

Abstract
Protease-activated receptor (PAR)-mediated vascular relaxations have been compared in coronary arteries of different diameters isolated from both humans and pigs.
Thrombin, trypsin, and the PAR1-activating peptide, TFLLR, all caused concentration-dependent relaxation of both large (epicardial; ∼2 mm internal diameter) and small (intramyocardial; ∼200 μm internal diameter) human coronary arteries. EC50 values for thrombin (0.006 u ml−1 in epicardial, 1.69 u ml−1 in intramyocardial) and trypsin (0.02 u ml−1 in epicardial, 1.05 u ml−1 in intramyocardial) were significantly (P<0.01) greater in intramyocardial arteries. By contrast, EC50 values for TFLLR were not different between epicardial (0.35 μM) and intramyocardial (0.43 μM) arteries.
In porcine coronary arteries, EC50 values for relaxations to thrombin (0.03 u ml−1 in epicardial 0.17 u ml−1 in intramyocardial) were also significantly (P<0.01) greater in the smaller arteries. EC50 values for both TFLLR and the PAR2-activating peptide, SLIGKV, were not different between the two different-sized pig coronary arteries.
PAR1-immunoreactivity was localized to the endothelium of human epicardial and intramyocardial arteries and both PAR1- and PAR2-immunoreactivity was observed in endothelial cells of equivalent porcine arteries.
These findings indicate that enzymatic activation of endothelial cell PARs in human (PAR1) and porcine (PAR1 and PAR2) coronary arteries is markedly reduced in intramyocardial arteries when compared with epicardial arteries, suggesting increased regulation of PAR-mediated vascular responses in resistance-type arteries.
Keywords: Protease-activated receptors, human coronary artery, smooth muscle relaxation, endothelium, thrombin, trypsin
Introduction
Protease-activated receptor-1 (PAR1) and PAR2 are serine protease-sensitive, G protein-coupled receptors which are thought to be widely expressed on vascular endothelial cells and as such may play important roles in the control of vascular tone and haemostasis (for review, see Coughlin, 1994; Déry et al., 1998; Cocks & Moffatt, 2000; Coughlin, 2000). PAR1 is essentially a thrombin receptor (Vu et al., 1991; Chen et al., 1994) but can be activated by other serine proteases at comparatively high concentrations, including trypsin (Kawabata et al., 1999; Altrogge & Monard, 2000). PAR2 is insensitive to thrombin cleavage and is activated most sensitively by trypsin (Nystedt et al., 1994; Böhm et al., 1996; Altrogge & Monard, 2000), but also by blood-borne trypsin-like enzymes such as mast cell tryptase (Fox et al., 1997; Mirza et al., 1997; Molino et al., 1997a) and coagulation factor VIIa (Camerer et al., 2000). For both PAR1 and PAR2, receptor activation can be mimicked pharmacologically by synthetic peptides corresponding to the distinct cleavage-derived tethered ligand sequences of each receptor (Vu et al., 1991; Böhm et al., 1996; Blackhart et al., 1996). The tethered ligand sequences of the cloned human PAR1 and PAR2 are SFLLRN (Vu et al., 1991) and SLIGKV (Böhm et al., 1996), respectively. However, with SFLLRN shown to activate PAR2 in addition to PAR1 (Blackhart et al., 1996; Al-Ani et al., 1999; Kawabata et al., 1999), a more selective PAR1-activating peptide, TFLLR (Blackhart et al., 1996; Hollenberg et al., 1997), has been developed.
PAR1- and PAR2-induced endothelium-dependent relaxation has been previously reported in a number of species. For example, activation of PAR1 (with thrombin or PAR1-activating peptides) or PAR2 (with trypsin or PAR2-activating peptides) induces endothelium-dependent relaxation of pig coronary artery (Hwa et al., 1996; Hamilton et al., 1999) and rat aorta (Magazine et al., 1996; Saifeddine et al., 1996; Hollenberg et al., 1997). One reported anomaly to such vascular PAR pharmacology is in human isolated coronary (Hamilton et al., 1998) and pulmonary (Hamilton et al., 2001b) arteries, in which thrombin, trypsin, and PAR1-activating peptides all cause endothelium-dependent relaxation via activation of a common PAR1-like receptor, with no role for PAR2.
The contribution of PARs to serine protease-induced changes in vascular reactivity have been well characterized in several species, including humans. However, such studies have been performed exclusively in large, conduit-type arteries. Although such arteries can provide some degree of blood flow control, it is the smaller arteries and resistance arterioles that contribute most to the regulation of flow (Epstein et al., 1985). For example, small arteries and arterioles account for more than 45% of peripheral resistance and thus are the major contributing vessels to systemic blood pressure (Heagerty & Izzard, 1995). In the heart, intramyocardial arteries of ∼300 μm internal diameter are estimated to contribute 20% of total coronary resistance (Chilian et al., 1986). Since several studies have reported diameter-dependent variations in a number of different receptor- and ion channel-dependent changes in vascular reactivity (Chilian et al., 1989; Lamping et al., 1989; Jones et al., 1995; Bowles et al., 1997; Gitterman & Evans, 2000), the aim of the present study was to determine whether resistance-like coronary arteries relax in response to PAR activation in a similar manner to larger, conduit-type coronary arteries. To this end, we have compared PAR-mediated relaxation in small, resistance-type intramyocardial arteries (∼200 μm internal diameter) with the well characterized responses in large, conduit-type epicardial arteries (∼2 mm internal diameter) from humans and pigs.
Methods
Tissue source
Human epicardial arteries (right main, left anterior descending, left circumflex) were dissected from the explanted hearts of patients undergoing transplantation at The Alfred Hospital (Melbourne, Australia). Heart tissue came from 11 patients (eight male, three female; 43±3 years, range 28 to 62 years) diagnosed with dilated congestive cardiomyopathy (n=6), restrictive cardiomyopathy (n=3), ischaemic heart disease (n=1), or septal defects (n=1).
Human intramyocardial arteries were dissected from distal segments of right atrial appendage which were obtained from 27 patients (22 male, five female; 62±2 years, range 41 to 75 years) undergoing coronary artery graft surgery at the Royal Melbourne Hospital (Melbourne, Australia). Intramyocardial arteries were also isolated from right atrial appendage segments of two of the heart transplant patients in the group described above. All human tissue was collected with informed patient consent and approval from the Ethics Committees of The University of Melbourne and either the Royal Melbourne Hospital or The Alfred Hospital.
Porcine epicardial and intramyocardial arteries were isolated from the heart of Large White pigs (either sex; 30–40 kg) obtained from a local abattoir. In order to obtain porcine arteries from a similar anatomical location and of a similar size as the human coronary arteries used, epicardial arteries were the right main artery and intramyocardial arteries were dissected from the distal portion of the right atrial appendage.
