

Abstract
Impairment in endothelial cell (EC) function plays a central role in vascular diseases (e.g. atherosclerosis, restenosis, diabetic angiopathies, microvascular angina, peripheral arterial disease). BRX-235 (a novel small molecule synthesized by Biorex, Hungary) has a potent vasculoprotective activity in different in vivo and in vitro studies. Since the importance of the p38 pathway in EC homeostasis and migration in particular is well documented, we have carried out studies to address the role of the p38 stress activated protein kinase (p38 SAPK) in the mode of action of BRX-235. In this study, Bovine aortic endothelial cells were used in a wounding migration assay (WMA) and for Western-blot analysis to study the effect and molecular mechanism of BRX-235-induced EC migration.
The bovine aortic endothelial (BAE) cells were shown to be good models for EC migration.
Both endothelial cell growth factor (ECGF)- and BRX-235-induced BAE cell migration were shown to be inhibited by SB 203580, a specific inhibitor of p38 SAPK.
It was also shown that, BRX-235 induces phosphorylation of p38 SAPK without affecting p38 SAPK protein levels. Thus, BRX-235 acts upstream of p38 SAPK.
In summary, we have shown that p38 SAPK is a potential pharmacological mediator for candidate drugs that target the endothelium.
Keywords: Bovine aortic endothelial cell; migration; wounding migration assay; p38 SAPK; phosphorylated-p38 SAPK; BRX-235 (Biorex, Hungary)
Introduction
Endothelial cells (EC) play a critical role in vascular homeostasis through the release of variety of autocrine and paracrine substances. They sense and integrate hemodynamic stimuli and regulate hemostasis and inflammation. Impairment in EC function plays a central role in many vascular diseases (e.g. reperfusion injury, atherosclerosis, restenosis, diabetic angiopathies, microvascular angina, peripheral arterial disease). Angioplasty denudes the vessel of endothelial cells that would normally generate paracrine inhibitors of vascular smooth muscle cell (VSMC) migration and proliferation.
Endothelial repair and angiogenesis involve EC migration and proliferation. Migration may in fact be responsible for subsequent proliferation by formation of regions of low cell density or by removal of inhibitory mechanisms due to cell–cell contact. EC migration is dependent on the p38 SAPK activation in basic-fibroblast growth factor (bFGF) (Tanaka et al., 1999) and VEGF (Rousseau et al., 2000) treated cells. Structural evidence has been published that endothelial cell growth factor (ECGF) is the precursor of fibroblast growth factor (FGF) (Burgess et al., 1986). In aortic vessels bFGF-induced EC proliferation is mediated by p38 (Rousseau et al., 2000). The importance of the p38 pathway is further accentuated by the findings showing that induction of endothelial transfer substance-1 (Ets-1), the major coordinator of gene expression during reendothelization after denuding injury, is mediated through p38 (Tanaka et al., 1998).
SB 203580 is one of the pyridinyl imidazole derivatives, which inhibits the activity of mitogen activated protein kinase (MAPK). In vivo it suppresses the activation of MAPK activated protein kinase-2 (MAPKAPK-2) and prevents the phosphorylation of the 27 kDa heat shock protein (Hsp27) in response to interleukin-1, cellular stress and bacterial endotoxin (Cuenda et al., 1995). Inhibition by SB 203580 implicated p38 SAPK in the migration of endothelial cells (Rousseau et al., 1977). Bimoclomol® (Biorex, Hungary) has been shown to affect the stress signal transduction pathways through stimulation of p38 SAPK (Balla et al., 1999; Kreuzer, 1999).
The stress response is a complex process and involves multiple signalling pathways. The ERK (extra-cellular signal-regulated kinase) pathway primarily transmits mitogenic and differentiation stimuli, whereas the JNK (c-Jun N-terminal kinase) and p38 pathways mainly transduce stress stimuli to nucleus. All three mitogen-activated protein kinases (MAPK) subfamilies play important roles in several functions. The expression of phosphorylated p38 SAPK in epithelial and myogenic cell lines (ECV and H9c2) increased by induction of H2O2 and was potentiated by Bimoclomol® (Balla et al., 1999). On the other hand, translocation of JNK and ERK was not affected either in human SMC (smooth muscle cells) (Kreuzer, 1999) or in EC (unpublished data).
BRX-235 is a member of a new family of non-toxic hydroxylamine-based compounds that originated with Bimoclomol® (Vígh et al., 1997; Nánási & Jednákovits, 2001). BRX-235 was recently developed in the research laboratories of Biorex R&D. Co. (Hungary) for treatment of atherosclerosis and the complications of atherosclerosis such as ischaemic heart disease, peripheral arterial disease and restenosis. The chemical structure of BRX-235 is 5,6-dihydro-5-(1-piperidinyl) methyl-3-(3-pyridil)-4H-1,2,4-oxadiazine (Figure 1).
Figure 1.
