

Abstract
The effects of ozone inhalation (90 min, 2.15±0.05 p.p.m.) and their modification by dexamethasone (20 mg kg−1) or the phosphodiesterase-4 inhibitor, rolipram (1 mg kg−1), administered (i.p.) 24 and 0.5 h before and 24 h after ozone exposure were examined in conscious guinea-pigs.
Ozone caused an early-phase bronchoconstriction (EPB) as a fall in specific airways conductance (sGaw) measured by whole body plethysmography, followed at 5 h by a late-phase bronchoconstriction (LPB) and increased respiratory rate. Rolipram did not alter this profile but dexamethasone inhibited the EPB.
Airway hyperreactivity to inhaled histamine (1 mM, 20 s) occurred at 0.5, 2, 12, 24 and 48 h after ozone inhalation, the 2 h change being abolished by rolipram and dexamethasone.
Bronchoalveolar lavage fluid (BALF) macrophages, eosinophils and neutrophils were significantly (P<0.05) elevated at 12, 24 and 48 h after ozone exposure, the 48 h influx being significantly attenuated (P<0.05) by rolipram and dexamethasone.
BALF nitric oxide (NO) metabolites decreased 0.5 h after ozone exposure by 52%, recovered at 2 h and significantly increased at 12 (101%) and 24 h (127%). The elevated NO was unaffected by rolipram or dexamethasone.
Lung oedema, measured from wet/dry weight differences, was significant 12, 24 and 48 h after ozone exposure, the latter being significantly attenuated (P<0.05) by rolipram and dexamethasone.
Ozone exposure of guinea-pigs produced features common to COPD. Although rolipram and dexamethasone did not affect the airway function changes, they inhibited the inflammation, airway hyperreactivity and oedema.
Keywords: Airway hyperreactivity, corticosteroid, leukocytes, oedema, ozone, phosphodiesterase-4 inhibitor, nitric oxide, lung function
Introduction
Oxidative stress can be defined as an increased exposure to oxidants (e.g. reactive oxygen species: ROS) and/or decreased anti-oxidant capacity, and is a widely recognized central feature of both asthma (Barnes et al., 1999) and chronic obstructive pulmonary disease (COPD) (Repine et al., 1997). Nearly 90% of all COPD patients are smokers (Repine et al., 1997). Considerable evidence now links the rich source of oxidants in cigarette smoke with pulmonary oxidative stress in COPD (Stockley, 1995; Repine et al., 1997). A release of ROS, by activated inflammatory cells in the airways, is a shared feature of both asthma and COPD. Both COPD and asthma are also associated with an exaggerated constrictor response of the airways to spasmogens (airway hyperreactivity: AHR), reduced air flow, dyspnoea (shortness of breath) and oedema (Vrugt & Aalbers, 1993). In COPD, the airway obstruction is characterized by a progressive and irreversible decline in FEV1 and an airways inflammation driven by neutrophils and macrophages (Postma & Kerstjens, 1998). In contrast, in asthma, the episodic obstruction, typically characterized by early- (EPB) and late- (LPB) phase bronchoconstrictions, is reversible by β-adrenoceptor agonists and the airways inflammation is driven by eosinophils (Bousquet et al., 2000).
Exposure of asthmatic subjects to antigen (Alving et al., 1993) and exacerbations in individuals with COPD (Bhowmik et al., 1998; Corradi et al., 1999) are associated with increases in levels of exhaled nitric oxide (NO). Derived from the nitric oxide synthase (NOS) conversion of L-arginine to L-citrulline, a basal level of the free radical, NO, maintains bronchodilatory airway tone, suppresses leukocyte activation and inhibits microvascular leakage (Barnes et al., 1999; Colasanti & Suzuki, 2000). However, excess airways NO, although having beneficial anti-bacterial and anti-viral activity, increases microvascular permeability to cause pulmonary oedema and promotes leukocyte influx and adhesion in the airways (Barnes et al., 1999). Under conditions of inflammatory-derived oxidative stress, excess NO also rapidly reacts with ROS (e.g. superoxide), to yield powerful nitrogen oxidants (e.g. peroxynitrite), which promote airway epithelial damage and AHR (Nijkamp & Folkerts, 1995; Giaid et al., 1998). Naturally occurring anti-oxidants and scavenging enzymes like superoxide dismutase, catalase and glutathione protect from such inflammatory cell-derived ROS and oxidative stress (Repine et al., 1997). In asthmatics and patients with COPD, exhaled NO also correlates with the degree of AHR, leukocyte influx and peroxynitrite formation, and negatively correlates with a decline in lung function (FEV1) (Saleh et al., 1998; Agusti et al., 1999; Ichinose et al., 2000).