Immunohistochemical studies
Epicardial arteries and segments of atrial appendage containing intramyocardial arteries, each isolated from both humans and pigs, were fixed in paraformaldehyde (4% w v−1 in phosphate buffered saline (PBS), pH 7.3) at 4°C overnight. Tissues were then washed in PBS, placed in a sucrose solution (15% w v−1 in PBS) at 4°C overnight, embedded in Tissue-Tek® OCT Compound, frozen on liquid nitrogen, and stored at −70°C until 14 μm sections were cut onto gelatin- (0.1% in water) and formalin- (0.2% in water) coated slides at −16°C using a cryostat.
Tissue sections were washed free of OCT using PBS and incubated with the primary antibody at room temperature for 48 h. PAR1 and PAR2 were localized using previously characterized IgG2a monoclonal antibodies produced in mice immunized with peptides corresponding to the amino-terminus of human PAR1 (S42FLLRNPNDKYEPF55, 1 : 100: (Brass et al., 1992; Hoxie et al., 1993) and human PAR2 (S37LIGKVDGTSHVTG50, 1 : 20: (Molino et al., 1997a,1997b). These primary antisera were sequentially labelled at room temperature with a biotinylated sheep anti-mouse antiserum (1 : 100, 1 h) and fluorescein isothiocyanate (FITC)-conjugated steptavidin (1 : 20, 1 h) before being washed with PBS and mounted in buffered glycerol. For epicardial arteries, fluorescence was observed under a microscope (Axioskop, Zeiss, Germany). For intramyocardial arteries, fluorescence was recorded using a confocal microscope (MRC 1000, Biorad, Australia).
Isometric tension studies: epicardial arteries
Three mm-long ring segments of human or porcine epicardial arteries were mounted in organ chambers for isometric tension measurement as previously described (Kilpatrick & Cocks, 1994; Hamilton & Cocks, 2000). Briefly, artery ring segments, which were only used if they were macroscopically free of atheromatous plaques, were mounted between two wire hooks – one attached to an adjustable support and the other to a force transducer – and immersed in 10 ml organ baths containing Krebs solution (composition (mmol/L): Na+ 144, Cl− 128.7, HCO3− 25, K+ 5.9, Ca2+ 2.5, Mg2+ 1.2, H2PO4− 1.2, SO42− 1.2 and glucose 11; pH 7.4) maintained at 37°C and continuously bubbled with 95% O2, 5% CO2.
Following a 60 min equilibration period, artery ring preparations were stretched to 5 g passive force and allowed to recover for 30 min before again being stretched to 5 g. After a further 30 min, tissues were exposed to an isotonic, high potassium Krebs solution (KPSS: composition in mM: K+ 124.9, Cl− 128.7, Na+ 25.0, HCO3− 25.0, Ca2+ 2.5, Mg2+ 1.2, SO42− 1.2, H2PO4− 1.2, glucose 6.1) to obtain a maximum contraction for each artery ring (KPSSmax: Kilpatrick & Cocks, 1994; Drummond & Cocks, 1996). The KPSS was then replaced with normal Krebs solution and the tissues allowed to return to their optimal passive force level over 60 min.
Tissues were contracted to ∼50% KPSSmax with titrated concentrations of the thromboxane A2 mimetic, U46619 (1–100 nM). Once the U46619-induced contraction had reached a stable plateau, cumulative half-log concentrations of the various PAR agonists were added to the organ bath. The maximum endothelium-dependent and -independent relaxation of each ring preparation was then determined with the addition of bradykinin (0.3 μM; pig) or substance P (10 nM; human), and isoprenaline (1 μM), respectively.
Isometric tension studies: intramyocardial arteries
Analogous to the organ bath set-up used for the larger epicardial arteries described above, 2 mm-long artery ring segments of human and porcine isolated intramyocardial arteries (internal diameter ∼200 μm) were mounted between two parallel wires in a Mulvany-Halpern myograph (model 410A, J.P. Trading, Denmark) containing 5 ml Krebs solution, heated to 37°C and continuously oxygenated with 95% O2, 5% CO2 (Mulvany & Halpern, 1977; Buus et al., 2000).
In an equilibration protocol similar to that used for the epicardial artery studies, porcine and human intramyocardial arteries were left to equilibrate at 37°C with zero tension for 60 min. The initial tension applied to each artery was then normalized to 90% of the internal arterial circumference (IC) if the artery was exposed to a transmural distending pressure of 100 mmHg (0.9.IC100: Mulvany & Halpern, 1977). After 30 min equilibration at 0.9.IC100 arteries were exposed to KPSS until the KPSSmax was obtained, before being reimmersed in Krebs to return the tissues to their baseline passive tension levels (0.9.IC100). All artery segments were contracted to ∼50% of their KPSSmax. For porcine coronary arteries, this was achieved with titrated concentrations of U46619 (typically 1–100 nM). U46619 at concentrations of up to 1 μM induced only small (<10%) contractions in human coronary arteries such that ACh (typically 0.03–1 μM) was used to contract these arteries to ∼50% KPSSmax. Importantly, ACh causes no endothelium-dependent relaxation of human coronary arteries (Kemp & Cocks, 1997). For all arteries, once the U46619- or ACh-induced contractions were stable, cumulative half-log concentrations of the various PAR agonists were added to the myograph chamber and when a maximum response was obtained to the PAR agonists, bradykinin (0.3 μM; pig) or substance P (30 nM; human) and then isoprenaline (1 μM) were added to determine the maximum endothelium-dependent and -independent relaxation of each ring preparation, respectively.
Statistical analyses
All relaxations were normalized as a per cent of each tissue's response to 1 μM isoprenaline and all data are expressed as means±s.e.mean. Normalized concentration-response curves were computer fitted to a sigmoidal regression curve (Graphpad Prism, Graphpad Software Inc.) to generate values for sensitivity (EC50). Differences in mean EC50 and maximum response (Rmax) were tested for significance by unpaired Student's t-tests. In all cases, differences were considered significant when P<0.05.
Drugs
The PAR1 and PAR2 antisera were kind gifts from Prof Lawrence Brass (University of Pennsylvania, PA, U.S.A.). Anti-rabbit antiserum and FITC-streptavidin were from Amersham Pharmacia Biotech (Bucks, U.K.) and OCT from Sakura Finetek (CA, U.S.A.). U46619 was from Sapphire Bioscience (NSW, Australia), trypsin (bovine pancreas) from Worthington Biochem (NJ, U.S.A.) and Hoe-140 from Hoechst (VIC, Australia). Endothelin-1, (−)-isoprenaline, substance P (acetate salt), and α-thrombin (bovine serum) were purchased from Sigma (MO, U.S.A.). All PAR-activating peptides were synthesized with carboxyl-terminal amidation and purified by reverse phase HPLC to ⩾95% by Auspep (VIC, Australia).
