

Abstract
The pharmacological profile of a series of (±)-2,5-dimethoxy-4-(X)-phenylisopropylamines (X=I, Br, NO2, CH3, or H) and corresponding phenylethylamines, was determined in Xenopus laevis oocytes injected with cRNA coding for rat 5-HT2A or 5-HT2C receptors. The efficacy and relative potency of these drugs were determined and compared to classical 5-HT2 receptor agonists and antagonists.
The rank order of agonist potency at the 5-HT2A receptor was: α-methyl-5-HT=5-HT>m-CPP>MK-212; at the 5-HT2C receptor the order was: 5-HT>α-methyl-5-HT>MK-212>m-CPP. All these compounds were full agonists at the 5-HT2C receptor, but α-methyl-5-HT and m-CPP showed lower efficacy at the 5-HT2A receptor.
4-(4-Fluorobenzoyl)-1-(4-phenylbutyl)piperidine (4F 4PP) was 200 times more potent as a 5-HT2A antagonist than at 5-HT2C receptors. Conversely, RS 102221 was 100 times more potent as a 5-HT2C antagonist, confirming their relative receptor selectivities.
The phenylisopropylamines were partial agonists at the 5-HT2A receptor, with Imax relative to 5-HT in the 22±7 to 58±15% range; the corresponding phenylethylamines had lower or undetectable efficacies. All these drugs had higher efficacies at 5-HT2C receptors; DOI was a full 5-HT2C agonist. 2C-I and the other phenylethylamines examined showed relative efficacies at the 5-HT2C receptor ranging from 44±10% to 76±16%.
2C-N was a 5-HT2 receptor antagonist; the mechanism was competitive at the 5-HT2A, but non-competitive at the 5-HT2C receptor. The antagonism was time-dependent at the 5-HT2C receptor but independent of pre-incubation time at the 5-HT2A receptor subtype.
The α-methyl group determines the efficacy of these phenylalkylamines at the 5-HT2A and 5-HT2C receptors.
Keywords: Hallucinogenesis, hallucinogenic phenylalkylamines, 5-HT2A/2C receptors agonists/antagonists, 5-HT2 receptor subtype-selective, 5-HT2 partial agonist, phenylisopropylamines/phenylethylamines
Introduction
Studies on the pharmacodynamics of phenylalkylamines have focused largely on the identification of the brain receptors involved in their psychotropic actions. An abundance of evidence for the participation of the 5-HT2 receptors in the action of hallucinogenic phenylalkylamines has accumulated (Nichols, 1997; Sanders-Bush & Mayer, 2001). Although radioligand displacement experiments show a correlation between 5-HT2 receptor affinity in rat brain membranes and the human hallucinogenic potency of several phenylisopropylamines (PIAs), these compounds are not generally subtype-selective (Glennon et al., 1992; Nelson et al., 1999).
The observation that ring-substituted psychotropic PIAs are more potent than the corresponding phenylethylamines (PEAs), that lack an α-methyl group, was the main impulse to study PIAs rather than PEAs. In humans, psychotropic ring-substituted PIAs are usually 2–10 times more potent than their PEA counterparts (reviewed by Shulgin & Shulgin, 1991), and similar results have been obtained in behavioural studies in rats (Nichols, 1997). More recently, the 2,5-dimethoxy-4-substituted PIAs have been the main subject of interest in this area because of their greater potency in rodents and in humans (Nichols, 1997). In particular, (±)-1-(2,5-dimethoxy-4-bromophenyl)-2-aminopropane (DOB) and (±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) (Figure 1) were quickly identified as potent hallucinogens (see review by Shulgin, 1981) and have since been extensively used as 5-HT2 receptor agonists (Baxter et al., 1995; Barnes & Sharp, 1999). Nevertheless, the influence of the α-methyl group on the direct interaction of these compounds at 5-HT2A receptors has not been sufficiently documented.
Figure 1.
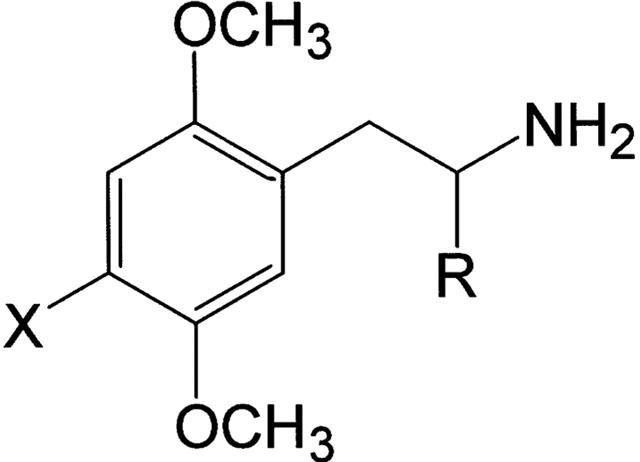
Structural formulae of the PIA/PEA pairs tested. PIAs: R=CH3; DOI, X=I; DOB, X=Br; DON, X=NO2; DOM, X=CH3; 2,5-DMA, X=H. PEAs: R=H; 2C-I, X=I; 2C-B, X=Br; 2C-N, X=NO2; 2C-D, X=CH3; 2C-H, X=H.
An interesting contribution by Glennon et al. (1992) was the observation that a few identically ring-substituted PIA/PEA pairs such as DOB/2C-B (Figure 1) have very similar relative affinities for the 5-HT2A receptor in rat brain membranes. This finding cannot account for the difference of in vivo hallucinogenic potency observed within PIA/PEA pairs. Shulgin (1981) proposed that the differences in potency might be a consequence of the greater metabolic lability of the PEAs compared to the corresponding PIAs. However, little progress has been made in this area within the past two decades, except for the observation that in some cases, the duration of action of PIAs is considerably longer than that of the corresponding PEAs (see Shulgin & Shulgin, 1991). Regarding their pharmacodynamics, several investigations suggest that activation of 5-HT2A/2C receptors, either as full or partial agonists, might underlie the psychotropic properties of the hallucinogenic phenylalkylamines (Nichols, 1997). The involvement of the 5-HT2B receptor in the hallucinogenic action of the PIAs is controversial. In radioligand displacement experiments, the affinities of a number of hallucinogenic PIAs are at least an order of magnitude lower at 5-HT2B than at 5-HT2A or 5-HT2C receptors (Nelson et al., 1999). However, two independent studies measuring agonist-elicited changes in intracellular calcium levels in two cell lines showed that the rank order of potency for DOI and DOB is 5-HT2A> 5-HT2B> 5-HT2C (Porter et al., 1999; Jerman et al., 2001). Although the 5-HT2B receptor is closely related in structure and pharmacology to the other receptor subtypes, its expression in brain is limited (Choi & Maroteaux, 1996); it has a clearly defined role in sympathetic outflow (Knowles & Ramage, 1999, 2000), but its only documented behavioural actions relate to anxiogenesis and hyperphagia (Kennett et al., 1996). At present, we cannot discard the involvement of the 5-HT2B receptor in the action of PIAs. The question of which 5-HT2 receptor subtypes participate in hallucinogenesis must await properly controlled studies using subtype-selective drugs or transgenic animals. It is obvious that neurochemical studies alone cannot account for the behavioural basis of drug action; combined in vivo studies with selective subtype receptor ligands are necessary for a complete understanding of hallucinogenesis.
