

Abstract
The contribution of nitric oxide (NO) to articular pain in arthritis induced by zymosan (1 mg, intra articular) in rats was assessed by measuring articular incapacitation (AI).
Systemic treatment with the non-selective NO synthase (NOS) inhibitor L-NAME (10–100 mg kg−1 i.p.) or with the selective iNOS inhibitors aminoguanidine (AG; 10–100 mg kg−1 i.p.) or 1400W (0.5–1 mg kg−1 s.c.) inhibited the AI induced by injection of zymosan 30 min later.
Local (intra articular) treatment with the NOS inhibitors (L-NAME or AG, 0.1–1 μmol; 1400W, 0.01 (μmol) 30 min before zymosan also inhibited the AI.
Systemic or local treatment with the NOS inhibibitors (L-NAME; AG, 100 mg kg−1 i.p. or 0.1 μmol joint−1; 1400W, 1 mg kg−1 s.c. or 0.01 μmol joint−1), 2 h after zymosan did not affect the subsequent AI.
Local treatment with the NO donors SNP or SIN-1, 2 h after zymosan did inhibit AI.
L-NAME and AG, given i.p. inhibited nitrite but not prostaglandin E2 (PGE2) levels in the joints. L-NAME (100 mg kg−1) but not AG (100 mg kg−1) increased mean arterial blood pressure. Neither L-NAME, AG nor the NO donor SIN-1 altered articular oedema induced by zymosan.
In conclusion, inhibitors of iNOS decrease pain in zymosan arthritis only when given before the zymosan. This was not due to inhibition of articular PGE2 release or oedema. NO donors also promoted antinociception in zymosan arthritis without affecting oedema.
Keywords: Arthritis, zymosan, nitric oxide, 3-morpholinosydnonimine, sodium nitroprusside, pain, oedema, inflammatory hyperalgesia
Introduction
Nitric oxide (NO) is produced by nitric oxide synthase (NOS) using L-arginine as substrate. Three isoforms of NOS have been described. Endothelial (NOS1) and neuronal (NOS3) isoforms are constitutive (cNOS) whereas NOS2 is the inducible isoform (iNOS). The cNOS produces NO in picomolar amounts for short periods, operating through a calcium dependent mechanism, whereas iNOS produces large and sustained amounts of NO after cell activation by inflammatory stimuli. Production of NO via cNOS has been linked to homeostasis, for instance, the regulation of arterial blood pressure, whereas NO produced after iNOS induction appears to be involved in pathophysiological phenomena (Moncada et al., 1991).
There is considerable evidence for the involvement of NO in experimental models of arthritis. Increased NO levels were associated with the development of streptococcal cell wall arthritis (McCartney-Francis et al., 1999) whereas administration of NOS inhibitors reversed inflammatory changes in experimental arthritis (Ialenti et al., 1993; Stefanovic-Racic et al., 1994; 1995tgci). Limitation of movement is a serious burden to patients presenting with inflammatory arthropathies. Although pain relief is frequently the main goal in the acute treatment of these conditions, little is known about the mechanisms involved in pain development in arthropathies. The rat knee-joint incapacitation test was designed to study articular incapacitation (AI), defined as the inability of a rat with an experimentally induced arthritis to walk normally (Tonussi & Ferreira, 1992). In this test, AI is assumed to be due to altered nociception following injection of an inflammatory stimulus into the joints. Using this test, we have, in a previous paper, demonstrated the development of gait disturbances (AI) in rats with zymosan (Zy)-induced arthritis (Rocha et al., 1999), with the animals developing AI from 2 h, reaching a maximal value between 3 and 4 h, after the injection of zymosan (Rocha et al., 1999).
There is also evidence supporting a role for NO in nociception. A NOS inhibitor, L-NMMA (L-NG-monomethylarginine), blocked acetylcholine induced peripheral analgesia (Duarte et al., 1990). Peripheral antinociception induced by morphine in rats with PGE2-induced hyperalgesia was shown to be mediated by NO release leading to cGMP activation (Ferreira et al., 1991). However, controversies regarding NO role in pain have arisen. For instance, injection of NO in humans produces pain, presumably through direct stimulation of local nociceptors (Holthusen & Arndt, 1994). Additionally, administration of L-NAME (L-NG-nitroarginine methyl ester), a non-selective NOS inhibitor, had antinociceptive effects in mice and reversed thermal hyperalgesia in rats with carrageenan arthritis (Lawand et al., 1997). More recently, it was shown that the NO donor 3-morpholinosydnonimine (SIN-1) produced either analgesia or nociception in rats, depending on the dose and the pain model studied (Sousa & Prado, 2001).
Considering that both the exact role of NO in pain development and the mediators involved in pain mechanisms during inflammatory arthropathies are still not fully defined, we decided to investigate NO participation in AI in zymosan-induced arthritis. The results reported herein show that administration of an NO donor in ongoing zymosan arthritis produced analgesia. On the other hand, NOS inhibitors, acting via iNOS inhibition, exerted antinociceptive effects in this model only when given prior to the injection of zymosan into the joint. Moreover, these anti-nociceptive effects were not secondary to an inhibition of oedema or of prostaglandin (PG) release into the affected joint.
Methods
Animals
Male Wistar rats (180–220 g) from our own animal facilities were used throughout the experiments. All efforts were made to minimize animal suffering and the number of animals used. Surgical procedures and animal treatments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (DHEW Publication, Bethesda, MD, U.S.A.).
Evaluation of articular incapacitation (AI)
During light ether anaesthesia, rats received a standard intra-articular (i.art.) injection of zymosan (1 mg in 50 μl total volume), dissolved in sterile saline, into their right knee joints. Control animals received saline. We used the rat knee joint incapacitation test, as described previously (Tonussi & Ferreira, 1992). Briefly, after zymosan injection, animals were put to walk on a steel rotary drum (30 cm wide×50 cm diameter), covered with a fine-mesh non-oxidizable wire screen, which rotates at 3 r.p.m. Specially designed metal gaiters were wrapped around both hind paws. After placement of the gaiters, the animals were allowed to walk freely to accustom themselves to the gaiters. The right paw was then connected via a simple circuit to a microcomputer data input/output port. The paw elevation time (PET) is the time that during a 60 s period the inflamed hind paw is not in contact with the cylinder. This is directly proportional to the articular incapacitation.
Evaluation of articular oedema and PGE2 release
The animals were anaesthetized (chloral hydrate (400 mg kg−1 i.p.), killed by cervical dislocation, and exsanguinated. The synovial cavity of the knee joints was then washed with 0.4 ml saline containing 5 U ml−1 heparin. The synovial exudates were collected by aspiration. After centrifugation (500×g, for 10 min−1), the supernatant was stored at −70°C and used for determination of PGE2 concentration using a commercially available ELISA kit (Cayman Chem. Co., Ann Arbor, U.S.A.) and also for assessing total nitrite released into the joints. After the synovial fluid was collected, the synovial membranes were surgically removed and the wet weight (g) was recorded. This material was dried (80°C) overnight and the dry weight (g) was recorded. The wet/dry (g) weight ratio was used as reflecting synovial oedema.