Results
Isometric tension studies: human coronary arteries
Thrombin caused concentration-dependent (EC50 0.006±0.001 u ml−1) and near-maximal (Rmax 90.8±2.2%) relaxations of contracted epicardial artery ring segments (n=9, from nine patients; Figure 1). In intramyocardial artery rings, thrombin caused relaxations with a similar Rmax (90.3±2.0%) but significantly (P<0.01) increased EC50 (1.69±0.45 u ml−1) when compared with relaxations of epicardial arteries (n=12, from 12 patients; Figure 1). Similarly, trypsin-induced relaxations of epicardial artery ring segments had a similar Rmax (89.9±2.3%) but significantly (P<0.01) decreased EC50 (0.02±0.003 u ml−1; n=9, from nine patients) when compared with relaxations in intramyocardial arteries (Rmax 77.9±4.4%, EC50 1.05±0.22 u ml−1; n=20, from 20 patients; Figure 1). In contrast to thrombin and trypsin, the selective PAR1-activating peptide, TFLLR, caused concentration-dependent relaxations of epicardial arteries (Rmax 91.7±3.3%, EC50 0.35±0.15 μM; n=8, from eight patients) which were not different to those in intramyocardial arteries (Rmax 93.4±1.3%, EC50 0.43±0.09 μM; n=5, from five patients; Figure 1). Neither the human PAR2- nor PAR4-activating peptides, SLIGKV and GYPGQV respectively, caused relaxations or contractions at concentrations of up to 300 μM in either epicardial or intramyocardial arteries (data not shown).
Figure 1.
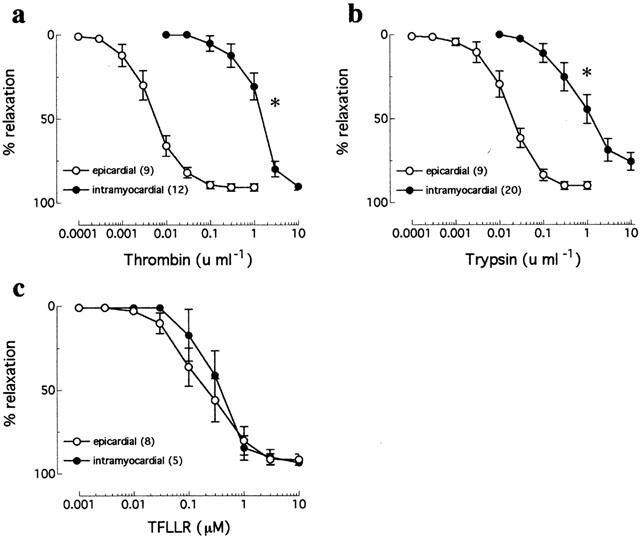
Cumulative concentration-response curves to (a) thrombin, (b) trypsin, and (c) TFLLR in human isolated epicardial and intramyocardial artery ring preparations contracted to ∼50% KPSSmax. Data are means±s.e.mean of 5–20 experiments (indicated in parentheses). *Denotes significantly different EC50 values (P<0.01).
In intramyocardial arteries, neither thrombin- nor trypsin-induced relaxations were affected by the B2 kinin receptor antagonist, Hoe-140 (1 μM; n=4, from four patients; Figure 2). By contrast, Hoe-140 caused a significant (P=0.001), ∼75 fold increase in EC50 to bradykinin but did not affect the Rmax (control=89.0±7.9% versus Hoe-140=76.9±10.8%) (n=4, from four patients; Figure 2). Thrombin- and trypsin-induced relaxations of intramyocardial arteries were similarly unaffected by the mixed ETA/ETB receptor antagonist, bosentan (10 μM; n=3, from three patients; Figure 3). Bosentan, however, inhibit both the initial contraction and subsequent relaxation caused by endothelin-1 in this tissue (Figure 3). Thrombin- and trypsin-induced relaxations of intramyocardial arteries were abolished by treatment of thrombin with D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (PPACK; 10 fold excess) and soybean trypsin inhibitor (10 mg ml−1), respectively (data not shown).
Figure 2.
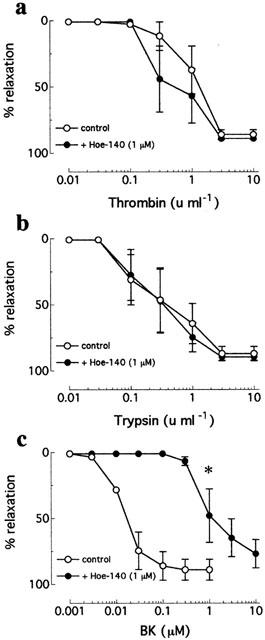
Cumulative concentration-response curves to (a) thrombin, (b) trypsin, and (c) bradykinin in human isolated intramyocardial artery rings contracted to ∼50% KPSSmax in the absence and presence of Hoe-140 (1 μM). Data are means±s.e.mean from four experiments. *Denotes EC50 value significantly different from the control (P=0.001).
Figure 3.
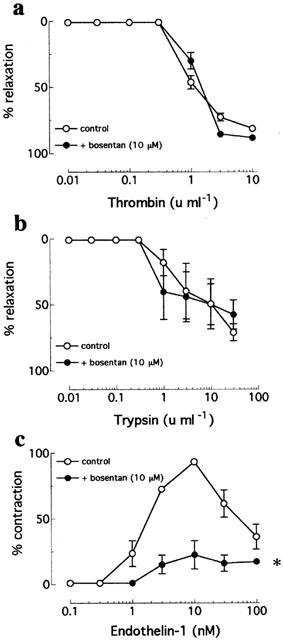
Cumulative concentration-response curves to (a) thrombin, (b) trypsin, and (c) endothelin-1 in human isolated intramyocardial artery rings contracted to ∼50% KPSSmax in the absence and presence of bosentan (10 μM). Data are means±s.e.mean from three experiments. *Denotes EC50 value significantly different from the control (P<0.001).
Immunohistochemical studies: human coronary arteries
Patchy PAR1-immunoreactivity was observed in endothelial cells of both epicardial and intramyocardial arteries, with less than half of the endothelial cells present staining with the PAR1-targeted antibody (Figure 4). However, PAR1-immunoreactivity appeared specific to the endothelium with little staining observed in the smooth muscle of either artery type (Figure 4).
Figure 4.