Several years ago we showed that a series of 3,4- and 2,4,5-ring substituted psychotropic PEAs are serotonergic agonists (Lobos et al., 1992; Sáez et al., 1994). Detailed studies comparing the profile of 2,5-dimethoxy-4-substituted PIA/PEA pairs have not been undertaken, with the exception of the 2,5-dimethoxy-4-trifluoromethyl-substituted phenylalkylamine pair (Nichols et al., 1994). On the basis of the study of this single PIA/PEA pair, they proposed that an α-methyl substituent increases the efficacy of phenylalkylamines at the 5-HT2A receptor but not at the 5-HT2C receptor. In view of the strong hydrophobic character of the 4-trifluoromethyl group in the PIA/PEA pair studied by Nichols et al. (1994) we were motivated to test additional pairs with a broad range of lipophilicities for the C-4 substituent. These compounds would allow testing of the hypothesis that the relatively weaker hallucinogenic potency of 2,5-dimethoxy-4-substituted PEAs, as compared to their PIA counterparts, might be a consequence, at least in part, of the differences in efficacy at the 5-HT2A receptor. With this aim, we compared the relative efficacy and potency of a series of 2,5-dimethoxylated PIA/PEA pairs with a variety of C-4 substituents (Figure 1) at rat 5-HT2A or 5-HT2C receptors, expressed in Xenopus laevis oocytes. The Xenopus oocyte was chosen as this cell allow the expression of a variety of non-endogenously present receptors, as the 5-HT2A or 5-HT2C receptor subtypes. Furthermore, this model system allows relatively simple determinations of drug potency and efficacy based on single cell recording of the currents elicited by 5-HT and related agonists. These receptors are coupled to the phospholipase C cascade, through Gq protein. The subsequent synthesis of IP3 releases intracellular calcium stores which, in this particular cell, activate a chloride channel, generating currents, which are easily detectable by the two-electrode configuration (Dascal et al., 1986; Moran & Marty, 1989). This model system has been used successfully to study the mechanism of action of important psychoactive drugs such as general anaesthetics (Yamakura et al., 2001), alcohol (Mihic et al., 1997), sedative hypnotics including barbiturates and the benzodiazepines (McKernan et al., 1995) as well as ALEPH-2, a 2,5-dimethoxy-4-substituted PIA which proved to be a partial 5-HT2A agonist but a full 5-HT2C receptor agonist (Acuña et al., 2000).
The results presented herein demonstrate that some psychoactive PEAs have weak or undetectable 5-HT2A receptor efficacies compared with their PIA counterparts. Furthermore, the lipophilicity of the C-4 substituent of these paired phenylalkylamines is not directly related to their intrinsic efficacy.
Methods
Oocyte harvesting, microinjection of rat 5-HT2A and5-HT2C receptor clones and characterization of5-HT responses
Xenopus laevis ovary lobes were surgically removed; stage V and VI oocytes were manually defolliculated and further treated with collagenase II as previously described (Acuña et al., 2000). Oocytes were then microinjected intracytoplasmatically with 10–20 ng of a cRNA for the rat 5-HT2A or 5-HT2C receptor clones. The oocytes were incubated for 36–48 h at 15°C in standard Barth's solution supplemented with 10 ui/l penicillin-streptomycin and 2 mM pyruvate.
The oocytes were impaled using the two-electrode technique in a voltage-clamp configuration using an OC-725C oocyte clamp (Warner Instrument Corp.); the membrane potential was fixed at −70 mV. To ascertain the expression of the receptors following transfection, the oocytes were challenged for 10 s with 100 nM 5-HT for the 5-HT2A and 10 nM 5-HT for the 5-HT2C receptor every 20–30 min. In case a significant current was recorded, the same concentration of 5-HT was repeated several times until a stable response was attained. 5-HT and related analogues were applied by superfusion at a constant flow rate of 2 ml/min−1. Uninjected oocytes did not respond to applications of 5-HT or of the drugs tested.
Agonist concentration-response protocols
Different concentrations of 5-HT and other analogues, including the PIAs and their corresponding PEAs, were used to perform concentration-response protocols. To minimize receptor desensitization, all agonists were added for 10 s at 20–30 min intervals, a procedure that provided the most reproducible results. In the majority of protocols, at least 6–7 concentrations of each compound were tested to ensure that the maximal response was attained. To compare the potency of the 5-HT analogues, care was taken in each oocyte to obtain a maximal 5-HT response, which was used to normalize the currents attained with the 5-HT agonists. This protocol allowed us to compare the relative potencies of agonists independently of the magnitude of the current evoked in each oocyte.
Median effective concentrations (EC50) and the maximal current generated (Imax) by each of the agonists examined relative to that elicited by 5-HT, including DOI, DOB, DON, DOM, 2,5-DMA, and their PEA counterparts 2C-I, 2C-B, 2C-N, 2C-D, and 2C-H, in both receptor subtypes, were derived from their respective concentration-response curves. These curves were always normalized according to the maximal 5-HT-evoked current attained in each single oocyte tested.
Studies with selective 5-HT receptor subtype antagonists; effect of pre-incubation
Two antagonists with different relative selectivities were used to assess the pharmacology of the 5-HT responses evoked by the 5-HT2A and the 5-HT2C receptors. 4F 4PP was chosen as a prototype drug with relative antagonist selectivity for the 5-HT2A receptor, while RS 102221 was studied as the prototype antagonist of the 5-HT2C receptor. Antagonism was assessed in two protocols, for both receptor subtypes: different concentrations of these antagonists were either co-applied with of 5-HT or they were pre-applied for 50 s and co-applied for the next 10 s with the test 5-HT concentration, a value close to the EC50 (100 nM for the 5-HT2A and 10 nM for the 5-HT2C receptor). Parallel experiments were also performed using methysergide and ketanserin, drugs which were co-applied with the 5-HT test concentration.
Pharmacological characterization of the antagonism of 5-HT2 receptors by 2C-N
Several protocols were designed to characterize the mechanisms of 2C-N-induced 5-HT receptor blockade. Sets of parallel experiments were performed in oocytes transfected with either the 5-HT2A or the 5-HT2C receptor subtypes.
Antagonist activity
To test whether 2C-N blocked the 10 s 5-HT-induced responses at either receptor subtype, the test concentration of 5-HT for each receptor subtype was co-applied with 0.1 nM–100 μM 2C-N. After each 2C-N application, the recovery of the 5-HT current was examined ensuring that the 5-HT response reached its standard value before the next concentration of 2C-N was evaluated.
Time-dependence
We next assessed whether the 2C-N-induced 5-HT2 receptor blockade was dependent on pre-application of this drug. For this purpose, the oocytes were pre-exposed to 2C-N for 10–300 s prior to the 5-HT testing concentrations used for each receptor subtype.
Mechanism of 2C-N antagonism
(a) 5-HT2A receptors. To examine the nature of the 2C-N blockade, a full 5-HT concentration-response curve was obtained prior to and following co-application of 0.04, 1 or 10 μM 2C-N. A first control 5-HT concentration-response curve was obtained, followed by a second, in the presence of each concentration of 2C-N (n=4 oocytes per curve). In all cases, the recovery of the 5-HT-evoked current was mandatory prior to application of the next concentration of 5-HT plus 2C-N. A Schild plot was derived according to Arunlakshana & Schild (1959). (b) 5-HT2C receptors. To test the antagonist properties of 2C-N, we pre-applied 50, 100 or 150 nM 2C-N for 3 min prior to each 5-HT concentration required to perform a concentration-response protocol (n=5–6, per curve). The same oocytes had previously been tested with a full 5-HT response curve. Care was always taken to ensure that a standard 5-HT-induced current recovered prior to the next 2C-N application.
Statistical analysis
Results are presented as mean±s.e.mean corresponding to experiments performed with 4–9 oocytes from at least two separate batches of cells. Based on standard deviation values, parallel experiments revealed that inter- was larger than intra-assay variability for 5-HT2 receptors, which in some cases may reach 25–40%.