Measurement of mean arterial pressure (MAP)
In an attempt to disclose microcirculatory blood flow alterations due to cNOS inhibition, MAP was measured. Briefly, rats were anaesthetized and a cannula placed in the carotid artery. The rats were allowed to recover and the MAP was recorded by connecting the arterial cannula to a pressure transducer and recording the blood pressure in a polygraph. MAP was measured 30 min after i.p. injection of either L-NAME 100 mg kg−1 i.p. or aminoguanidine 100 mg kg−1 i.p. and then subsequently at hourly intervals for a period of 6 h and compared to the baseline values.
Determination of NO production
Production of NO was determined by measuring total nitrite/nitrate (NO2−/NO3−) by the Griess reaction. Total NO2−/NO3− levels were determined with the NO3− in the samples (0.08 ml) converted to NO2− by incubation of 0.01 ml nitrate reductase from Aspergillus species (1 u ml−1) and 0.01 ml NADPH (1 mM) for 30 min at 37°C. NO2− levels were determined spectrophotometrically at 540 nm by measuring the absorbance of test samples (0.1 ml) after adding 0.1 ml Griess reagent (sulphanilic acid (1% w v−1) and N-(1-naphythyl)ethylenediamine (0.1 w v−1) in 5% phosphoric acid) and comparing these values with those from standard solutions of NaNO2 (1–100 μM).
Drug treatments
Prophylactic interventions
In order to analyse the effect of NOS inhibitors injected systemically and administered prior to induction of zymosan arthritis, groups of rats received L-NAME (10–100 mg kg−1 i.p.), aminoguanidine (10–100 mg kg−1 i.p.) or 1400W (0.5–1 mg kg−1 s.c.) 30 min prior to zymosan injection intra-articularly (i.art.). To analyse the prophylatic effect of NOS inhibitors administered locally either L-NAME 0.1–1 μmol joint−1 (0.15–1.39 mg kg−1), aminoguanidine 0.1–1 μmol joint−1 (0.06–0.61 mg kg−1), or 1400W 0.01 μmol/joint (0.014 mg kg−1) were administered i.art. 30 min prior to arthritis induction. Two other groups received either D-NAME (100 mg kg−1 i.p.) or L-arginine (1 g kg−2 p.o.) 30 min prior to L-NAME (100 mg kg−1) followed by zymosan i.art., 30 min later.
Therapeutic interventions
In order to evaluate the effects of NOS inhibitors injected systemically on existing zymosan arthritis, other groups of rats received either L-NAME (100 mg kg−1 i.p.), aminoguanidine (100 mg kg−1 i.p.) or 1400W (1 mg kg−1 s.c.) 2 h after injection of zymosan i.art. In order to evaluate the effect of NOS inhibitors injected locally on existing zymosan-arthritis, groups of rats received either L-NAME (0.3 μmol joint−1), aminoguanidine (0.1 μmol joint−1) or 1400W (0.01 μmol joint−1) 2 h after injection of zymosan i.art.
Therapeutic effect of NO donors
In order to evaluate the local therapeutic effect of exogenous NO addition, either of two NO donors, sodium nitroprusside (SNP 100–500 μg i.art.) or 3-morpholinosydnonimine (SIN-1; 1–10 μg i.art) were given 2 h after the injection of zymosan.
Control groups
Non-treated (NT) groups consisted of rats with zymosan-arthritis that received 0.9% w v−1 sterile saline either systemically or locally. The control groups of rats received only 0.9% w v−1 sterile saline i.art.
Drugs and reagents
All drugs were purchased from Sigma Chemical Co., St. Louis, U.S.A., except for SIN-1 that was purchased from Cayman Chemical Co., Ann Arbor, U.S.A.
Statistics
Results are expressed as mean±s.e.mean. Statistically significant differences between groups were analysed using one-way ANOVA followed by the Bonferroni test. P<0.05 was considered significant.
Results
Effect of systemic treatment with NOS inhibitors on articular NO production
The time course of release of nitrite into the joint exudates of rats with zymosan-arthritis is shown in Figure 1A. The release started 1 h after zymosan injection and reached a maximum value at 6 h. In rats whose joints were injected only with saline (SAL), the nitrite concentrations in the joint exudates were low and remained at these levels throughout the experimental period.
Figure 1.
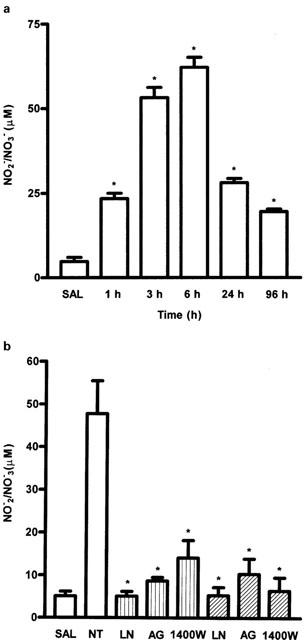
(A) Time–course of the release of NO into the joint exudate of rats with zymosan arthritis. Zymosan was injected intra-articularly (i.art.) and the NO release in exudates (as NO2−/NO3−) was measured at different times, as shown, after zymosan. Control (SAL) animals received only saline i.art. There was a rapid increase in NO release after zymosan, which was still raised at 96 h. Injection of saline into the joints did not increase NO release throughout the experiment. Results are expressed as the mean±s.e.mean of values for each group of six animals. *P<0.05 compared to control rats. (B) Effect of systemic prophylactic (dotted bars) or therapeutic (hatched bars) administration of NOS inhibitors on nitrite release into the joint exudates in zymosan arthritis. Either L-NAME (LN) (100 mg kg−1 i.p.), AG (100 mg kg−1 i.p.) or 1400W (1 mg kg−1 s.c.) were injected 30 min before (prophylactic) or 2 h after (therapeutic) zymosan. Non-treated (NT) rats were given saline i.p. 30 min prior to zymosan. Control (SAL) animals received only saline i.art. Release of NO was measured at 6 h after zymosan. All three inhibitors reduced the NO release to almost control levels at this time. Results are expressed as the mean±s.e.mean of values for each group of six animals. *P<0.05 compared to NT rats.
Systemic prophylactic (30 min before zymosan) or therapeutic (2 h after zymosan) administration of the NOS inhibitors (L-NAME, AG, and 1400W) reduced this nitrite release into the joint exudates. At 6 h after zymosan, this reduction was to almost control (saline injected) levels at the highest doses of inhibitors used (Figure 1B). Similar data were obtained when these compounds were injected locally, whether administered in a prophylactic or therapeutic strategy (data not shown).