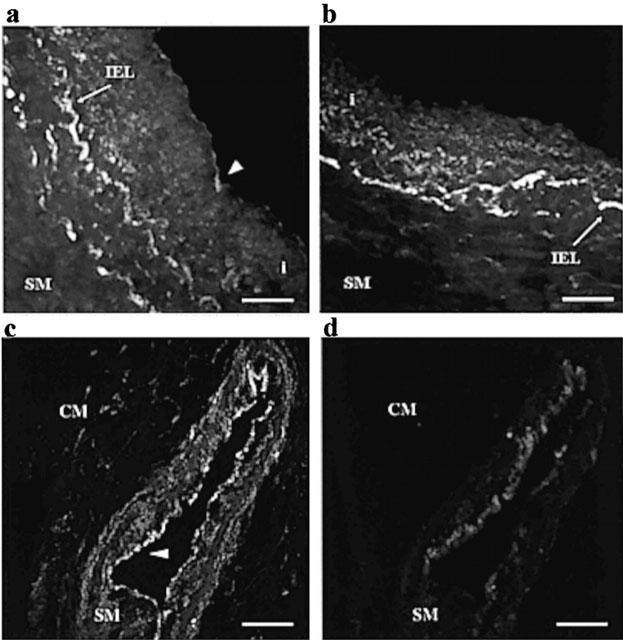
Digitized photographs of 14 μm transverse sections of (a, b) epicardial and (c, d) intramyocardial human coronary arteries showing immunoreactivities for (a, c) PAR1 and (b, d) in the absence of the primary antibody. Note staining in sections of the endothelium (arrow heads). Note also the autofluorescence of the internal elastic lamina (IEL) of the epicardial artery which persists in the absence of the primary antibody (b). Photographs are representative of results from the arteries of three patients. In each case the scale bar represents 40 μm. SM=smooth muscle, CM=cardiac muscle, i=intima.
Isometric tension studies: porcine coronary arteries
Thrombin caused concentration-dependent relaxations of epicardial arteries (EC50 0.03±0.01 u ml−1, Rmax 87.0±3.0%; n=6; Figure 5). In animal-matched intramyocardial arteries, thrombin-induced relaxations had a significantly (P<0.01) greater EC50 (0.17±0.07 u ml−1) but similar Rmax (93.0±1.0%) (n=6; Figure 5). Similarly, relaxations to trypsin in epicardial arteries (EC50 0.01±0.002 u ml−1, Rmax 90.4±2.4%; n=5) had a significantly (P<0.001) greater EC50 (0.42±0.28 u ml−1), but similar Rmax (89.0±3.8%) in intramyocardial arteries from the same animals (n=5; Figure 5).
Figure 5.

Cumulative concentration-response curves to (a) thrombin, (b) trypsin, (c) TFLLR, and (d) SLIGKV in porcine isolated epicardial and intramyocardial artery ring preparations contracted to ∼50% KPSSmax. Data are means±s.e.mean from 4–6 experiments. *Denotes significantly different EC50 values (P<0.01). Note that in all experiments, epicardial and intramyocardial arteries were animal-matched.
In contrast to thrombin and trypsin, relaxations to the PAR1-selective activating peptide, TFLLR, in epicardial arteries (EC50 0.28±0.05 μM, Rmax 95.3±1.0%) were not different to those in animal-matched intramyocardial arteries (EC50 0.18±0.33 μM, Rmax 89.9±5.1%) (n=5; Figure 5). Also, relaxations to the PAR2-selective activating peptide, SLIGKV, in epicardial arteries (EC50 0.51±0.12 μM, Rmax 94.3±1.9%) were not different to those in animal-matched intramyocardial arteries (EC50 0.78±0.20 μM, Rmax 80.9±6.5%) (n=4; Figure 5). As in the human intramyocardial arteries, relaxations to thrombin and trypsin in porcine intramyocardial arteries were abolished by PPACK treatment (10 fold excess) and soybean trypsin inhibitor (10 mg ml−1), respectively (data not shown). Also, the PAR4-activating peptide, GYPGQV, caused neither contraction nor relaxation at concentrations of up to 300 μM (not shown).
Immunohistochemical studies: porcine coronary arteries
PAR1- and PAR2-immunoreactivity was observed in the endothelial cell layer of both epicardial and intramyocardial coronary arteries (Figure 6). In contrast to the observations with PAR1 in the human arteries, significant PAR1- and PAR2-immunoreactivity was detected in the smooth muscle layer of the intramyocardial arteries (Figure 6).
Figure 6.
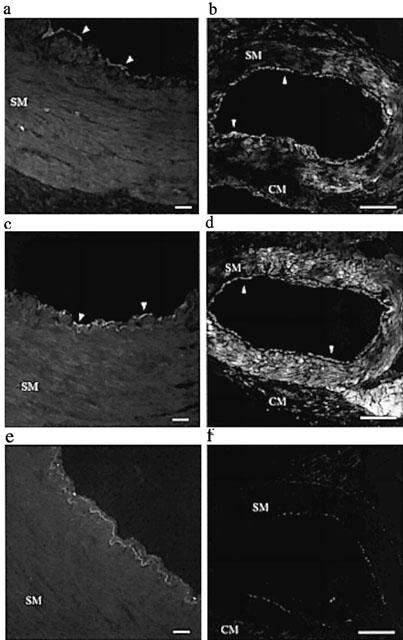
Digitized photographs of 14 μm transverse sections of (a, c, e) epicardial and (b, d, f) intramyocardial porcine isolated coronary arteries showing immunoreactivities for (a, b) PAR1, (c, d) PAR2, and (e, f) in the absence of a primary antibody. Note staining in sections of the endothelium (arrow heads). Photographs are representative of results from the arteries of three animals. In each case the scale bar represents 40 μm. SM=smooth muscle, CM=cardiac muscle.
Discussion
To our knowledge, this study is the first to report PAR localization in resistance-like arteries and is the first to directly examine PAR-mediated vascular relaxation of such arteries. The major finding of the present study, however, was that enzyme-induced PAR-mediated relaxation of both porcine and human isolated coronary arteries was markedly dependent on artery diameter.