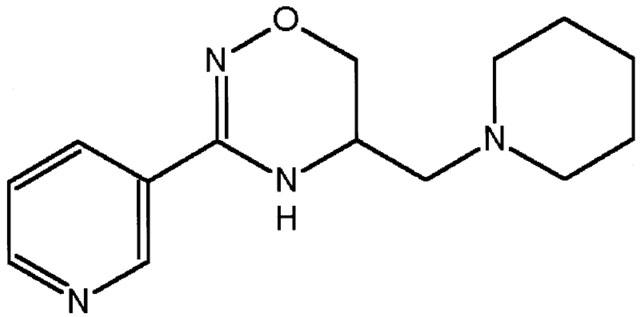
Structure of BRX-235: 5,6,dihydro-5-(1-piperidinyl)methyl-3-(3-pyridil)-4-H-1,2,4-oxadiazine.
Here we show that BRX-235 is a potent inducer of BAE cell migration, an effect that is believed to be behind the EC repair capability of BRX-235. We also show that BRX-235 mediates this action via p38 SAPK phosphorylation.
Methods
Reagents
Chemicals were obtained from Sigma Chemicals Co. (St. Louis, MO, U.S.A.) unless otherwise indicated. The pyridinyl imidazole (SB 203580), a specific inhibitor of p38 SAP kinase activity (Lee et al., 1994), was obtained from Upstate Biotech. Inc. (La Jolla, CA, U.S.A.).
Isolation and cultivation of bovine aortic endothelial (BAE) cells
A section of bovine aorta was placed into sterile PBS-solution containing an antibiotic mixture and was incubated at room temperature for 60 min. Trypsin-EDTA solution (Sigma) was layered on the inner surface of a piece of aortic vessel and endothelial cells were digested at 37°C for 15 min, washed and resuspended in RPMI 1640 (Gibco-BRL, U.S.A.) containing 20% FCS (Sigma). The isolated and stained cells were von Willebrand (Factor VIII) positive endothelial cells. BAE cells were seeded in a 96-well plate for wounding migration assay (WMA). The cell count was adjusted to 1–2×106 cells ml−1 and 1 ml of cell suspension was layered in a 6-well plate (Costar, U.S.A.) for phosphorylation studies.
Wounding migration assay
The assay was carried out as described previously (Yamamura et al., 1996). BAE cells were seeded in the 96-well plate and, at approximately 90% confluency, the monolayer was wounded with a scraper (width=1 mm) along a horizontal line. Wells were rinsed with completed RPMI 1640 medium with 2% FCS and incubated at 37°C in 5% CO2. Cells migrating to the wounded area were counted 0, 4, 24, and 48 h after wounding using a computerized image analysis software (KSK2 software; Pictron, Hungary). The number of cells in the wounded area was expressed as the number of occupied pixels. For monitoring the effects of BRX-235 (Biorex, Hungary) and MAP Kinase inhibitor (SB 203580) on migration of BAE cells, reagents were diluted in the medium and added to the cells after wounding.
Western blot analysis
For protein blotting BAE cells were cultivated in 6-well plates (Costar, U.S.A.). The confluent monolayers were wounded along 10 lines at the start of the experiment. At indicated times cells were scraped off using a rubber policeman (Costar) with 10 ml ice-cold PBS (pH=7.4) and collected by centrifugation at 1000×g for 4 min at 4°C. Pellets were homogenized by ultrasound homogenizer (Branson Sonifer 450) for 15 s at 0°C in 100–150 μl lysis buffer containing (mM): Tris HCl 50 (pH=8), EDTA 5, NaCl 150, phenylmethylsulphonyl fluoride 1, benzamidine 1, amino-caproic acid 1, ocadaic acid 10, NaF 50, Na-o-vanadate 5, 0.1% sodium dodecyl sulphate and 1% Triton X-100. Lysates were centrifuged at 16,000×g for 5 min at 4°C and the supernatants were placed on ice. The protein concentration was determined according to Bradford (1976) and samples were stored at −80°C until further use.
Twenty micrograms of protein was electrophoresed on a 12% SDS-polyacrylamide gel (SDS–PAGE) using a mini-slab gel apparatus (Bio-Rad, U.S.A.). The separated proteins were transferred onto a polyvinylidenefluoride (PVDF) membrane (Immobilon P, Millipore, U.S.A.) blocked with 5% skim milk in Tris buffer salt (TBS, pH=7.4) containing 0.1% Tween 20, for 1 h at room temperature. Membranes were probed with anti-phospho-p38 antibody by commercially available kits (PhosphoPlus p38 MAP kinase Antibody Kit, New England Biolabs, U.S.A.) overnight at 4°C. Analysis of threonine and tyrosine phosphorylation of p38 SAPK was performed according to the manufacturer's instructions. After probing with the secondary peroxidase-labelled antibody at room temperature for 1 h, proteins were visualized using enhanced chemiluminescence (ECL) and exposing them to ECL Hyperfilm (Amersham, U.K.). In order to show the amounts of p38 SAPK, blots were stripped and reprobed using phosphorylation-state independent p38 SAPK-specific anti-p38 antibody to determine total p38 SAPK levels.
