

Abstract
Sepsis induced by S. aureus was used to investigate whether neutrophil migration failure to infectious focus correlates with lethality in Gram-positive bacteria-induced sepsis in mice.
By contrast with the sub-lethal (SL-group), the lethal (L-group) intraperitoneal inoculum of S. aureus caused failure of neutrophil migration (92% reduction), high CFU in the exudate, bacteremia and impairment of in vitro neutrophil chemotactic activity.
Pre-treatments of L-group with adequate doses of aminoguanidine prevented the neutrophil migration failure and improved the survival of the animals (pre-treated: 43%; untreated: 0% survival). Thus, the impairment of neutrophil migration in the L-group appears to be mediated by nitric oxide (NO).
The injection of S. aureus SL-inoculum in iNOS deficient (−/−) or aminoguanidine-treated wild-type mice (pre- and post-treatment), which did not present neutrophil migration failure, paradoxically caused severe peritonitis and high mortality. This fact is explainable by the lack of NO dependent microbicidal activity in migrated neutrophils.
In conclusion, although the NO microbicidal mechanism is active in neutrophils, the failure of their migration to the infectious focus may be responsible for the severity and outcome of sepsis.
Keywords: Nitric oxide, failure of neutrophil migration, S. aureus, sepsis
Introduction
With mortality rates ranging from 30 to 50%, septic shock, despite aggressive surgical intervention, adequate antibiotic therapy and intensive life support, is one of the leading causes of mortality in intensive care units throughout the world (Sands et al., 1997). Nowadays, the incidence of sepsis due to Gram-positive bacteria is similar to that of Gram-negative bacteria (Sriskandan & Cohen, 1999; Cohen, 2000), therefore highlighting the importance of a better understanding of mechanisms involved in Gram-positive septicaemia. Staphylococcus aureus (S. aureus) is the major cause of nosocomial Gram-positive infections and septicaemia in humans. S. aureus is also the major contributor to osteomyelitis, invasive endocarditis and septic arthritis (Bannan et al., 1999).
The pathophysiological events of sepsis are not a consequence of direct effects of the bacteria and/or their by-products (endotoxin and exotoxins), but result from an intense systemic inflammatory response syndrome (SIRS), which occurs when the host fails to restrict the pathogens to a localized area (Lynn & Cohen, 1995). Hence, the production of pro-inflammatory mediators, mainly the cytokines tumour necrosis factor (TNF)-α, interleukin (IL)-1β and interferon (IFN-γ)-γ, in the infectious focus is important in the recruitment and activation of leukocytes, key events in restricting the spread of infection (Hinshaw et al., 1992; 1995; Zhao et al., 1998). However, the overproduction of these mediators in the circulation results in SIRS, which culminates in morbidity and/or mortality of the individual (Walley et al., 1996; Bone et al., 1997; Martineau & Shek, 2000).
This dual effect of cytokines is mediated mainly by nitric oxide (NO; Cunha et al., 1994; Bhagat et al., 1999). The NO that participates in the protective effects is produced by the inducible NO synthase expressed in leukocytes present in the infectious focus and stimulated by the cytokines described above. NO is a key mediator of the neutrophil microbicidal activity against the majority of pathogens, including Gram-positive and Gram-negative bacteria, an event crucial in the elimination of the infection and, consequently, the restoration of tissue integrity (Conlan, 1997). However, the overproduction of NO, stimulated by circulating cytokines, has been implicated in several disorders observed in sepsis. There is evidence that NO mediates the vascular relaxation associated with hypotension (Wolkow, 1998), refractoriness to vasopressor catecholamines (Silva-Santos & Assreuy, 1999), platelet aggregation (Hoehn & Krause, 2001; Broeders et al., 2001), and organ lesions (Evans & Stefanovic-Racic, 1996; Numata et al., 1998). In fact, inducible nitric oxide synthase (iNOS) inhibitors were found to increase systemic vascular resistance and the responsiveness to catecholamines in experimental endotoxemia (Galanos & Freudenberg, 1993) and in patients with septic shock (Petros et al., 1994).
Early studies from our laboratory demonstrated that failure of neutrophil migration to an inflammatory site is observed in sepsis induced by endotoxemia (Tavares-Murta et al., 1998) and by caecal-ligation and puncture (CLP; Benjamim et al., 2000). The outcome of the animals with CLP correlated with the failure of neutrophil migration to the infectious focus (Benjamim et al., 2000). NO mediates the neutrophil migration failure, since two inhibitors of NOS, aminoguanidine and L-NMMA, protected animals from impairment of neutrophil migration induced by CLP surgery and endotoxemia, respectively (Tavares-Murta et al., 1998; Benjamim et al., 2000). The involvement of NO in the failure of neutrophil migration is in accordance with the literature that shows that NOS inhibitors increase the adhesion of leukocytes to endothelial cells both in vitro and in vivo (Hickey & Kubes, 1997; Hickey et al., 1997; Hickey, 2001). We also observed that the protective effect of aminoguanidine on animals undergoing CLP depends on the dose of the drug administered in the pre-treatment of the animals. Doses of up to 30 mg Kg−1 prevented neutrophil migration failure and protected the CLP-animals from death. However, a high dose of aminoguanidine (90 mg Kg−1) did not improve survival in the CLP-animals, in spite of preventing the failure of neutrophil migration. It was suggested that the high dose of aminoguanidine inhibits NO production for a longer period, thus inhibiting the microbicidal ability of neutrophils which had migrated to the infectious focus (Benjamim et al., 2000).
In the present study we investigated whether the failure of neutrophil migration is observed in sepsis induced by Gram-positive bacteria, and also whether NO mediates this process.
Methods
Staphylococcus aureus preparation
Staphylococcus aureus was obtained from ATCC (American Type Culture Collection, U.S.A.) number 25923. Lyophilized preparations were cultured in brain heart infusion (BHI) medium to establish purity. Thereafter, large amounts of lyophilized bacteria were obtained. Eighteen hours before each experiment samples of lyophilized bacteria were cultured again in BHI medium at 37°C. The bacterial suspension was centrifuged and the pellet was resuspended in sterile phosphate-buffered saline (PBS) for administration to the animals. The number of colony forming units (CFU) of the bacterial suspension was determined through serial log dilution and plating on Mueller-Hinton agar dishes (Difco Laboratories, Detroit, U.S.A.); colony-forming units were counted after 18 h, and the results were expressed as the number of CFU per experimental animal cavity or CFU per ml of blood.
Induction of sepsis
Animals used in this study were C57BL/6 (wild-type) mice, bred at the Faculty of Medicine of Ribeirão Preto - USP, and C57BL/6 iNOS−/− mice, purchased from Jackson Laboratories (Bar Harbor, Maine, U.S.A.); both groups comprised males weighing between 18 and 22 g. The animals were housed under the same conditions in a sterile laminar flow cabinet until the time of the experiment and received water and food ad libitum. The genotype of iNOS−/− mice was confirmed by DNA PCR. Sepsis was induced through intraperitoneal injection of Staphylococcus aureus. Briefly, bacterial suspensions were injected into the peritoneal cavity in a volume of 500 μl. The quantity of bacteria injected was: 0.01; 0.08; 0.5; 3 and 16×109 CFU/cavity. The survival rate of mice and the number of neutrophils migrated into the peritoneal cavity were determined at various times after bacterial inoculation. After this set of experiments two bacterial inoculums (0.5×109 CFU/cavity and 16×109 CFU/cavity) were selected for further study, these were named sub-lethal (SL) and lethal (L) inoculums, respectively. All mice undergoing lethal inoculation develop early clinical signs of sepsis, including lethargy, piloerection, and tachypnea. The animals injected with SL inoculum also develop early clinical signs, although the manifestation disappears within the first 24 h. The animals injected with sub-lethal and lethal inoculums were analysed for the following parameters: survival rate, assessed daily for 7 days; bacteria in the blood (bacteremia) and the number of bacteria in the peritoneal cavity; neutrophil migration into the peritoneal cavity; cytokine levels in the peritoneal exudate and in sera. The effect of treatment of the septic animals with the NO inhibitor, aminoguanidine, on the parameters described above was analysed.
Bacteremia and number of bacteria in peritoneal cavities
At given time points (2, 4, 12 and 24 h after bacterial inoculation), mice were killed, and the peritoneal cavity was washed with sterile saline. For peritoneal lavage, the skin of the abdomen was cut open in the midline after thorough disinfection and without injury to the muscle. Sterile PBS (2 ml) was injected into and aspirated out of the peritoneal cavity. Aliquots of serial dilutions of these peritoneal lavage fluids were plated on Mueller-Hinton agar dishes and incubated at 37°C; colony-forming units were analysed after 18 h. The results were expressed as the number of CFU per peritoneal cavity. Blood was collected at the same time points under sterile conditions. Five microliters of blood were plated on Mueller-Hinton agar dishes and incubated at 37°C; colony-forming units were analysed after 18 h, and the results were expressed as the number of CFU per ml of blood.
Leukogram
Animals were anaesthetized in an ether chamber, and a sample of blood (diluted in 15% EDTA K3) was collected from the caudal vein at determined time points (2, 4, 12 and 24 h). Total counts were made in a cell counter (Coulter Ac T Series Analyser; Coulter Corp., Miami, U.S.A.), and differential counts were made on slides stained by the May – Grunwald – Giemsa method. The results were expressed as the number of cells×106 per ml of blood.