Since there have been no previous reports of PARs in resistance-like arteries, for the purpose of comparison we chose to investigate such responses in two species, human and pig, in which PAR-mediated relaxations of corresponding large conduit-type arteries have been well characterized. Thus, in human isolated epicardial arteries, thrombin and trypsin cause endothelium-dependent relaxation via a common, PAR1-like receptor, with no role for PAR2 (Hamilton et al., 1998). By contrast, thrombin and trypsin cause endothelium-dependent relaxation of porcine isolated epicardial arteries predominantly via activation of PAR1 and PAR2, respectively (Hwa et al., 1996; Hamilton & Cocks, 2000). The observations of the present study indicate that similar PAR subtypes are responsible for thrombin- and trypsin-induced relaxation of intramyocardial arteries from these two species. As was observed in human epicardial arteries (Hamilton et al., 1998), thrombin and trypsin caused relaxations of human intramyocardial arteries which were entirely dependent on the catalytic activity of each enzyme and were mimicked by the selective PAR1-activating peptide, TFLLR, but not by the PAR2- or PAR4-activating peptides, SLIGKV and GYPGQV respectively. This contrasts with porcine intramyocardial arteries that relaxed in response to TFLLR and SLIGKV, but not GYPGQV, suggesting that PAR1 and PAR2 are capable of causing relaxation of these arteries. Although the requirement of a functional endothelial layer for PAR-induced relaxations in intramyocardial arteries was not examined directly in the present study, immunohistochemical localization of PAR1 (human) and both PAR1 and PAR2 (porcine) to the endothelium of intramyocardial arteries suggests that, similar to the equivalent epicardial arteries of each species, the observed relaxations of intramyocardial arteries were due to activation of endothelial cell PARs.
Given the high concentrations of thrombin and trypsin required to elicit relaxation of human intramyocardial arteries, we wished to ensure that the observed responses were in fact due to PAR activation. To our knowledge, three other pathways exist by which the enzymes used in our study could induce vascular responses independently of PARs but still dependent on their catalytic activity. First, trypsin can cleave high molecular weight kininogen from human vascular endothelial cells to generate bradykinin (Nishikawa et al., 1992; van iwaarden et al., 1988). Also trypsin, like kallikrein and cathepsin G, has been shown to directly activate the human bradykinin B2 receptor most likely by cleavage of the receptor without exposure of a tethered ligand (Hecquet et al., 2000). The third possible activity-dependent mechanism by which PAR-activating enzymes may cause vasodilator responses is that reported for thrombin whereby it stimulates a matrix metalloprotease-2-dependent release of endothelin-1 (Fernandez-Patron et al., 2000). Neither of the non-PAR trypsin mechanisms was likely to have contributed to enzyme-mediated relaxation of human intramyocardial arteries since both trypsin- and thrombin-induced responses persisted in the presence of B2 kinin receptor blockade with Hoe-140 (Hock et al., 1991), which blocks the non-PAR responses to trypsin. Also, a role for ET-1 was equally unlikely since relaxations to both thrombin and trypsin were insensitive to the ETA/ETB receptor antagonist bosentan. Therefore, if thrombin or trypsin cause relaxation of human intramyocardial arteries independently of PARs, this occurs via a novel mechanism(s).
Although the PAR subtypes involved in relaxations to thrombin and trypsin appeared to be common between epicardial and intramyocardial arteries, we observed a marked decrease in sensitivity to PAR-activating enzymes in intramyocardial arteries. This phenomenon was not species- or receptor-specific since it was observed in both human (PAR1 only) and pig (PAR1 and PAR2) arteries. In addition, the decreased sensitivity to enzymatic PAR activation was unlikely to be due to fewer receptors or decreased coupling in the smaller arteries since the selective PAR-activating peptides, TFLLR in human arteries and both TFLLR and SLIGKV in porcine arteries, were equipotent in the two different-sized vessels. Furthermore, experiments in porcine coronary arteries were performed using animal-matched epicardial and intramyocardial arteries to avoid potential inter-animal variations. Experiments on human epicardial and intramyocardial arteries were performed on vessels from the same subject on two occasions and both times the sensitivities to thrombin and trypsin, but not TFLLR, were less in the small arteries when compared with the large arteries (EC50: thrombin (u ml−1): large=0.01 and 0.01, small=5.01 and 1.26; trypsin (u ml−1): large=0.03 and 0.01, small=0.50 and 0.79; TFLLR (μM): large=0.63 and 0.63, small=0.40 and 0.40). Therefore the selective decrease in sensitivity to the enzyme PAR agonists observed in the human arteries is unlikely to be due to patient or surgery-related differences.
The reason for the observed decrease in sensitivity to thrombin and trypsin in intramyocardial arteries remains unresolved. One explanation is a greater proportion of precleaved PARs in the small arteries when compared with their larger epicardial counterparts since previous studies have shown that PAR1 activation by SFLLRN persists following desensitization to thrombin in various PAR1 expression systems (Ishii et al., 1993; Brass et al., 1994; Hammes & Coughlin, 1999). A similar TFLLR-induced relaxation may occur in human intramyocardial arteries previously exposed to thrombin (or trypsin). If this is the case, an increased level of ‘tonic' PAR activation in intramyocardial arteries when compared with epicardial arteries may explain the maintained sensitivity to peptide PAR agonists but decreased sensitivity to enzyme PAR agonists in these arteries. Blood-borne thrombin is unlikely to account for such ‘tonic' PAR activation in the present study since artery preparations were examined in the absence of blood. Moreover, exposure of arteries to thrombin or trypsin in vitro generally occurred ⩾2 h after the artery preparations had been washed free of blood. If activating PAR cleavage had occurred in vivo (for example, by thrombin), previous studies have shown that 2 h is adequate time to regenerate sufficient intact cell surface PARs to regain complete sensitivity to activating enzymes (Brass et al., 1994; Hein et al., 1994; Ellis et al., 1999). Therefore, if previously cleaved cell surface PARs account for the decreased sensitivity to PAR-activating enzymes, the mechanism(s) by which the precleaved PARs are formed in the absence of blood in vitro is unknown.
Perhaps a more plausible explanation for the decreased sensitivity to thrombin and trypsin in intramyocardial arteries is that the expression of endogenous serine protease inhibitors (serpins: for review, see Potempa et al., 1994; Wright, 1996) is higher in intramyocardial arteries compared with epicardial arteries. For example, antithrombin III is located on human vascular endothelial cells (Chan & Chan, 1981; Bartha et al., 1987) and is the main inhibitor of thrombin in human plasma (Bonniec et al., 1995; de boor et al., 1995). Also, α1-antitrypsin regulates trypsin activity in the gut (Potempa et al., 1994) and airways (Cichy et al., 1997) and is a major protease inhibitor in human plasma (Davíla et al., 1999). The results of the present study may reflect an increased activity of these, or similar protease inhibitors, in intramyocardial arteries. If this is the case, such serpin expression would have to increase as the diameter of the coronary artery decreases. In support of this hypothesis, a recent study reported that PAR2 activation protected the myocardium from ischaemia-reperfusion injury in the rat heart (Napoli et al., 2000). Intriguingly, this protective effect was observed following infusion of a PAR2-activating peptide, SLIGRL, but not of trypsin (Napoli et al., 2000). Such an effect of infused PAR agonists presumably occurs at the level of small, intramyocardial arteries, such as those examined in vitro in the present study. In a separate study, combined inhibition of α1-antitrypsin and α1-antichymotrypsin activity provided similar protection against ischaemia-reperfusion injury of the rat pancreas (von dobschuetz et al., 1999). Since the protective effect in the rat pancreas was predominantly attributed to a preservation of post-ischaemia capillary perfusion it is possible that it was mediated by increased serine protease-induced activation of endothelial PAR2 in the absence of the inhibitory regulation of the activating enzyme(s). These previous studies provide support, albeit indirect, for an increased regulation of the enzymatic activators of PARs in vivo as the artery diameter decreases, similar to that which we propose accounts for the decreased sensitivity to these enzymes in intramyocardial arteries, when compared with epicardial arteries, in vitro.