The currents generated by 5-HT or the other agonists tested were plotted against concentration using Graph-Pad Software (Graph-Pad Inc., San Diego, CA, U.S.A.) to obtain the median effective concentration (EC50), relative maximal current (Imax) (Acuña et al., 2000). In the case of antagonists, each median effective concentration (IC50) was interpolated from an antagonist concentration plot, which was also analysed using the same software. Nonparametric analysis, Kruskal-Wallis, Mann-Whitney and Friedman & Quade tests were also used for statistical analysis. In all cases, significance was set at a P value <0.05.
Drugs and chemicals used
Most of the (±)-PIA (DOI, DOB, DOM, DON and 2,5-DMA) and their PEA counterparts (2C-I, 2C-B, 2C-D, 2C-N and 2C-H) (Figure 1) were synthesized as detailed by Shulgin & Shulgin (1991), and prepared as the hydrochloride salts. DON and 2C-N were synthesized by direct nitration of 2,5-DMA or 2C-H, respectively, in aqueous solution, and were used as the nitrates. 2C-B was obtained by bromination of 2C-H in acetic acid with Br2 and used as the hydrobromide. 5-HT, α-methyl-5-HT, 1-(3-chlorophenyl)piperazine dihydrochloride (m-CPP), ketanserin tartrate and methysergide maleate were purchased from RBI (Natick, MA, U.S.A.). 6-Chloro-2-(1-piperazinyl)pyrazine (MK-212), 4-(4-fluorobenzoyl)-1-(4-phenylbutyl) piperidine (4F 4PP), and 8-[5-(2,4-dimethoxy-5- (4-trifluoromethylphenylsulphonamido)phenyl-5-oxopentyl]-1,3,8-triazaspiro [4,5]decane-2,4-dione (RS 102221) were from Tocris (Ballwin, MO, U.S.A.).
Results
Potency of 5-HT and related synthetic agonists
5-HT was five times more potent at the 5-HT2C than at the 5-HT2A receptor subtype. The rank order of potency of several agonists for the 5-HT2A receptor was: α-methyl-5-HT>5-HT>m-CPP>MK-212. In contrast, at the 5-HT2C receptor, the relative rank order of potency of these compounds was: 5-HT>α-methyl-5-HT>MK-212>m-CPP. Their corresponding EC50s and Imax are detailed in Table 1. The concentration-response curves of all these agonists were parallel (Figure 2). Full agonism was observed with all these drugs at the 5-HT2C receptor, while only partial agonism was attained with α-methyl-5-HT and m-CPP at the 5-HT2A receptor (Table 1 and Figure 2).
Table 1.
Activity of 5-HT and related agonists evaluated at 5-HT2A and 5-HT2C receptors; summary of curve-derived parameters for each agonist
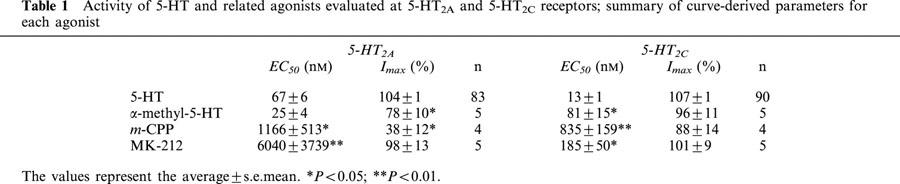
Figure 2.
Relative potency and efficacy of 5-HT and related agonists at 5-HT2 receptors. Agonist concentration-response curves were obtained using oocytes injected with either the 5-HT2A (A) or 5-HT2C (B) receptor cRNAs. The agonist curves were normalized with respect to the maximal response attained by 5-ht in each oocyte. 5-HT (n=83–90); α-methyl-5-HT (n=5), MK-212 (n=5); m-CPP (n=4).
Selective receptor subtype antagonism
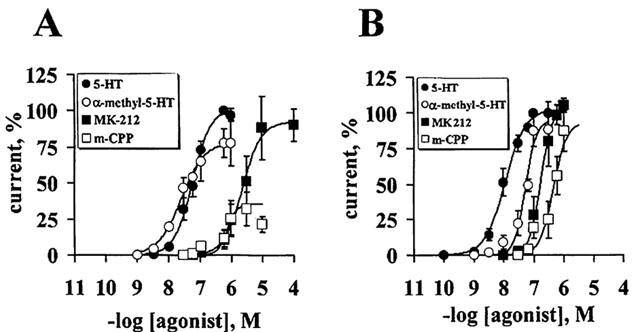
4F 4PP was more potent than RS 102221 as a 5-HT2A receptor antagonist, whereas RS 102221 was more potent than 4F 4PP in reducing the 5-HT evoked responses at the 5-HT2C receptor. The potency of these antagonists increased at least 50 fold following 1 min pre-incubation as compared to their co-application with 5-HT, and indication that these antagonists have a rather slow onset of action. 4F 4PP is a weak 5-HT2C antagonist; its potency does not increase significantly after pre-incubation for 1 min. The IC50 values of these antagonists co- and pre-applied are detailed in Table 2. Representative tracings of 5-HT-evoked currents and their selective blockade by these drugs are shown in Figure 3A,D.
Table 2.
Selective antagonism of 4F 4PP and RS 102221 on 5-HT-evoked current at 5-HT2A and 5-HT2C receptors

Figure 3.
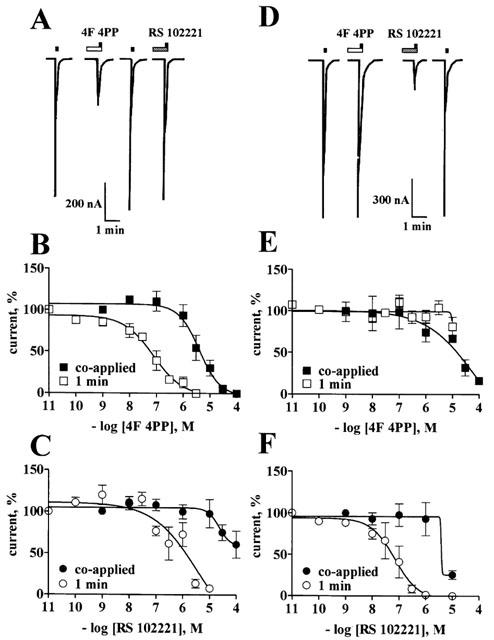
4F 4PP and RS 102221 selectively block 5-HT responses at 5-HT2A and 5-HT2C receptors. Upper panels: Representative tracings from a single oocyte showing the selective inhibition of the currents induced by 100 nM 5-HT at 5-HT2A receptors (A) and 10 nM 5-HT at 5-HT2C receptors (D) after 1 min pre-incubation with either 100 nM 4F 4PP (open rectangle) or 100 nM RS 102221 (shaded rectangle). Applications of 5-HT are represented by the closed squares. Lower panels: Blockade of the currents induced by 100 nM 5-HT at 5-HT2A receptors by different concentrations of 4F 4PP (B), or RS 102221 (C), co-applied or pre-incubated for 1 min prior to 5-HT. Blockade of the currents induced by 10 nM 5-HT at 5-HT2C receptors by different concentrations of 4F 4PP (E), or RS 102221 (F) co-applied or pre-incubated for 1 min prior to 5-HT. A complete antagonist concentration-response curve was obtained per oocyte at the 5-HT2A (n=7) and at the 5-HT2C receptor (n=5–6). Symbols indicate the mean values; bars s.e.mean.