Measurement of blood pressure
The blood pressures of septic animals were monitored by a modification of a method described elsewhere (Franchini et al., 2000; Inoue et al., 2000). All surgical procedures were performed under aseptic conditions. Mice were anaesthetized with a mixture of ketamine (70 mg Kg−1, i.p.) and diazepam (6 mg Kg−1, i.p.) and placed on a temperature-controlled surgical table to maintain body temperature at 37°C. One hour after femoral artery catheterization, the animals received sub-lethal and lethal bacterial inoculums. Pulsatile arterial pressure was monitored 4 h after inoculation for a period of 1 h.
Neutrophil migration into the peritoneal cavity
Neutrophil migration was quantified at 2, 4, 12 and 24 h after bacterial inoculation. The animals were killed in an ether chamber and the cells present in the peritoneal cavity were collected by injecting 2 ml of PBS containing 1 mM EDTA. Total counts were made in a cell counter (Coulter) and differential cell counts were made on centrifuge slides (Cytospin 3, Shandon Southern Products, Atsmoore, U.K.) stained by the May – Grunwald – Giemsa method. The results were expressed as the number of neutrophils per cavity.
Treatment of inoculated animals with the NOS inhibitor, aminoguanidine
Mice were treated s.c. with aminoguanidine according to the following schemes: (a) 10, 20, 30 mg Kg−1, 30 min before lethal bacterial inoculation; (b) 30 mg Kg−1 before lethal bacterial inoculation and 5 mg Kg−1 at 6 h after inoculation; (c) 30 mg Kg−1 before sub-lethal inoculation followed by the same dose 2 h after inoculation. In all treatments the neutrophil migration, CFU in the exudate and in blood and survival rate were analysed as described above.
In vitro neutrophil chemotaxis
Purified viable neutrophils were obtained 3 h after sub-lethal and lethal inoculation and from lethal inoculated animals pre-treated with aminoguanidine (30 mg Kg−1, 30 min before bacterial inoculation). Briefly, blood was obtained through retro-orbital puncture and neutrophils were purified using ‘Ficol Hypaque' modified medium (NIM™·2) according to the manufacturer's instructions. Chemotaxis was studied in 48-well chambers (Neuroprobe Inc., Cabin John, MD, U.S.A.) separated by 5 μm pore size polyvinylpyrrolidone-free polycarbonate membranes. Twenty-eight microlitres of formyl-Met-Leu-Phe (fMLP) (10−6 and 10−7 M) and leukotriene B4 (LTB4)(10−7 M) diluted in RPMI 1640 containing bovine serum albumin 0.01% (RPMI-BSA) were placed in the bottom of each well and 50 μl of the polymorphonuclear cell (PMN) suspension (106 cells ml−1) were added to the top of each well. The chamber was then incubated for 1 h at 37°C with 5% CO2, after which it was removed and the cells were fixed and stained with a Diff-Quick stain kit. The number of neutrophils which had migrated to the lower side of the filter was counted (100×objective) in five random fields. The results are representative of two separate experiments performed in triplicate for each sample, and are expressed as the number of neutrophils per field. Neutrophils obtained from normal mice migrating towards fMLP or LTB4 served as a positive control.
Cytokine measurements
The concentrations of TNF-α, IL-1β and IL-10 in the sera and peritoneal exudates were determined by a double-ligand ELISA. Briefly, flat-bottomed 96-well microtiter plates were coated with 100 μl per well of antibody specific to one of the above cytokines at a dilution of 2 μg ml−1 (TNF-α and IL-1β) and 1 μg ml−1 (IL-10) in coating buffer and incubated overnight at 4°C. Next, the plates were washed and non-specific binding was blocked for 120 min at 37°C with 1% bovine serum. Samples (undiluted) and standards were loaded on to plates. Recombinant murine TNF-α, IL-1β and IL-10 standard curves were used to calculate the cytokine concentrations. The plates were thoroughly washed, and the appropriate biotinylated polyclonal or monoclonal anti-cytokine antibody was added. The plates were washed 1 h later, avidin-peroxidase (diluted 1 : 5000) was added to each well for 15 min, and each plate was thoroughly washed again. Next, substrate (0.4 mg of OPD +0.4 μl of H2O2 in 1 ml of substrate buffer) was added and the reaction was stopped with H2SO4 (1 M); finally, the O.D. was measured on an ELISA plate scanner (Spectra Max 250 - Molecular Device) at 490 nanometres. The results were expressed as ng of TNF-α, IL-10 or IL-1β ml−1 in the supernatant or sera, comparing the optical density in the samples with the standard curves.
Drugs, reagents, and antibodies
The following materials were obtained from the sources indicated. rmIL-1β (lot 63/668; specific activity, 100,000 IU/0.1 μg ampoule), rmTNF-α (lot 99/532; specific activity, 200,000 IU/1 μg ampoule), purified anti-mouse IL-1β, purified anti-mouse TNF-α, biotinylated anti-mouse TNF-α (lot 250697), and biotinylated anti-mouse IL-1β (lot 250997) were gifts of Dr S. Poole (National Institute for Biological Standards and Control, London, U.K.). rmIL-10 (417-ML), anti-mouse IL-10 monoclonal antibody (MAB417), and biotinylated anti-mouse IL-10 (BAF417) were purchased from R&D Laboratories (Minneapolis, MN, U.S.A.). Aminoguanidine was purchased from Research Biochemicals International (Natick, MA, U.S.A.). BHI was purchased from Oxoid (CM225). All other reagents were purchased from Sigma.
Statistical analysis
The data (except survival curves) are reported as mean±s.e. and are representative of two or three separate experiments. Analysis of variance was used to compare the means of different treatments. If significance was identified, individual comparisons were subsequently made by Bonferroni's t-test for unpaired values. Statistical significance was set at P<0.05. Survival rates were expressed as percentages, and a log rank test (x2 test) was used to examine differences between survival curves.
Results
Mortality and neutrophil migration following bacterial inoculation
Figure 1A illustrates the survival curve of mice injected with different S. aureus inoculums (0.01; 0.08; 0.5; 3 and 16×109 CFU/cavity). All of the animals inoculated with 0.01, 0.08, and 0.50×109 CFU/cavity survived for 7 days after bacterial inoculation. The animals inoculated with 3×109 CFU/cavity showed 50% mortality at day 2, whereas animals inoculated with 16×109 CFU/cavity showed 100% mortality on the first day of observation. Figure 1B shows that the administration of S. aureus as the inoculums described above induced a bell-shape curve for neutrophil migration. The neutrophil migration into the peritoneal cavity increased after intraperitoneal (i.p.) injection of the S. aureus, peaking with the inoculum of 0.5×109 CFU/cavity, and declining with the higher CFU inoculations (3 and 16×109 CFU/cavity). The neutrophil migration observed after injection of 16×109 CFU/cavity was not statistically different from that observed following administration of PBS. In the subsequent experiments, the investigated parameters were determined after injection of 0.5×109 and 16×109 CFU/cavity, which were designated sub-lethal (SL-group) and lethal (L-group) groups, respectively.
Figure 1.
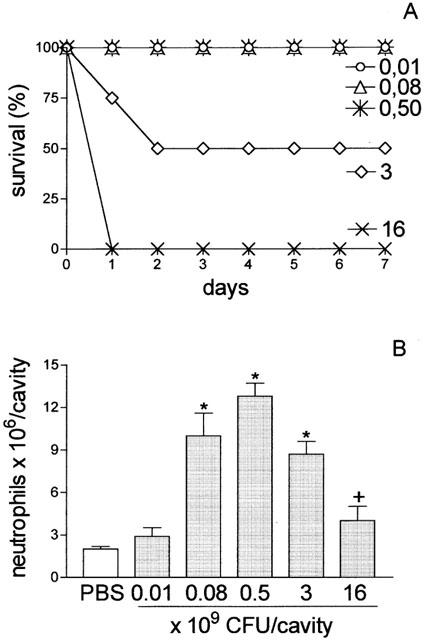
Survival curve and quantification of neutrophil migration into the peritoneal cavity after S. aureus inoculation. Groups of animals received different bacterial inoculum (0.01, 0.08, 0.5, 3, 16×109 CFU/cavity; 500 μl; i.p.). (A) Survival was determined daily until 7 days after bacterial inoculation. Results are expressed as per cent survival and are representative of three different experiments (P<0.001, Mantel-cox log rank test). (B) Neutrophil migration into the peritoneal cavity was quantified 4 h after bacterial inoculation or PBS injection. Results are expressed as mean±s.e. neutrophils per cavity and are representative of three independent experiments (n=4). *P<0.01 compared with control group (PBS). +P<0.001 compared with 0.5×109 CFU/animal (analysis of variance, followed by Bonferroni's test).
