyme (Oki et al., 2000; Sette & Conti, 1996; Sette et al., 1994a).
Whilst many analyses have been done on PDE4D3 as a target for PKA-mediated phosphorylation and activation, it has been pointed out (Sette & Conti, 1996; Souness & Rao, 1997) that an identical PKA consensus sequence, namely Arg-Arg-Glu-Ser-Phe, is found in the UCR1 of long isoforms from all four PDE4 sub-families. However, no information is available as to whether long isoforms of other PDE4 sub-families can be phosphorylated and activated by PKA or if this property is unique to PDE4D3. Certainly UCR1 and UCR2 are linked to each other and to the catalytic unit by regions that have very different primary sequences (Houslay et al., 1998), which may alter accessibility of PKA to the site within UCR1, as well as affect any conformational change consequent upon PKA phosphorylation. In addition to this, there are distinct differences in primary sequence of a number of the helices that form the catalytic unit of each of these sub-families (Xu et al., 2000). These differences undoubtedly underpin sub-family variation in sensitivity to the action of certain inhibitors and catalytic activity, for example (Houslay, 2001; Houslay et al., 1998). Thus, there is no a priori reason to expect that long PDE4 isoforms from other sub-families may either be phosphorylated by PKA or will be activated as a consequence of any such phosphorylation event.
This study demonstrates that examples of long isoforms from the three other PDE4 sub-families are capable of acting as PKA substrates both in vitro and in intact cells and that this modification leads to enzyme activation. In all cases studied, PKA phosphorylation occurs at a single serine residue in UCR1. For the first time we have developed phospho-specific antisera against this site which allows for the detection of phosphorylated PDE4 long forms from all four PDE4 sub-families. In addition, we show that decreased mobility on SDS – PAGE electrophoresis reported for the PKA-phosphorylated form of PDE4D3 (Oki et al., 2000; Sette & Conti, 1996) is likely to be unique to this isoform and cannot be taken as a universal indicator of PKA phosphorylation of PDE4 long isoforms.
Methods
Materials
[3H]-cAMP and enhanced chemiluminescence (ECL) reagent were from Amersham Bioscience (Amersham, U.K.). Dithiothreitol, Triton X-100 and N-{1-(2,3-dioleoyloxy)propyl}-N,N,N,-trimethylammonium methylsulfate (DOTAP) and protease inhibitor tablets were obtained from Boehringer Mannheim (Mannheim, Germany). Bradford reagent was from Bio-Rad (Herts, U.K.). Rolipram was a kind gift from Schering AG (Berlin, Germany). All other materials were from Sigma (Poole, U.K.).
Deletion mutagenesis and point mutations
Site-directed mutagenesis was performed using a QuickChange DNA mutagenesis kit (Stratagene, La Jolla, CA U.S.A.) according to the manufacturer's instructions. All mutagenesis and deletion constructs were confirmed by DNA sequencing as described previously (McPhee et al., 1999). The cDNA clones and expression vectors used for the various PDE4 isoforms have been described previously (Bolger et al., 1996; 1997; Huston et al., 1997; Owens et al., 1997).
Antibodies
The various antisera specific for each of the four PDE4 sub-families have been described elsewhere by us (Bolger et al., 1997; Huston et al., 1997; MacKenzie & Houslay, 2000; McPhee et al., 1999). Briefly, these were generated against fusion proteins formed between glutathione-S-transferase (GST) and portions of the extreme C-terminal regions that are unique to each of the PDE4 sub-families. Such regions are found in common to all active isoforms within each particular sub-family and thus antisera detect all isoforms within a particular sub-family.
This study describes a novel polyclonal antiserum (PS54-UCR1-A1) able to detect the (protein kinase A) phospho-serine form of the Arg-Arg-Glu-Ser-Phe motif found in the conserved UCR1 region of all long isoforms. This was generated using a phosphopeptide whose sequence was SQRRES*FLYRSDSDYDLSP. The phosphorylated serine residue is indicated (S*) as well as the PKA consensus sequence, which is underlined. This sequence reflects amino acids 49 – 67, inclusively, in PDE4D3.
We also generated a novel polyclonal antiserum (PS13-4D3-A1) able to detect the Ser13 PKA-phosphorylated form of PDE4D3. This was generated using a phosphopeptide whose sequence was FRRHS*WISFDVDNGTSAGR. The phosphorylated serine residue is indicated (S*) as well as the PKA consensus sequence, which is underlined. This sequence reflects amino acids 9 – 27, inclusively, in PDE4D3, except for position 16, where a serine residue was substituted for a cysteine residue to increase peptide stability.
In each case the peptides were synthesized with a phosphorylated seryl amino acid residue corresponding to positions 13 or 54, and an additional tyrosine amino acid was added at the C-terminus to allow conjugation to keyhole limpet haemocyanin (KLH). Peptides were synthesized, conjugated to KLH, and used for antisera production by Neosystems (Strasbourg, France). Female, pathogen-free New Zealand rabbits (weight 2 – 2.5 kg; Neosystems) were immunized four times over a three-month period, using 200 μg of coupled antigen in Freunds adjuvant. A portion of the recovered antisera was purified by peptide affinity chromatography at Cambridge Research Biochemicals (Cambridge, U.K.). No difference in results was obtained using affinity-purified antisera. We believe that this was because these peptides are not particularly immunogenic in their non-phosphorylated state. Indeed, we have been singularly unsuccessful in making antisera directed against the non-phosphorylated versions of these peptides (data not shown).
SDS – PAGE and Western blotting
Eight per cent acrylamide gels were used and the samples boiled for 5 min after being resuspended in Laemmli buffer (Laemmli, 1970). Gels were run at 8 mA gel−1 overnight or 50 mA gel for 4 – 5 h with cooling. For detection of transfected PDE by Western blotting, 2 – 50 μg protein samples were separated by SDS – PAGE and then transferred to nitrocellulose before being immunoblotted using the indicated specific antisera. Labelled bands were identified using peroxidase linked to anti-rabbit IgG and the Amersham ECL Western blotting kit was used as a visualization protocol.
Transient expression of PDE4 isoforms in COS1 cells
The generation of expression plasmids encoding the various PDE isoforms has been described in detail previously (Bolger et al., 1996; 1997; Hoffmann et al., 1999; Huston et al., 1997; McPhee et al., 1995; 1999; Owens et al., 1997). Transfection was done using the COS1 SV40-transformed monkey kidney cell line maintained at 37°C in an atmosphere of 5% CO2/95% air in complete growth medium containing DMEM supplemented with 0.1% penicillin/streptomycin (10000 units ml−1), glutamine (2 mM) and 10% FCS. We have described this before in some detail (Hoffmann et al., 1999). Briefly, however, COS1 cells were transfected using DEAE dextran. The DNA to be transfected (5 μg) was mixed, and incubated for 15 min with 250 μl of 10 mg ml−1 DEAE-dextran in PBS to give a DNA-dextran mix. When cells reached 70% confluence, in 100 mm dishes, the medium was removed and the cells were given 10 ml of fresh DMEM containing 0.1 mM chloroquine and the DNA-dextran mix (250 μl). The cells were then incubated for 4 h at 37°C. After this period the medium was removed and the cells shocked with 10% DMSO in PBS. After PBS washing, the cells were returned to normal growth medium and left for a further 2 days before use. For determination of PDE activity the cells were homogenized in KHEM buffer ((mM) KCl 50, EGTA 10, MgCl2 1.92, dithiothreitol 1, HEPES 50, final pH 7.2,) containing ‘complete' protease inhibitors (Boehringer – Mannheim) of final concentrations 40 μg ml−1 PMSF, 156 μg ml−1 benzamine, 1 μg ml−1 aprotonin, 1 μg ml−1 leupeptin, 1 μg ml−1 pepstatin A and 1 μg ml−1 antipain. As described previously (Baillie et al., 2000; MacKenzie et al., 2000), in such transfected cells then >98% of the total PDE activity was due to the recombinant PDE4 isoenzyme. In some instances the transfected COS1 cells were plated onto six-well plates for use in experiments and then serum-starved over night before being treated with the indicated ligands for the stated lengths of time.