Statistical evaluation
The number of migrated cells was given in pixels (150,000 pixel points=3 mm2 surface) and values were also expressed in percentages. The means±s.e.mean were calculated. Comparisons between groups were assessed by Student's t-test. Statistical significance was defined as *P<0.05; **P<0.01; ***P<0.005.
Results
Migration of bovine aortic endothelial cells is mediated by the p38 SAPK pathway
In this study the migration of BAE cells was assessed. Migration was induced by mechanical wounding of the BAE cell layer. By 48 h post-wounding a significant increase in migration was recorded (Figure 2a). This spontaneous migration was significantly inhibited by submicromolar (at 10, 100 and 1000 nM) concentration of SB 203580. At 1 μM concentration the spontaneous migration was inhibited by 60%. This correlates well with data published on human umbilical venous endothelial cells (HUVEC) (Rousseau et al., 1997).
Figure 2.
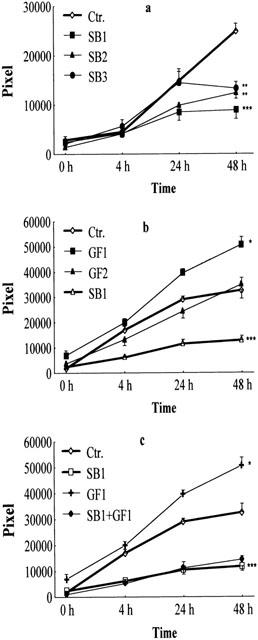
Effect of a specific p38 inhibitor (SB 203580) or endothelial cell growth factor and the combination of the two materials on migration of BAE cells. The number of migrated cells in pixels was measured at 0, 4, 24, 48 h after incubation with diluted ECGF or SB 203580. (a) Migration of BAE cells was measured in the presence of SB 203580 at 1000 nM (SB1), 100 nM (SB2) and 10 nM (SB3) respectively, or without inhibitor (Ctr.) using spontaneously migrating cells as control (n =12). (b) BAE cells had been treated with ECGF diluted to 1 : 25 (GF1), diluted to 1 : 100 (GF2) or without growth factor (Ctr.) using unstimulated cells as control. (c) Inhibition of SB 203580 on ECGFs-activated BAE cell migration. SB 203580 (1 μM, SB1) reduces the migration activity of BAE cells that had been treated with ECGFs (25 fold diluted, GF1) for 48 h. The number of migrated cells in pixels was determined after 0, 4, 24, and 48 h post-treatment. ***P<0.005 compared to non-stimulated, spontaneously migrated BAE cells (Ctr.), evaluated by one-tailed Student's t-test.
To validate our system we studied the effect of the endothelial cell growth supplement (ECGFs) on BAE cell migration. ECGF is a rich source of endothelial cell growth factor (Maciag et al., 1982), a potent mitogen and an inducer of EC migration. ECGFs at a dilution of 1 : 25 significantly stimulated the migration of BAE cells, while the 1 : 100 fold dilution had only a slight effect (Figure 2b).
SB 203580 at 1 μM potently inhibited endothelial cell growth factor supplement (ECGFs)-induced BAE cell migration compared to the control value (Figure 2c).
Induction of the migration of BAE cells by BRX-235
BRX-235 dose-dependently stimulated BAE cell migration at 10, 100 and 1000 nM (26.5, 63.3 and 73.5% respectively) (Figure 3).
Figure 3.
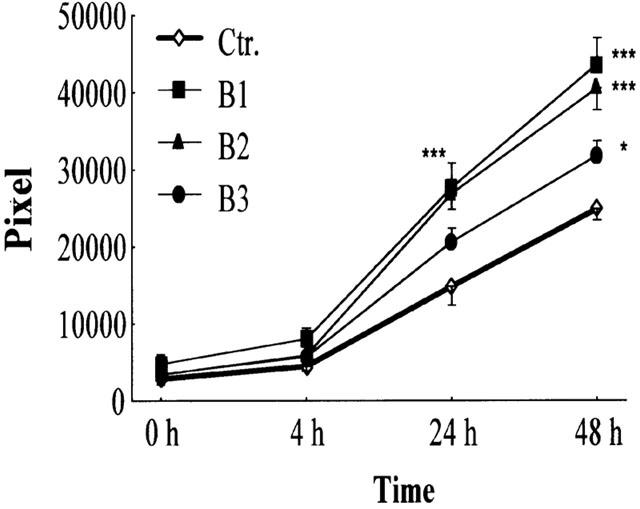
BRX-235 stimulates BAE cell migration. The number of migrated BAE cells with BRX-235 (1000 nM, B1; 100 nM, B2; and 10 nM, B3) was determined for the times indicated. Results are expressed as mean±s.e.mean (n=12).