Kinetics of neutrophil migration, bacteremia and peritoneal exudate bacterial infection
SL- and L-groups were analysed for neutrophil migration, bacteremia and bacterial number in the peritoneal exudate 2, 4, 12 and 24 h after inoculation. Significant neutrophil migration was observed in the SL-group at 4, 12 and 24 h after bacterial inoculation, when compared to PBS. The migration seen at 4 h after bacterial injection was sustained at 12 h, before declining at 24 h. With regard to the L-group, an impairment of neutrophil migration was observed at all time points, when compared to SL-group (Figure 2A). The SL-group showed fewer bacteria (CFU) in the peritoneal exudates and did not exhibit bacteremia at 2, 4, 12 and 24 h after bacterial inoculation. However, the L-group exhibited a greater number of bacteria in the peritoneal exudate, principally at 12 h after bacterial inoculation, and a massive bacteremia was observed at all time points investigated (Figure 2B,C).
Figure 2.
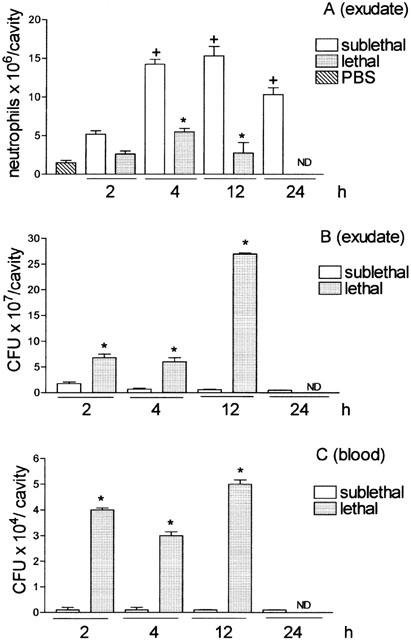
Quantification of neutrophil migration into the peritoneal cavity (A), CFU in the exudate (B), and in the blood (C). Groups of animals received i.p. sublethal (0.5×109 CFU in 500 μl cavity−1) or lethal (16×109 CFU in 500 μl cavity−1) bacterial inoculations. (A) Neutrophil migration into the peritoneal cavity was determined 2, 4, 12 and 24 h after bacterial inoculation. Results are expressed as mean±s.e. neutrophils per cavity. (B and C) The CFU was determined in the exudate and blood collected at the times indicated. The exudate and blood were diluted, spread-plated and incubated at 37°C, and the CFU were determined 18 h later. Results are expressed as CFU per cavity (exudate) or as CFU per ml of blood and are representative of three different experiments (n=3 – 4 per experiment). *P<0.01 compared with PBS group. +P<0.05 compared with sublethal group. (Analysis of variance, followed by Bonferroni's test).
Despite the failure of neutrophil migration into the peritoneal cavity, massive neutrophil accumulation and oedema were observed in the pulmonary tissue of the L-group, examined 12 h later (data not shown in figure).
Leukogram
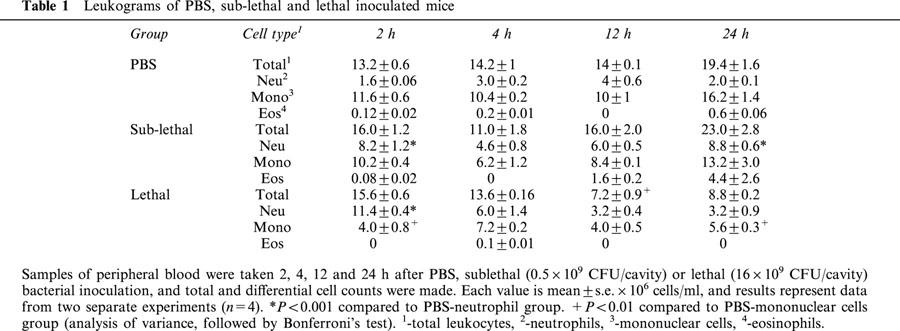
To determine whether the reduction in the neutrophil migration was due to neutropenia, we measured the numbers of this cell type in septic syndrome induced by S. aureus. Table 1 shows the leukogram (total number of leukocytes, neutrophils, eosinophils, and mononuclear cells) of PBS-, SL- and L-groups at 2, 4, 12 and 24 h after inoculation. Contrary to our expectations, a neutrophilia was observed in SL- and L-group at 2 h after bacterial inoculation and no significant difference was observed when compared with the PBS-group at time points of 4, 12 and 24 h after bacterial inoculation. It was also observed that the number of mononuclear cells in the L-group was reduced at all time points studied, when compared to the PBS-group. The number of eosinophils was similar in all experimental groups at each time point investigated. The significance for the outcome of sepsis of the monocytopenia observed in the L-group was not addressed in the present investigation and this requires further study.
Table 1.
Leukograms of PBS, sub-lethal and lethal inoculated mice
Mean blood pressure
To determine whether the reduced neutrophil migration into the peritoneal cavity was due to blood pressure changes, we monitored the blood pressure of the animals following bacterial inoculation. Neither SL- nor L-groups showed changes in the mean blood pressure 4 h after bacterial administration (Table 2).
Table 2.
Blood pressure of mice inoculated with S. aureus

Peritoneal exudate and serum cytokine levels following bacterial inoculation
Figure 3 shows the concentration of TNF-α, IL-1β, and IL-10 in the peritoneal exudates and sera, 4 h after bacterial inoculation. The TNF-α concentration in the exudates and sera increased significantly in SL- and L-groups compared with PBS. Between the SL- and L-groups there was no significant difference in the concentration of TNF-α, either in exudates or in sera. The pre-treatment of the L-group with aminoguanidine (30 mg Kg−1, 30 min before lethal inoculation) did not alter TNF-α production either in exudates or in sera (Figure 3A,B). The IL-1β levels in exudates of SL-, L- and Ag-groups were significantly higher than that observed in the PBS-group but were not different from each other (Figure 3C). In sera, the level of this cytokine in the SL-group was not significantly different to the PBS group. However, in the L-group, the IL-1β level was significantly higher when compared with PBS- or with SL- groups. The treatment with aminoguanidine did not significantly change the level of this cytokine (Figure 3D). As observed for IL-1β, levels of IL-10 in the exudates of SL-, L- and Ag-groups were significantly higher than that observed in the PBS group and were not different from each other (Figure 3E). In sera, the levels of IL-10 in SL-, L- and Ag-groups were also significantly higher than control. However, in this case, the aminoguanidine treatment of L-group promoted a significant increase in IL-10 production (Figure 3F).
Figure 3.
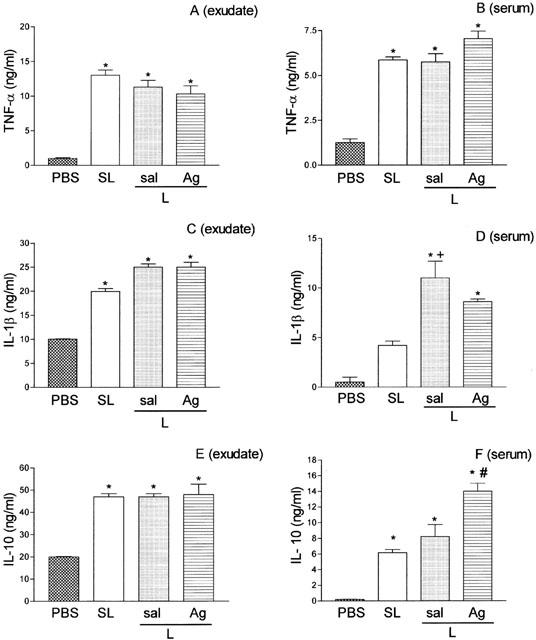
Concentration of TNF-α (A and B), IL-1β (C and D), and IL-10 (E and F) in the peritoneal exudate and in sera of mice injected with PBS, sub-lethal or lethal inoculums of S. aureus pre-treated or not with aminoguanidine. The cytokine concentrations in peritoneal exudate (left panels) and in sera (right panels) were determined at 4 h after PBS injection or inoculation of sub-lethal (SL, 0.5×109 CFU/animal), or lethal (L, 16×109 CFU/animal) inoculums of S. aureus pre-treated with PBS or with aminoguanidine (Ag, 30 mg Kg−1, 30 min before bacterial inoculation). Results are expressed as mean±s.e. nanograms of cytokine per millilitre of exudate or sera (n=4). *P<0.01 compared with PBS group. +P<0.05 compared with sublethal inoculum. #P<0.05 compared with lethal inoculum (analysis of variance, followed by Bonferroni's test).
Effect of aminoguanidine treatment on neutrophil migration, bacteremia, bacteria in the peritoneal fluid and survival rate
Having established that the failure of neutrophil migration towards the infectious focus in L-group correlated with the high mortality of the animals, we investigated whether the pre-treatment of the animals with aminoguanidine might prevent the failure of neutrophil migration, decrease the number of bacteria in the peritoneal exudate as well as the bacteremia, and, as a consequence, increase the survival rate of the animals. As shown in Figure 4A, pre-treatment of the L-group with aminoguanidine, s.c., at a dose of 10 – 30 mg Kg−1, 30 min before lethal inoculation (amino-group), caused a dose-dependent inhibition of the failure of neutrophil migration observed in L-group. The dose of 30 mg Kg−1 was almost 100% effective in preventing such failure (Figure 4A). The prevention of the neutrophil migration failure was accompanied by a significant decrease in the number of bacteria in the peritoneal exudate (Figure 4B) and the bacteremia (Figure 4C), compared with these parameters observed in L-group. Interestingly, the pretreatment of the L-group with aminoguanidine at a dose of 30 mg Kg−1 did not prevent the death of the animals. The survival rate of the amino-group was similar to that observed in the non-pre-treated L-group (Figure 4D). The phagocytic ability of the neutrophils recruited into the peritoneal cavity was also analysed. We observed that the phagocytic index in SL-group was lower than that in the L-group and the aminoguanidine pre-treatment did not change the phagocytic ability of the neutrophils in the latter (SL-group: 35%; L-group: 90%; amino-group: 95%; n=8).