Parallel studies examined the antagonism by ketanserin or methysergide, classical 5-HT2 receptor antagonists. Different concentrations of the antagonists were co-applied with 100 nM 5-HT. Their Ic50 from these protocols were 4.2±1.3 and 5.1±1.7 μM, respectively (n=4). The blockade caused by these antagonists was not easily reversible, in spite of prolonged drug washouts. We did not examine the interaction of these compounds with 5-HT2C receptors.
Comparison of the agonist potencies and efficacies of PIA/PEA pairs
At the 5-HT2A receptor, the efficacies of all the PIAs were significantly greater than those of their corresponding PEAs; in contrast, the efficacies were not significantly different at the 5-HT2C receptor, with the exception of the DOI/2C-I pair. Each PEA was much less efficacious at the 5-HT2A than at the 5-HT2C receptor. However, the PIAs did not show very marked differences in efficacy between both receptor subtypes. Only DOI and DOM were almost full agonists at the 5-HT2C subtype, while their efficacies at the 5-HT2A receptor relative to 5-HT were about 50%, like DOB and 2,5-DMA (but not the less efficacious DON). The EC50 values and the relative Imax for all these compounds are summarized in Table 3. In contrast to the practically full agonism of DOI at the 5-HT2C receptor, 2C-I was a partial agonist at both subtypes, showing a significantly lower efficacy at the 5-HT2A receptor (Table 3 and Figure 4). Representative tracings showing the partial agonism of DOI at the 5-HT2A receptor and full agonism at the 5-HT2C receptor are shown in Figure 4A,C.
Table 3.
Activity of 5-HT and the 2,5-dimethoxy-4-substituted phenylalkylamines evaluated at 5-HT2A and 5-HT2C receptors
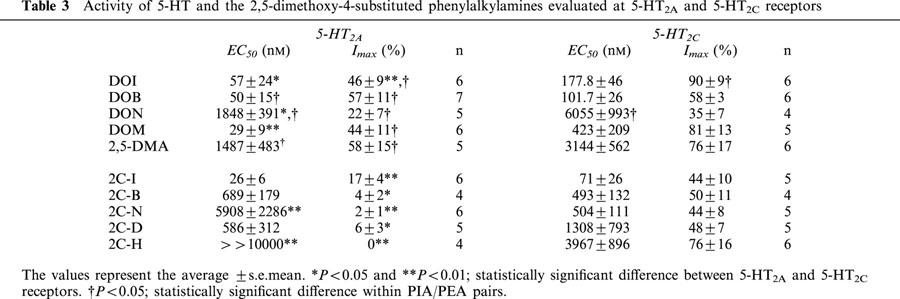
Figure 4.
Concentration-response curves for DOI and 2C-I at 5-HT2 receptors. Upper panels: Representative tracings show partial DOI agonism at 5-HT2A (A) and full agonism at 5-HT2C receptors (C). Oocytes were challenged with 5-HT concentrations eliciting the maximal current (600 nM at 5-HT2A receptors and 100 nM at 5-HT2C receptors) to compare with partial DOI agonism. 5-HT applications are represented by the closed squares; DOI by open squares. Lower Panels: DOI and 2C-I concentration-response curves in oocytes expressing 5-HT2A (B) and 5-HT2C (D) receptors. The DOI or 2C-I currents generated in each oocyte were normalized to the maximal 5-HT current attained in each oocyte (n=12 for 5-HT2A; n=11 for 5-HT2C receptor), DOI (n=6), 2C-I (n=5–6) for each receptor subtype. Symbols represent mean values; bars, s.e.mean.
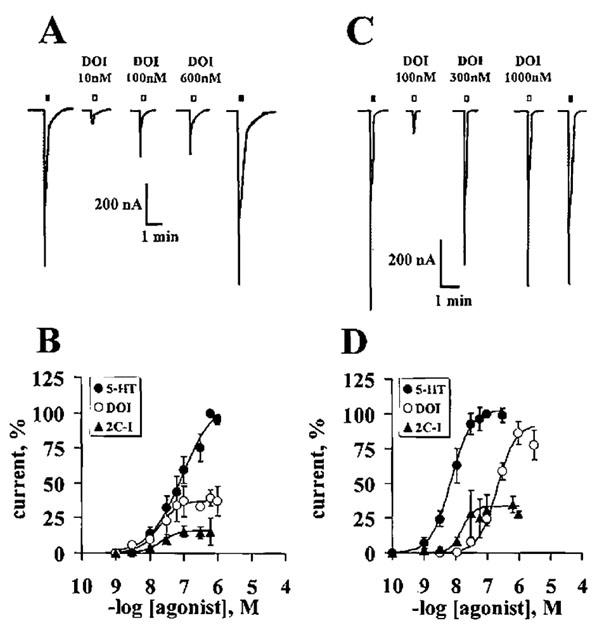
DON is a partial agonist at both receptor subtypes, but 2C-N showed an almost complete lack of efficacy at 5-HT2A receptors, while retaining partial agonism at the 5-HT2C receptor (Figure 5 and Table 3). See representative tracings of the effects of 2C-N at 5-HT2A and at 5-HT2C receptors in Figure 5A,C.
Figure 5.
DON and 2C-N concentration-response curve at 5-HT2 receptors. Upper panels: Representative tracings show the lack of efficacy of 2C-N at the 5-HT2A receptor (A) as compared to its partial agonism at the 5-HT2C receptor (C). Open squares represent the 10 s 2C-N applications; closed squares indicate the additions of 600 or 100 nM 5-HT, which elicited the maximal 5-HT current at 5-HT2A and 5-HT2C receptors respectively. Lower panels: 5-HT, DON and 2C-N concentration-response curves at 5-HT2A (B) or 5-HT2C receptors (D). The currents generated by DON or 2C-N were normalised using the maximal 5-HT-evoked current in each oocyte. Four to six separate oocytes were studied per agonist for each receptor subtype. Symbols represent the mean values; bars, s.e.mean.
Comparing the 5-HT2C/5-HT2A potency ratios of the PIAs, Table 3 shows that in most cases these were only 2 to 3, but the corresponding ratio for DOM was about 15, suggesting that a methyl group at C-4 may be unfavourable for 5-HT2C receptor affinity.
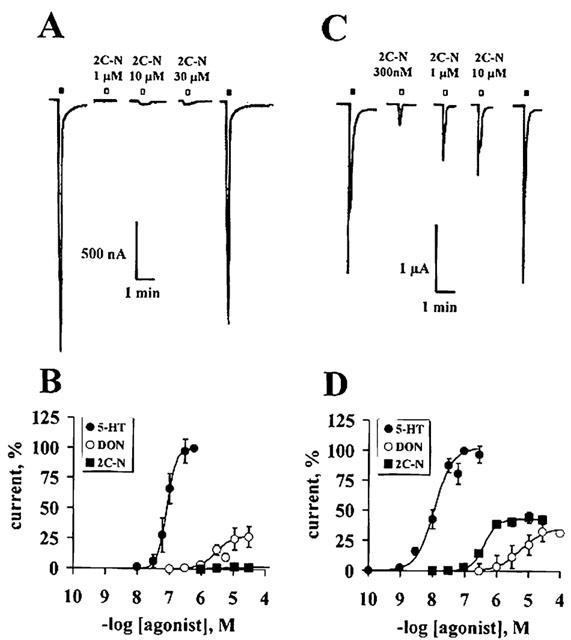
2C-N is a competitive 5-HT2A receptor antagonist
Co-application of increasing concentrations of 2C-N plus 100 nM 5-HT, led eventually to the total suppression of the 5-HT2A receptor-gated current (Figure 6A). The blockade was independent of the pre-exposure time of 2C-N, since 50 nM 2C-N induced 50% inhibition, which did not vary with increasing pre-exposure times up to 5 min (Figure 6B). Co-incubation of increasing concentrations of 2C-N with 5-HT led to parallel shifts of the concentration-response curve to the right (Figure 6C). The Schild plot pA2 was 8.14±0.51 (inset), corresponding to a Kb of 7.02±3.12 nM (r=0.97±0.01 with a slope of 0.48±0.1).