Anti-inflammatory steroids are the mainstay of severe asthma treatment (Saleh et al., 1998; Bousquet et al., 2000). Although recent studies suggest that there is little clinical benefit from administering steroids in COPD, other than improving the patient's perceived quality of life and the incidence of disease exacerbation (Burge, 1999; Calverley, 2000). Recent studies suggest that inhibiting the adenosine 3′, 5′ monophosphate (cAMP)-selective phosphodiesterase (PDE)-4 isozyme, is a potential therapeutic target for improving COPD and severe asthma (Torphy et al., 1999). PDE4 inhibitors elevate cAMP levels, by inhibiting its degradation via PDE4, and induce airway smooth muscle relaxation (bronchodilation), inhibit allergen- and lipopolysaccharide (LPS)-induced AHR and suppress excess NO generation and immunocompetent cell activation and migration (Danahay & Broadley, 1997; Torphy et al., 1999; Toward & Broadley, 2001a).
Ozone (O3) is an unstable allotropic species of oxygen that spontaneously decomposes into molecular (O2) and ionized (O−) oxygen. Anti-oxidant and scavenger enzymes normally sequester increased levels of ROS after ozone exposure, however, oxidative stress occurs if the level of ozone exceeds the capacity of the anti-oxidative mechanisms (Cardile et al., 1995). In animals and humans, inhaled ozone (O3) induces AHR to various spasmogens (Schultheis, et al., 1994; Seltzer et al., 1986; Arakida et al., 2000), decreases expiratory flow and volume (Hazucha, 1987; Joad et al., 1994) and bronchial biopsies and lavage exhibit evidence of leukocyte influx, oedema and epithelial damage (Murlas & Roum, 1985; Aris et al., 1993). Thus, oxidant stress arising after ozone inhalation shares many features associated with COPD and asthma. Previous studies from this laboratory have examined the effects of ozone inhalation on AHR, leukocyte influx into the airways and oedema in guinea-pigs (Johnson et al., 1998). However, as with many previous studies investigating the airways response to ozone (Murlas et al., 1993; Schultheis et al., 1994; Holbrook et al., 1996; Inoue et al., 2000; Nakano et al., 2000), anaesthesia was utilized to determine AHR, which may influence vagal tone or sensory reflexes. In the current study, we assessed both AHR and airway function changes in conscious guinea-pigs following ozone exposure. This technique has the advantage of preserved neural reflexes and the ability to monitor the time-course of airway function (changes in airway calibre and respiratory rate) over several days, with pared analysis before and after ozone exposure in the same animal. Thus, the first aim of this study was to examine the hypothesis that changes in respiratory function over time correlate with airways hyperreactivity, leukocyte infiltration, oedema and NO generation. NO metabolites in bronchoalveolar lavage fluid (BALF) have previously been shown to be elevated at 5 h after ozone exposure in guinea-pigs (Inoue et al., 2000), but in the present study we monitored changes at intervals up to 48 h. To validate the ozone model further as a suitable paradigm of COPD and for evaluating potential drugs for its treatment, we also determined the profile of activity of two well-studied standard classes of anti-inflammatory agent – the corticosteroid, dexamethasone, and the PDE4 inhibitor, rolipram. Preliminary findings from this study have been presented to the British Pharmacology Society (Toward & Broadley, 2000a).
Methods
Animals
Groups of six male Dunkin-Hartley guinea-pigs, weighing 300 – 400 g were used throughout. Animals received food and water ad libitum, and room temperature (22±2°C) and lighting (maintained on a 12 h cycle) was regulated. This work complied with the guidelines for care and use of laboratory animals according to the Animals (Scientific Procedures) Act 1986 and GlaxoSmithKline policy.