In conclusion, the results presented here suggest that the PAR subtypes linked to relaxation of small resistance-like coronary arteries are the same as those in large conduit-type coronary arteries in both the human and pig. These PARs, which we have localized to the endothelial cell layer, exhibit markedly decreased sensitivity to proteolytic but not non-proteolytic activation as the artery diameter decreases. We hypothesize that, as a result of increased activity of inhibitory endothelial serpins, PAR-mediated endothelial activation is tightly controlled in the microvasculature. It is tempting to speculate that such a tightly regulated system is important in the settings of clinically prevalent microvasculature coagulopathies such as disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, or sepsis-induced microvascular thrombosis. Microvascular endothelial cells are noted for an antithrombotic surface under ‘normal' conditions. However, each of the microvasculature coagulopathies mentioned above is associated with significant procoagulant activity, including a decrease in circulating antithrombin III (Lorente et al., 1993). Furthermore, a concomitant inflammatory component to such conditions results in cytokine production which can, in turn, increase PAR expression in vascular endothelial cells (Nystedt et al., 1996; Hamilton et al., 2001a). Increased PAR signalling in activated endothelial cells suggest that these receptors may represent novel therapeutic targets against various forms of vascular inflammatory disease.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia. We thank the staff of the Cardiothoracic Surgery Unit at the Royal Melbourne Hospital and the Heart and Lung Transplant Unit at The Alfred Hospital for their generous assistance and co-operation in the collection of human tissue for this study and also J.D. Sumner for assistance during the course of this work.
Abbreviations
- FITC
fluorescein isothiocyanate
- HUVEC
human umbilical vein endothelial cells
- IC
internal circumference
- KPSS
high potassium physiological Krebs solution
- PAR
protease-activated receptor
- PBS
phosphate buffered saline
- Rmax
maximum response
References
- AL-ANI B., SAIFEDDINE M., KAWABATA A., RENAUX B., MOKASHI S., HOLLENBERG M.D. Proteinase-activated receptor 2 (PAR2): Development of a ligand-binding assay correlating with activation of PAR2 by PAR1- and PAR2-derived peptide ligands. J. Pharm. Exp. Ther. 1999;290:753–760. [PubMed] [Google Scholar]
- ALTROGGE L.M., MONARD D. An assay for high-sensitivity detection of thrombin activity and determination of proteases activating or inactivating protease-activated receptors. Anal. Biochem. 2000;277:33–45. doi: 10.1006/abio.1999.4356. [DOI] [PubMed] [Google Scholar]
- BARTHA K., KOVACS T., LERANT I., PAPP B., CSONKA E., KOLEVSKA P., MACHOVICH R. Interaction of thrombin-antithrombin III complex with cultured aortic endothelial cells. Thromb. Res. 1987;47:541–552. doi: 10.1016/0049-3848(87)90359-8. [DOI] [PubMed] [Google Scholar]
- BLACKHART B.D., EMILSSON K., NGUYEN D., TENG W., MARTELLI A.J., NYSTEDT S., SUNDELIN J., SCARBOROUGH R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996;271:16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- BÖHM S.K., KONG W., BRÖMME D., SMEEKENS S.P., ANDERSON D.C., CONNOLLY A., KAHN M., NELKEN N.A., COUGHLIN S.R., PAYAN D.G., BUNNETT N.W. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONNIEC B.F.C., GUINTO E.R., STONE S.R. Identification of thrombin residues that modulate its interactions with antithrombin III and α1-antitrypsin. Biochemistry. 1995;34:12241–12248. doi: 10.1021/bi00038a019. [DOI] [PubMed] [Google Scholar]
- BOWLES D.K., HU Q., LAUGHLIN M.H., STUREK M. Heterogeneity of L-type calcium current density in coronary smooth muscle. Am. J. Physiol. 1997;273:H2083–H2089. doi: 10.1152/ajpheart.1997.273.4.H2083. [DOI] [PubMed] [Google Scholar]
- BRASS L.F., PIZARRO S., AHUJA M., BELMONTE E., BLANCHARD N., STADEL J.M., HOXIE J.A. Changes in the structure and function of the human thrombin receptor during activation, internalization and recycling. J. Biol. Chem. 1994;269:2943–2952. [PubMed] [Google Scholar]
- BRASS L.F., VASSALO R.R.J., BELMONTE E., AHUJA M., CICHOWSKI K., HOXIE J.A. Structure and function of the human platelet thrombin receptor: studies using monoclonal antibodies directed against a defined domain within the receptor N terminus. J. Biol. Chem. 1992;267:13795–13798. [PubMed] [Google Scholar]
- BUUS N.H., SIMONSEN U., PILEGAARG H.K., MULVANY M.J. Nitric oxide, prostenoid and non-NO, non-prostenoid involvement in acetylcholine relaxation of isolated human arteries. Br. J. Pharmacol. 2000;129:184–192. doi: 10.1038/sj.bjp.0703041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMERER E., HUANG W., COUGHLIN S.R. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN T.K., CHAN V. Antithrombin III, the major modulator of intravascular coagulation, is synthesised by human endothelial cells. Thromb. Haemost. 1981;46:504–506. [PubMed] [Google Scholar]
- CHEN J., ISHII M., WANG L., ISHII K., COUGHLIN S.R. Thrombin receptor activation: confirmation of the intramolecular tethered liganding hypothesis and discovery of an alternative liganding mode. J. Biol. Chem. 1994;269:16041–16045. [PubMed] [Google Scholar]
- CHILIAN W.M., EASTHAM C.L., MARCUS M.L. Microvascular distribution of coronary vascular resistance in beating left ventricle. Am. J. Physiol. 1986;251:H779–H788. doi: 10.1152/ajpheart.1986.251.4.H779. [DOI] [PubMed] [Google Scholar]
- CHILIAN W.M., LAYNE S.M., EASTHAM C.L., MARCUS M.L. Heterogeneous microvascular coronary alpha-adrenergic vasoconstriction. Circ. Res. 1989;64:376–388. doi: 10.1161/01.res.64.2.376. [DOI] [PubMed] [Google Scholar]