Phosphorylation and dephosphorylation in vitro ofPDE4 isoenzymes
This was done as described previously (Hoffmann et al., 1998, 1999). The indicated PDE4 species from 5×105 transfected COS1 cells was immunoprecipitated as a complex with protein G-sepharose. This was incubated for 30 min at 4°C with one volume of phosphorylation buffer (100 mM-Tris-HCl, pH 7.5, 10 mM-MgCl2, 30 mM beta-mercaptoethanol, 10%-glycerol) containing 0.1 mM-γ-[32P]-ATP (100 Mbq mMol−1). The reaction was started by introduction of one unit of the free active catalytic unit of PKA (Upstate Biotechnology) and allowed to continue for a period of 30 min at 30°C. The sepharose was washed four times with 1 ml PDE-buffer and resuspended in PDE-buffer for analysis. Mock phosphorylation reactions were performed as described above but PKA catalytic subunit was omitted from the reaction mixtures. After such in vitro phosphorylation samples were subjected to SDS – PAGE with visualization by phosphorimager.
Dephosphorylation of PDE4D3 was done by taking lysate (90 μl) from COS1 cells transfected to express PDE4D3 and then adding to this 10 μl of 10× alkaline phosphatase reaction mixture (10 mM-TrisHCl, 1 mM-MgCl2, 0.1 mM-ZnCl2, 50 mM-KCl and 50% glycerol) with five units (1 U μl−1) of alkaline phosphatase. This was incubated at 30°C for 20 min after which a further five units of alkaline phosphatase was added and then the mixture incubated for a further 10 min at 30°C. The reaction was then stopped by the addition of 5× Laemmli sample buffer with boiling prior to analysis by SDS – PAGE.
Assay of intracellular cAMP
This was done as described previously by us (Baillie et al., 2001; Heyworth & Houslay, 1983; Tang & Houslay, 1992).
Assay of cAMP PDE activity
PDE activity was determined by a modification of the two-step procedure of Thompson & Appleman (1971) as described previously by us (Marchmont & Houslay, 1980) in 10 mM MgCl2, 20 mM TrisHCl buffer final pH 7.4. All assays were conducted at 30°C with initial rates taken from linear time-courses. Activity was linear with added protein concentration. Untransfected and mock (vector only)-transfected COS1 cells exhibited a PDE activity of 9±3 pmol min−1 mg protein−1.
Protein analysis
Protein concentration was determined using BSA as standard (Bradford, 1976). SDS – PAGE was done as described by Laemmli (Laemmli, 1970).
Results
Forskolin treatment activates long PDE4 isoforms expressed in transfected COS1 cells
COS-1 cells were transfected so as to express various PDE4 isoforms. The expression of these species was determined by immunoblotting using specific antisera (Figure 1). As noted previously by us (see e.g. Baillie et al., 2000; Huston et al., 1997; 2000; MacKenzie et al., 2000; McPhee et al., 1999), in all such transfected cells >98% of the total PDE activity was due to the activity of the recombinant PDE4 isoform.
Figure 1.
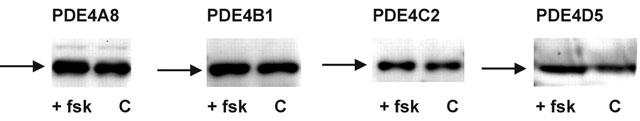
Expression of PDE4 isoforms in transfected COS1 cells. Shows immunoblots of COS1 cells transfected to express PDE4A8, PDE4B1, PDE4C2 and PDE4D5 long isoforms. Detection was performed using antisera specific for the particular PDE4 sub-class. In each instance a single immunoreactive species (arrowed), of the appropriate molecular weight, was identified using transfected cells. These were 98 kDa for PDE4A8, 104 kDa for PDE4B1, 80 kDa for PDE4C2 and 105 kDa for PDE4D5. Data are shown here for the indicated transfected cells that either had (+fsk) or had not (C) been treated with forskolin (100 μM) together with the non-selective, low affinity PDE inhibitor, IBMX (100 μM) for 15 min prior to harvesting. The data shown are typical of experiments done at least three times with different transfections.
Transfected COS-1 cells were challenged with the adenylyl cyclase activator, forskolin (100 μM) together with the non-selective PDE inhibitor IBMX (100 μM), in order to increase the intracellular concentration of cAMP (Tobias et al., 1997) (Table 1). Such a treatment led (Figure 2) to a time-dependent increase in the activity of examples of long isoforms from all four PDE4 subfamilies. This increase in PDE activity was, however, ablated (Figure 2) by co-challenge with the PKA inhibitor, H89 (0.5 μM).
Table 1.
PDE4 transfected COS1 cells
Figure 2.
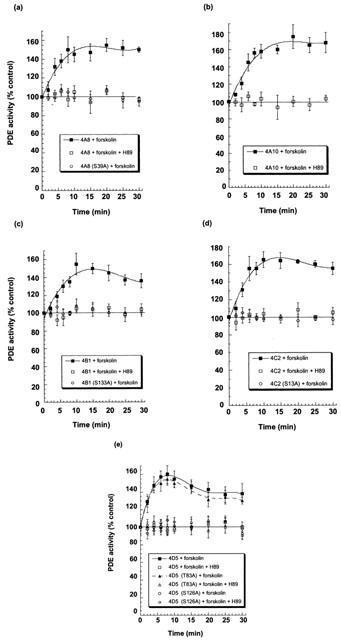
Activation of PDE4 long isoforms in forskolin-challenged cells. COS1 cells, transfected to express the indicated PDE4 isoforms, were challenged with the adenylyl cyclase activator, forskolin (100 μM), together with the non-specific PDE inhibitor, IBMX (100 μM). They were then harvested at the indicated time for analysis of PDE4 activity. COS1 cells were transfected with PDE4A8 (a), PDE4A10 (b), PDE4B1 (c), PDE4C2 (d) and PDE4D5 (e). In such cells then >98% of the total PDE activity was due to the transfected species whose activities are given in Table 1. Note that the IC50 value for inhibition of the various PDE4 isoforms by IBMX is around 30 μM. Thus, subsequent to cell harvesting, disruption and dilution of extract into the assay, a functionally insignificant level of IBMX (<1 nM) will be carried over into the PDE assay. Where indicated, the PKA selective inhibitor H89 (0.5 μM) was also added with forskolin and IBMX to the cells. As indicated, experiments were also performed using COS1 cells that were transfected to express various mutant PDE4 isoforms where the putative serine target in UCR1 for phosphorylation by PKA had been replaced with an alanine residue. These were Ser89Ala-PDE4A8, Ser133Ala-PDE4B1, Ser13Ala-PDE4C2 and Ser126Ala-PDE4D5. In addition to this the Thr83Ala-PDE4D5 mutant was also evaluated in order to delete a potential PKA phosphorylation site within the unique N-terminal region of PDE4D5. Activities are shown as a percentage relative to that of the control, untreated form. Data are given as means±s.d. for n=3 experiments done using separate transfections.