Effect of systemic treatment with NOS inhibitors on articular incapacitation (AI)
The effect of treatment with the NOS inhibitors (L-NAME, aminoguanidine or 1400W), given before the zymosan, on AI is shown in Figure 2A. Injection of zymosan into the articular joints of rats provoked an increase in paw elevation time (PET), starting 2 h, and peaking between 3–4 h, after zymosan injection. Increase in PET is assumed to reflect articular inflammatory pain and is due to stimulation of periarticular nociceptors (Rocha et al., 1999). All three inhibitors of NOS dose-dependently reduced AI, measured between 3–4 h after zymosan. The reduction of AI observed after L-NAME or AG was about equal at the highest dose used, whereas the NOS inhibitor 1400W appeared more potent and to reduce AI almost to control level, i.e., to completely reverse the AI induced by zymosan. However, as shown in Figure 2B, when the NOS inhibitors were given 2 h after the zymosan, at the highest doses, none of them modified AI.
Figure 2.
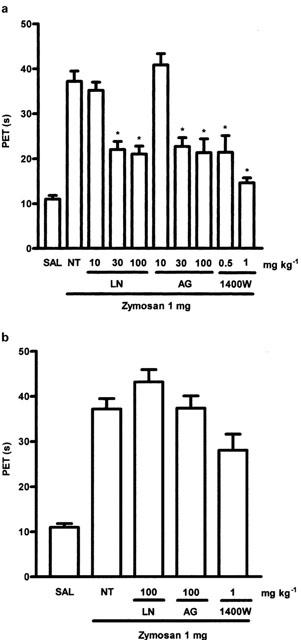
(A) Dose-dependent effects of systemic prophylactic administration of NOS inhibitors on articular incapacitation (AI) in zymosan arthritis. The AI was measured hourly over 4 h after zymosan injection as the increase in paw elevation time (PET) assessed using the rat knee joint incapacitation test (see text for details). Either L-NAME (LN - 10, 30, 100 mg kg−1 i.p.), aminoguanidine (AG - 10, 30, 100 mg kg−1 i.p.) or 1400W (0.5–1 mg kg−1 s.c.) were injected 30 min prior to zymosan 1 mg injection i.a. Non-treated (NT) rats were given saline i.p. followed by zymosan. Control (SAL) animals received saline i.art. There was a dose dependent reduction of the AI response after treatment with any of the inhibitors. Results are expressed as the mean±s.e.mean of maximal PET obtained during 3–4 h of arthritis; n=6 animals for each group. *P<0.05 compared to NT rats. (B) Effect of systemic therapeutic administration of L-NAME (LN), aminoguanidine (AG) or 1400W on AI in zymosan arthritis. The AI was measured hourly over 4 h after zymosan. LN (100 mg kg−1 i.p.), AG (100 mg kg−1 i.p.) or 1400W (1 mg kg−1 s.c.) was given 2 h after zymosan. Non-treated (NT) rats were given saline (i.p.), 2 h after zymosan. Control rats (SAL) received saline i.art. The inhibitors were no longer effective in reducing AI when given after the zymosan. Results are expressed as the mean±s.e.mean of maximal PET measured 3–4 h after the zymosan; n=6 animals for each group. *P<0.05 compared to NT rats.
Figure 3 shows that administration of D-NAME (100 mg kg−1 i.p.), the inactive enantiomer of L-NAME, did not alter AI and also that combined administration of the NOS substrate L-arginine (1 g kg−1 p.o.) and L-NAME (100 mg kg−1 i.p.) reversed the inhibitory effect of L-NAME on AI. Note that all these treatments were given before the zymosan.
Figure 3.
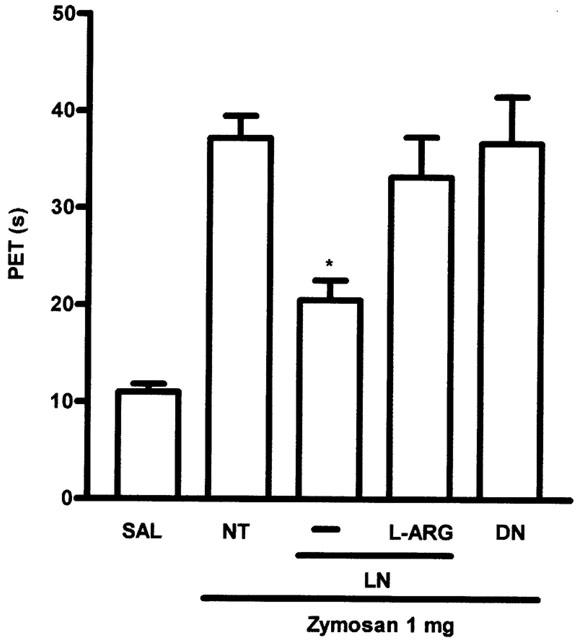
Effect of systemic prophylactic combined administration of L-arginine (L-ARG) and L-NAME (LN) and D-NAME (DN) on AI in zymosan arthritis. The AI was measured as previously described. LN or DN (100 mg kg−1 i.p.) was given 30 min before zymosan. L-ARG (1 g kg−1) was given p.o., 30 min before the L-NAME. Non-treated (NT) rats received saline i.p. followed by zymosan. Control (SAL) animals received saline i.art. Results are expressed as the mean±s.e.mean of maximal PET obtained during 3–4 h of arthritis; n=6 animals for each group. *P<0.05 compared to NT rats.
Effect of local treatment with NOS inhibitors on AI
In a further series of experiments, the NOS inhibitorsL-NAME 0.1–1 μmol joint−1 (0.15–1.39 mg kg−1, AG 0.1–1 μmol joint−1 (0.06–0.61 mg kg−1) or 1400W 0.01 μmol joint−1 (0.014 mg kg−1) were given locally, i.e., by injection into the joint. Given 30 min before the zymosan, these inhibitors markedly decreased the AI (Figure 4A). However, as noted with systemic treatment, giving the inhibitors locally, 2 h after zymosan injection, had no effect on the AI (Figure 4B).
Figure 4.