Next we studied the potential role of p38 SAPK in the mechanism of BRX-235-stimulated BAE cell migration using SB 203580. SB 203580 was used at 1 μM concentration that has been shown to inhibit efficiently the BAE cell migration both on spontaneous (Figure 2a) and ECGFs-induced cells (Figure 2c). However, SB 203580 potently inhibited migration induced by 10 and 100 nM BRX-235 (90 and 76%, respectively) (Figure 4). Inhibition was less profound (38%) when BRX-235 was used at 1 μM). Thus, involvement of the p38 SAPK pathway is a common theme in the mechanism of spontaneous, ECGFs- and BRX-235-induced BAE cell migration.
Figure 4.
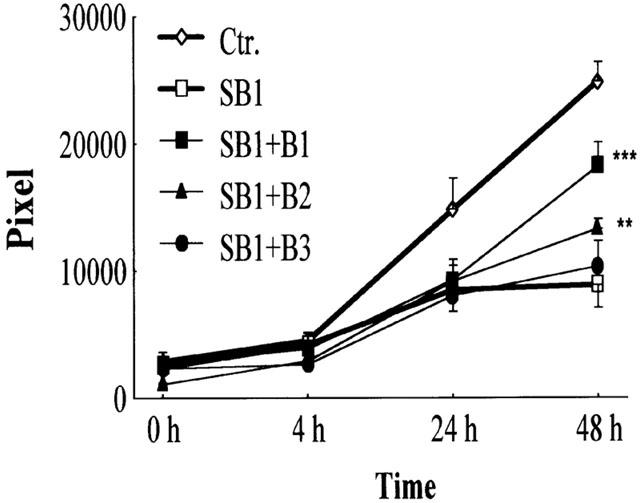
BRX-235 enhances the migration of endothelial cells inhibited by SB 203580. Migration of BAE cells was evaluated in a WMA system. Cells were co-cultivated with SB 203580 (1 μM, SB1) and BRX-235 (1000 nM, B1; 100 nM, B2; 10 nM, B3) for 48 h and the number of pixels was evaluated at 0, 4, 24 and 48 h of incubation. Data were compared to the values of migrated BAE cells attenuated by SB 1. (**P<0.01 and ***P<0.005; n=12).
Phosphorylation of p38 SAPK by BRX-235
The increases in threonine- and tyrosine-phosphorylation of p38 SAPK reflect the activation state of p38 SAPK. Thus, the threonine- and tyrosine-phosphorylation of p38 SAPK upon BRX-235 treatment was examined. BAEC-s were stimulated with BRX-235 (1 μM) for 10, 20, 30 and 240 min and lysates were immunoblotted for p38 SAPK. Following treatment with BRX-235 the amount of phosphorylated p38 SAPK (pp38 SAPK) increased 1.78 fold after 10 min, 1.98 fold after 20 min, and declined after 240 min (Figure 5). ECGF induced p38 SAPK phosphorylation by about the same extent as BRX-235 but with different kinetics (Figure 5). The amount of p38 SAPK did not change following BRX-235 treatment, indicating that the BRX-235 acts on the p38 SAPK signalling pathway upstream of the kinase.
Figure 5.
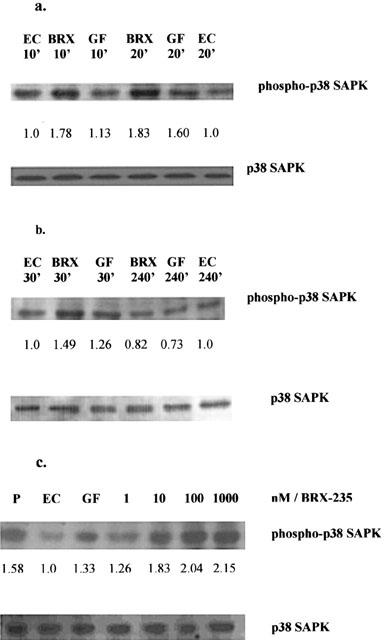
BRX-235 induces p38 SAPK phosphorylation. (a) BAE cells were stimulated with BRX-235 (BRX, 1 μM), and with ECGFs (GF, 25 fold diluted) or without BRX-235 (EC) for 10 and 20 min as indicated. The lysates were run on a 12% SDS–PAGE. After probing, the membranes were developed with anti-pp38 SAPK (upper panel); Membranes were stripped and reprobed using a phosphorylation state-independent p38 SAPK-specific antibody (lower panel). (b) The amount of pp38 SAPK from BAE cells were studied after stimulation with BRX-235 (BRX, 1 μM), and ECGFs (GF, 25 fold diluted) or without BRX-235 (EC) for 30 and 240 min as indicated. The amounts of pp38 SAPK were quantified with an image analytical system (Multi-Analyst software, Bio-Rad, CA, U.S.A.). The increase over control is given. Blots are representative of three experiments. (c) Dose-dependent activation of p38 SAPK by BRX-235. BAE cells were stimulated with BRX-235 (lanes 4, 5, 6, 7; 1, 10, 100 and 1000 nM respectively), ECGFs (GF 25 fold diluted, lane 3), vehicle (EC, lane 2) and lysate prepared from anisomycin stimulated C-6 glioma cells as positive control (P, lane 1) for 15 min. Lysates were run on a 12% SDS–PAGE. After probing the membrane with anti-pp38 SAPK (upper panel), membranes were stripped and reprobed using a phosphorylation state-independent p38 MAPK-specific antibody. The amounts of pp38 SAPK were quantified with an image analysis system (Multi-Analyst software, Bio-Rad, CA, U.S.A.). The increase over control is given. In the figure the blots illustrate the data and results of three experiments.