However, in marked contrast to the wild-type enzymes, forskolin challenge failed to elicit the activation (Figure 2) of long PDE4 isoforms where the putative serine target for PKA phosphorylation in UCR1 was changed to alanine. These mutant species were Ser89Ala-PDE4A8, Ser133Ala-PDE4B1, Ser13Ala-PDE4C2 and Ser126Ala-PDE4D5 (Figure 2).
Forskolin+IBMX challenge activated the PDE4A10 long isoform to a similar degree to that seen for PDE4A8 and this effect was also ablated by H89 (Figure 2). However, we were unable to generate the cognate Ser84Ala mutant form of PDE4A10. This is undoubtedly because the extremely GC-rich 5′ region of the cDNA encoding this isoform has been shown to be severely detrimental to PCR-based operations on this cDNA (Rena et al., 2001).
As well as having a PKA phosphorylation site in UCR1, PDE4D5 has a further potential site located within its unique N-terminal region, namely Thr83. This is found within the consensus sequence Arg-Arg-Phe-Thr83-Val. However, in forskolin-challenged COS1 cells the Thr83Ala mutant form of PDE4D5 was activated to a similar degree to wild-type PDE4D5 (Figure 2).
Protein kinase A can phosphorylate recombinant PDE4 isoforms
Lysates from COS1 cells transfected to express various PDE4 isoforms were treated with the free, activated form of the catalytic unit of PKA under phosphorylation conditions with 32P[ATP] (Figure 3). This showed that the PDE4A8, PDE4B1, PDE4C2 and PDE4D5 long isoforms all became radiolabelled (phosphorylated) upon such treatment (Figure 3). The phosphorylation of these proteins was, however, only observed in the presence of added activated PKA (Figure 3). Furthermore, phosphorylation was not observed when we used mutant forms of these various PDE4 isoforms where the putative serine target in UCR1 was changed to alanine (Figure 3).
Figure 3.
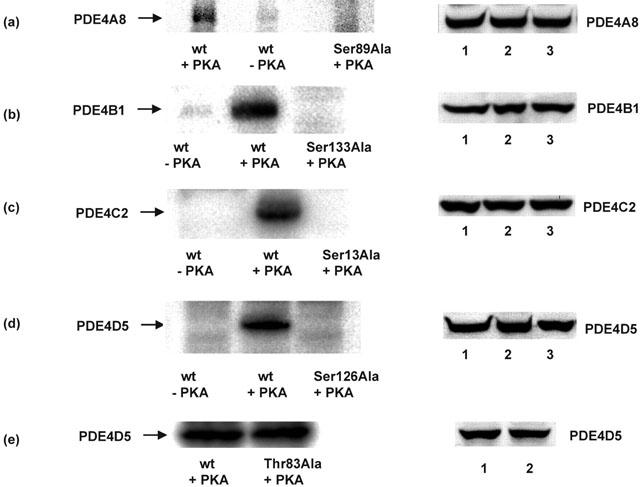
Phosphorylation of long PDE4 isoforms by PKA. COS1 cells were transfected to express various long PDE4 isoforms. Lysates containing the indicated recombinant wild-type (wt) forms were then treated with a phosphorylation mixture containing radiolabelled 32P-ATP in either the presence or absence of the activated catalytic unit of PKA. They were then subjected to SDS – PAGE with detection using a phosphorimager. In the presence of PKA then a radiolabelled species of the appropriate size, namely 98 kDa for PDE4A8 (a), 104 kDa for PDE4B1 (b), 80 kDa for PDE4C2 (c) and 105 kDa for PDE4D5 (d), was obtained in each instance. However, no such species were apparent for samples that had not been treated with PKA catalytic unit. Ser89Ala-PDE4A8 (a), Ser133Ala-PDE4B1 (b), Ser13Ala-PDE4C2 (c) and Ser126Ala-PDE4D5 (d) PDE4 isoforms were also used, as indicated, where the putative serine target site in UCR1 for phosphorylation by PKA had been mutated to alanine. In these instances data are shown for experiments where active PKA catalytic unit was added. Also shown (e) is data for the treatment, with activated PKA, of the Thr83Ala-PDE4D5 mutant form where the potential PKA phosphorylation site in the unique N-terminal region of PDE4D5 was mutated to alanine. In each case, equal immunoreactive amounts of the various PDE4 constructs were used in these experiments, as demonstrated by the immunoblots shown in the right hand panels using antisera specific for the indicated PDE4 sub-family analysed. In the immunoblots tracks 1, 2, 3 analyse, respectively, experiments of wt+PKA, wt – PKA and Ala mutant+PKA as in the left hand panel. The data shown are typical of experiments done at least three times with enzymes from different transfections.
The Thr83Ala-PDE4D5 mutant was phosphorylated by PKA to a similar extent to the wild-type enzyme (Figure 3). This indicates that this potential PKA phosphorylation site in the unique N-terminal region of PDE4D5 did not serve as a substrate for phosphorylation by PKA in the intact PDE4D5 enzyme. This may be due to steric hindrance due to the conformation of this region.
Challenge of transfected COS1 cells with forskolin causes the phosphorylation of PDE4 isoforms at a serine residue within UCR1
We wished to determine whether challenge of transfected COS cells with forskolin did indeed cause the PKA-mediated phosphorylation of the serine target within UCR1 of the various PDE4 isoforms in intact cells. To assess this we generated a novel antiserum to a phosphopeptide that spanned the target serine for PKA phosphorylation in UCR1 of PDE4D long isoforms. The sequence of this peptide was SQRRES*FLYRSDSDYDLSP, with the phosphorylated serine PKA target residue indicated (S*) and the PKA consensus sequence underlined. The cognate region to this is totally conserved in PDE4B long forms, and differs by only a single residue (shown in lower case) for long PDE4A forms, being SQRRES*FLYRSDSDYDmSP, and for PDE4C long forms, being SQRRES*FLYRSDSDYeLSP. The antiserum generated to be specific for the PKA phosphorylated serine target (S*) within UCR1 was called PS54-UCR1-A1.
Using the PS54-UCR1-A1 antiserum, we failed to observe (Figure 4a,b) any immunoreactive species in COS1 cells transfected to express the long PDE4D3 isoform unless cells had been challenged with forskolin+IBMX. This immunoreactive species was not detected if the phospho-peptide used to generate the PS54-UCR1-A1 antiserum was added to the immunoblotting mixture (Figure 4a). PDE4D3 is phosphorylated at both Ser13 and Ser54 by PKA (Sette & Conti, 1996; Sette et al., 1994a). The PS54-UCR1-A1 antiserum failed to identify an immunoreactive species in forskolin+IMBX treated COS1 cells transfected to express the Ser54Ala-PDE4D3 but did in cells expressing the Ser13Ala-4D3 mutant (Figure 4b). No immunoreactive species was evident when cells expressed a mutant with both sites mutated (Ser13Ala : Ser54Ala-PDE4D3; Figure 4b).
Figure 4.