Measurement of respiratory function (sGaw and breaths min−1)
Whole-body plethysmography was used to monitor respiratory function in the conscious guinea-pig. Changes in airway calibre (bronchoconstriction and bronchodilation) were recorded as specific airways conductance (sGaw) and respiratory rate was measured as breaths per minute (breaths min−1). The method was as described by Griffiths-Johnson et al., 1988, although a computerized data acquisition system replaced the original oscilloscope and angle resolver (Toward & Broadley, 2000b). The guinea-pig was placed in a restrainer and a face mask was fitted over the snout of the animal. The restrained guinea-pig was then sealed inside the plethysmography chamber. As the animal breathed, a computerized Biopac data acquisition system (Biopac systems Inc., Santa Barbara, CA, U.S.A.) acquired and stored data (Acknowledge software) referring to air flow across a pneumotachograph (Mercury FIL). The resulting change in box volume (pressure) was simultaneously measured. Two UP pressure transducers (Pioden Controls Ltd, Canterbury, Kent, U.K.) measured changes in air flow (UP1) and box pressure (UP2). The resultant waveforms could then be rapidly analysed by comparing the gradients of the flow and the box pressure waves at a point where flow tended towards zero, i.e. in the first 30 ms of expiration. A function of these parameters, correcting for ambient pressure and the weight of the animal, determined a value for sGaw. The breathing rate over the 5 s reading was multiplied by 20 to determine an integer value for respiratory rate (breaths min−1). At least five breaths were analysed for each animal at each time point. Before each experiment the animals were handled and familiarized with the equipment to reduce stress.
Ozone exposure
Groups of six guinea-pigs were box-exposed for 90 min to ozone generated via medi-air (0.35 l.min−1, 18.8 – 23.5% oxygen in nitrogen) passed through an ozonizer (model 100; Sander, Frankfurt, Germany). The level of ozone was regulated by a variable voltage supply. The ozone level within the exposure chamber (620×300×420 mm) was monitored every 15 min using a colourmetric indicator tube (ozone 0.05/b) and pump (Accuro) system (Dragger Sicherheitstechnik Gmbh, Germany) and regulated accordingly to maintain a constant level of ozone (2.15±0.05 p.p.m.). Murlas & Roum (1985) have previously shown that this level of ozone exposure was effective at causing AHR and mild-inflammation in guinea-pig airway, without fatality. Control animals were exposed for 90 min to medi-air. The average of two baseline determinations of respiratory function (sGaw and breaths min−1) was obtained prior to exposure to ozone or medi-air and then at regular intervals (0, 15, 30 min and hourly) after the exposure.
Spasmogen exposures
Airway reactivity to nebulized histamine (1 mM, 20 s), delivered as a nose-only exposure, was assessed 24 h before and at 0.5, 2, 24, 48 or 72 h after box-exposure. This dose of histamine caused a small threshold bronchoconstriction in naïve animals, compared with 3 mM which was required to produce significant bronchoconstriction (−22.6±6.7% decrease from baseline sGaw value). Histamine via inhalation has been shown to be at least a 100-fold more efficacious on functional changes in the guinea-pig lung compared to the nasal cavity (Finney & Forsberg, 1994). A nasal component of the changes in sGaw was considered negligible compared to those in the lung. Measurements of sGaw were taken before and at 0, 5 and 10 min after exposure to the spasmogen.
Administration of anti-inflammatory compounds
In other animals, the phosphodiesterase-4 (PDE4) inhibitor, rolipram (1 mg kg−1), the corticosteroid, dexamethasone (20 mg kg−1), or vehicle were administered (i.p.) 24 and 0.5 h before exposure to ozone and 24 h afterwards. In these animals, AHR was assessed at 2 h after the completion of ozone exposure and BALF was removed at 48 h. These doses were selected based upon data from other studies using models of inflammation and that were without adverse effects (Toward & Broadley, 2001a,2001b). No animal appeared to be in respiratory distress, or exhibit other signs of discomfort during the exposure regimes or during any of the other procedures described.
Bronchoalveolar lavage
Within 20 min of assessing airway reactivity to histamine, 48 h after medi-air exposure or 48 h after ozone exposure with or without rolipram or dexamethasone treatment, the guinea-pigs were overdosed with pentobarbitone sodium (400 mg kg−1, i.p., Euthatal®). The thoracic cavity was opened to allow access to the lungs, and the trachea was exposed. A ligature was placed around the right large-bronchi, to prevent fluid from entering the right side of the lung. The trachea was then cannulated and the animals underwent a bronchoalveolar lavage of the left lung lobes. Saline (0.9% NaCl: 0.5 ml.100 g−1 body weight) was flushed through the cannula into the lungs, and recovered 3 min later. This was repeated and a total cell count (cells.ml−1) of the pooled bronchoalveolar fluid (BALF) sample was determined using a haemocytometer (Neubauer). A Cytospin smear (Shandon Centrifuge: 1000 r.p.m., 7 min) of the sample was differentially stained (Leishman's: 1.5% in methanol, 6 min), and a minimum of 500 cells (macrophages, eosinophils and neutrophils) were counted. Lymphocytes were not identified or counted. In all the groups studied there was no significant difference in the volumes of reclaimed BALF (typically, 70 – 80% recovery).