Figure 4.
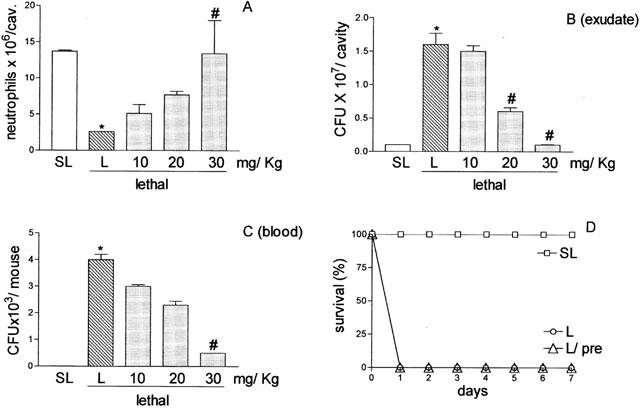
Effect of aminoguanidine pre-treatment of mice inoculated with S. aureus on neutrophil migration into the peritoneal cavity, CFU in exudate and in the blood, and survival. Lethal (16×109 CFU/cavity) inoculated mice were pre-treated with PBS or with 10, 20 or 30 mg Kg−1 aminoguanidine, subcutaneously, 30 min before S. aureus inoculation. Animals inoculated with the sub-lethal dose (0.5×109 CFU/cavity) (SL) were pre-treated with PBS. Neutrophil migration into the peritoneal cavity (A), CFU in exudate (B) and in the circulation (C) were determined 4 h after bacterial inoculation. Results are expressed as mean±s.e. of neutrophils per cavity, or of CFU per cavity (exudate) or CFU per ml of blood. Results are representative of three independent experiments (n=4). +P<0.01 compared with sublethal inoculation. #P<0.05 compared with lethal inoculation pre-treated with PBS (analysis of variance, followed by Bonferroni's test). In D survival is expressed as per cent survival and was determined daily for 7 days (P<0.001 Mantelcox log rang test).
In an attempt to explain why the aminoguanidine pre-treatment prevented the failure of neutrophil migration at 4 h after the bacterial injection, but did not prevent the death of the L-group animals, we analysed the effect of aminoguanidine pre-treatment on the neutrophil migration failure in the L-group at later time points. This analysis is supported by the fact that aminoguanidine has a short half-life (Bowman et al., 1996). It was observed that aminoguanidine at a dose of 30 mg Kg−1, administered in a single dose, did not affect the failure observed 12 h later. Consistent with the lack of effect on neutrophil migration failure, this scheme of aminoguanidine treatment did not prevent bacterial proliferation in the peritoneal exudate nor the bacteremia at this time point. However, when a second small dose (5 mg Kg−1) of aminoguanidine was administered 6 h after the lethal inoculation in the aminoguanidine pre-treated animals (pre+post), the failure of neutrophil migration was not observed at 12 h after bacterial injection (Figure 5A). This scheme of treatment also reduced the CFU in the exudate (Figure 5B), and abolished the bacteremia at this time point (Figure 5C). Consistent with these results, the pre+post scheme of aminoguanidine treatment increased the survival rate of the L-group (Figure 5D). However, under the scheme of two injections of aminoguanidine, if the second dose was 30 mg Kg−1, all the animals in the L-group died, despite the prevention of the failure of neutrophil migration (data not shown in the figure). Later, we investigated the effect of the treatment of the SL-group, which did not present failure of neutrophil migration, with two different schemes of aminoguanidine doses: (a) 30 mg Kg−1, 30 min before SL inoculation (pre), and (b) 30 mg Kg−1, 30 min before SL inoculation plus 30 mg Kg−1, 2 h after SL inoculation (pre+post). As shown in Figure 6A the neutrophil migration into the peritoneal cavity of the SL-group was unaffected by both aminoguanidine treatments. However, contrary to the result obtained with a single dose of aminoguanidine (pre-treatment, pre) that also did not affect the growth of the bacteria in the exudate or the blood of SL-group, the scheme of two injections of 30 mg Kg−1 of aminoguanidine (pre+post) caused an increase in the CFU in the peritoneal exudate and of bacteremia (Figure 6B,C). Consistent with these results, the scheme of two doses of aminoguanidine reduced the survival rate in the SL-group (Figure 6D).
Figure 5.
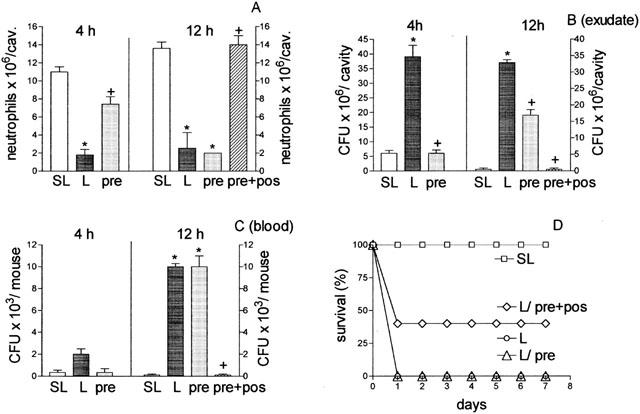
Effect in mice injected with a lethal inoculum of S. aureus of different schemes of aminoguanidine treatment on neutrophil migration into the peritoneal cavity (A) CFU in exudate (B), and in the sera (C), and on the survival of the animals (D). Lethal (L, 16×109 CFU/cavity) inoculated mice were pre-treated with PBS or with 30 mg Kg−1 of aminoguanidine, subcutaneously, 30 min before bacterial inoculation (pre) or 30 mg Kg−1 aminoguanidine, subcutaneously, 30 min before bacterial inoculation and 5 mg Kg−1, 6 h after (pre+post). The mice inoculated with the sub-lethal dose (SL, 0.5×109 CFU/cavity) were pre-treated with PBS. Neutrophil migration into the peritoneal cavity (A) CFU in exudate (B) and in the blood (C) were determined 4 and 12 h after bacterial inoculation. Results are expressed as mean±s.e.+P<0.001 compared with sublethal group. #P<0.05 compared with lethal group treated with PBS (analysis of variance, followed by Bonferroni's test). In D survival of the animals is expressed as per cent and was determined daily for 7 days for all experimental groups (P<0.001 Mantel-cox log rang test). Results are representative of two independent experiments (n=4).
Figure 6.
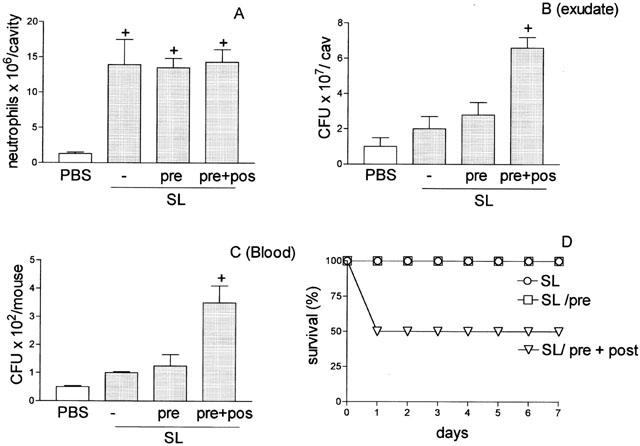
Effect of aminoguanidine pre-treatment of mice injected with sub-lethal dose of S. aureus on neutrophil migration into the peritoneal cavity (A), CFU in the exudate (B) and in the blood (C), and on survival of the animals (D). Sub-lethal (0.5×109 CFU cavity−1) (SL) inoculated mice were pre-treated with PBS (−) or 30 mg Kg−1 of aminoguanidine, subcutaneously, 30 min before bacterial inoculation (pre) or 30 mg Kg−1 aminoguanidine, subcutaneously, 30 min before and 2 h after bacterial inoculation (pre+post). Neutrophil migration into the peritoneal cavity (A), CFU in the exudate (B) and in blood (C) were determined 4 h after bacterial inoculation. Results are expressed as mean±s.e. and are representative of three independent experiments (n=4). *P<0.05 compared with animals pre-treated with PBS. +P<0.01 when compared with SL-group. #P<0.01 when compared with L-group after pre-treatment (pre) (B and C) (analysis of variance, followed by Bonferroni's test). In (D) survival is expressed as % and was determined daily for 7 days (P<0.001 Mantel-cox log rang test).