In order to examine the effect of BRX-235 induced p38 SAPK phosphorylation we performed Western blots utilizing an anti-pp38 SAPK antibody that detects active forms of the kinase. Cells were incubated with BRX-235 (1, 10, 100 and 1000 nM) for 15 min, and the amounts of phosphorylated p38 SAPK were determined. Phosphorylated p38 SAPK increased after BRX-235 treatment in a dose-dependent manner (Figure 5c, upper panel). After a 15 min exposure, BRX-235 (at 1 nM) slightly activated p38 SAPK. The lower panel of Figure 5c shows that, regardless of culture conditions, equal amounts of p38 SAPK protein were immunoblotted using a phosphorylation state-independent p38 SAPK-specific antibody.
Phosphorylated-p38 SAPK activities with SB 203580 and BRX-235
The inhibitor, SB 203580 has not modulated the phosphorylation status of p38 SAPK neither in untreated nor in BRX-235 (1 μM, line B1) or ECGFs-stimulated cells (Figure 6, line GF). The phosphorylation status (pp38/p38) only decreased slightly upon exposure to SB 203580 in BRX-235 stimulated cells (1.67 vs 1.82) and in ECGFs-treated cells (1.58 vs 1.62).
Figure 6.

Effect of BRX-235 with SB 203580 on phosphorylated-p38 SAPK. BAE cells were treated with SB 203580 (SB 1 μM, line 1) or with p38 inhibitor and another agent, either BRX-235 (SB+B 1 μM each, line 3) or ECGFs (SB+GF 1 μM and 25 fold diluted, line 4) for 30 min. The lysates of BAE cells after incubation (for 30 min at 37°C in air containing 5% CO2) were run on a 12% SDS–PAGE either exposed with agents or cells without treatment (EC, line 2), transferred to membranes, and probed with a specific antibody directed against the phosphorylated threonine and tyrosine of p38 SAPK (phospho-p38 SAPK; upper panel). These blots were reprobed using a phosphorylation state independent p38 SAPK specific antibody. The amounts of p38 SAPK are visualized in the lower strip (p38 SAPK; lower panel). In SB 203580-treated BAE cells the amounts of phosphorylated p38 SAPK decreased. Amounts of pp38 SAPK were higher in cells treated with SB 203580 and with either BRX-235 (SB+B1) or ECGFs (SB+GF) than in untreated (EC) control cells. The phosphorylated status of p38 SAPK have been expressed (pp38 SAPK/p38 SAPK) and depicted on figure.
Discussion
Both spontaneous and growth factor-induced migration of BAE cells is mediated by the p38 SAPK pathway (Figure 2). This correlates well with data from studies on HUVEC (Rousseau et al., 1977; 2000). We also presented evidence that BRX-235, a drug candidate to treat endothelial dysfunction, is a potent inducer of EC migration (Figure 3). Earlier studies have shown that BRX-235 significantly reduced the intima thickness in Apo-E deficient mice, improved endothelial dysfunction and induced EC recovery in spontaneously hypertensive animals (unpublished data). Using SB 203580, a p38 SAPK specific inhibitor, we demonstrated that the stimulation of EC migration by BRX-235 is also mediated by p38 SAPK (Figure 4). Western blots have shown that BRX-235 stimulates phosphorylation of p38 SAPK at pharmacological dose ranges (Figure 5). The transient nature of this stimulation supports the notion that BRX-235 modifies stress signalling. Therefore BRX-235 acts upstream of p38 SAPK to induce BAE cell migration. Our data show (Figure 6c) that SB 203580 does not interfere with either the basal or the BRX-235 or ECGF-induced phosphorylation of p38 SAPK confirming that SB 203580 primarily acts on activated p38 SAPK (Kumar et al., 1997; 1999), although contradicting reports have also been published (Armstrong et al., 1999).
Bimoclomol® potentiates the activation and the accumulation of all major classes of heat shock proteins, including Hsp60, Hsp70, Hsp90 and GRP94 (glucose regulated protein 94) (Vígh et al., 1998) and BRX-235 has been shown to have a similar, or a slightly lower effect in connection with Hsp70 at pharmacologically equivalent doses in several cell-lines (3T3, ECV, A715, H9C2, PC12, etc.) (unpublished data).