Figure 6.
2C-N is a competitive antagonist at the 5-HT2A receptor. (A) The co-application of different concentrations of 2C-N plus 100 nM 5-HT blocked the 5-HT-induced currents in a concentration-dependent manner; the sole application of 2C-N did not elicit responses (n=5–6). (B) The inhibition of the 100 nM 5-HT-induced currents by 50 nM 2C-N was time-independent (n=4). (C) Rightward displacements of the control 5-HT concentration-response curve by the co-application of 5-HT plus 0.04 μM (n=4); 1 μM (n=4) or 10 μM 2C-N (n=2). The insert shows a Schild plot. Symbols represent mean values; bars, s.e.mean.
2C-N is a non-competitive 5-HT2C receptor antagonist
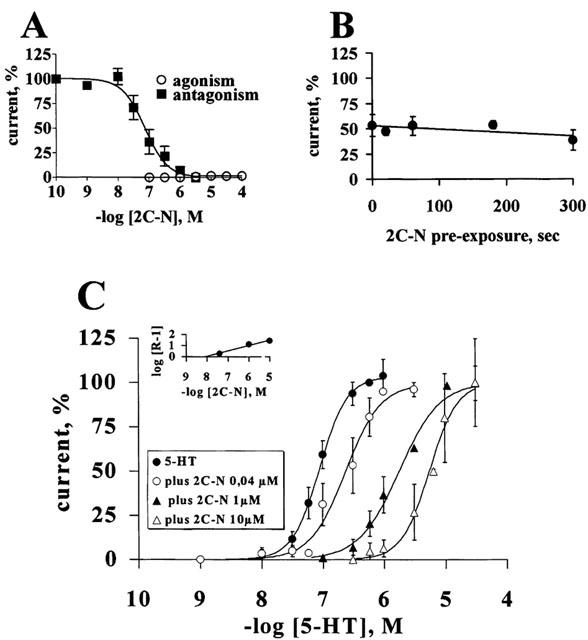
Increasing concentrations of 2C-N co-applied with 10 nM 5-HT only partially reduced the 5-HT2C receptor-induced currents, reaching at 30 μM a maximal inhibition of 67±7%. 2C-N alone generated a current that reached at 10 μM a relative Imax of 44±8%. (Figure 7A and Table 3). The blockade caused by pre-application of 50 nM 2C-N was time-dependent, reaching 100% inhibition after 5 min (Figure 7B). 2C-N applied for 3 min shifted the 5-HT concentration-response curves downwards, in a concentration-dependent manner, suggesting a non-competitive antagonism (Figure 7C). 50 nM 2C-N did not generate a current per se (Figure 7A), while effectively blocking 5-HT-evoked currents. 150 nM 2-CN, which caused a minor and transient current not exceeding ≈10% of the maximal 5-HT response, further blocked the 5-HT-evoked currents (Figure 7D).
Figure 7.
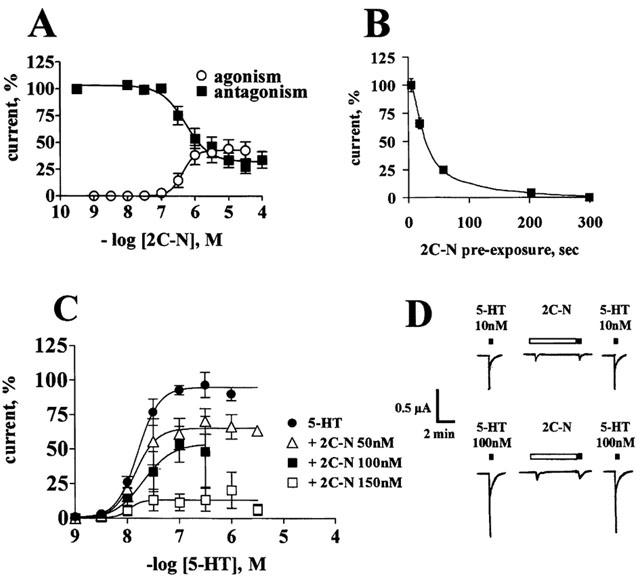
2C-N is a non-competitive 5-HT2C receptor antagonist. (A) Co-application of different concentrations of 2C-N plus 10 nM 5-HT caused only a partial reduction of the 5-HT-induced currents; 2C-N alone evoked partial agonism (n=4–6 per concentration). (B) The 50 nM 2C-N-induced blockade of the 10 nM 5-HT currents is time-dependent (n=4). (C) Non-competitive blockade of the 5-HT concentration-response curve: control 5-HT concentration-response curve (n=17); following a 3 min pre-incubation with 50 nM (n=6); 100 nM (n=6) or 150 nM 2C-N (n=5). Symbols indicate mean values; bars, s.e.mean. (D) Representative tracings show the 150 nM 2C-N agonist/antagonist effects. Open rectangle represents a 3 min 2C-N application, closed squares represent 10 s 5-HT applications.
Discussion
Using the Xenopus laevis oocytes as an assay to determine the efficacies and relative potencies of a series of 2,5-dimethoxy-4-substituted phenylalkylamines at 5-HT2A and 5-HT2C receptors we demonstrate that psychotropic drugs such as 2C-I, 2C-B or 2C-D have a weak or practically undetectable intrinsic efficacy at 5-HT2A receptors. In agreement with Nichols (1997), the corresponding PIAs, are full or partial agonists at these receptors, validating the use of this working model. The present results encompassing 2,5-dimethoxy-4-substituted PIA/PEA pairs, set the stage for a general pharmacological discussion of the activity of PIA/PEA pairs at 5-HT2 receptor subtypes. We are aware that the present findings only describe the pharmacodynamic properties of these drugs acting on 5-HT2A and 5-HT2C receptors. Parallel behavioural studies using selective 5-HT receptor subtype ligands are required to complement the understanding of the putative mechanisms involved in the psychotropic activity of 2,5-dimethoxy-4-substituted PIA/PEA pairs and other drugs.
The importance of the α-methyl group for drug activity in the phenylalkylamines was recognized early in the pharmacology of PIAs as compared to PEA analogues (Piness et al., 1930; Alles, 1933; Alles & Prinzmetal, 1933), but no clear interpretation has been put forth at the molecular level. The present study is the first focused on the comparison of serotonergic actions of a series of hallucinogenic and non-hallucinogenic PIA/PEA pairs. Nichols et al. (1994), using the phosphatidylinositol turnover assay to characterize the efficacy of the 2,5-dimethoxy-4-trifluoromethyl analogues DOCF3 and 2C-CF3, had observed that these compounds are practically equipotent partial agonists at 5-HT2C receptors. DOCF3 is a full 5-HT2A receptor agonist while 2C-CF3 is only a partial agonist. Their results were also interpreted to indicate the crucial influence of the α-methyl group in the 5-HT2A receptor.
The investigation of drug pairs with identical ring substitution patterns, combined with our assays of a series of related phenylalkylamines at 5-HT2A and 5-HT2C receptors, allows us to make novel proposals regarding the potency and efficacy of these compounds. In general, the presence of the α-methyl group is associated with significantly greater drug potency and efficacy at the 5-HT2A receptor, while at the 5-HT2C receptor the results are less clear-cut and may go in the opposite direction. Therefore, we suggest that the 5-HT2A, but not the 5-HT2C receptor may have a hydrophobic pocket near the anionic binding site, which preferentially accommodates the α-methyl group of one of the PIA enantiomers.