Later, we investigated the pattern of neutrophil migration and the presence of bacteria in the peritoneal fluids and in the blood of animals genetically deficient in inducible NOS (iNOS−/−) after injection with S. aureus. The neutrophil migration determined 4 and 12 h after the injection of a sublethal inoculum of S. aureus was similar in iNOS+/+ and iNOS−/− mice (Figure 7A). However, contrary to the result obtained in iNOS+/+, the iNOS−/− presented a higher number of CFU in the exudate harvested 12 h after SL-injection (Figure 7B). Consistent with the higher level of bacteria in the exudate, the iNOS−/− mice injected with SL- inoculum exhibited higher mortality when compared to their respective controls. The iNOS−/− injected with S. aureus lethal inoculum already presented a higher mortality 6 h after bacterial inoculation, whereas the iNOS+/+ exhibited significant lethality only 24 h after (Figure 7C). We did not determine the neutrophil migration and the number of bacteria in the iNOS−/− lethal group because they died within the first hour after bacterial inoculation after presenting a rapid onset of more severe clinical signs such as piloerection, lethargy and tachypnea.
Figure 7.
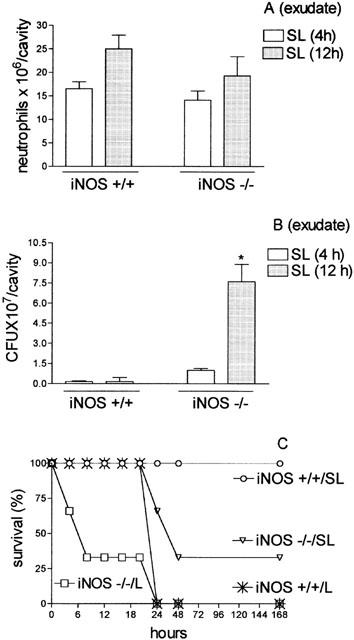
Quantification of neutrophil migration into the peritoneal cavity (A), CFU in the peritoneal exudate (B) and survival of iNOS Knockout mice (C) injected with sub-lethal or lethal inoculums of S. aureus. Groups of control animals (iNOS+/+) or iNOS−/− received a sub-lethal (SL, 0.5×109 CFU/cavity) i.p. inoculation of S. aureus. In (A) neutrophil migration into the peritoneal cavity was quantified 4 (SL or L) and 12 h (SL 12 h) after bacterial inoculation. Results are expressed as mean±s.e. neutrophils per cavity. In (B) exudate was collected 4 or 12 h after bacterial injection and the CFU in the exudate were assessed by dilution and spread-plating followed by counting 18 h later. Results are expressed as mean±s.e. CFU per cavity (exudate). *P<0.01 compared with iNOS−/− SL-inoculated mice (analysis of variance, followed by Bonferroni's test). In (D) survival of the animals is expressed as per cent survival and was determined daily for 7 days.
In vitro neutrophil migration
Chemotaxis to fMLP and LTB4 was investigated in blood neutrophils from PBS-, SL- and L-groups and in the L-group pre-treated with aminoguanidine. As shown in Figure 8 the neutrophil chemotaxis of L-group induced by fMLP and LTB4 was significantly reduced, when compared with chemotaxis observed in neutrophils from the control group (PBS), or from the SL-group. The reduction of chemotaxis in the lethal group was prevented by the pre-treatment of the animals with aminoguanidine (30 mg Kg−1), 30 min before lethal inoculation (Figure 8).
Figure 8.
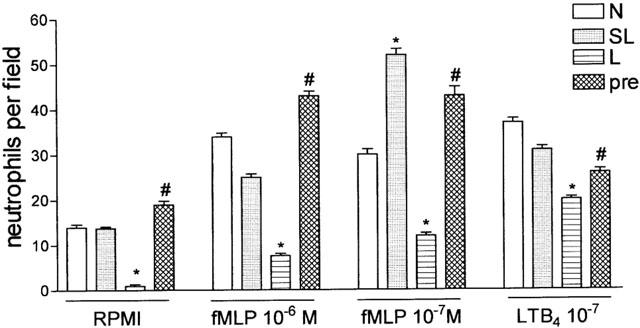
In vitro migration of neutrophils obtained from normal, sub-lethal and lethal inoculated mice pre-treated with PBS or with aminoguanidine. Blood neutrophils were collected and purified from normal mice (N), sub-lethal (SL, 0.5×109 CFU/animal), or lethal (L, 16×109 CFU/animal) inoculated mice pre-treated with PBS or with aminoguanidine (pre, 30 mg Kg−1, s.c., 30 min. before lethal inoculation). Neutrophils (5×104/well) were assayed for fMLP- and LTB4-induced chemotaxis (see Methods). Results are expressed as mean±s.e. neutrophils per field and are representative of two independent experiments (n=4). *P<0.001 compared with sub-lethal inoculation. #P<0.001 compared with lethal inoculation pre-treated with PBS (analysis of variance, followed by Bonferroni's test).
Discussion
S. aureus is the leading cause of Gram-positive bacterial sepsis in humans and the fact that some S. aureus strains are becoming resistant to specific antibiotics indicates the importance of the understanding the pathogenesis of sepsis induced by this bacterium (Bannan et al., 1999; Cohen, 2000). Lethal outcome in septicaemic shock is in general thought to be associated with cardiovascular collapse and organ failure resultant of an intense systemic inflammatory response syndrome (SIRS; Walley et al., 1996). In this context it has recently been suggested that the failure of neutrophil defence mechanisms may be a major factor responsible for the host's failure to restrict the pathogen to a localized area, allowing spreading of the micro-organism, culminating in SIRS, septicaemic shock and death (Benjamim et al., 2000). In the present study, we demonstrated that the septicaemic lethal outcome after S. aureus injection was associated with the failure of neutrophil migration to the infectious site. The mortality rate depended on the number of bacteria injected. The lethal group (16×109 CFU/cavity) was characterized by the failure of neutrophil migration and a high number of bacteria in the peritoneal cavity and circulation. The incapacity of the L-group animals to restrict the bacteria to the infected site was not due to a reduction in emigrated neutrophil phagocytic capacity, since the phagocytic index in this group was even higher than that observed in SL-group. Despite the fact that NO reduces the phagocytic capacity of neutrophils in vitro (Forslund & Sundqvist, 1997), it does not affect such capacity in S. aureus injected mice. The phagocytic index in the L-group, in which NO production seems to be elevated, was found to be higher than in the SL-group. Moreover, the inhibition of NO production by the treatment of the animals with aminoguanidine did not affect the phagocytic index.
Failure of neutrophil migration has been also described in Gram-negative septicaemia induced by E. coli endotoxin or by CLP surgery, a polymicrobial model with a predominance of Gram-negative bacteria (Baker et al., 1983; Rocha & Ferreira, 1986; Tavares-Murta et al., 1998; Benjamim et al., 2000). Thus, despite different etiological agents, the severity of sepsis in animal models correlates well with the degree of neutrophil migration impairment. The failure of neutrophil migration into the peritoneal cavity in the lethal group was not due to neutropenia because, as shown here, levels of circulating neutrophils in these animals are higher than those observed in PBS- and SL-groups. Haemodynamic changes also cannot be responsible for this failure, since the blood pressure of animals in the L-group was similar to that observed in the SL-group. Another cause of migration failure could be a deficiency in the release of chemotactic cytokines, including TNF-α and IL-1β, in the inflammatory focus (Dinarello, 1997; Oberzolzer et al., 2000). There was no difference, however, in the amounts of TNF-α, IL-1β and IL-10 in the exudates of L- and SL- groups, ruling out the hypothesis that the failure of neutrophil migration observed in L-group is due to a reduction in IL-1β and TNF-α production at the infectious focus. However, the results do not exclude the possibility that the failure of neutrophil migration is due to a reduction in the release of other neutrophil chemotactic mediators, such as the CXC chemokines and leukotriene B4 (Murdoch & Finn, 2000; Canetti et al., 2001).
During an infection the high production of pro-inflammatory cytokines in the circulation may result in bacterial dissemination, hypotension and multiorgan dysfunction (Walley et al., 1996). Previous studies have demonstrated that the impairment of neutrophil migration observed in endotoxemic animals is mediated by systemic release of the cytokines TNF-α, IL-8, or macrophage-derived neutrophil chemotactic factor (MNCF). Intravenous administration of these cytokines inhibits the neutrophil migration induced by inflammatory stimuli in normal animals. On the other hand, in endotoxemic animals, intravenous administration of antibodies against TNF-α partially restores neutrophil migration failure (Tavares-Murta et al., 1998). In the present study, however, neither the circulating levels of TNF-α nor IL-10 could be solely responsible for the migration failure in L-group, since serum concentrations of these molecules were similar in L- and SL- groups. On the other hand, the serum concentration of IL-1β, a cytokine apparently devoid of inhibitory effect on cell migration, was significantly higher in animals from the L- compared to the SL-group. This finding stresses the importance of investigating the involvement of IL-1β in the process. In this context, the ability of IL-1β to induce iNOS is well recognized (Cunha et al., 1994) and, as discussed below, NO mediates the neutrophil migration failure. Although IL-10 does not mediate the failure of neutrophil migration observed in the L-group, the fact that aminoguanidine treatment caused an increase in its circulating levels suggests that IL-10 could mediate, at least in part, the protective effect of aminoguanidine treatment. This conclusion is supported by the fact that a specific antibody against IL-10 reversed the protective effect of treatment with the NOS inhibitor, L-NAME, in mice that had undergone lethal CLP. It was also reported that the treatment of CLP animals with L-NAME increased the CLP-induced release of IL-10 (Hogaboam et al., 1998). Moreover, IL-10 has been described as an anti-inflammatory cytokine (Oberzolzer et al., 2002).