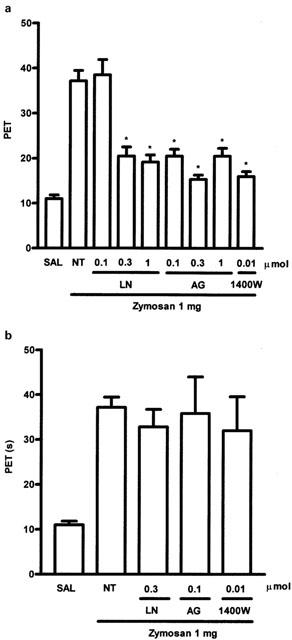
Dose-dependent effect of local prophylactic administration of NOS inhibitors on AI in zymosan arthritis in rats. Either L-NAME (LN) 0.1, 0.3 and 1.0 μmol joint−1), AG (0.1, 0.3 and 1.0 μmol joint−1) or 1400W (0.01 μmol joint−1) were injected i.art. 30 min prior to zmosan injection. Non-treated (NT) rats were given saline (50 μl) i.art. followed by zymosan. Control (SAL) rats received saline i.art. All three inhibitors given locally were highly effective in reducing AI. Results are expressed as the mean±s.e.mean of maximal PET obtained during 3–4 h of arthritis; n=6 animals for each group. *P<0.05 compared to NT rats. (B) Effect of local therapeutic administration of L-NAME (LN), aminoguanidine (AG) or 1400W on AI. The AI was measured hourly over 4 h. LN (0.3 μmol joint−1), AG (0.1 μmol joint−1) or 1400W (0.01 μmol joint−1) were given i.art. 2 h after zymosan. Non-treated (NT) rats were given saline i.p., 2 h after zymosan. Control (SAL) rats received saline i.art. Local application of the NOS inhibitors, at the highest doses used prophylactically (see A), did not reduce AI. Results are expressed as the mean±s.e.mean of maximal PET measured 3–4 h after zymosan; n=6 animals for each group. *P<0.05 compared to NT rats.
Effect of local therapeutic administration of two NO donors, SNP and SIN-1, on AI
To assess the effect of exogenous NO in our model of articular inflammatory pain, we used two NO donors, sodium nitroprusside (SNP:) or 3-morpholynosydnonimine (SIN-1). The sources of NO were given after the induction of the arthritis, locally, 2 h after zymosan. Figure 5 shows that, over a range of doses, both SNP (100–500 μg i.art.) and SIN-1 (10–100 μmol, i.art.) inhibited AI, to a degree comparable with that obtained with the local application of NOS inhibitors, when given before zymosan (Figure 2A).
Figure 5.
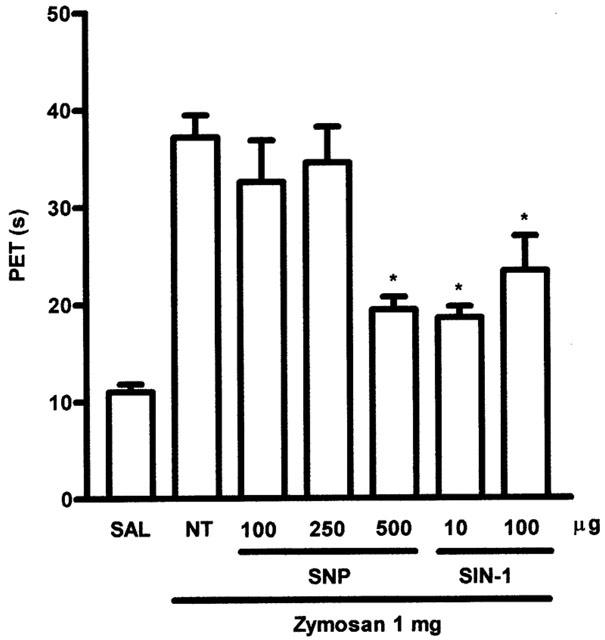
Effect of local therapeutic administration of sodium nitroprusside (SNP) or 3-morpholinosydnonimine (SIN-1) on AI. SNP (100–500 μg) or SIN-1 (10–100 μg) were injected i.art. 2 h after zymosan. Non-treated (NT) rats were given saline (i.art). 2 h after zymosan. Control (SAL) animals received saline i.art. Local injection of the NO donors after zymosan was able to reduce AI, showing a direct anti-nociceptive effect of NO in the inflamed joint. Results are expressed as the mean±s.e.mean of maximal PET measured 3–4 h after zymosan; n=6 animals for each group. *P<0.05 compared to NT rats.
Effect of NOS inhibitors on PGE2 release, synovial oedema, and mean arterial pressure (MAP)
Because of the interaction between NOS and PG synthesis (Salvemini et al., 1993) and because PG-induced oedema could affect the incapacitation we were measuring, we assayed for PGE2 in the joint exudates and for articular oedema. Since the maximal inhibition of AI was achieved with L-NAME or AG, at 100 mg kg−1 i.p., we decided to use these doses in these further experiments. As shown in Table 1, neither L-NAME nor AG, when administered prophylactically, modified the increase in PGE2 content in the affected joint at 6 h. The marked synovial oedema induced by zymosan was also unaffected by NOS inhibition. Treatment after zymosan (2 h) with systemic L-NAME or AG (which did not affect AI; see Figure 2B) or the nitric oxide donor SIN-1 locally (which reduced AI; see Figure 5) also did not modify articular oedema. Table 1 also shows that L-NAME (100 mg kg−1 i.p.) but not AG (100 mg kg−1 i.p.) administration caused a marked increase in MAP.
Table 1.
Effect of NOS inhibitors on mean arterial pressure (MAP) and prostaglandin E2 (PGE2) release, and articular oedema in zymosan-induced arthritis

Discussion
Our experiments have shown that inhibitors of NOS decrease articular inflammatory pain when given early, before induction of the arthritis, but not when given late, after the arthritis had been induced. However, treatment with NO donors after induction of arthritis was effective in reducing pain. These results appear paradoxical but we would conclude that, overall, NO has a direct anti-nociceptive effect on articular inflammatory pain in zymosan arthritis. The anti-nociceptive activity observed when NOS inhibitors were given prophylactically, before the zymosan, is probably due to suppression of the synovitis induced by zymosan, thus preventing the subsequent development of the inflammatory reactions leading to pain.
Administration of D-NAME, the inactive enantiomer of L-NAME, as well as combined administration of L-arginine and L-NAME did not alter articular incapacitation, suggesting that the anti-nociceptive effect of L-NAME was most likely to be due to inhibition of NO synthesis. The isoform of NOS most relevant here would appear to be the inducible iNOS. Thus, aminoguanidine (AG), which is currently used as a selective iNOS inhibitor, was effective in reducing AI, our measure of articular pain, suggesting that both iNOS and cNOS inhibition produced antinociception in our model. There are many reports showing that AG is a selective iNOS inhibitor (Griffiths et al., 1993). AG was shown to reduce plasma leakage in the mouse skin (Fuji et al., 1996) and to reduce rat paw inflammation induced by carrageenan (Salvemini et al., 1996b), effects that were assumed to be due to iNOS inhibition. However, AG, in the presence of calcium, calmodulin, and other cofactors can also inhibit cNOS (Wolff & Lubeskie, 1995a, 1995b; Laszlo et al., 1995). We therefore used 1400W, which is approximately 500 times more potent and specific as an iNOS inhibitor than AG, and currently one of the most selective iNOS inhibitors available (Garvey et al., 1997). Our results showed that 1400W also dose-dependently inhibited AI, reinforcing our suggestion that the anti-nociceptive effects observed with L-NAME and AG were due to the inhibition of iNOS.