- CICHY J., POTEMPA J., TRAVIS J. Biosynthesis of α1-proteinase inhibitor by human lung-derived epithelial cells. J. Biol. Chem. 1997;272:8250–8255. doi: 10.1074/jbc.272.13.8250. [DOI] [PubMed] [Google Scholar]
- COCKS T.M., MOFFATT J.D. Protease-activated receptors: sentries for inflammation. Trends Pharmacol. Sci. 2000;21:103–108. doi: 10.1016/s0165-6147(99)01440-6. [DOI] [PubMed] [Google Scholar]
- COUGHLIN S.R. Protease-activated receptors start a family. Proc.Natl. Acad. Sci. U.S.A. 1994;91:9200–9202. doi: 10.1073/pnas.91.20.9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUGHLIN S.R. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- DAVÍLA F.M., PORTELA M.D., ROJO M.G., GARCIA J.G., RULLAN A.M.P., PEREZ R.P., VINCENTE M.C. Coronary artery dissection in alpha-1-antitrypsin deficiency. Histopath. 1999;34:376–378. doi: 10.1046/j.1365-2559.1999.0669d.x. [DOI] [PubMed] [Google Scholar]
- DE BOOR H.C., PREISSNER K.T., BOUMA B.N., DE GROOT P.G. Internalization of vitronectin-thrombin-antithrombin complex by endothelial cells leads to deposition of the complex into the subendothelial matrix. J. Biol. Chem. 1995;270:30733–30740. doi: 10.1074/jbc.270.51.30733. [DOI] [PubMed] [Google Scholar]
- DÉRY O., CORVERA C.U., STEINHOFF M., BUNNETT N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- DRUMMOND G.R., COCKS T.M. Evidence for mediation by endothelium-derived hyperpolarizing factor of relaxation to bradykinin in the bovine isolated coronary artery independently of voltage-operated Ca2+ channels. Br. J. Pharmacol. 1996;117:1035–1040. doi: 10.1111/j.1476-5381.1996.tb16693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELLIS C.A., MALIK A.B., GILCHRIST A., HAMM H., SANDOVAL R., VOYNO-YASENETSKAYA T., TIRUPPATHI C. Thrombin induces proteinase-activated receptor-1 gene expression in endothelial cells via activation of Gi-linked Ras/mitogen-activated protein kinase pathway. J. Biol. Chem. 1999;274:13718–13727. doi: 10.1074/jbc.274.19.13718. [DOI] [PubMed] [Google Scholar]
- EPSTEIN S.E., CANNON R.O.I., TALBOT T.L. Hemodynamic principles in the control of coronary blood flow. Am. J. Cardiol. 1985;56:4E–10E. doi: 10.1016/0002-9149(85)91169-5. [DOI] [PubMed] [Google Scholar]
- FERNANDEZ-PATRON C., RADOMSKI M.W., DAVIDGE S.T. Role of matrix metalloproteinase-2 in thrombin-induced vasorelaxation of rat mesenteric arteries. Am. J. Physiol. 2000;278:H1473–H1479. doi: 10.1152/ajpheart.2000.278.5.H1473. [DOI] [PubMed] [Google Scholar]
- FOX M.T., HARRIOTT P., WALKER B., STONE S.R. Identification of potential activators of proteinase-activated receptor-2. FEBS Lett. 1997;417:267–269. doi: 10.1016/s0014-5793(97)01298-2. [DOI] [PubMed] [Google Scholar]
- GITTERMAN D.P., EVANS R.J. Properties of P2X and P2Y receptors are dependent on artery diameter in the rat mesenteric bed. Br. J. Pharmacol. 2000;131:1561–1568. doi: 10.1038/sj.bjp.0703760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILTON J.R., CHOW J.M., COCKS T.M. Protease-activated receptor-2 turnover stimulated independently of receptor activation in porcine coronary endothelial cells. Br. J. Pharmacol. 1999;127:617–622. doi: 10.1038/sj.bjp.0702583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILTON J.R., COCKS T.M. Heterogeneous mechanisms of endothelium-dependent relaxation to thrombin and peptide activators of protease-activated receptor-1 in porcine isolated coronary artery. Br. J. Pharmacol. 2000;130:181–188. doi: 10.1038/sj.bjp.0703146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILTON J.R., FRAUMAN A.G., COCKS T.M. Increased expression of protease-activated receptor-2 (PAR2) and PAR4 in human coronary artery by inflammatory stimuli unveils endothelium-dependent relaxations to PAR2 and PAR4 agonists. Circ. Res. 2001a;89:92–98. doi: 10.1161/hh1301.092661. [DOI] [PubMed] [Google Scholar]
- HAMILTON J.R., MOFFATT J.D., FRAUMAN A.G., COCKS T.M. Protease-activated receptor 1 (PAR1) but not PAR2 or PAR4 mediates endothelium-dependent relaxation to thrombin and trypsin in human pulmonary arteries. J. Cardiovasc. Pharmacol. 2001b;38:108–119. doi: 10.1097/00005344-200107000-00012. [DOI] [PubMed] [Google Scholar]
- HAMILTON J.R., NGUYEN P.B., COCKS T.M. Atypcial protease-activated receptor mediates endothelium-dependent relaxation of human coronary arteries. Circ. Res. 1998;82:1306–1312. doi: 10.1161/01.res.82.12.1306. [DOI] [PubMed] [Google Scholar]
- HAMMES S.R., COUGHLIN S.R. Protease-activated receptor-1 can mediate responses to SFLLRN in thrombin-desensitized cells: evidence for a novel mechanism for preventing or terminating signaling by PAR1's tethered ligand. Biochemistry. 1999;38:2486–2493. doi: 10.1021/bi982527i. [DOI] [PubMed] [Google Scholar]
- HEAGERTY A.M., IZZARD A.S. Small-artery changes in hypertension. J. Hypertens. 1995;13:1560–1565. [PubMed] [Google Scholar]
- HECQUET C., TAN F., MARCIC B.M., ERDOS E.G. Human bradykinin B2 receptor is activated by kallikrein and other serine proteases. Mol. Pharmacol. 2000;58:828–836. doi: 10.1124/mol.58.4.828. [DOI] [PubMed] [Google Scholar]
- HEIN L., ISHII K., COUGHLIN S.R., KOBILKA B.K. Intracellular targeting and trafficking of thrombin receptors: A novel mechanism for resensitization of a G protein-coupled receptor. J. Biol. Chem. 1994;269:27719–27726. [PubMed] [Google Scholar]
- HOCK F.J., WIRTH K., ALBUS U., LINZ W., GERHARDS H.J., WIEMER G., HENKE S.T., BREIPOHL G., KÖNIG W., KNOLLE J., SCHÖLKENS B.A. HOE 140 a new potent and long-acting bradykinin antagonist: in vitro studies. Br. J. Pharmacol. 1991;102:769–773. doi: 10.1111/j.1476-5381.1991.tb12248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLLENBERG M.D., SAIFEDDINE M., BAHJAT A., KAWABATA A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can. J. Physiol. Pharmacol. 1997;75:832–841. [PubMed] [Google Scholar]