The failure of neutrophil migration induced by intravenous administration of TNF-α, IL-8 or MNCF, as well as that observed in endotoxemia or in CLP-sepsis, is mediated by NO. The impairment of neutrophil migration observed in these experimental conditions was prevented by selective iNOS inhibitors and adequate doses of aminoguanidine prevented animal death in the CLP model (Tavares-Murta et al., 1998; Benjamim et al., 2000; Tavares-Murta et al., 2001). In the present series of experiments the pre-treatment of L-group with a single dose of aminoguanidine prevented the neutrophil migration failure in a dose-dependent manner. Thus, the failure of neutrophil migration observed 4 h after S. aureus inoculation was also mediated by NO. In a previous study, it was observed that the doses of aminoguanidine used here are able to inhibit NO production. Doses of 30 mg Kg−1 of this drug inhibited the LPS-induced increase in NO production by more than 90% (Tavares-Murta et al., 2001). Failure of neutrophil migration 12 h after bacterial inoculation, which was not prevented by a single dose of aminoguanidine due to its short half-life (2 h; Bowman et al., 1996), could be prevented by an additional administration of aminoguanidine (5 mg Kg−1) 6 h after bacterial injection. In such instance prevention of neutrophil migration impairment was accompanied by a decreased number of bacteria in the peritoneal fluid and in blood as well as an improved survival rate.
The reduction of neutrophil migration in sepsis could be a consequence of the reduction in neutrophil chemotaxis and/or adhesion to the endothelium. We confirmed that the chemotactic ability of the neutrophils is reduced and that this is mediated by NO during sepsis, since L-group derived neutrophils presented a significant reduction in chemotaxis induced in vitro by fMLP and LTB4, which was avoided by pre-treatment of the animals with aminoguanidine. This reduction in chemotaxis is in line with the earlier observation of lower expression of receptors for fMLP and C5a in neutrophils incubated with S. aureus supernatants (Veldkamp et al., 2000). Failure of neutrophil migration in vitro has also been demonstrated in septic patients and was correlated with the outcome of sepsis (Tavares-Murta et al., 2002). The fact that the treatment of animals with aminoguanidine restored the chemotactic ability of the neutrophils suggests that NO interferes with the chemotactic apparatus of the neutrophil. In fact, it has been shown that NO influences ADP-ribosylation of actin monomers in neutrophils and thereby inhibits polymerisation into F-actin (Rochon & Frojmovic, 1992; Granger & Kubes, 1994; Forslund et al., 2000). Furthermore, NO inhibits the function of β2 integrins with a consequent reduction in neutrophil adhesion mediated by this group of molecules (Banick et al., 1997, Benjamim et al., 2002).
Our results, however, do not exclude the possibility that the adhesion of neutrophils to endothelial cells is also reduced in S. aureus-induced sepsis. In fact, this may well be occurring since a reduction in leukocyte rolling and adhesion has been seen in post-capillary venules of the mesentery during CLP-sepsis; this reduction was avoided by pre-treatment of the animals with an iNOS inhibitor and did not occur in iNOS−/− mice (Benjamim et al., 2002). In vivo and in vitro observations of the effects of NOS inhibitors describe an increase in neutrophil rolling on and, later, adhesion to endothelial cells (Hickey et al., 1997; Hickey & Kubes, 1997; Hickey, 2001). Delivery of exogenous NO inhibits the leukocyte adhesion to endothelial cells stimulated by inflammatory mediators, and leukocyte transmigration to inflammatory sites (Sundqvist et al., 1994; Lopez-Neblina et al., 1996, Guidot et al., 1996; Kosonen et al., 1999; Iatenti et al., 2000). It is interesting to note that accumulation of neutrophils in the lung seems to be independent of neutrophil rolling (Wickel et al., 1998), and it occurs during experimental sepsis as well as in septic patients (Ware & Matthay, 2000; Nagase et al., 2000); in addition, this may constitute an event which further contributes to the lethal outcome. In the present investigation, there was clear neutrophil accumulation and oedema in the pulmonary tissue of the L-group despite the inhibition of neutrophil migration to the infectious focus.
It should be pointed out that in our experiments, when a rather high additional dose of aminoguanidine (30 mg Kg−1) was used, all the animals died despite the prevention of neutrophil migration failure. Thus, intense and prolonged iNOS inhibition may abolish the microbicidal activity of emigrated neutrophils, thereby aggravating the infection due to bacterial proliferation. In this study, the importance of NO microbicidal activity was further emphasized by the fact that in animals from the SL-group treated with 2 doses of aminoguanidine (30 mg Kg−1+30 mg Kg−1) neutrophil migration was preserved but high mortality still occurred due to disseminated infection (increased number of CFU in peritoneal fluids and blood). This high mortality with increased CFU in the exudate also occurred in iNOS−/− mice injected with a SL-inoculum of S. aureus, in spite of normal neutrophil migration to the peritoneal cavity. This latter result thus confirms the importance for neutrophil microbicidal activity of the activation of the inducible isoform of NOS (Kaplan et al., 1996; Fierro et al., 1999). NO microbicidal activity of host cells, including neutrophils, is well established (Sakiniene et al., 1997; Fierro et al., 1999). The relevance of this protective mechanism is illustrated in our experiments by the very early death (within a few hours) of lethal inoculated iNOS−/− mice.
Failure of neutrophil migration has also been described in other diseases, including diabetes, cirrhosis and acquired immunological deficiency syndrome (AIDS), all pathologies associated with high susceptibility to infection (Pereira et al., 1987; Ellis et al., 1998; Fiuza et al., 2000). However, it has not yet been investigated whether NO mediates the observed failure of neutrophil migration in these diseases.
In conclusion, we have demonstrated that NO produced by iNOS has a dual effect in Gram-positive S. aureus sepsis. On the one hand, it is an important mediator of the microbicidal activity of neutrophils present at the infectious site. However, on the other hand, it also impairs the recruitment of neutrophils towards the infectious site. Therapeutic strategies should be developed to specifically prevent the inhibitory effect of NO on neutrophil migration, whilst maintaining its contribution to the microbicidal activity of the leukocytes. The present investigation should also stimulate further clinical studies on the role of NO in neutrophil chemotaxis and bactericidal activity in human sepsis.
Acknowledgments
We thank Jamilson Conceição Alves, Fabiola Leslie Mestriner, Giuliana Bertozi, Ana Katia dos Santos, and Heleni Tamburus for technical assistance. Financial support received from CNPq, FAPESP, PRONEX, Brazil.
Abbreviations
- AIDS
acquired immunological deficiency syndrome
- BHI
brain heart infusion
- BSA
bovine serum albumin
- CFU
colony forming units
- CLP
caecal ligature and puncture
- fMLP
formyl-Met-Leu-Phe
- IFN-γ
interferon-γ
- IL-1β
interleukin 1β
- iNOS
inducible nitric oxide synthase
- iNOS−/−
genetically deficient in inducible NOS
- i.p.