The anti-inflammatory properties of L-arginine analogues have been linked to local microcirculatory vasoconstriction that could be reversed by vasodilators (Ridger et al., 1997). Although there is marked heterogeneity of the influence of NO in the regional microvasculature among different tissues (Greenblatt et al., 1993) MAP is influenced by alterations in peripheral vascular resistance occurring in the microcirculation. Thus, significant microcirculatory vasoconstriction resulting from cNOS inhibition should lead to an increase in MAP. In the present study, animals treated with the highest effective dose of L-NAME did exhibit an increase in MAP. However, MAP was not altered in animals that received the highest effective dose of AG. Hence, in the present study, local vasoconstriction and increased MAP was not necessary for the anti-nociceptive actions of AG.
Is is possible that articular oedema, rather than articular pain, may have caused the lack of joint mobility that we recorded as AI. This possibility is less likely in our experiments since the prophylactic administration of the iNOS inhibitors or the therapeutic injection of SIN-1, though being able to inhibit AI, did not alter local oedema. In concordance with these data, intra-articular injection of dextran, that promotes non-inflammatory oedema, did not elicit AI, as measured by the rat-knee joint as in the present study (Tonussi & Ferreira, 1992 ). L-NAME has been shown to reduce the oedema induced by bradykinin, histamine, and zymosan-activated plasma, carragenin, or dextran (Teixeira et al., 1993; Salvemini et al., 1996a, 1996b; Ialenti et al., 1992). However, L-NAME and haemoglobin, which acts as an NO scavenger, reduced paw oedema in rats only in the initial phase (1 h) after stimulus injection (Cuzzocrea et al., 1997) whereas we measured articular oedema 6 h after zymosan. Controversies have also arisen concerning the contribution of NO to oedema development. Systemic administration of L-NAME or 1400W inhibited the oedema formation in experimental colitis (Kankuri et al., 2001). On the other hand, L-NAME promoted an increase in vascular permeability in the isolated perfused rat lung (Mundy & Dorrington, 2000) and administration of L-arginine, the NOS substrate, prevented the increase in vascular permeability induced by contrast-media in the rat lung (Sendo et al., 2000). More recently, local (intra-pleural) and systemic administration of NOS inhibitors increased and reduced oedema in carrageenin-induced pleuritis, respectively (Paul-Clark et al., 2001). Another recent study showed that administration of a selective iNOS inhibitor did not reduce the acute inflammatory response and exacerbated the chronic inflammation in rats subjected to streptococcal cell wall arthritis (McCartney-Francis et al., 2001). As a whole, these results suggest that NO may have different effects on oedema development depending on the route of administration and also on the tissue studied.
Anti-nociception has previously been associated with inhibition of NOS. L-NAME provoked dose-dependent anti-nociception in the formalin-induced paw-licking model in mice (Moore et al., 1991). Intrathecal administration of AG and 2-amino-5,6-dihydro-methylthiazine, another selective iNOS inhibitor, decreased carrageenan-induced thermal hyperalgesia in rats (Osborne & Coderre, 1999). In joints, local administration of either L-NAME or 7-nitro-indazole, a selective neuronal NOS inhibitor, prevented radiant heat hyperalgesia in rats subjected to carrageenan-kaolin arthritis (Lawand et al., 1997). However, to our knowledge, the present study is the first to evaluate the role of NO in articular inflammatory pain.
A particularly relevant interaction in inflammatory models is that between NO and cyclo-oxygenase, the rate-limiting enzyme in PG biosynthesis (Salvemini et al., 1993) but the outcome of this interaction varies from model to model. Thus, NO either promoted the release of prostaglandins through iNOS activation (Salvemini et al., 1995a, 1995b) or decreased prostaglandin production (Swierkosz et al., 1995). This could be caused by a biphasic effect of NO that was demonstrated on a macrophage cell line of mice (Tetsuka et al., 1994; Milano et al., 1995). In our model of arthritis, neither L-NAME nor AG administration altered PGE2 release into the synovial fluid, showing that the anti-nociception following iNOS blockade was not due to inhibition of PGE2 release. This is in concordance with a recent study showing that, in carrageenin-induced pleurisy, S-(2-aminoethyl) isothiorea (AE-ITU), a specific iNOS inhibitor, did not alter PGE2 release (Paul-Clark et al., 2001).
In our experiments, inhibition of iNOS was anti-nociceptive. However, in the hyperalgesia provoked by local injection of either carrageenan or PGE2 in the rat hind paw it was reported that the anti-nociceptive effect of acetylcholine was due to NO release (Duarte et al., 1990). This apparent discrepancy could perhaps be explained by the fact that these authors studied the effect of local administration and used different pain models. However, in our experiments, all three NOS inhibitors L-NAME, AG, or 1400W were effective in inhibiting AI when administered locally (intra-articularly). Thus, systemic as well as local iNOS inhibition, given prophylactically, was equally anti-nociceptive in our model.
In zymosan-induced arthritis, AI peaks between 3–4 h after injection of zymosan (Rocha et al., 1999) and in order to assess the effect of NOS inhibition on established AI, we gave L-NAME or AG, 2 h after zymosan. In contrast with the results of giving NOS inhibitors before the zymosan, this later administration did not affect AI, irrespective of systemic or local application. Because of this failure, we decided to evaluate the effect of NO donors, also given therapeutically, i.e., 2 h after zymosan. Our results have clearly shown that local administration of two NO donors (SNP or SIN-1) significantly reduced AI. We must emphasize that these NO donors were given as therapeutic interventions when the arthritis was already well established, pointing to a direct analgesic effect for NO donors in articular inflammatory pain.
At least two points of discussion emerge from the results. First, the apparent paradox of both NOS inhibitors and NO donors providing anti-nociceptive effects. This we believe may be resolved by postulating that the anti-nociceptive effect of NOS inhibitors given prophylactically is due to prevention of synovial inflammation. Once the synoviocytes are stimulated by zymosan, leading to the release of pro-inflammatory algesic mediators, the subsequent administration of NOS inhibitors (therapeutic intervention) cannot prevent the activation of nociceptors by the algesic mediators already present. However, NO donors were effective in the presence of algesic mediators because NO has a direct anti-nociceptive effect.