Detection of phosphorylated long PDE4 isoforms using a novel antiserum specific for the PKA phosphorylated serine residue in UCR1. These data show the results of immunoblots done with the PS54-UCR1-A1 antiserum. COS-1 cells were transfected with the indicated PDE4 isoform and then harvested and analysed by Western blotting. In (a) cells were transfected with PDE4D3 and then either challenged or not, as indicated, with forskolin (100 μM) together with IBMX (100 μM) for 15 min. In one instance the antiserum was treated with a 10-fold excess of the peptide used to generate it in order to block interaction. The single immunoreactive species identified in the second lane migrated with immunoreactive PDE4D3 as identified by stripping and re-probing the blots using a PDE4D-specific antiserum (data not shown). In (b) cells were transfected with the indicated PDE4D3 mutant isoform and then either treated (fsk) or not with forskolin (100 μM) together with IBMX (100 μM), followed by harvesting and Western blotting. The mutants used were serine to alanine replacements as indicated. In (c) cells were transfected to express the indicated PDE4 long isoforms, either as wild-type or with the putative PKA target serine in UCR1 mutated to alanine. Cells were then treated or not with forskolin (100 μM) together with IBMX (100 μM), as indicated, for 15 min prior to harvesting and Western blotting. In some instances cells were additionally treated with H89 (0.5 μM) together with forskolin and IBMX. The single immunoreactive species identified in forskolin-treated cells transfected to express either PDE4A8 or PDE4B1 or PDE4C2 or PDE4D5 migrated as expected for these isoforms (see legend to Figure 1). In each comparative experiment we ensured that equal amounts of the indicated PDE4 long isoform was present. This was gauged by immunoblotting lysates with a PDE4 subfamily-specific antiserum in each case. This was then confirmed when the blots were stripped and re-probed for the appropriate isoform (data not shown). The data shown are representative of at least three separate experiments using different transfections.
Using the PS54-UCR1-A1 antiserum little or no immunoreactive species was observed (Figure 4c) for COS1 cells transfected to express the long PDE4A8, PDE4B1, PDE4C2 and PDE4D5 isoforms unless the cells had first been challenged with forskolin+IBMX. The appearance of such immunoreactive species was ablated by co-incubation of cells with H89 (Figure 4c). Additionally, no immunoreactive species were evident in forskolin+IBMX-challenged COS1 cells expressing PDE4 isoforms where the serine target site in UCR1 for PKA phosphorylation had been mutated to alanine (Figure 4c).
The altered mobility state associated with PKA-phosphorylated PDE4D3 is not a universal characteristic of PKA phosphorylated PDE4 isoforms
At least in certain cells, PKA phosphorylation of PDE4D3 appears to elicit a decrease in its mobility upon SDS – PAGE (Oki et al., 2000; Sette et al., 1994a). We show this here for PDE4D3 in transfected COS1 cells challenged with forskolin+IBMX (Figure 5a). Such a mobility shift could be reversed by treatment of PDE4D3, from forskolin+IBMX challenged cells, with alkaline phosphatase (Figure 5b). Intriguingly, we did not observe (Figure 1) any change in the mobility of PDE4A8, PDE4B1, PDE4C2 and PDE4D5 in transfected COS1 cells challenged with forskolin+IBMX to elicit the PKA phosphorylation of these isoforms.
Figure 5.

The PKA-mediated mobility shift of PDE4D3 requires phosphorylation at both Ser13 and Ser54. In (a) COS-1 cells were transfected to express the indicated wild-type or mutant forms of PDE4D3. They were then either treated (+fsk) or not (C) with forskolin (100 μM) together with IBMX (100 μM) for 10 min prior to harvesting. They were then identified using a PDE4D specific antiserum. In (b) is shown an experiment where cells were treated with forskolin together with IBMX for 10 min prior to harvesting and then samples were either treated with alkaline phosphatase (+ptase) or not (C) as described in Methods. The data shown are representative of at least three separate experiments using different transfections.
Unlike wild-type PDE4D3, neither the Ser54Ala- nor the Ser13Ala-PDE4D3 single mutant forms, nor the Ser13Ala : Ser54Ala-PDE4D3 double mutant, showed any major change in mobility on SDS – PAGE subsequent to challenge of cells with forskolin+IBMX (Figure 5a).
Figure 6a follows the time-course of altered mobility and phosphorylation of PDE4D3 in forskolin+IBMX-challenged COS1 cells. Immunoblotting, using a vesicular stomatis virus (VSV) glycoprotein specific monoclonal antibody, allowed us to follow time-dependent changes in the mobility of a transfected VSV epitope-tagged form of PDE4D3 in COS1 cells challenged with with forskolin+IBMX (Figure 6a; upper panel). At the start of the experiment, in the absence of forskolin, PDE4D3 migrated as a single ‘high mobility' species. However, a doublet was evident 1 – 2 min after forskolin challenge due to the appearance of a band of reduced mobility (Figure 6a; upper panel). If these blots were then stripped and reprobed with the PS54-UCR1-A1 antiserum, a very similar pattern was evident, except that no immunoreactive, phosphorylated species was evident at zero time (Figure 6a; middle panel). This antiserum detected two immunoreactive species (Figure 6a; middle panel) that migrated identically to the two species identified using the VSV monoclonal antibody (Figure 6a; upper panel).
Figure 6.
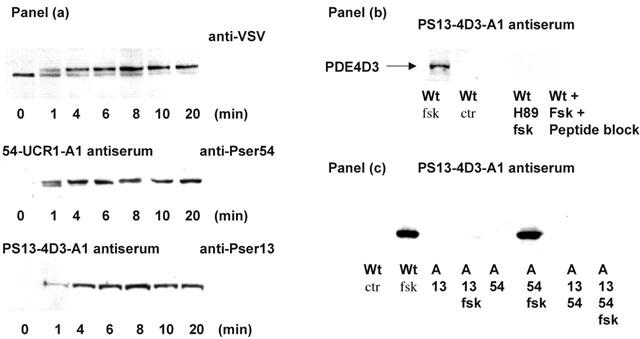
Time dependence of the PKA-induced mobility shift of PDE4D3 determined using novel antisera specific for the PKA phosphorylated serine residue in UCR1 and for Ser13 in the unique N-terminal region of PDE4D3. Panel (a) shows a time-course of the effect of challenging COS-1 cells transfected to express PDE4D3 with forskolin (100 μM) together with IBMX (100 μM). At the indicated times cells were harvested and then immunoblotted with antiserum specific for PDE4D (upper lane), with the PS54-UCR1-A1 antiserum (middle lane) and also with the PS13-4D3-A1 antiserum (lower lane). Lanes show data for equal loading of immunoreactive PDE4D3 in all experiments. The data shown are representative of at least three separate experiments using different transfections. Panel (b) shows the use of the novel polyclonal antiserum, PS13-4D3-A1, to detect the PKA-phosphorylated form of Ser13 in PDE4D3. Shown are lysates of transfected COS cells immunoblotted using this antiserum. COS1 cells transfected to express wild type PDE4D3 (wt) were either challenged (+fsk) or not (ctr) with forskolin together with IBMX for 10 min prior to harvesting. The single immunoreactive band identified in ‘+fsk' treated cells was identified as PDE4D3 by stripping and re-probing the blots with PDE4D-specific antiserum (data not shown). Also shown are lysates from ‘+fsk' treated PDE4D3 transfected COS1 cells where the polyclonal antiserum, PS13-4D3-A1, had been pre-absorbed with a 10-fold excess of the peptide used to generate it. Panel (c) shows immunoblots, done using antiserum PS13-4D3-A1, of COS1 cells transfected to express the indicated PDE4D3 mutants which were either treated (fsk) or not, as indicated, with forskolin together with IBMX for 10 min prior to harvesting. Used were wild-type PDE4D3 (wt), the Ser13Ala mutant, the Ser54Ala mutant and the Ser13Ala : Ser54Ala double mutant of PDE4D3. The data shown are representative of at least three separate experiments using different transfections.