intraperitoneal
- L-NMMA
L-NG-monomethyl arginine
- LTB4
leukotriene B4
- MNCF
macrophage-derived neutrophil chemotactic factor
- NO
nitric oxide
- PBS
phosphate buffered saline
- PMN
polymorphonuclear cells
- S. aureus
Staphylococcus aureus
- SIRS
systemic inflammatory response syndrome
- TNF-α
tumour necrosis factor alpha
References
- AJUEBOR M.N., VIRÀG I., FLOWER R.J., PERRETTI M., SZABO C. Role of inducible nitric oxide synthase in the regulation of neutrophil migration in zymosan-induced inflammation. Immunology. 1998;95:625–630. doi: 10.1046/j.1365-2567.1998.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAKER C.C., CHAUDRY I.H., GAINES H.O., BAUE A.E. Evaluation of factors affecting mortality rate after sepsis in a murine caecal ligation and puncture model. Surgery. 1983;94:331–335. [PubMed] [Google Scholar]
- BANNAN J., VISVANATHAN K., ZABRIESKIE J.B. Structure and function of streptococcal and staphylococcal superantigens in septic shock. Infect. Dis. Clin. North Am. 1999;13:387–396. doi: 10.1016/s0891-5520(05)70081-7. [DOI] [PubMed] [Google Scholar]
- BANICK P.D., CHEN Q., XU Y.A., THOM S.R. Nitric oxide inhibits neutrophil β2 integrin function by inhibition of membrane-associated cyclic GMP synthesis. J. Cell. Physiol. 1997;172:12–24. doi: 10.1002/(SICI)1097-4652(199707)172:1<12::AID-JCP2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- BENJAMIM C.F., FERREIRA S.H., CUNHA F.Q. Role of nitric oxide in the failure of neutrophil migration in sepsis. J. Infect. Dis. 2000;182:214–223. doi: 10.1086/315682. [DOI] [PubMed] [Google Scholar]
- BENJAMIM C.F., SILVA J.S., FORTES Z.B., OLIVEIRA M.A., FERREIRA S.H., CUNHA F.Q. Inhibition of leukocyte rolling by nitric oxide during sepsis leads to reduced migration of microbicidal-active neutrophils. Infec. Immun. 2002;70:1–9. doi: 10.1128/IAI.70.7.3602-3610.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHAGAT K., HINGORANI A.D., PALACIOS M., CHARLES I.G., VALLANCE P. Cytokine-induced venodilatation in human in vivo: eNOS masquerading as iNOS. Cardiovascular Research. 1999;41:754–764. doi: 10.1016/s0008-6363(98)00249-1. [DOI] [PubMed] [Google Scholar]
- BONE R.C., GRODZIN C.J., BALK R.A. Sepsis: a new hypothesis for pathogenesis of the disease process. Chest. 1997;112:235–243. doi: 10.1378/chest.112.1.235. [DOI] [PubMed] [Google Scholar]
- BOWMAN M.A., SIMELL O.G., PECK A.B., CORNELIUS J., LUCHETTA R., LOOK Z., MACLAREN N.K., ATKINSON M.A. Pharmacokinetics of aminoguanidine administration and effects on the diabetes frequency in nonobese diabetic mice. J. Pharmacol. Exp. Ther. 1996;279:790–794. [PubMed] [Google Scholar]
- BROEDERS M.A., TANGELDER G.J., SLAAF D.W., RENEMAN R.S., EGBRINK M.G. Endogenous nitric oxide and prostaglandins synergistically counteract thromboembolism in arterioles but not in venules. Arterioscler. Thromb. Vasc. Biol. 2001;21:163–169. doi: 10.1161/01.atv.21.1.163. [DOI] [PubMed] [Google Scholar]
- CANETTI C., SILVA J.S., FERREIRA S.H., CUNHA F.Q. Tumor necrosis factor-alpha and leukotriene B4 mediate the neutrophil migration in immune inflammation. Br. J. Pharmacol. 2001;134:1619–1628. doi: 10.1038/sj.bjp.0704403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN M. Changing patterns of infectious disease. Nature. 2000;406:762–767. doi: 10.1038/35021206. [DOI] [PubMed] [Google Scholar]
- CONLAN J.W. Critical roles of neutrophils in host defense against experimental systemic infection of mice by Listeria monocytogenes, Salmonella typhimurium, and Yersinia enterocolitica. Infect. Immun. 1997;65:630–635. doi: 10.1128/iai.65.2.630-635.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNHA F.Q., ASSREUY J., MOSS D.W., REES D., LEAL L.M., MONCADA S., CARRIER M., O'DONNELL C.A., LIEW F.Y. Differential induction of nitric oxide synthase in various organs of the mouse during endotoxemia: role of TNF-α and IL-1β. Immunology. 1994;81:211–215. [PMC free article] [PubMed] [Google Scholar]
- CUZZOCREA S., MAZZON E., CALABRO G., DUGO L., DE SARRO A., VAN DE LOO F.A.J., CAPUTI A.P. Inducible nitric oxide synthase-knockout mice exhibit resistance to pleurisy and lung injury caused by carrageenan. Am. J. Respir. Care Med. 2000;162:1859–1866. doi: 10.1164/ajrccm.162.5.9912125. [DOI] [PubMed] [Google Scholar]
- DINARELLO P.A. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest. 1997;112:321S–329S. doi: 10.1378/chest.112.6_supplement.321s. [DOI] [PubMed] [Google Scholar]
- ELLIS M., GRUPTA S., GALANT S., HAKIN S., VANDEVEN C., TOY C., CAIRO M.S. Impaired neutrophil function in patients with AIDS or AIDS-related complex: a comprehensive evaluation. J. Infect. Dis. 1998;158:1268. doi: 10.1093/infdis/158.6.1268. [DOI] [PubMed] [Google Scholar]
- EVANS C.H., STEFANOVIC-RACIC M. Nitric oxide in arthritis. Methods. 1996;10:38–42. doi: 10.1006/meth.1996.0076. [DOI] [PubMed] [Google Scholar]
- FIERRO I.M., NASCIMENTO-DA SILVA V., ARRUDA M.A.B., FREITAS M.S., PLOTKOWSKI M.N., CUNHA F.Q., BARJA-FIDALGO C. Induction of NOS in rat blood PMN: in vivo and in vitro modulation by tyrosine kinase and involvement in bactericidal activity. J. Leuk. Biol. 1999;65:508–514. doi: 10.1002/jlb.65.4.508. [DOI] [PubMed] [Google Scholar]
- FIUZA C., SALCEDO M., CLEMENT G., TELLADO J.M. In vivo neutrophil dysfunction in cirrhotic patients with advanced liver disease. J. Infect. Dis. 2000;182:526–533. doi: 10.1086/315742. [DOI] [PubMed] [Google Scholar]
- FRANCHINI K.G., TORSINI A.S., SOARES H.A., SAAD M.J.A. Early activation of the multicomponent signaling complex associated with focal adhesion kinase induced by pressure overload in rat heart. Circ. Res. 2000;87:558–565. doi: 10.1161/01.res.87.7.558. [DOI] [PubMed] [Google Scholar]
- FRANCO-PENTEADO C.F., DESOUZA I., TEIXEIRA S.A., RIBEIRO-DASILVA G., DENUCCI G., ANTUNES E. Role of nitric oxide on the increased vascular permeability and neutrophil accumulation induced by staphylococcal enterotoxin B into the mouse paw. Biochem. Pharmacol. 2001;61:1305–1311. doi: 10.1016/s0006-2952(01)00573-1. [DOI] [PubMed] [Google Scholar]
- FORSLUND T., NILSSON H.M., SUNDQVIST T. Nitric oxide regulates the aggregation of stimulated human neutrophils. Biochem. Biophys. Res. Comm. 2000;274:482–487. doi: 10.1006/bbrc.2000.3156. [DOI] [PubMed] [Google Scholar]
- FORSLUND T., SUNDQVIST T. Nitric oxide-releasing particles inhibit phagocytosis in human neutrophils. Biochem. Biophys. Res. Com. 1997;233:492–495. doi: 10.1006/bbrc.1997.6490. [DOI] [PubMed] [Google Scholar]
- GALANOS C., FREUDENBERG M.A. Mechanisms of endotoxin shock and endotoxin hypersensitivity. Immunobiology. 1993;187:346–356. doi: 10.1016/S0171-2985(11)80349-9. [DOI] [PubMed] [Google Scholar]
- GRANGER D.N., KUBES P. The microcirculation and inflammation: modulation of leukocyte-endothelial cell adhesion. J. Leukocyte Biol. 1994;55:662–675. [PubMed] [Google Scholar]
- GUIDOT D.M., HYBERTSON B.M., KITLOWSKI R.P., REPINE J.E.Inhaled NO prevents IL-1-induced neutrophil accumulation and associated acute edema in isolated rat lungs Am. J. Physiol. 19962L225–L229.Pt 1 [DOI] [PubMed] [Google Scholar]
- HICKEY M.J. Role of inducible nitric oxide synthase in the regulation of leukocyte recruitment. Clinical Science. 2001;100:1–12. [PubMed] [Google Scholar]
- HICKEY M.J., KUBES P. Role of nitric oxide in regulation of leukocyte-endothelial cell interactions. Exp. Physiol. 1997;82:339–348. doi: 10.1113/expphysiol.1997.sp004029. [DOI] [PubMed] [Google Scholar]
- HICKEY M.J., SHARKEY K.A., SIHOTA E.G., REINHARDT P.H., MACMICKING J.D., NATHAN C., KUBES P. Inducible nitric oxide synthase-deficient mice have enhanced leukocyte-endothelium interactions in endotoxemia. FASEB J. 1997;11:955–964. doi: 10.1096/fasebj.11.12.9337148. [DOI] [PubMed] [Google Scholar]
- HINSHAW L.B., EMERSON T.E., JR, TAYLOR F.B., JR, CHANG A.C., DUERR M., PEER G.T., FLOURNOY D.J., WHITE G.L., KOSANKE S.D., MURRAY C.K. Lethal Staphylococcus aureus-induced shock in primates: prevention of death with anti-TNF antibody. J. Trauma. 1992;33:568–573. [PubMed] [Google Scholar]
- HINSHAW L.B., EMERSON T.E., JR, TAYLOR F.B., JR, CHANG A.C., DUERR M., PEER G.T., FLOURNOY D.J., WHITE G.L., KOSANKE S.D., MURRAY C.K. Mice deficient in IL-1beta converting enzyme are deficient in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- HOEHN T., KRAUSE M.F. Response to inhaled oxide in premature and term neonates. Drugs. 2001;61:27–39. doi: 10.2165/00003495-200161010-00004. [DOI] [PubMed] [Google Scholar]