p38 SAPK stabilizes the microfilaments in cells by activation of Hsp27. This leads to phosphorylation regulated F-actin polymerization and changes of the actin cytoskeleton that result in the motility of endothelial cells (Rousseau et al., 1997). Bimoclomol®, a close analogue of BRX-235, with pharmacologically similar profile activates p38 phosphorylation without affecting ERK or JNK phosphorylation (Balla et al., 1999; Kreuzer, 1999). p38 SAPK but not JNK and ERK regulates the activation of cytoskeletal alpha-actin in mechanical stress (Lew et al., 1999). Because of this notion, the activation of ERK and JNK by BRX-235 has not been specifically checked. Several authors have found parallelism or contradictory effect on the activation of p38 and JNK in various models (Schiaffonati et al., 2001; Recio & Merlino, 2002; Chaturvedi et al., 2002; Montaner & Perez-Thomas, 2002; Dougherty et al., 2002; Fleisher et al., 2001; Surapisitchat et al., 2001). Bimoclomol® potentiates stress-induced Hsp induction without affecting the overall heat shock factor 1 (HSF1) phosphorylation and it is known that JNK phosphorylation is followed by HSF1 phosphorylation (Park & Liu, 2001 and unpublished data). Additional experiments related to ERK and JNK activation are in progress. On the other hand SB 203580 inhibited the migration of BAE cells at concentration as low as 10 nM (Figure 2a). At this concentration we believe SB 203580 is specific for p38 SAPK.
Four isoforms of p38 MAPKs have been identified in mammalian cells, which are all phosphorylated and activated by MKK6 (MAPK kinase 6). In addition, MMK3 activates p38 SAPKα, p38 SAPKγ, and p38 SAPKδ, whereas MKK4 can only activate p38 SAPKα isomer (Nebreda & Porras, 2000). These kinases are specific for p38 SAPK (Igarashi et al., 2000). Thus, BRX-235 can target MKK6/3 to activate the p38 SAPK pathway. Alternatively, BRX-235 might directly bind p38 SAPK making it a better substrate for upstream kinases.
SB 203580 has been shown to be specific for p38 SAPKα and p38 SAPKβ isoforms (Wang et al., 1998). Our data showing that SB 203580 efficiently inhibits EC migration is correlated with data of the literature and confirm that p38α and p38β isoforms play a dominant role in EC migration (Azuma et al., 2001; Hedges et al., 1999; Rousseau et al., 1997).
Vascular endothelial growth factor (VEGF) and the basic fibroblast growth factor (bFGF) are being developed to treat a variety of vascular diseases due to their stimulatory effect on EC migration and proliferation (Gibbons, 1996). Both VEGF (Rousseau et al., 2000) and bFGF induce p38 activation. p38 SAPK is thought to orchestrate re-endothelization after angioplasty. It has been shown that re-endothelization limits neointima formation and subsequent restenosis (Ohashi et al., 2000). p38 SAPK has also been shown to mediate myocardial adaptation to ischaemia (Maulik et al., 1998). Nuclear factor κB (Maulik et al., 1998), Hsp27 (Armstrong et al., 1999; Craig et al., 2000) and mitochondrial K(ATP) channels (Baines et al., 1999) have all been shown as downstream effectors of p38 SAPK to mediate ischaemic preconditioning. Although re-endothelization and angiogenesis are distinct phenomena with respect to regulation and function, the mechanisms are partly overlapping. However, angiogenesis is a double-edged sword so we studied the angiogenic effect of BRX-235 using the chicken chorioallantoic membrane system. The compound showed no angiogenic effect at any of the pharmacological and supra pharmacological doses tested (unpublished data).
In summary, BRX-235-induced endothelial repair is mediated through the p38 SAPK pathway. p38 SAPK-mediated EC migration certainly contributes favourably to the therapeutic effects of BRX-235 in atherosclerosis and restenosis.