Although the Xenopus oocyte model bears no direct relationship to the classical biochemical or behavioural approaches used in the research of serotonergic mechanisms, this system possesses two important advantages. First, it is suited to test the action of drugs acting on defined receptor subtypes, allowing the determination of relative drug potencies and their intrinsic efficacies. Second, the Xenopus laevis model system allows the performance of strictly controlled protocols, comparing in a single oocyte a complete drug concentration-response curve with its respective control, thus reducing variability. We are aware nevertheless that the density of the 5-HT2 receptors expressed per oocyte is variable, obliging us to normalize each protocol. Standard deviations are at times larger than desired. On the other hand, drug applications are limited to 10 s, minimizing receptor desensitization. This is particularly relevant when comparing our results on PIAs with those measuring the accumulation of intracellular IP3. The latter technique requires a 15–30 min agonist application (Newton et al., 1996; Fitzgerald et al., 1999), a condition causing considerable 5-HT2 receptor desensitization. Therefore, caution must be exercised in the interpretation of IP3 accumulation protocols to determine serotonergic drug efficacies. Further validating the use of this experimental model in the study of serotonergic drugs, our results show that the rank potencies of the PIAs are consistent with the reported 5-HT2A and 5-HT2C affinities obtained in radioligand displacement assays using [125I]-DOI (Nelson et al., 1999). Moreover, in the cases of the three hallucinogenic PIAs studied by us for which these affinities are available (DOI, DOB and DON), the 2C/2A affinity ratios parallel the potency ratios reported in the present work. Similarly, α-methyl-5-HT, m-CPP, MK-212, as 5-HT receptor agonists, and methysergide, ketanserin, 4F 4PP and RS 102221 as 5-HT receptor antagonists, follow the published rank order of potencies for these receptors (Bonhaus et al., 1997; Herndon et al., 1992; Martin & Humphrey, 1994; Baxter et al., 1995; Porter et al., 1999; Jerman et al., 2001).
Considering that the psychotropic activity of PIAs is believed to rely mainly on 5-HT2A and to some extent 5-HT2C activity, our present research was focussed on these receptors, since a possible role of 5-HT2B receptors in the actions of psychotropic PIAs derivatives has not yet been established. Nelson et al. (1999) showed that the three hallucinogenic PIAs representative of our series have 30–40 times lower affinities at 5-HT2B than at 5-HT2A receptors in agonist radioligand displacement experiments and their corresponding 2B/2C affinity ratios are 10 to 20. Nevertheless, the potencies of DOI and DOB are slightly greater at 5-HT2B than at 5-HT2C receptors in cell lines in which agonist-elicited calcium increases were determined (Porter et al., 1999; Jerman et al., 2001). The present study does not exclude the participation of 5-HT2B receptors in the action of these PIA/PEA pairs. In future studies we intend to characterize our drugs at 5-HT2B receptors, combining the use of a molecular approach with behavioural studies.
Regarding the influence of the C-4 substituents on the pharmacodynamics of the 2,5-dimethoxy PIAs, it is evident that iodine and bromine increase, and nitro decreases drug potency as compared to hydrogen. This confirms a previously observed trend relating the lipophilicity of the C-4 substituents to the affinities of these compounds at 5-HT2A/2C receptors (Seggel et al., 1990; Nelson et al., 1999), although the expectation that DOI should be more potent than DOB does not hold. Also, DOM is unexpectedly potent at the 5-HT2A receptor. Regarding the PEAs, 2C-I is much more potent than 2C-B at both receptor subtypes, as predicted. 2C-N is as potent as 2C-B as a partial agonist at the 5-HT2C receptor. As with DOM, the potency of 2C–D is greater at the 5-HT2A receptor. Thus, a C-4 methyl group favours 5-HT2A potency beyond predictions based on its lipophilicity. A nitro group at C-4 decreases the potency of these drugs except at the 5-HT2C receptor. The lack of a uniform tendency accounting for drug potency, indicates that the hydrophobic character of C-4 substituents with similar bulk, and therefore able to occupy a common hydrophobic pocket in the receptor, is not the only factor determining the potency of these PIAs. Taken together, our results suggest that additional factors such as the presence of an area of negative charge around the C-4 substituents, could play a role in agonist potency at these receptors. This proposal, however, would have to be assessed using a combination of molecular modelling and quantum-chemical approaches. Notwithstanding this caveat, the present results allow us to conclude that efficacy depends more strongly on the presence of an α-methyl group than on the physical chemical properties of the C-4 substituents, while potency is a complex function involving both the chiral centre and substitution at C-4. The latter suggestion reinforces the need of examining the binding domain of 5-HT2 receptors as a whole, rather than considering individual interactions with specific aminoacid side chains or subdomains.
Our finding that 2C-N is a competitive 5-HT2A receptor antagonist and a non-competitive 5-HT2C receptor antagonist highlights the potential of the Xenopus laevis oocytes as a model that can differentiate between the structural requirements of each receptor subtype. The antagonist property of 2C-N was an interesting confirmation of the work of Sáez et al. (1994), who reported that 2C-N blocked both norepinephrine and serotonin vasoconstrictions in the isolated rat thoracic aorta. However, the novelty of the present results is the demonstration of the different mechanisms of 5-HT2A versus 5-HT2C antagonism. Perhaps a slower dissociation rate at the 5-HT2C receptor subtype might partially account for the non-competitive blockage, while the competitive nature at the 5-HT2A receptor might reflect a faster off rate. Moreover, we have found that 2C-I antagonizes the 5-HT2A but not the 5-HT2C receptor-induced currents (Huidobro-Toro & Cassels, 2001). Villalobos et al. (personal communication), further corroborated that 2C-I antagonizes the 5-HT-induced vasomotor response in rings of human chorionic vessels; these biopsies are rich in 5-HT2 receptors and lack adrenergic receptors. These results extend the oocyte findings to functional studies highlighting the potential of these PEAs as 5-HT2 antagonists.
The present results clearly illustrate that 2C-B and 2C-D, two well-established hallucinogens (Shulgin & Shulgin, 1991), are practically devoid of 5-HT2A receptor efficacy in the Xenopus oocyte model, and the similarly hallucinogenic 2C-I also has rather low efficacy at these receptors. LSD is recognized as a partial 5-HT2A and 5-HT2C receptor agonist, while lisuride, a non-hallucinogenic congener of LSD, is also an agonist at 5-HT2A and 5-HT2C receptors (Egan et al., 1998), indicating that not all 5-HT2A/2C receptor agonists trigger hallucinogenic effects. 5-HT and the 5-HT2A/2C receptor agonists DOB and LSD produce a biphasic modulation of NMDA responses. This result led to the proposal that phenylalkylamine and indoleamine hallucinogens may interact with different pre- and postsynaptic 5-HT2A receptor signalling pathways to modulate NMDA receptor-mediated sensory, perceptual, affective and cognitive processes (Arvanov et al., 1999). Although some psychotropic ring-substituted PIAs are devoid of dopaminergic receptor affinity, indirect dopaminergic mechanisms in the action of these drugs cannot be discarded (Benloucif et al., 1993). Likewise, the possibility that some of these 2,5-dimethoxy-4-substituted phenylalkylamines could act in vivo releasing bioamines, in conjunction with their direct actions at defined receptors, might establish the final neuronal pathways leading to hallucinogenesis. In addition, the recent report that novel G-protein coupled receptors which are selectively activated by tyramine and β-phenylethylamine, tryptamine and possibly octopamine, must also be considered when evaluating the in vivo actions of these phenylalkylamines (Borowsky et al., 2001). Consequently, the role of non-serotonin receptors and the possibility of different transduction pathways within the 5-HT2 receptor type must be explored in order to understand the mechanisms of hallucinogenesis by phenylalkylamines.