- HOGABOAM C.M., STEINHAUSER M.L., SCHOCK H., LUKACS N., STRIETER R.M., STANDIFORD T., KUNKEL S.T. Therapeutic effects of nitric oxide inhibition during experimental fecal peritonitis: role of interleukin-10 and monocyte chemoattractant protein 1. Infect. Immun. 1998;2:650–655. doi: 10.1128/iai.66.2.650-655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IALENTI A., IANARO A., MAFFIA P., DI ROSA M. Nitric oxide inhibits leucocyte migration in carragenin-induced rat pleurisy. Inflamm. Res. 2000;8:411–417. doi: 10.1007/s000110050609. [DOI] [PubMed] [Google Scholar]
- INOUE R.Y., GONTIJO J.A.R., FRANCHINI K.G. Hemodilution mediates hemodynamic changes during acute expansion in unanesthetized rats. Am. J. Reg. Inter. Comp. Physiol. 2000;279:R2243–R2251. doi: 10.1152/ajpregu.2000.279.6.R2243. [DOI] [PubMed] [Google Scholar]
- KAPLAN S.S., LANCASTER J.R., JR, BRASFORD R.E., SIMMONS R.L. Effect of nitric oxide on staphylococcal killing and interactive effect with superoxide. Infect. Immun. 1996;64:69–76. doi: 10.1128/iai.64.1.69-76.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOSONEN O., KANKAANRANTA H., MALO-RANTA M., MOILANEN E. Nitric oxide-releasing compounds inhibit neutrophil adhesion to endothelial cells. Eur. J. Pharmacol. 1999;2:111–117. doi: 10.1016/s0014-2999(99)00581-6. [DOI] [PubMed] [Google Scholar]
- LELAMALY K., WANG W., GENGARO P., EDELSTEIN C., SCHRIEIR R.W. Effects of nitric oxide and peroxynitrite on endotoxin-induced leukocyte adhesion to the endothelium. J. Cell Physiol. 2001;3:337–342. doi: 10.1002/jcp.1128. [DOI] [PubMed] [Google Scholar]
- LOPEZ-NEBLINA F., TOLEDO-PEREYRA L.H., MIRMIRAN R., PAEZ-ROLLYS A.J. Time dependence of Na-nitroprusside administration in the prevention of neutrophil infiltration in the rat ischemic kidney. Transplantation. 1996;2:179–183. doi: 10.1097/00007890-199601270-00002. [DOI] [PubMed] [Google Scholar]
- LYNN W.A., COHEN J. Adjunctive therapy for septic shock: a review of experimental approaches. Clin. Infect. Dis. 1995;20:143–158. doi: 10.1093/clinids/20.1.143. [DOI] [PubMed] [Google Scholar]
- McCARTNEY-FRANCIS N.L., SONG X-Y., MIZEL D.E., WAHL S.M. Selective inhibition of inducible nitric oxide synthase exacerbates erosive joint disease. J. Immunol. 2001;166:2734–2740. doi: 10.4049/jimmunol.166.4.2734. [DOI] [PubMed] [Google Scholar]
- MARTINEAU L., SHEK P.N. Peritoneal cytokine concentration and survival outcome in an experimental bacterial infusion model of peritonitis. Crit. Care Med. 2000;28:788–794. doi: 10.1097/00003246-200003000-00030. [DOI] [PubMed] [Google Scholar]
- MURDOCH C., FINN A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;10:3032–3043. [PubMed] [Google Scholar]
- NAGASE T., UOZUMI N., ISHII S., KUME K., IZUMI T., OUCHI Y., SHIMIZU T. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat. Immunol. 2000;1:42–46. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- NUMATA M., SUZUKI S., MIYAZAWA N., MIYASHITA A., NAGASHIMA Y., INOUE S., KANEKO T., OKUBO T. Inhibition of inducible nitric oxide synthase prevents LPS-induced acute lung injury in dogs. J. Immunol. 1998;160:3031–3037. [PubMed] [Google Scholar]
- OBERZOLZER A., OBERZOLZER C., MOLDAWER L.L. Cytokine signaling-regulation of the immune response in normal and critically ill patients. Crit. Care Med. 2000;28:N3–N12. doi: 10.1097/00003246-200004001-00002. [DOI] [PubMed] [Google Scholar]
- OBERZOLZER A., OBERZOLZER C., MOLDAWER L.L. Interleukin-10: a complex role in the pathpgenesis of sepsis syndromes and its potencial as an anti-inflammatory drug. Crit. Care Med. 2002;30 suppl 1:58–63. [PubMed] [Google Scholar]
- PAUL-CLARK M.J., GILROY D.W., WILLIS D., WILLOUGHBY D.A., TOMLINSON A. Nitric oxide synthase inhibitors have opposite effects on acute inflammation depending on their route of administration. J. Immunol. 2001;2:1169–1177. doi: 10.4049/jimmunol.166.2.1169. [DOI] [PubMed] [Google Scholar]
- PEREIRA M.A.A., SANNOMYA P., LEME J.G. Inhibition of leukocyte chemotaxis by factor in alloxan-induced diabetic rat plasma. Diabetes. 1987;36:1307–1314. doi: 10.2337/diab.36.11.1307. [DOI] [PubMed] [Google Scholar]
- PETROS A., LAMB G., LEONE A., MONCADA S., BENNETT D., VALLANCE P. Effects of a nitric oxide synthase inhibitor in humans with septic shock. Cardiovasc. Res. 1994;28:34–39. doi: 10.1093/cvr/28.1.34. [DOI] [PubMed] [Google Scholar]
- ROCHA N.P., FERREIRA S.H. Restoration by levamisole of endotoxin-inhibited neutrophil migration, oedema and increased permeability induced by carrageenin. Eur. J. Pharmacol. 1986;122:87–92. doi: 10.1016/0014-2999(86)90162-7. [DOI] [PubMed] [Google Scholar]
- ROCHON Y.P., FROJMOVIC M.M. A model for the recruitment of neutrophils at sites of inflammation. Physiological relevance of in vivo neutrophil aggregation. Med. Hypoth. 1992;38:132–138. doi: 10.1016/0306-9877(92)90086-r. [DOI] [PubMed] [Google Scholar]
- SAKINIENE E., BREMELL T., TARKOWISK A. Inhibition of nitric oxide synthase (NOS) aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin. Exp. Immunol. 1997;110:370–377. doi: 10.1046/j.1365-2249.1997.4431456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANDS K.E., BATES D.W., LANKEN P.N., GRAMAN P.S., HIBBERD P.L., KAHN K.L., PARSONNET J., PANZER R., ORAV E.J., SNYDMAN D.R. Epidemiology of sepsis syndrome in 8 academic medical centers. JAMA. 1997;278:234–240. [PubMed] [Google Scholar]
- SILVA-SANTOS J.E., ASSREUY J. Long-lasting changes of rat blood pressure to vasoconstrictors and vasodilators induced by nitric oxide donor infusion: involvement of potassium channels. J. Pharmacol. Exp. Therap. 1999;290:380–387. [PubMed] [Google Scholar]
- SRISKANDAN S., COHEN J. Gram-positive sepsis. Mechanisms and differences from gram-negative sepsis. Infect. Dis. North Am. 1999;13:397–412. doi: 10.1016/s0891-5520(05)70082-9. [DOI] [PubMed] [Google Scholar]
- SUNDQVIST T., FORSLUND T., BENGTSSON T., AXESSON K.L. S-nitroso-N-acetylpenicillamine reduces leukocyte adhesion to type I collagen. Inflammation. 1994;6:625–631. doi: 10.1007/BF01535260. [DOI] [PubMed] [Google Scholar]
- TAVARES-MURTA B.M., CUNHA F.Q., FERREIRA S.H. The intravenous administration of tumor necrosis factor-alpha, interleukin-8 and macrophage-derived neutrophil chemotactic factor inhibits neutrophil migration by stimulating nitric oxide production. Br. J. Pharmacol. 1998;124:1369–1374. doi: 10.1038/sj.bjp.0701965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAVARES-MURTA B.M., MACHADO J.S., FERREIRA S.H., CUNHA F.Q. Nitric oxide mediates the inhibition of neutrophil migration induced by systemic administration of LPS. Crit. Care Med. 2001;30:1–6. doi: 10.1023/a:1010927921018. [DOI] [PubMed] [Google Scholar]
- TAVARES-MURTA B.M., ZAPAROLLI M., FERREIRA R.B., VERGARA M.L.S., CUNHA F.Q., FERREIRA S.H. Failure of neutrophil migration chemotactic function in septic patients. Inflammation. 2002;4:247–253. doi: 10.1097/00003246-200205000-00017. [DOI] [PubMed] [Google Scholar]
- VELDKAMP K., HEEZIUS H.C.J.M., VERHOEF J., VAN STRIJP J.A.G., VAN KESSEL K.P.M. Modulation of neutrophil migration chemokine receptors by Staphylococcus aureus supernatant. Infect. Immun. 2000;68:5908–5913. doi: 10.1128/iai.68.10.5908-5913.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLEY R.K., LUKACS N.W., STANDIFORD T.J., STRIETER R.M., KUNKEL S.L. Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect. Immun. 1996;64:4733–4738. doi: 10.1128/iai.64.11.4733-4738.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARE L.B., MATTHAY M.A. The acute respiratory distress syndrome. NEJM. 2000;4:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- WICKEL D.J., MERCER-JONES M., PEYTON J.C., SHROTRI M.S., CHEADKE W.G. Neutrophil migration into peritoneum is P-selectin dependent, but sequestration in lungs is selectin independent during peritonitis. Shock. 1998;4:265–269. doi: 10.1097/00024382-199810000-00006. [DOI] [PubMed] [Google Scholar]
- WOLKOW P.P. Involvement and dual effects of nitric oxide in septic shock. Inflamm. Res. 1998;47:152–166. doi: 10.1007/s000110050309. [DOI] [PubMed] [Google Scholar]
- ZHAO Y.E., NILSSON I.M., TARKOWISKY A. The role of interferon-gamma in experimental Staphylococcus aureus septicaemia versus arthritis. Immunology. 1998;93:80–85. doi: 10.1046/j.1365-2567.1998.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]