Abbreviations
- BAE cell
bovine aortic endothelial cell
- bFGF
basic fibroblast growth factor
- EC
endothelial cell
- ECGF
endothelial cell growth factor
- ECGFs
endothelial cell growth factor supplement
- ECL
enhanced chemiluminescence
- ERK
extra-cellular signal-regulated kinase
- Ets-1
endothelial transfer substance-1
- GRP94
glucose regulated protein 94
- HUVEC
human umbilical venous endothelial cells
- MAPKK
mitogen activated kinase kinase
- SAPK
stress activated protein kinase
- SMC
smooth muscle cell
- VEGF
vascular endothelial growth factor
- WMA
wounding migration assay
References
- ARMSTRONG S.C., DELACEY M., GANOTE C.E. Phosphorylation state of hsp27 and p38 MAPK during preconditioning and protein phosphatase inhibitor protection of rabbit cardiomyocytes. J. Mol. Cell. Cardiol. 1999;31:555–567. doi: 10.1006/jmcc.1998.0891. [DOI] [PubMed] [Google Scholar]
- AZUMA N., AKASAKA N., KITO H., IKEDA M., GAHTAN V., SASAJIMA T., SUMPIO B.E. Role of p38 MAP kinase in endothelial cell alignment induced by fluid shear stress. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H189–H197. doi: 10.1152/ajpheart.2001.280.1.H189. [DOI] [PubMed] [Google Scholar]
- BAINES C.P., COHEN M.V., DOWNEY J.M. Signal transduction in ischemic preconditioning: the role of kinases and mitochondrial K(ATP) channels. J. Cardiovasc. Electrophysiol. 1999;10:741–754. doi: 10.1111/j.1540-8167.1999.tb00251.x. [DOI] [PubMed] [Google Scholar]
- BALLA A., JÓZSA I., VÍGH L., KRAJCSI P. Effect of Bimoclomol on stress signalling. Fundam. Clin. Pharmacol. 1999;13 Suppl. 1:359s. [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- BURGESS W.H., MEHLMAN T., MARSHAK D.R., FRASER B.A., MACIAG T. Structural evidence that endothelial cell growth factor beta is the precursor of both endothelial cell growth factor alpha and acidic fibroblast growth factor. Proc. Natl. Acad. Sci. U.S.A. 1986;83:7216–7220. doi: 10.1073/pnas.83.19.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHATURVEDI L.S., KOUL S., SEKHON A., BHANDARI A., MENON M., KOUL H.K. Oxalate selectively activates p38 MAPK and c-JNK signal transductin pathways in renal epithelial cells. J. Biol. Chem. 2002;1:31. doi: 10.1074/jbc.M108203200. [DOI] [PubMed] [Google Scholar]
- CRAIG R., LARKIN A., MINGO A.M., THUREAUF D.J., ANDREWS C., MCDONOUGH P.M., GLEMBOTSKI C.C. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in cardiac myocyte model system. J. Biol. Chem. 2000;275:23814–23824. doi: 10.1074/jbc.M909695199. [DOI] [PubMed] [Google Scholar]
- CUENDA A., ROUSE J., DOZA Y.N., MEIER R., COHEN P., GALLAGHER T.F., YOUNG P.R., LEE J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- DOUGHERTY C.J., KUBASIAK I.A., PRENTICE H., ANDREKA P., BISHOPRIC N.H., WEBSTER K.A. Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress. Biochem. J. 2002;362:561–567. doi: 10.1042/0264-6021:3620561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLEISHER F., DABEW R., GOKE B., WAGNER A. Stress kinase inhibition acute experimental pancreatitis. World J. Gastroenterol. 2001;7:259–265. doi: 10.3748/wjg.v7.i2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIBBONS G.H. VEGF and bFGF variety of vascular diseases and enhanced EC migration, proliferation. Science. 1996;272:689–693. doi: 10.1126/science.272.5262.689. [DOI] [PubMed] [Google Scholar]
- HEDGES J.C., DECHERT M.A., YAMBOLIEV I.A., MARTIN J.L., HICKEY E., WEBER L.A., GERTHOFFER W.T. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J. Biol. Chem. 1999;274:24211–24219. doi: 10.1074/jbc.274.34.24211. [DOI] [PubMed] [Google Scholar]
- IGARASHI M., YAMAGUCHI H., HIRATA A., DAIMON M., TOMINAGA M., KATO T. Insulin activates p38 mitogen-activated (MAP) kinase via a MAP kinase kinase (MKKK) 3/MKK6 pathway in vascular smooth muscle cells. Eur. J. Clin. Invest. 2000;30:668–677. doi: 10.1046/j.1365-2362.2000.00671.x. [DOI] [PubMed] [Google Scholar]
- KREUZER J. Expression of heat shock protein in human smooth muscle cells in vitro after treatment with Bimoclomol. Fundam. Clin. Pharmacol. 1999;13 Suppl. 1:S83. [Google Scholar]
- KUMAR S., JIGNA M.S., ADAMS J.L., LEE J.C. Pyridinylimidazole compound SB 203580 inhibits the activity but not the activation of p38 mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 1999;263:825–831. doi: 10.1006/bbrc.1999.1454. [DOI] [PubMed] [Google Scholar]
- KUMAR S., MCDONNAL P.C., GUM R.J., HAND A.T., LEE J.C., YOUNG P.R. Novel homologues of CeBP/p38 MAP kinase: activation, substrate, specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem. Biophys. Res. Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- LEE J.C., LAYDON J.T., MCDONNELL P.C., GALLAGHER T.F., KUMMER S., GREEN D., MCNULTY D., BLUMENTHAL M.J., HEYS J.R., LANDVATTER S.W., STRICKLER J.E., MCLAIGHLIN M.M., SIEMENS I.R., FISHER S.M., LIVI G.P., WHITE J.R., ADAMS J.L., YOUNG P. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- LEW A.M., GLOGANER M., MCMULLOCH C.A. Specific inhibition of skeletal alpha-actin gene transcription by applied mechanical forces through integrins and actin. Biochem. J. 1999;341:647–653. [PMC free article] [PubMed] [Google Scholar]