In summary, the study of this series of 2,5-dimethoxy-4-substituted PIA/PEA pairs has allowed us to identify structural features that separately determine potency and efficacy at 5-HT2 receptors. The present results highlight our finding that activation of the 5-HT2A receptor is not a prerequisite to account for the hallucinogenic effects of psychotropic PEAs. The Xenopus laevis oocyte model, previously used to study the mechanism of central depressant drugs, has now been extended to the research of psychostimulants acting directly on brain neurotransmitter receptors. Our results demonstrate that certain 2,5-dimethoxy-4-substituted PEAs with weak intrinsic activity effectively antagonise 5-HT2A receptors. The different nature of their antagonism at 5-HT2C receptors also suggests that they may serve as prototypes for the development of novel subtype-selective drugs.
Acknowledgments
This research was partially funded by the Centre of Cell Regulation and Pathology (FONDAP 13980001), the Millennium Institutes for Fundamental and Applied Biology (MIFAB) and Advanced Studies in Cell Biology and Biotechnology (CBB), and DICYT grant 020091SB. The authors thank Dr P. Bull for assistance in the replication of the 5-HT receptor clones and Prof G. Owen for editorial assistance.
Abbreviations
- 2C-B
4-bromo-2,5-dimethoxyphenylethylamine
- 2C-CF3
2,5-dimethoxy-4-trifluoromethylphenylethylamine
- 2C-D
2,5-dimethoxy-4-methylphenylethylamine
- 2C-H
2,5-dimethoxyphenylethylamine
- 2C-I
2,5-dimethoxy-4-iodophenylethylamine
- 2C-N
2,5-dimethoxy-4-nitrophenylethylamine
- 2,5-DMA
(±)-1-(2,5-dimethoxy)-2-aminopropane
- DOB
(±)-1-(4-bromo-2,5-dimethoxyphenyl)-2-aminopropane
- DOCF3
(±)-1-(2,5-dimethoxy-4-trifluoromethylphenyl)-2-aminopropane
- DOI
(±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
- DOM
(±)-1-(2,5-dimethoxy-4-methylphenyl)-2-aminopropane
- DON
(±)-1-(2,5-dimethoxy-4-nitrophenyl)-2-aminopropane
- Imax
maximal current generated
- 4F 4PP
4-(4-fluorobenzoyl)-1-(4phenylbutyl) piperidine
- LSD
d-lysergic acid diethylamide
- m-CPP
1-(3-chlorophenyl)piperazine dihydrochloride
- MK-212
6-chloro-2-(1-piperazinyl)pyrazine
- PEA
phenylethylamine
- PIA
phenylisopropylamine
- RS 102221
8-[5-(2,4-dimethoxy-5-(4-trifluoromethylphenylsulphonamido)phenyl-5-oxopentyl]-1,3,8-triazaspiro[4,5]decane-2,4-dione
References
- ACUÑA C., SCORZA C., REYES-PARADA M., CASSELS B.K., HUIDOBRO-TORO J.P. Aleph-2, a suspected anxiolytic and putative hallucinogenic phenylisopropylamine derivative, is a 5-HT2A and 5-HT2C receptor agonist. Life Sci. 2000;67:3241–3247. doi: 10.1016/s0024-3205(00)00906-1. [DOI] [PubMed] [Google Scholar]
- ALLES G.A. The comparative physiological actions of dl-β-phenylisopropylamine. II. Bronchial effect. J. Pharmacol. Exp. ther. 1933;48:161–174. [Google Scholar]
- ALLES G.A., PRINZMETAL M. The comparative physiological actions of dl-β-phenylisopropylamine. I. Pressor effect and toxicity. J. Pharmacol. Exper. Therap. 1933;47:339–354. [Google Scholar]
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARVANOV V.L., LIANG X., RUSSO A., WANG R.Y. LSD and DOB: interaction with 5-HT2A receptors to inhibit NMDA receptor-mediated transmission in the rat prefrontal cortex. Eur. J. Neurosci. 1999;11:3064–3072. doi: 10.1046/j.1460-9568.1999.00726.x. [DOI] [PubMed] [Google Scholar]
- BARNES N.M., SHARP T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- BAXTER G., KENNETT G., BLANEY F., BLACKBURN T. 5-HT2 receptor subtypes: a family re-united. Trends. Pharmacol. Sci. 1995;16:105–110. doi: 10.1016/s0165-6147(00)88991-9. [DOI] [PubMed] [Google Scholar]
- BENLOUCIF S., KEEGAN M., GALLOWAY M. Serotonin-facilitated dopamine release in vivo: pharmacological characterization. J. Pharmacol. Exp. ther. 1993;265:373–377. [PubMed] [Google Scholar]
- BONHAUS D., WEINHARDT K., TAYLOR M., DESOUZA A., MCNEELEY P., SZCZEPANSKI K., FONTANA D., TRINH J., ROCHA C., DAWSON M., FLIPPIN L., EGLEN R. RS-102221: A novel high affinity and selective, 5-HT2C receptor antagonist. Neuropharmacology. 1997;36:621–629. doi: 10.1016/s0028-3908(97)00049-x. [DOI] [PubMed] [Google Scholar]
- BOROWSKY B., ADHAM N., JONES K., RADDATZ R., ARTYMYSHYN R., OGOZALEK K., DURKIN M., LAKHANI P., BONINI J., PATHIRANA S., BOYLE N., PU X., KOURANOVA E., LICHTBLAU H., OCHOA Y., BRANCHEK T., GERALD C. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Nat. Acad. Sci. U.S.A. 2001;98:8966–8971. doi: 10.1073/pnas.151105198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI D.S., MAROTEAUX L. Immunohistochemical localisation of the serotonin 5-HT2B receptor in mouse gut, cardiovascular system, and brain. FEBS Lett. 1996;391:45–51. doi: 10.1016/0014-5793(96)00695-3. [DOI] [PubMed] [Google Scholar]
- DASCAL N.C., IFUNE R., HOPKINS T.P., SNUTCH H., LUBBERT N., DAVIDSON M.I. Involvement of a GTP-binding protein in mediation of serotonin and acetylcholine response in Xenopus oocytes injected with rat brain messenger RNA. Mol. Brain Res. 1986;11:201–209. doi: 10.1016/0169-328x(86)90026-4. [DOI] [PubMed] [Google Scholar]
- EGAN C.T, , HERRICK-DAVIS K., MILLER K., GLENNON R.A., TEITLER M. Agonist activity of LSD and lisuride at cloned 5HT2A and 5HT2C receptors. Psychopharmacology. 1998;136:409–414. doi: 10.1007/s002130050585. [DOI] [PubMed] [Google Scholar]
- FITZGERALD L.W., CONKLIN D.S., KRAUSE C.M., MARSHALL A.P., PATTERSON J.P., TRAN D.P., IYER G., KOSTICH W.A., LARGENT B.L., HARTIG P.R. High-affinity agonist binding correlates with efficacy (intrinsic activity) at the human serotonin 5-HT2A and 5-HT2C receptors: evidence favoring the ternary complex and two-state models of agonist action. J. Neurochem. 1999;72:2127–2134. doi: 10.1046/j.1471-4159.1999.0722127.x. [DOI] [PubMed] [Google Scholar]