This direct effect of NO provides the second point of discussion, as there are several reports that NO donors may produce either hyperalgesia or anti-nociception depending on the model, route of administration and species studied. In the rat paw pressure test, high dose SIN-1 was hyperalgesic (Aley et al., 1998) whereas another study showed that SIN-1 reduced PGE2 induced hyperalgesia (Ferreira et al., 1991). Local intradermal or intravenous NO injection was shown to evoke pain (Holthusen & Arndt, 1994; Kindgen-Milles & Arndt, 1996) whereas dermal nitroglycerin patches decreased postoperative pain in humans (Lauretti et al., 1999). With respect to joint pain, to our knowledge there is only one study showing that transdermal nitroglycerin provided partial symptomatic pain relief in patients with painful shoulders (Berrazueta et al., 1996). However, prolonged or extensive use of nitroglycerin is unlikely, as its vadodilating activity induces side effects such as headache and hypotension. Our study extends this earlier observation and suggests that NO donors other than nitroglycerin may also provide analgesia in human articular inflammatory pain.
The mechanisms to explain how NO is affecting pain transduction pathways are probably multiple. In the present study, NO donors could be analgesic through down-regulation of peripheral nociceptors in the joint, operating through activation of cGMP. As we mentioned above, in the prostaglandin E2-induced hyperalgesia in the rat hind paw, NO promoted analgesia that was blocked by a cGMP inhibitor (Duarte et al., 1990). In agreement with this proposal, the anti-nociceptive effect of sildenafil in the abdominal writhings test in mice was recently shown to be mediated by the inhibition of the phosphodiesterase 5 enzyme, thus increasing NO and cGMP production. This effect of sildenafil was potentiated by the combined administration of sodium nitroprusside and L-arginine (Jain et al., 2001). Also, the antinociceptive activity of the non-steroidal antiinflammatory drug (NSAID) ketorolac was shown to involve the activation of the NO–cGMP pathway, followed by opening of ATP-sensitive potassium channels (Lazaro-Ibanez et al., 2001). In addition to these effects, reaction of NO with thiol groups could also promote analgesia, through down-regulation of NMDA receptors, by inhibiting calcium influx. This mechanism could be important in neuropathic pain, where excessive activation of NMDA receptors occurs (Lipton & Stamler, 1994).
The NO donors effects may also be indirectly mediated, through the inhibition of the release of other pain mediators. For instance, NO donors were shown to inhibit the in vitro release of substance P from dorsal horn neurons, an effect that was associated with an increase in cGMP levels (Kamisaki et al., 1995). The production of cytokines and nerve growth factor (NGF) has also been associated with pain development during inflammatory conditions (Tal, 1999; Pezet et al., 2001). In addition, tumour necrosis factor-α induced interleukin-1β and NGF production were associated with the acute hyperalgesia provoked by the intraplantar injection of Freund's adjuvant in rats (Woolf et al., 1997). Reduction of pro-inflammatory cytokines production by NO-naproxen was reported to be due to the addition of NO to the NSAID naproxen (Cicala et al., 2000). Based on these data, we cannot exclude the possibility that the NO donors antinociceptive effect in zymosan-induced arthritis is related to decreased NGF release, secondary to an inhibition of pro-inflammatory cytokines production.
In conclusion, the results presented in this study show that local administration of an NO donor was anti-nociceptive in zymosan arthritis, by reducing articular inflammatory pain. Additionally, we have also shown that prophylactic administration of NOS inhibitors also reduced this inflammatory pain. The latter effect reflected inhibition of the iNOS isoform and probably prevention of the inflammatory condition but did not depend on inhibition of articular oedema or of PGE2 release into the joints.
Acknowledgments
This work was supported by CAPES, CNPq, FAPESP, and FUNCAP.
Abbreviations
- 1400W
N-(3-(aminomethyl)benzyl)acetamide
- AG
aminoguanidine
- AI
articular incapacitation
- ANOVA
one-way analysis of variance
- cGMP
guanosine 3′5′ cyclic monophosphate
- D-NAME
D-NG-nitroarginine methyl ester
- ELISA
enzyme-linked immunosorbent assay
- i.art.
intra-articular
- i.p.
intra-peritoneal
- iNOS
inducible nitric oxide synthase
- L-NAME
L-NG-nitroarginine methyl ester
- L-NMMA
(L-NG-monomethylarginine)
- MAP
mean arterial pressure
- NGF
nerve growth factor
- NMDA
N-methyl-D-aspartate
- NO
nitric oxide
- NOS
nitric oxide synthase
- NSAID
non-steroidal antiinflammatory drug
- NT
non-treated
- p.o.
per os
- PET
paw elevation time
- PGE2
prostaglandin E2
- s.c.
subcutaneous
- SAL
saline
- SIN-1
(3-morpholinosydnonimine)
- SNP
sodium nitroprusside
- Zy
zymosan
References
- ALEY K.O., MCCARTER G., LEVINE J.D. Nitric oxide signaling in pain and nociceptor sensitization in the rat. J. Neurosci. 1998;18:7008–7014. doi: 10.1523/JNEUROSCI.18-17-07008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERRAZUETA J.R., LOSADA A., POVEDA J., OCHOTECA A., RIESTRA A., SALAS E., AMADO J.A. Successful treatment of shoulder pain syndrome due to supraspinatus tendinitis with transdermal nitroglycerin. A double blind study. Pain. 1996;66:63–67. doi: 10.1016/0304-3959(96)03021-7. [DOI] [PubMed] [Google Scholar]
- CICALA C., IANARO A., FIORUCCI S., CALIGNANO A., BUCCI M., GERLI R., SANTUCCI L., WALLACE J.L., CIRINO G. NO-naproxen modulates inflammation, nociception and down-regulates T cell response in rat Freund's adjuvant arthritis. Br. J. Pharmacol. 2000;130:1399–1405. doi: 10.1038/sj.bjp.0703449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUZZOCREA S., ZINGARELLI B., CALAPAI G., NAVA F., CAPUTI A.P. Zymosan-activated plasma induces paw oedema by nitric oxide and prostaglandin production. Life Sci. 1997;60:215–220. doi: 10.1016/s0024-3205(96)00618-2. [DOI] [PubMed] [Google Scholar]