- HOXIE J.A., AHUJA M., BELMONTE E., PIZARRO S., PARTON R., BRASS L.F. Internalization and recycling of activated thrombin receptors. J. Biol. Chem. 1993;268:13756–13763. [PubMed] [Google Scholar]
- HWA J.J., GHIBAUDI L., WILLIAMS P., CHINTALA M., ZHANG R., CHATTERJEE M., SYBERTZ E. Evidence for the presence of a proteinase-activated receptor distinct from the thrombin receptor in vascular endothelial cells. Circ. Res. 1996;78:581–588. doi: 10.1161/01.res.78.4.581. [DOI] [PubMed] [Google Scholar]
- ISHII K., HEIN L., KOBILKA B., COUGHLIN S.R. Kinetics of thrombin receptor cleavage on intact cells: relation to signaling. J. Biol. Chem. 1993;268:9780–9786. [PubMed] [Google Scholar]
- JONES C.J., KUO L., DAVIS M.J., CHILIAN W.M. Regulation of coronary blood flow: coordination of heterogeneous control mechanisms in vascular microdomains. Cardiovasc. Res. 1995;29:585–596. [PubMed] [Google Scholar]
- KAWABATA A., SAIFEDDINE M., AL-ANI B., LEBLOND L., HOLLENBERG M.D. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J. Pharmacol. Exp. Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- KEMP B.K., COCKS T.M. Evidence that mechanisms dependent and independent of nitric oxide mediate endothelium-dependent relaxation to bradykinin in human small resistance-like coronary arteries. Br. J. Pharmacol. 1997;120:757–762. doi: 10.1038/sj.bjp.0700928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KILPATRICK E.V., COCKS T.M. Evidence for differential roles of nitric oxide (NO) and hyperpolrization in endothelium-dependent relaxation of pig isolated coronary artery. Br. J. Pharmacol. 1994;112:557–565. doi: 10.1111/j.1476-5381.1994.tb13110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMPING K.G., KANATSUKA H., EASTHAM C.L., CHILIAN W.M., MARCUS M.L. Nonuniform vasomotor responses of the coronary microcirculation to serotonin and vasopressin. Circ. Res. 1989;65:343–351. doi: 10.1161/01.res.65.2.343. [DOI] [PubMed] [Google Scholar]
- LORENTE J.A., GARCIA-FRADE L.J., LANDIN L., DE PABLO R., TORRADO C., RENES E., GARCIA-AVELLO A. Time course of hemostatic abnormalities in sepsis and its relation to outcome. Chest. 1993;103:1536–1542. doi: 10.1378/chest.103.5.1536. [DOI] [PubMed] [Google Scholar]
- MAGAZINE H.I., KING J.M., SRIVASTAVA K.D. Protease activated receptors modulate aortic vascular tone. Int. J. Cardiol. 1996;53:S75–S80. doi: 10.1016/0167-5273(96)02569-7. [DOI] [PubMed] [Google Scholar]
- MIRZA H., SCHMIDT V.A., DERIAN C.K., JESTY J., BAHOU W.F. Mitogenic responses mediated through the proteinase-activated receptor-2 are induced by expressed forms of mast cell α- or β-tryptases. Blood. 1997;90:3914–3922. [PubMed] [Google Scholar]
- MOLINO M., BARNATHAN E.S., NUMEROF R., CLARK J., DREYER M., CUMASHI A., HOXIE J.A., SCHECHTER N., WOOLKALIS M., BRASS L.F. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J. Biol. Chem. 1997a;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- MOLINO M., WOOLKALIS M.J., REAVEY-CANTWELL J., PRATICO D., ANDRADE-GORDON P., BARNATHAN E.S., BRASS L.F. Endothelial cell thrombin receptors and PAR-2: Two protease-activated receptors located in a single cellular environment. J. Biol. Chem. 1997b;272:11133–11141. doi: 10.1074/jbc.272.17.11133. [DOI] [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneous and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- NAPOLI C., CICALA C., WALLACE J.L., DE NIGRIS F., SANTAGADA V., CALIENDO G., FRANCONI F., IGNARRO L.J., CIRINO G. Protease-activated receptor-2 modulates myocardial ischemia-reperfusion injury in the rat heart. Proc. Nat. Acad. Sci. U.S.A. 2000;97:3678–3683. doi: 10.1073/pnas.97.7.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISHIKAWA K., SHIBAYAMA Y., KUNA P., CALCATERRA E., KAPLAN A.P., REDDIGARI S.R. Generation of vasoactive peptide bradykinin from human umbilical vein endothelium-bound high molecular weight kininogen by plasma kallikrein. Blood. 1992;80:1989–1988. [PubMed] [Google Scholar]
- NYSTEDT S., EMILSSON K., WAHLESTEDT C., SUNDELIN J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYSTEDT S., RAMAKRISHNAN V., SUNDELIN J. The proteinase-activated receptor 2 is induced by inflammatory mediators in human endothelial cells. Comparison with the thrombin receptor. J. Biol. Chem. 1996;271:14910–14915. doi: 10.1074/jbc.271.25.14910. [DOI] [PubMed] [Google Scholar]
- POTEMPA J., KORZUS E., TRAVIS J. The serpin family of proteinase inhibitors: structure, function and regulation. J. Biol. Chem. 1994;269:15957–15960. [PubMed] [Google Scholar]
- SAIFEDDINE M., AL-ANI B., CHENG C.-H., WANG L., HOLLENBERG M.D. Rat proteinase-activated receptor-2 (PAR-2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br. J. Pharmacol. 1996;118:521–530. doi: 10.1111/j.1476-5381.1996.tb15433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN IWAARDEN F., DE GROOT P.G., BOUMA B.N. The binding of high molecular weight kininogen to cultured human endothelial cells. J. Biol. Chem. 1988;263:4698–4703. [PubMed] [Google Scholar]
- VON DOBSCHUETZ E., HOFFMANN T., MESSMER K. Inhibition of neutrophil proteinases by recombinant serpin Lex032 reduces capillary no-reflow in ischemia/reperfusion-induced acute pancreatisis. J. Pharmacol. Exp. Ther. 1999;290:782–788. [PubMed] [Google Scholar]
- VU T.-K.H., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- WRIGHT H.T. The structural puzzle of how serpin serine proteinase inhibitors work. BioEssays. 1996;18:453–464. doi: 10.1002/bies.950180607. [DOI] [PubMed] [Google Scholar]