PDE4D3 is also phosphorylated by PKA at Ser13 (Sette & Conti, 1996). To follow phosphorylation at this site we generated a further phospho-specific antiserum, in this case called PS13-4D3-A1. This detected an appropriately sized immunonoreactive species in PDE4D3-transfected COS1 cells that had been challenged with forskolin+IBMX but not in untreated cells (Figure 6b,c). The generation of this species was ablated with the PKA selective inhibitor, H89 and its detection was prevented by use of the blocking antigen phospho-peptide (Figure 6b). Such an immunoreactive species was not evident in forskolin+IBMX-challenged COS1 cells expressing either the Ser13Ala- or Ser13Ala : Ser54Ala-PDE4D3 mutant forms but was seen for the Ser54Ala-PDE4D3 mutant (Figure 6c). Additionally, incubation with the peptide used to generate the PS54-UCR1-A1 antiserum did not ablate detection of an immunoreactive species in forskolin+IBMX-treated COS1 cells expressing wild-type PDE4D3 (data not shown). The PS13-4D3-A1 antiserum thus appears to detect specifically the PKA-mediated phosphorylation of Ser13 in PDE4D3.
Indeed, as this phosphorylation site is found only in PDE4D3, such an antiserum will in fact uniquely detect such a phosphorylated PDE4D3 form. This proved to be the case as, using the PS13-4D3-A1 antiserum, we failed to identify immunoreactive species in forskolin-challenged COS1 cells transfected to express the various PDE4A, PDE4B and PDE4C forms analysed here, as well as the PDE4D5 isoform (data not shown).
We used the PS13-4D3-A1 antiserum to re-probe the blots from COS1 cells transfected to express PDE4D3 that had been challenged with forskolin for various periods of time (Figure 6a; lower panel). In the absence of forskolin, no immunoreactive species was evident (Figure 6a; lower panel). However, for all time periods studied, this phospho-Ser13-specific antiserum detected but a single immunoreactive species. This migrated with upper, reduced mobility form detected with both the anti-VSV mAb for recombinant PDE4D3 and the phospho-Ser54-specific antiserum (Figure 6a). The intensity of signal detected by the phospho-Ser13-specific antiserum increased over time, being consistent with an increased amount of Ser13-phosphorylated form of PDE4D3. However, unlike with the two other antisera, we never observed a doublet using the PS13-4D3-A1 antiserum (Figure 6a).
Rolipram inhibition of PKA-phosphorylated PDE4 isoforms
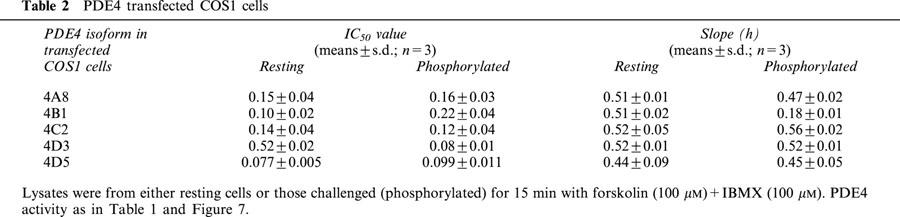
The susceptibility of the activity of the various recombinant PDE4 isoforms, expressed in transfected COS1 cells, to inhibition by the PDE4 selective rolipram was assessed using 1 μM cAMP as substrate (Figure 7). As reported before (Alvarez et al., 1995; Hoffmann et al., 1998; Sette & Conti, 1996; Sette et al., 1994a), we noted that PKA-activated PDE4D3 showed a heightened sensitivity to inhibition by rolipram (Figure 7f). In contrast to this, using PDE4 isoforms from the other three sub-families, we did not see any increase in their susceptibility to inhibition by rolipram (Figure 7; Table 2). Neither did we see increased sensitivity to rolipram inhibition for PDE4D5 (Figure 7e). The only apparent difference noted was for PDE4B1, for which the shallower slope of the inhibition plot implied heightened sensitivity to rolipram inhibition at lower concentration, for the phosphorylated form (Figure 7c).
Figure 7.
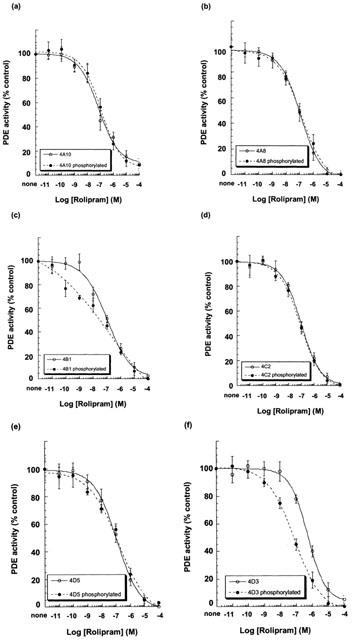
Rolipram inhibition of PKA-phosphorylated long PDE4 isoforms. High speed supernatant/soluble (S2) fractions were made from COS1 cells transfected to express the indicated PDE4 isoforms that either had or had not been treated with forskolin (100 μM) together with IBMX (100 μM) for 10 min prior to harvesting. The inhibitory effect of rolipram on the PDE activity was assessed using 1 μM cAMP as substrate. The indicated enzymes were (a) PDE4A10, (b) PDE4A8, (c) PDE4B1, (d) PDE4C2, (e) PDE4D5 and (f) PDE4D3. Activity of the various PDE4 species in transfected COS cells was as indicated in Table 1 and in Figure 2. The data presented give mean±s.d. of three separate experiments using different transfections.
Table 2.
PDE4 transfected COS1 cells
Discussion
It is now well established that the long PDE4D3 isoform provides a target for phosphorylation by PKA (Alvarez et al., 1995; Hoffmann et al., 1998; 1999; Oki et al., 2000; Sette & Conti, 1996; Sette et al., 1994a, 1994b) at both Ser13 located within its unique N-terminal region and Ser54 located within UCR1 (Hoffmann et al., 1998; Sette & Conti, 1996). PKA phosphorylation of PDE4D3 leads to a decrease in its mobility on SDS – PAGE (Oki et al., 2000; Sette & Conti, 1996) and enhanced sensitivity to inhibition by the PDE4 selective inhibitor, rolipram (Alvarez et al., 1995). It also generates an activated form of the enzyme that can be entirely attributed to phosphorylation of Ser54 (Hoffmann et al., 1998; Sette & Conti, 1996). However, three other PDE4 gene families generate a range of other long PDE4 isoforms. These all contain an identical PKA consensus sequence that is identical to that found in the UCR1 of PDE4D3, namely Arg-Arg-Glu-Ser-Phe. We have been able to show here that examples of long isoforms from each of the three other PDE4 sub-families can also be phosphorylated by PKA at the cognate serine within this site and that such a modification leads to enzyme activation. We ascertained that PKA caused the phosphorylation of this cognate serine residue using two independent approaches. Firstly, we generated various mutant PDE4 isoforms where the serine target for PKA phosphorylation in UCR1 was replaced by alanine. Such mutant forms could not be phosphorylated in vitro by treatment with activated PKA. Secondly, we generated a novel antiserum able to detect specifically the phosphorylated form of this serine residue in UCR1. This antiserum was then employed to detect the PKA-mediated phosphorylation of these various PDE4 long isoforms in intact cells. In addition, we showed that phosphorylation in intact cells treated with forskolin and IBMX to increase cAMP levels was ablated by the PKA inhibitor, H89. Thus it would seem that PKA phosphorylation of a cognate serine residue in UCR1, causing enzyme activation, is a common characteristic of long PDE4 isoforms from all four sub-families.
Here we have used forskolin to stimulate adenylyl cyclase activity and IBMX to reversibly inhibit the elevated PDE levels in PDE4 isoform-transfected cells so as to ensure that cAMP levels were sufficiently increased to allow PKA to phosphorylate these isoforms. This approach has allowed us to show that long isoforms from all four PDE4 sub-families can be phosphorylated and activated by PKA. As such, these analyses can be expected to serve as a paradigm for PDE4 expressed in cells where PKA is activated.