- MACIAG T., HOOVER G.A., WEINSTEIN R. High and low molecular weight forms of endothelial cell growth factor. J. Biol. Chem. 1982;257:5333–5336. [PubMed] [Google Scholar]
- MAULIK N., SATO M., PRICE B.D., DAS D.K. An essential role of NFκB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett. 1998;429:365–369. doi: 10.1016/s0014-5793(98)00632-2. [DOI] [PubMed] [Google Scholar]
- MONTANER B., PEREZ-THOMAS R. The cytotoxic induces phosphorylation of p38-MAPK but not of SAPK/JNK. Toxicol. Lett. 2002;129:93–98. doi: 10.1016/s0378-4274(01)00477-5. [DOI] [PubMed] [Google Scholar]
- NÁNÁSI P.P., JEDNÁKOVITS A. Multilateral in vivo and in vitro protective effect of the novel heat shock protein coinducer, Bimoclomol: results of preclinical studies. Cardiovasc. Drug Rev. 2001;19:133–151. doi: 10.1111/j.1527-3466.2001.tb00060.x. [DOI] [PubMed] [Google Scholar]
- NEBREDA A.R., PORRAS A. P38 MAP kinase: beyond the stress response. Trends Biochem. Sci. (TIBS) 2000;25:257–259. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- OHASHI N., MATSUMORI A., FURUKAWA Y., ONO K., OKADA M., IWASAKI A., MIYAMOTO T., NAKANO A., SASAYAMA A. Role of p38 mitogen-activated protein kinase in neointimal hyperplasia after vascular injury. Arterioscler. Thromb. Vasc. Biol. 2000;12:2521–2526. doi: 10.1161/01.atv.20.12.2521. [DOI] [PubMed] [Google Scholar]
- PARK J., LIU A.Y. JNK phosphorylates the HSF1 transcriptional activation domain: role of JNK in the α-regulation of the heat shock response. J. Cell. Biochem. 2001;82:326–338. doi: 10.1002/jcb.1163. [DOI] [PubMed] [Google Scholar]
- RECIO J.A., MERLINO G. Hepatocytes growth factor/scatter factor activates proliferation in melanoma cells through p38 MAPK, ATF2 and cyclin D1. Oncogene. 2002;21:1000–1008. doi: 10.1038/sj.onc.1205150. [DOI] [PubMed] [Google Scholar]
- ROUSSEAU S., HOULE F., KOTANIDES H., WITTE L., WALTEMBERGER J., LANDRY J., HOUT J. Vascular endothelial growth factor (VEGF)-driven actin based motility is med#iated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J. Biol. Chem. 2000;275:10661–10672. doi: 10.1074/jbc.275.14.10661. [DOI] [PubMed] [Google Scholar]
- ROUSSEAU S., HOULE F., LANDRY J., HOUT J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- SCHIAFFONATI L., MARONI P., BENDINELL P., TIBERIO L., PICOLETTI R. Hyperthermia induces gene expression of HSP70 and phosphorylation of mitogen protein kinase in the rat cerebellum. Neurosci. Lett. 2001;312:75–78. doi: 10.1016/s0304-3940(01)02182-6. [DOI] [PubMed] [Google Scholar]
- SURAPISITCHAT J., HOEFEN R.J., PI X., YOSHIZUMI M., YAN C., BERK B.C. Fluid stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in HUVEK: Inhibitory crosstalk among MAPK family member. PNAS. 2001;98:6476–6481. doi: 10.1073/pnas.101134098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANAKA K., ABE M., SATO Y. Roles of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in the signal transduction of basic fibroblast growth factor in endothelial cell during angiogenesis. Jpn. J. Cancer. Res. 1999;90:647–654. doi: 10.1111/j.1349-7006.1999.tb00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANAKA K., ODA N., IWASAKA C., ABE M., SATO Y. Induction of Ets-1 in endothelial cells during reendothelialization after denuding injury. J. Cell. Physiol. 1998;176:235–244. doi: 10.1002/(SICI)1097-4652(199808)176:2<235::AID-JCP2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- VÍGH L., LITERÁTI P.N., HORVÁTH I., TÖRÖK ZS., BALOGH G., GLATZ A., KOVÁCS E., BOROS I., FERDINÁNDY P., FARKAS B., JASZLITS L., JEDNÁKOVITS A., KORÁNYI L., MARESCA B. Bimoclomol: A nontoxic, hydroxylamine derivate with stress protein-inducing activity and cytoprotective effects. Nat. Med. 1997;3:1150–1154. doi: 10.1038/nm1097-1150. [DOI] [PubMed] [Google Scholar]
- VÍGH L., MARESCA B., HARWOOD J.L. Does the membrane's physical state control the expression of heat shock and other genes. Trends Biochem. Sci. (TIBS) 1998;23:369–374. doi: 10.1016/s0968-0004(98)01279-1. [DOI] [PubMed] [Google Scholar]
- WANG Y., HUANG S., SAH V.P., ROSS J., JR, BROWN J.H., HAN J., CHIEN K. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J. Biol. Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- YAMAMURA S., NELSON P.R., KENT K.C. Role of protein kinase C in attachment, spreading, and migration of endothelial cells. J. Surg. Res. 1996;63:349–354. doi: 10.1006/jsre.1996.0274. [DOI] [PubMed] [Google Scholar]