- DUARTE I.D.G., LORENZETTI B.B., FERREIRA S.H. Peripheral analgesia and activation of the nitric oxide-cyclic GMP pathway. Eur. J. Pharmacol. 1990;186:289–293. doi: 10.1016/0014-2999(90)90446-d. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H., DUARTE I.D., LORENZETTI B.B. The molecular mechanism of action of peripheral morphine analgesia: stimulation of the cGMP system via nitric oxide release. Eur. J. Pharmacol. 1991;201:121–122. doi: 10.1016/0014-2999(91)90333-l. [DOI] [PubMed] [Google Scholar]
- FUJI E., IRIE K., OGAWA K., MURAKI T. Role of nitric oxide and prostaglandins in lipopolysaccharide-induced increase in vascular permeability in mouse skin. Eur. J. Pharmacol. 1996;297:257–263. doi: 10.1016/0014-2999(95)00758-x. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., FURFINE E.S., KIFF R.J., LASZLO F., WHITTLE B.J.R., KNOWLES R.G. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- GREENBLATT E.P., LOEB A.L., LONGNECKER D.E. Marked regional heterogeneity in the magnitude of EDRF/NO-mediated vascular tone in awake rats. J. Cardiovasc. Res. 1993;21:235–240. doi: 10.1097/00005344-199302000-00008. [DOI] [PubMed] [Google Scholar]
- GRIFFITHS M.J.D., MESSENT R.J. , MACALLISTER R.J., EVBANS T.W. Aminoguanidine selectively inhibits inducible nitric oxide synthase. Br. J. Pharmacol. 1993;110:963–968. doi: 10.1111/j.1476-5381.1993.tb13907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLTHUSEN H., ARNDT J.O. Nitric oxide evokes pain in humans on intracutaneous injection. Neurosci. Lett. 1994;165:71–74. doi: 10.1016/0304-3940(94)90712-9. [DOI] [PubMed] [Google Scholar]
- IALENTI A., IANARO A., MONCADA S., DI ROSA M. Modulation of acute inflammation by endogenous nitric oxide. Eur. J. Pharmacol. 1992;211:177–182. doi: 10.1016/0014-2999(92)90526-a. [DOI] [PubMed] [Google Scholar]
- IALENTI A., MONCADA S., DI ROSA M. Modulation of adjuvant arthritis by endogenous nitric oxide. Br. J. Pharmacol. 1993;110:701–706. doi: 10.1111/j.1476-5381.1993.tb13868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAIN N.K., PATIL C.S., SINGH A., KULKAMI S.K. Sildenafil-induced peripheral analgesia and activation of the nitric oxice–cyclic GMP pathway. Brain Res. 2001;909:170–178. doi: 10.1016/s0006-8993(01)02673-7. [DOI] [PubMed] [Google Scholar]
- KAMISAKI Y., NAKAMOTO K., WADA K., ITOH T. Nitric oxide regulates substance P release from rat spinal cord synaptosomes. J. Neurochem. 1995;65:2050–2056. doi: 10.1046/j.1471-4159.1995.65052050.x. [DOI] [PubMed] [Google Scholar]
- KANKURI E., VAALI K., KNOWLES R.G., LAHDE M., KORPELA R., VAPAATALO H., MOILANEN E. Suppression of acute experimental colitis by a highly selective inducible nitric-oxide synthase inhibitor, N-[3-(aminomethyl)benzyl]acetamidine. J. Pharmacol. Exp. Ther. 2001;298:1128–1132. [PubMed] [Google Scholar]
- KINDGEN-MILLES D., ARNDT J.O. Nitric oxide as a chemical link in generation of pain from veins in humans. Pain. 1996;64:139–142. doi: 10.1016/0304-3959(95)00081-X. [DOI] [PubMed] [Google Scholar]
- LASZLO F., EVANS S.M., WHITTLE B.J.R. Aminoguanidine inhibits both constitutive and inducible nitric oxide synthase isoforms in rat intestinal microvasculature in vivo. Eur. J. Pharmacol. 1995;272:169–175. doi: 10.1016/0014-2999(94)00637-m. [DOI] [PubMed] [Google Scholar]
- LAURETTI G.R., LIMA I.C., REIS M.P., PRADO W.A., PEREIRA N.L. Oral ketamine and transdermal nitroglycerin as analgesic adjuvants to oral morphine therapy for cancer pain management. Anesthesiology. 1999;90:1528–1533. doi: 10.1097/00000542-199906000-00005. [DOI] [PubMed] [Google Scholar]
- LAWAND N.B., WILLIS W.D., WESTLUND K.N. Blockade of joint inflammation and secondary hyperalgesia by L-NAME, a nitric oxide inhibitor. Neuroreport. 1997;8:895–899. doi: 10.1097/00001756-199703030-00016. [DOI] [PubMed] [Google Scholar]
- LAZARO-IBANEZ G.G., TORRES-IOPEZ J.E., GRANADOS-SOTO V. Participation fo the nitric oxide-cyclic GMP-ATP-sensitive K(+) channel pathway in the antinociceptive action of ketorolac. Eur. J. Pharmacol. 2001;426:39–44. doi: 10.1016/s0014-2999(01)01206-7. [DOI] [PubMed] [Google Scholar]
- LIPTON S.A., STAMLER J.S. Actions of redox congeners of nitric oxide at the NMDA receptor. Neuropharmacology. 1994;33:1229–1233. doi: 10.1016/0028-3908(94)90021-3. [DOI] [PubMed] [Google Scholar]
- MCCARTNEY-FRANCIS N.L., SONG X.Y., MIZEL D.E., WAHL C.L., WAHL S.M. Hemoglobin protects from streptococcal cell wall-induced arthritis. Arthritis Rheum. 1999;42:1119–1127. doi: 10.1002/1529-0131(199906)42:6<1119::AID-ANR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- MCCARTNEY-FRANCIS N.L., SONG X.Y., MIZEL D.E., WAHL S.M. Selective inhibition of inducible nitroc oxide synthase exacerbates erosive joint disease. J. Immunol. 2001;166:2734–2740. doi: 10.4049/jimmunol.166.4.2734. [DOI] [PubMed] [Google Scholar]
- MILANO S., ARCOLEO F., DIELI M., D'AGOSTINO P., DE NUCCI G., CILLARI E. Prostaglandin E2 regulates inducible nitric oxide in the murine macrophage cell line J774. Prostaglandins. 1995;49:105–115. doi: 10.1016/0090-6980(94)00004-g. [DOI] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M.J., HIGGS E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. J. Pharm. Exp. Ther. 1991;43:109–141. [PubMed] [Google Scholar]
- MOORE P.K., OLUYOMI R.C., BABBEDGE R.C., WALLACE P., HART S.L. L-NG-nitro-arginine-methyl-ester exhibits antinociceptive activity in the mouse. Br. J. Pharmacol. 1991;102:198–202. doi: 10.1111/j.1476-5381.1991.tb12153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUNDY A.L., DORRINGTON K.L. Inhibition of nitric oxide synthesis augments pulmonary oedema in isolated perfused rabbit lung. Br. J. Anaesth. 2000;85:570–576. doi: 10.1093/bja/85.4.570. [DOI] [PubMed] [Google Scholar]