It would appear, however, that the PDE4D3 isoform exhibits certain unique attributes regarding its regulation by PKA. It has been shown previously to be phosphorylated at two distinct sites by PKA (Sette & Conti, 1996), unlike other long forms which we show here are phosphorylated solely at the common site in UCR1. Furthermore, we show that, of the various long PDE4 isoforms evaluated here, including PDE4D5 from the same sub-family, PDE4D3 is the only species where PKA phosphorylation engenders increased sensitivity to inhibition by rolipram and altered mobility on SDS – PAGE. In addition, where PDE4D3 appears to be activated some 2 – 3-fold by PKA phosphorylation (Hoffmann et al., 1998; Sette & Conti, 1996) this is somewhat greater than the activation we observe for other long PDE4 isoforms including PDE4D5. This suggests that the unique N-terminal region of PDE4D3 may serve to confer these unique attributes. In this regard, there is evidence that the N-terminal regions of PDE4A (Bolger et al., 1996; Shakur et al., 1993; 1995), PDE4B (Huston et al., 1997) and PDE4D (Bolger et al., 1997) families can affect functional aspects of these enzymes.
Our generation of antisera specific for the two PKA phosphorylation sites in PDE4D3 have allowed a unique insight into the regulation of this enzyme. Use of the PS54-UCR1-A1 antiserum identified that two populations of Ser54-phosphorylated PDE4D3 existed 1 min after challenge with forskolin+IBMX, one of which exhibited a mobility akin to that of dephosphorylated PDE4D3 while the other had a reduced mobility (Figure 6). In contrast to this the PS13-4D3-A1 antiserum only ever detected a single species of reduced mobility (Figure 6). This suggests to us that the phosphorylation of both sites is needed to attain the low mobility species and that the phosphorylation of Ser54 might even precede that of Ser13 in COS cells treated with forskolin+IBMX. Consistent with the notion that phosphorylation of both sites is needed to attain the low mobility state were our observations that mutation of either Ser13 or Ser54 to alanine prevented the PKA-mediated mobility shift in PDE4D3 (Figure 5).
Whilst the degree of activation by PKA of the various PDE4 isoforms analysed here may not seem at first site to be profound, Oki et al. (2000) have shown, for PDE4D3, that such elevated rates of hydrolysis of cAMP can exert a cumulative acceleration in the degradation of cAMP. Indeed, there is a growing body of evidence that PDE4 isoforms are anchored to specific sites in cells (Houslay, 2001; Houslay & Milligan, 1997; Houslay et al., 1998) and thus activation by PKA may be expected to provide a change in the steady-state levels of cAMP and activation of PKA, at least in a localized environment. This may be of particular importance as it is now well-established that anchored populations of PKA serve to sample and act on changes in gradients of cAMP within the cell interior (Colledge & Scott, 1999; Klauck & Scott, 1995). Indeed, it is now known (Colledge & Scott, 1999) that there is a very large family of PKA anchor proteins (AKAPs) that control specific intracellular events. Recently, it has been shown that PDE4D3 can interact with various AKAPs where it regulates localized PKA activity with feedback control provided by PKA phosphorylation (Dodge et al., 2001; Tasken et al., 2001). It is possible that other PKA-regulated PDE4 isoforms may act similarly with other AKAPs. Thus one might predict that the various PDE4 isoforms may have distinct functional roles in cells and thus sub-family selective PDE4 inhibitors might have rather different functional effects which could be exploited therapeutically.
Acknowledgments
M.D. Houslay thanks the Medical Research Council and Novartis for financial support of this study. T. McSorley thanks the MRC and Novartis for a Collaborative Studentship (M.D. Houslay, G. van Heeke).
Abbreviations
- AKAP
PKA anchor protein
- ECL
enhanced chemiluminescence
- GST
glutathione-S-transferase
- KLH
keyhole limpet haemocyanin
- PDE
cyclic nucleotide phosphodiesterase
- PDE4
rolipram-inhibited, cAMP-specific PDE
- PKA
cAMP-dependent protein kinase
- STR
serine target residue for phosphorylation by PKA located in UCR1
- UCR1
upstream conserved region 1
- UCR2
upstream conserved region 2
- (VSV)
vesicular stomatis virus.
References
- ALVAREZ R., SETTE C., YANG D., EGLEN R.M., WILHELM R., SHELTON E.R., CONTI M. Activation and selective inhibition of a cyclic AMP-specific phosphodiesterase, PDE-4D3. Mol. Pharmacol. 1995;48:616–622. [PubMed] [Google Scholar]
- BAILLIE G., MACKENZIE S.J., HOUSLAY M.D. Phorbol 12-myristate 13-acetate triggers the protein kinase A-Mediated phosphorylation and activation of the PDE4D5 cAMP phosphodiesterase in human aortic smooth muscle cells through a route involving Extracellular Signal Regulated Kinase (ERK) Mol. Pharmacol. 2001;60:1100–1111. doi: 10.1124/mol.60.5.1100. [DOI] [PubMed] [Google Scholar]
- BAILLIE G.S., MACKENZIE S.J., MCPHEE I., HOUSLAY M.D. Sub-family selective actions in the ability of Erk2 MAP kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br. J. Pharmacol. 2000;131:811–819. doi: 10.1038/sj.bjp.0703636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNETTE M.S. Phosphodiesterase 4 (PDE4) inhibitors in asthma and chronic obstructive pulmonary disease (COPD) Prog. Drug. Res. 1999;53:193–229. doi: 10.1007/978-3-0348-8735-9_5. [DOI] [PubMed] [Google Scholar]
- BEARD M.B., OLSEN A.E., JONES R.E., ERDOGAN S., HOUSLAY M.D., BOLGER G.B. UCR1 and UCR2 domains unique to the cAMP-specific phosphodiesterase (PDE4) family form a discrete module via electrostatic interactions. J. Biol. Chem. 2000;275:10349–10358. doi: 10.1074/jbc.275.14.10349. [DOI] [PubMed] [Google Scholar]
- BEAVO J.A. Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol. Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- BOLGER G. Molecular biology of the cyclic AMP-specific cyclic nucleotide phosphodiesterases: a diverse family of regulatory enzymes. Cell. Signal. 1994;6:851–859. doi: 10.1016/0898-6568(94)90018-3. [DOI] [PubMed] [Google Scholar]
- BOLGER G., MICHAELI T., MARTINS T., ST JOHN T., STEINER B., RODGERS L., RIGGS M., WIGLER M., FERGUSON K. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol. Cell. Biol. 1993;13:6558–6571. doi: 10.1128/mcb.13.10.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLGER G.B., ERDOGAN S., JONES R.E., LOUGHNEY K., SCOTLAND G., HOFFMANN R., WILKINSON I., FARRELL C., HOUSLAY M.D. Characterisation of five different proteins produced by alternatively spliced mRNAs from the human cAMP-specific phosphodiesterase PDE4D gene. Biochem. J. 1997;328:539–548. doi: 10.1042/bj3280539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLGER G.B., MCPHEE I., HOUSLAY M.D. Alternative splicing of cAMP-specific phosphodiesterase mRNA transcripts: characterization of a novel tissue-specific isoform, RNPDE4A8. J. Biol. Chem. 1996;271:1065–1071. doi: 10.1074/jbc.271.2.1065. [DOI] [PubMed] [Google Scholar]