- GLENNON R.A., RAGHUPATHI R., BARTYZEL P., TEITLER M., LEONHARDT S. Binding of phenylalkylamine derivatives at 5-HT1C and 5-HT2 serotonin receptors: evidence for a lack of selectivity. J. Med. Chem. 1992;35:734–740. doi: 10.1021/jm00082a014. [DOI] [PubMed] [Google Scholar]
- HERNDON J., ISMAIEL A., INGHER S., TEITLER M., GLENNON A. Ketanserin analogues: structure-affinity relationships for 5-HT2 and 5-HT1C serotonin receptor binding. J. Med. Chem. 1992;35:4903–4910. doi: 10.1021/jm00104a017. [DOI] [PubMed] [Google Scholar]
- HUIDOBRO-TORO J.P, CASSELS B.K. Tales of 1001 nights, the α-methyl arabesque. Biol. Res. 2001;34:R-19. [Google Scholar]
- JERMAN J.C., BROUGH S.J., GAGER T., WOOD M., COLDWELL M.C., SMART D., MIDDLEMISS D.N. Pharmacological characterisation of human 5-HT2 receptor subtypes. Eur. J. Pharmacol. 2001;414:23–30. doi: 10.1016/s0014-2999(01)00775-0. [DOI] [PubMed] [Google Scholar]
- KENNETT G.A., BRIGHT F., TRAIL B., BAXTER G.S., BLACKBURN T.P. Effects of the 5-HT2B receptor agonist, BW 723C86, on three rat models of anxiety. Br. J. Pharmacol. 1996;117:1443–1448. doi: 10.1111/j.1476-5381.1996.tb15304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOWLES I.D., RAMAGE A.G. Evidence for a role of central 5-HT2B as well as 5-HT2A receptors in cardiovascular regulation in anaestetized rats. Br. J. Pharmacol. 1999;128:530–542. doi: 10.1038/sj.bjp.0702822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOWLES I.D., RAMAGE A.G. Evidence that activation of central 5-HT(2B) receptors causes renal sympathoexcitation in anaestetized rats. Br. J. Pharmacol. 2000;129:177–183. doi: 10.1038/sj.bjp.0703011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOBOS M., BORGES Y., GONZÁLEZ E., CASSELS B.K. The action of the psychoactive drug 2C-B on isolated rat thoracic aorta. Gen. Pharmacol. 1992;23:1139–1142. doi: 10.1016/0306-3623(92)90301-y. [DOI] [PubMed] [Google Scholar]
- MCKERNAN R.M., WAFFORD K., QUIRK K., HADINGHAM K.L., HARLEY E.A., RAGAN C.I., WHITING P.J. The pharmacology of the benzodiazepine site of the GABA-A receptor is dependent on the type of gamma-subunit present. J. Recept. Signal Transduct. Res. 1995;15:173–183. doi: 10.3109/10799899509045215. [DOI] [PubMed] [Google Scholar]
- MARTIN G.R., HUMPHREY P.P. Receptors for 5-hydroxytryptamine: current perspectives on classification and nomenclature. Neuropharmacology. 1994;33:261–273. doi: 10.1016/0028-3908(94)90058-2. [DOI] [PubMed] [Google Scholar]
- MIHIC J., YE Q., WICK M., KOLTCHINES V., KRASOWSKI M., FINN S., MASCIA M.P., VALENZUELA F., HANSON K., GREENBLATT E., HARRIS A., HARRISON N. Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- MORAN O., MARTY A. Protein kinase C modulates neurotransmitter response in Xenopus oocytes injected with rat brain RNA. Mol. Brain Res. 1989;5:193–202. doi: 10.1016/0169-328x(89)90035-1. [DOI] [PubMed] [Google Scholar]
- NELSON D.L., LUCAITES V.L., WAINSCOTT D.B., GLENNON R.A. Comparisons of hallucinogenic phenylisopropylamine binding affinities at cloned human 5-HT2A, 5-HT2B and 5-HT2C receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;359:1–6. doi: 10.1007/pl00005315. [DOI] [PubMed] [Google Scholar]
- NEWTON R.A., PHIPPS S.L., FLANIGAN T.P., NEWBERRY N.R., CAREY J.E., KUMAR C., MCDONALD B., CHEN C., ELLIOT J.M. Characterisation of human 5-Hydroxytryptamine2A and 5-Hydroxytryptamine2C receptors expressed in the human neuroblastoma cell line SH-SY5Y: Comparative stimulation by hallucinogenic drugs. J. Neurochem. 1996;67:2521–2531. doi: 10.1046/j.1471-4159.1996.67062521.x. [DOI] [PubMed] [Google Scholar]
- NICHOLS D.E.Role of Serotonergic Neurons and 5-HT Receptors in the Action of Hallucinogens Handbook of Experimental Pharmacology 1997129Berlin–Heidelberg: Springer; 563–585.ed. Baumgarten, H.G. & Göthert, M. [Google Scholar]
- NICHOLS D.E., FRESCAS S., MARONA-LEWICKA D., HUANG X., ROTH B.L., GUDELSKY G.A., NASH J.F. 1-(2,5-Dimethoxy-4-(trifluoromethyl)-phenyl)-2-aminopropane: A potent serotonin 5-HT2A/2C agonist. J. Med. Chem. 1994;37:4346–4351. doi: 10.1021/jm00051a011. [DOI] [PubMed] [Google Scholar]
- PINESS G., MILLER H., ALLES G.A. Clinical observations on phenylaminoethanol sulfate. J. Am. MA. 1930;94:790–791. [Google Scholar]
- PORTER R.H.P., BENWELL K.R., LAMB H., MALCOLM C.S., ALLEN N.H., REVELL D.F., ADAMS D.R., SHEARDOWN M.J. Functional characterization of agonists at recombinant human 5-HT2A, 5-HT2B and 5-HT2C receptors in CHO-K1 cells. Br. J. Pharmacol. 1999;128:13–20. doi: 10.1038/sj.bjp.0702751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SÁEZ P., BORGES Y., GONZÁLEZ E., CASSELS B.K. α-Adrenergic and 5-HT2-serotonergic effects of some β-phenylethylamines on isolated rat thoracic aorta. Gen. Pharmacol. 1994;25:211–216. doi: 10.1016/0306-3623(94)90035-3. [DOI] [PubMed] [Google Scholar]
- SANDERS-BUSH E., MAYER S.5-Hydroxytryptamine (serotonin): receptor agonists and antagonists Goodman & Gilman's The Pharmacological Basis of Therapeutics 2001New York, USA: McGraw-Hill; 269–290.10th. edn., ed. Hardman, J., Limbird, L. & Gilman, A.G. pp [Google Scholar]
- SEGGEL R., YOUSIF Y., LYON A., TITELER M., ROTH L., SUBA A., GLENNON A. A structure-affinity study of the binding of 4-substituted analogues of 1-(2,5-dimethoxyphenyl)-2-aminopropane at 5-HT2 serotonin receptors. J. Med. Chem. 1990;33:1032–1036. doi: 10.1021/jm00165a023. [DOI] [PubMed] [Google Scholar]
- SHULGIN A.T.Hallucinogens Burger's Medicinal Chemistry 19813New York: Wiley; 1109–1137.4th. edn., ed. Wolff, M.E. [Google Scholar]
- SHULGIN A.T., SHULGIN A. IHKAL–A Chemical Love Story. Berkeley, CA: Transform Press; 1991. [Google Scholar]
- YAMAKURA T., BERTACCINI E., TRUDELL J., HARRIS A. Anesthetics and ion channels: molecular models and sites of action. Annu. Rev. Pharmacol. Toxicol. 2001;41:23–51. doi: 10.1146/annurev.pharmtox.41.1.23. [DOI] [PubMed] [Google Scholar]