- OSBORNE M.G., CODERRE T.J. Effects of intrathecal administration of nitric oxide synthase inhibitors on carrageenan-induced thermal hyperalgesia. Br. J. Pharmacol. 1999;126:1840–1846. doi: 10.1038/sj.bjp.0702508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAUL-CLARK M.J., GILROY D.W., WILLIS D., WILLOUGHBY D.A., TOMLINSON A. Nitric oxide synthase inhibitors have opposite effects on acute inflammation depending on their route of administration. J. Immunol. 2001;166:1169–1177. doi: 10.4049/jimmunol.166.2.1169. [DOI] [PubMed] [Google Scholar]
- PEZET S., ONTENIENTE B., JULLIEN J., JUNIER M.P., GRANNEC G., RUDKIN B.B., CALVINO B. Differential regulation of NGF receptors in primary sensory neurons by adjuvant-induced arthritis in the rat. Pain. 2001;90:113–125. doi: 10.1016/s0304-3959(00)00393-6. [DOI] [PubMed] [Google Scholar]
- RIDGER V.C., PETTIPHER E.R., BRYANT C.E., BRAIN S.D. Effect of the inducible nitric oxide synthase inhibitors aminoguanidine and L-N6-(-iminoethyl)lysine on zymosan induced plasma extravasation in rat skin. J. Immunol. 1997;159:383–390. [PubMed] [Google Scholar]
- ROCHA F.A.C., ARAGÃO A.G.M., Jr, DE OLIVEIRA R.C., VALE M.R., RIBEIRO R.A. Periarthritis promotes articular incapacitation in zymosan-induced arthritis in rats. Inflam. Res. 1999;48:485–490. doi: 10.1007/s000110050491. [DOI] [PubMed] [Google Scholar]
- SALVEMINI D., MANNING P.T., ZWEIFEL B.S., SEIBERT K., CONNOR J., CURRIE M.G., NEEDLEMAN P., MASFERRER J.L. Dual inhibition of nitric oxide and prostaglandin production contributes to the antiinflammatory properties of nitric oxide synthase inhibitors. J. Clin. Invest. 1995a;96:301–308. doi: 10.1172/JCI118035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVEMINI D., MISKO T.P., MASFERRER J.L., SEIBERT K., CURRIE M.G., NEEDLEMAN P. Nitric oxide activates cyclo-oxygenase enzymes. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7140–7144. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVEMINI D., SETTLE S.L., MASFERRER J.L, , SEIBERT K., CURRIE M., CURRIE M., NEEDLEMAN P. Regulation of prostaglandin by nitric oxide: an in vivo analysis. Br. J. Pharmac. 1995b;114:1171–1178. doi: 10.1111/j.1476-5381.1995.tb13330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVEMINI D., WANG Z.Q., BOURDON D.M., STERN M.K., CURRIE M.G., MANNING P.T. Evidence of peroxynitrite involvement in the carrageenan-induced rat paw edema. Eur. J. Pharmacol. 1996a;303:217–220. doi: 10.1016/0014-2999(96)00140-9. [DOI] [PubMed] [Google Scholar]
- SALVEMINI D., WANG Z.Q., WYATT P.S., BOURDON D.M., MARINO M.H., MANNING P.T., CURRIE M.G. Nitric oxide: a key mediator in the early and late phase of carrageenan-induced rat paw inflammation. Br. J. Pharmacol. 1996b;118:829–838. doi: 10.1111/j.1476-5381.1996.tb15475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENDO T., KATAOKA Y., TAKEDA Y., FURUTA W., OISHI R. Nitric oxide protects against contrast media-increased pulmonary vascular permeability in rats. Invest. Radiol. 2000;35:472–478. doi: 10.1097/00004424-200008000-00003. [DOI] [PubMed] [Google Scholar]
- SOUSA A.M., PRADO W.A. The dual effect of a nitric oxide donor in nociception. Brain Res. 2001;897:9–19. doi: 10.1016/s0006-8993(01)01995-3. [DOI] [PubMed] [Google Scholar]
- STEFANOVIC-RACIC M., MEYERS K., MESCHTER C., COFFEY J.W., HOFFMAN R.A., EVANS C.H. N-monomethyl arginine, an inhibitor of nitric oxide synthase, suppresses the development of adjuvant arthritis in rats. Arthritis Rheum. 1994;37:1062–1069. doi: 10.1002/art.1780370712. [DOI] [PubMed] [Google Scholar]
- STEFANOVIC-RACIC M., MEYERS K., MESCHTER C., COFFEY J.W., HOFFMAN R.A., EVANS C.H. Comparison of the nitric oxide synthase inhibitors methylarginine and aminoguanidine as prophylactic and therapeutic agents in rat adjuvant arthritis. J. Rheumatol. 1995;22:1922–1928. [PubMed] [Google Scholar]
- SWIERKOSZ T.A., MITCHELL J.A., WARNER T.D., BOTTING R.M., VANE J.R. Co-induction of nitric oxide and cyclo-oxygenase: interactions between nitric oxide and prostanoids. Br. J. Pharmacol. 1995;114:1335–1342. doi: 10.1111/j.1476-5381.1995.tb13353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAL M. A role for inflammation in chronic pain. Curr. Rev. Pain. 1999;3:440–446. doi: 10.1007/s11916-999-0071-4. [DOI] [PubMed] [Google Scholar]
- TEIXEIRA M.M., WILLIAMS T.J., HELLEWELL P.G. Role of prostaglandins and nitric oxide in acute inflammatory reactions in guinea-pig skin. Br. J. Pharmacol. 1993;110:1515–1521. doi: 10.1111/j.1476-5381.1993.tb13994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TETSUKA T., DAPHNA-IKEN D., SRIVASTAVA S.K., BAIER L.D., DUMAINE J., MORRISON A.R. Cross-talk between cyclooxygenase and nitric oxide pathways prostaglandin E2 negatively modulates induction of nitric oxide synthase by interleukin-1. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12168–12172. doi: 10.1073/pnas.91.25.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TONUSSI C.R., FERREIRA S.H. Rat knee-joint incapacitation test: an objective screen for central and peripheral analgesics. Pain. 1992;48:421–427. doi: 10.1016/0304-3959(92)90095-S. [DOI] [PubMed] [Google Scholar]
- WOLFF D.J., LUBESKIE A. Aminoguanidine is an isoform-selectikve, mechanism-based inactivator of nitric oxide synthase. Arch. Biochem. Biophys. 1995a;316:290–301. doi: 10.1006/abbi.1995.1040. [DOI] [PubMed] [Google Scholar]
- WOLFF D.J., LUBESKIE A. Inactivation of nitric oxide synthase isoforms by diaminoguanidine and NG-amino-L-arginine. Arch. Biochem. Biophys. 1995b;325:227–234. doi: 10.1006/abbi.1996.0028. [DOI] [PubMed] [Google Scholar]
- WOOLF C.J., ALLCHORNE A., SAFIEH-GARABEDIAN B., POOLE S. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumor necrosis factor alpha. Br. J. Pharmacol. 1997;121:417–424. doi: 10.1038/sj.bjp.0701148. [DOI] [PMC free article] [PubMed] [Google Scholar]