- BRADFORD M. Protein determination in biological samples. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CAVALLA D., FRITH R. Phosphodiesterase IV inhibitors: structural diversity and therapeutic potential in asthma. Curr. Med. Chem. 1995;2:561–572. [Google Scholar]
- COLLEDGE M., SCOTT J.D. AKAPs: from structure to function. Trends Cell Biol. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- CONTI M., JIN S.L.C. The molecular biology of cyclic nucleotide phosphodiesterases. Prog. Nucl. Acid Res. 1999;63:1–38. doi: 10.1016/s0079-6603(08)60718-7. [DOI] [PubMed] [Google Scholar]
- DODGE K.L., KHOUANGSATHIENE S., KAPILOFF M.S., MOUTON R., HILL E.V., HOUSLAY M.D., LANGEBERG L.K., SCOTT J.D. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERDOGAN S., HOUSLAY M.D. Challenge of human Jurkat T-cells with the adenylate cyclase activator forskolin elicits a net increase in cyclic AMP phosphodiesterase activity by upregulating PDE3 and inducing PDE4D1 and PDE4D2 splice variants as well as downregulating a novel PDE4A splice variant. Biochem J. 1997;321:165–175. doi: 10.1042/bj3210165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIEMBYCZ M.A. Phosphodiesterase 4 inhibitors and the treatment of asthma: where are we now and where do we go from here. Drugs. 2000;59:193–212. doi: 10.2165/00003495-200059020-00004. [DOI] [PubMed] [Google Scholar]
- HE W., HUANG F.C., HANNEY B., SOUNESS J., MILLER B., LIANG G., MASON J., DJURIC S. Novel cyclic compounds as potent phosphodiesterase 4 inhibitors. J. Med. Chem. 1998;41:4216–4223. doi: 10.1021/jm970575f. [DOI] [PubMed] [Google Scholar]
- HEYWORTH C.M., HOUSLAY M.D. Insulin exerts actions through a distinct species of guanine nucleotide regulatory protein: inhibition of adenylate cyclase. Biochem. J. 1983;214:547–552. doi: 10.1042/bj2140547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFFMANN R., BAILLIE G.S., MACKENZIE S.J., YARWOOD S.J., HOUSLAY M.D. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase, HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999;18:893–903. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFFMANN R., WILKINSON I.R., MCCALLUM J.F., ENGELS P., HOUSLAY M.D. cAMP-specific phosphodiesterase HSPDE4D3 mutants which mimic activation and changes in rolipram inhibition triggered by protein kinase A phosphorylation of Ser-54: Generation of a molecular model. Biochem. J. 1998;333:139–149. doi: 10.1042/bj3330139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOUSLAY M.D. PDE4 cAMP-specific phosphodiesterases. Prog. Nucleic Acid Res. Mol. Biol. 2001;69:249–315. doi: 10.1016/s0079-6603(01)69049-4. [DOI] [PubMed] [Google Scholar]
- HOUSLAY M.D., MILLIGAN G. Tailoring cAMP signalling responses through isoform multiplicity. Trends Biochem. Sci. 1997;22:217–224. doi: 10.1016/s0968-0004(97)01050-5. [DOI] [PubMed] [Google Scholar]
- HOUSLAY M.D., SULLIVAN M., BOLGER G.B. The multi-enzyme PDE4 cyclic AMP specific phosphodiesterase family: intracellular targeting, regulation and selective inhibition by compounds exerting anti-inflammatory and anti-depressant actions. Adv. Pharmacol. 1998;44:225–342. doi: 10.1016/s1054-3589(08)60128-3. [DOI] [PubMed] [Google Scholar]
- HUSTON E., BEARD M., MCCALLUM F., PYNE N.J., VANDENABEELE P., SCOTLAND G., HOUSLAY M.D. The cAMP-specific phosphodiesterase PDE4A5 is cleaved downstream of its SH3 interaction domain by caspase-3: consequences for altered intracellular distribution. J. Biol. Chem. 2000;275:28063–28074. doi: 10.1074/jbc.M906144199. [DOI] [PubMed] [Google Scholar]
- HUSTON E., LUMB S., RUSSELL A., CATTERALL C., ROSS A.H., STEELE M.R., BOLGER G.B., PERRY M., OWENS R., HOUSLAY M.D. Molecular cloning and transient expression in COS7 cells of a novel human PDE4B cyclic AMP specific phosphodiesterase, HSPDE4B3. Biochem. J. 1997;328:549–558. doi: 10.1042/bj3280549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLAUCK T.M., SCOTT J.D. The postsynaptic density: a subcellular anchor for signal transduction enzymes. Cell. Signal. 1995;7:747–757. doi: 10.1016/0898-6568(95)02003-9. [DOI] [PubMed] [Google Scholar]
- KOVALA T., LORIMER I.A., BRICKENDEN A.M., BALL E.H., SANWAL B.D. Protein kinase A regulation of cAMP phosphodiesterase expression in rat skeletal myoblasts. J. Biol. Chem. 1994;269:8680–8685. [PubMed] [Google Scholar]
- LAEMMLI U.K. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature (London) 1970;222:680–682. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LIM J., PAHLKE G., CONTI M. Activation of the cAMP-specific phosphodiesterase PDE4D3 by phosphorylation: identification and function of an inhibitory domain. J. Biol. Chem. 1999;274:19677–19685. doi: 10.1074/jbc.274.28.19677. [DOI] [PubMed] [Google Scholar]
- MACKENZIE S.J., BAILLIE G.S., MCPHEE I., BOLGER G.B., HOUSLAY M.D. ERK2 MAP kinase binding, phosphorylation and regulation of PDE4D cAMP specific phosphodiesterases: the involvement of C-terminal docking sites and N-terminal UCR regions. J. Biol. Chem. 2000;275:16609–16617. doi: 10.1074/jbc.275.22.16609. [DOI] [PubMed] [Google Scholar]
- MACKENZIE S.J., HOUSLAY M.D. The action of rolipram on specific PDE4 cAMP phosphodiesterase isoforms and on the phosphorylation of CREB and p38 MAP kinase in U937 monocytic cells. Biochem. J. 2000;347:571–578. doi: 10.1042/0264-6021:3470571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANGANIELLO V.C., MURATA T., TAIRA M., BELFRAGE P., DEGERMAN E. Diversity in cyclic nucleotide phosphodiesterase isoenzyme families. Arch. Biochem. Biophys. 1995a;322:1–13. doi: 10.1006/abbi.1995.1429. [DOI] [PubMed] [Google Scholar]
- MANGANIELLO V.C., TAIRA M., DEGERMAN E., BELFRAGE P. Type III cGMP-inhibited cyclic nucleotide phosphodiesterases (PDE 3 gene family) Cell. Signal. 1995b;7:445–455. doi: 10.1016/0898-6568(95)00017-j. [DOI] [PubMed] [Google Scholar]
- MARCHMONT R.J., HOUSLAY M.D. An intrinsic and a peripheral protein constitute the cyclic AMP phosphodiesterase activity of rat liver plasma membranes. Biochem. J. 1980;187:381–392. doi: 10.1042/bj1870381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCPHEE I., POOLEY L., LOBBAN M., BOLGER G., HOUSLAY M.D. Identification, characterization and regional distribution in brain of RPDE-6 (RNPDE4A5), a novel splice variant of the PDE4A cyclic AMP phosphodiesterase family. Biochem. J. 1995;310:965–974. doi: 10.1042/bj3100965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCPHEE I., YARWOOD S.J., HUSTON E., SCOTLAND G., BEARD M.B., ROSS A.H., HOUSLAY E.S., HOUSLAY M.D. Association with the src family tyrosyl kinase lyn triggers a conformational change in the catalytic region of human cAMP-specific phosphodiesterase HSPDE4A4B: consequences for rolipram inhibition. J. Biol. Chem. 1999;274:11796–11810. doi: 10.1074/jbc.274.17.11796. [DOI] [PubMed] [Google Scholar]
- OKI N., TAKAHASHI S.I., HIDAKA H., CONTI M. Short term feedback regulation of cAMP in FRTL-5 thyroid cells: role of PDE4D3 phosphodiesterase activation. J. Biol. Chem. 2000;275:10831–1087. doi: 10.1074/jbc.275.15.10831. [DOI] [PubMed] [Google Scholar]
- OWENS R.J., LUMB S., REES-MILTON K., RUSSELL A., BALDOCK D., LANG V., CRABBE T., BALLESTEROS M., PERRY M.J. Molecular cloning and expression of a human phosphodiesterase 4C. Cell. Signal. 1997;9:575–585. doi: 10.1016/s0898-6568(97)00072-7. [DOI] [PubMed] [Google Scholar]
- RENA G., BEGG F., ROSS A., MACKENZIE C., MCPHEE I., CAMPBELL L., HUSTON E., SULLIVAN M., HOUSLAY M.D. Molecular cloning and characterization of the novel cAMP specific phosphodiesterase, PDE4A10. Mol. Pharmacol. 2001;59:996–1011. doi: 10.1124/mol.59.5.996. [DOI] [PubMed] [Google Scholar]
- ROGERS D.F., GIEMBYCZ M.A. Asthma therapy for the 21st century. Trends Pharmacol. Sci. 1998;19:160–164. doi: 10.1016/s0165-6147(98)01198-5. [DOI] [PubMed] [Google Scholar]
- SCHUDT C., TENOR H., HATZELMANN A. PDE isoenzymes as targets for anti-asthma drugs. Eur. Respir. J. 1995;8:1179–1183. doi: 10.1183/09031936.95.08071179. [DOI] [PubMed] [Google Scholar]
- SETTE C., CONTI M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase: involvement of serine 54 in the enzyme activation. J. Biol. Chem. 1996;271:16526–16534. doi: 10.1074/jbc.271.28.16526. [DOI] [PubMed] [Google Scholar]
- SETTE C., IONA S., CONTI M. The short-term activation of a rolipram-sensitive, cAMP-specific phosphodiesterase by thyroid-stimulating hormone in thyroid FRTL-5 cells is mediated by a cAMP-dependent phosphorylation. J. Biol. Chem. 1994a;269:9245–9252. [PubMed] [Google Scholar]
- SETTE C., VICINI E., CONTI M. The ratPDE3/IVd phosphodiesterase gene codes for multiple proteins differentially activated by cAMP-dependent protein kinase. J. Biol. Chem. 1994b;269:18271–18274. [PubMed] [Google Scholar]
- SEYBOLD J., NEWTON R., WRIGHT L., FINNEY P.A., SUTTORP N., BARNES P.J., ADCOCK I.M., GIEMBYCZ M.A. Induction of phosphodiesterases 3B, 4A4, 4D1, 4D2, and 4D3 in Jurkat T- cells and in human peripheral blood T-lymphocytes by 8-bromo-cAMP and Gs-coupled receptor agonists: potential role in beta2-adrenoreceptor desensitization. J. Biol. Chem. 1998;273:20575–20588. doi: 10.1074/jbc.273.32.20575. [DOI] [PubMed] [Google Scholar]
- SHAKUR Y., PRYDE J.G., HOUSLAY M.D. Engineered deletion of the unique N-terminal domain of the cyclic AMP-specific phosphodiesterase RD1 prevents plasma membrane association and the attainment of enhanced thermostability without altering its sensitivity to inhibition by rolipram. Biochem. J. 1993;292:677–686. doi: 10.1042/bj2920677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAKUR Y., WILSON M., POOLEY L., LOBBAN M., GRIFFITHS S.L., CAMPBELL A.M., BEATTIE J., DALY C., HOUSLAY M.D. Identification and characterization of the type-IVA cyclic AMP- specific phosphodiesterase RD1 as a membrane-bound protein expressed in cerebellum. Biochem. J. 1995;306:801–809. doi: 10.1042/bj3060801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SODERLING S.H., BEAVO J.A. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr. Opin. Cell. Biol. 2000;12:174–179. doi: 10.1016/s0955-0674(99)00073-3. [DOI] [PubMed] [Google Scholar]
- SOUNESS J.E., RAO S. Proposal for pharmacologically distinct conformers of PDE4. Cell. Signal. 1997;9:227–236. doi: 10.1016/s0898-6568(96)00173-8. [DOI] [PubMed] [Google Scholar]
- SPINA D., LANDELLS L.J., PAGE C.P. The role of phosphodiesterase enzymes in allergy and asthma. Adv. Pharmacol. 1998;44:33–89. doi: 10.1016/s1054-3589(08)60125-8. [DOI] [PubMed] [Google Scholar]
- SWINNEN J.V., JOSEPH D.R., CONTI M. The mRNA encoding a high-affinity cAMP phosphodiesterase is regulated by hormones and cAMP. Proc. Natl. Acad. Sci. U.S.A. 1989;86:8197–8201. doi: 10.1073/pnas.86.21.8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG E.K.Y., HOUSLAY M.D. Glucagon, vasopressin and angiotensin all elicit a rapid, transient increase in hepatocyte protein kinase C activity. Biochem. J. 1992;283:341–346. doi: 10.1042/bj2830341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TASKEN K.A., COLLAS P., KEMMNER W.A., WITCZAK O., CONTI M., TASKEN K. Phosphodiesterase 4D and protein kinase A type II constitute a signaling unit in the centrosomal area. J. Biol. Chem. 2001;276:21999–2002. doi: 10.1074/jbc.C000911200. [DOI] [PubMed] [Google Scholar]
- THOMPSON W.J. Cyclic nucleotide phosphodiesterases: pharmacology, biochemistry and function. Pharmacol. Ther. 1991;51:13–33. doi: 10.1016/0163-7258(91)90039-o. [DOI] [PubMed] [Google Scholar]
- THOMPSON W.J., APPLEMAN M.M. Multiple cyclic nucleotide phosphodiesterase activities from rat brain. Biochem. 1971;10:311–316. [PubMed] [Google Scholar]
- TOBIAS E.S., ROZENGURT E., CONNELL J.M., HOUSLAY M.D. Co-transfection with protein kinase D confers phorbol-ester-mediated inhibition on glucagon-stimulated cAMP accumulation in COS cells transfected to overexpress glucagon receptors. Biochem. J. 1997;326:545–551. doi: 10.1042/bj3260545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORPHY T.J. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am. J. Respir. Crit. Care Med. 1998;157:351–370. doi: 10.1164/ajrccm.157.2.9708012. [DOI] [PubMed] [Google Scholar]
- VERGHESE M.W., MCCONNELL R.T., LENHARD J.M., HAMACHER L., JIN S.-L.C. Regulation of distinct cyclic AMP-specific phosphodiesterase (PDE4) isozymes in human monocytic cells. Mol. Pharmacol. 1995;47:1164–1171. [PubMed] [Google Scholar]
- VICINI E., CONTI M. Characterization of an intronic promoter of a cyclic adenosine 3′,5′- monophosphate (cAMP) – specific phosphodiesterase gene that confers hormone and cAMP inducibility. Mol. Endocrinol. 1997;11:839–850. doi: 10.1210/mend.11.7.9941. [DOI] [PubMed] [Google Scholar]
- XU R.X., HASSELL A.M., VANDERWALL D., LAMBERT M.H., HOLMES W.D., LUTHER M.A., ROCQUE W.J., MILBURN M.V., ZHAO Y., KE H., NOLTE R.T. Atomic structure of PDE4: insights into phosphodiesterase mechanism and specificity. Science. 2000;288:1822–1825. doi: 10.1126/science.288.5472.1822. [DOI] [PubMed] [Google Scholar]