Measurement of nitric oxide (NO) production
The remaining BALF sample was then centrifuged (1200 r.p.m., 6 min) and the supernatant was removed and frozen (−20°C). A spectrophotometric assay was used to determine the stable decomposition products of NO (nitrite and nitrate) in BALF. The assay was based on the Griess reaction (Grisham et al., 1996) as previously described (Toward & Broadley, 2000b). Briefly, 100 μl samples of the BALF were incubated (37°C) for 30 min with HEPES buffer (50 mM, pH 7.4), FAD (5 μM), NADPH (0.1 mM), distilled water (290 μl) and nitrate reductase (0.2 U.ml−1) for the conversion of nitrate to nitrite. In an identical set of tubes, nitrate reductase was omitted for determining nitrite only. Any unreacted NADPH in the solution (500 μl) was then oxidised by incubating (25°C, 10 min) with potassium ferricyanide (1 mM). The samples were then incubated (25°C) with 1 ml of the Griess reagent (NED: 0.2% (w v−1), sulfanilamide: 2% (w v−1), solubilized in double distilled water (95%) and phosphoric acid (5%) (v v−1)) for 10 min and the absorbance measured at 543 nm. Levels of nitrite (NO metabolite) in the BALF samples were calibrated against Griess reagent absorbance from a sodium nitrite (0 – 150 μM) standard curve. To maintain standard conditions, all the samples to be compared were assayed at the same time. The maximum linear limit of detection for the assay was 1 mM.
Measurement of wet/dry lung weight
Changes in lung oedematous fluid after exposure to medi-air, ozone or ozone and rolipram or dexamethasone treatment, were quantified as the difference in the wet/dry lung weights. An increase in lung wet weight after ozone exposure, compared to naïve lung, was considered to be the result of oedema (Hwang et al., 2001). After removing BALF from the left side of the lung, the right lung lobes were removed below the ligature, by dissecting through the right large-bronchi. The lung lobes were blotted dry, weighted in a pre-weighted tube and placed in an oven (80°C) with silica crystals. They remained in the oven until there was no further reduction in lung weight (without charring), normally a period of 24 – 48 h. The difference between the wet and oven-dried lung weight was a measure of fluid in the lung. The dry lung weight was not significantly different in any of the groups studied.
Drugs and solutions
Dexamethasone-21-phosphate disodium salt, dimethylsulphoxide (DMSO), flavin adenine dinucleotide (FAD) disodium salt, histamine diphosphate salt, N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid] (HEPES) free acid, nicotinamide adenine dinucleotide phosphate (NADPH: reduced form), naphthyleneethylenediamine (NED), sodium nitrite, phosphoric acid, potassium ferricyanide, rolipram and sulfanilamide were purchased from Sigma (Poole, Dorset, U.K.), Euthatal® from Rhone Meriux (Harlow, Essex, U.K.) and Aspergillus nitrate reductase (NADPH: nitrate oxidoreductase, EC 1.6.6.2) from Boehringer Mannheim (Indianapolis, U.S.A.). Rolipram and dexamethasone were dissolved in 50% DMSO, 50% saline (0.9% NaCl: Baxter Healthcare, Thetford, Norfolk, U.K.) and introduced as 1.0 ml volumes made up with sterile saline. The final volume of DMSO was less than 5% in the vehicle and the treatment. This vehicle has previously been shown to have no affect in naïve animals, or on lung function and the inflammatory response in other inflammatory models (Toward & Broadley, 2001a, 2001b). Unless otherwise indicated, all other solutions were made up in sterile saline.
Data analysis
To reduce inter-subject variability, changes in sGaw from the baseline sGaw values taken before a procedure, are presented as a percentage of the mean baseline value preceding ozone, medi-air or histamine challenge. Absolute values of baseline sGaw are stated in the figure legends. Changes in respiratory rate are presented as the number of breaths per minute (breaths min−1). The BALF cell counts, the combined NO metabolites and the wet/dry lung weights were compared using analysis of variance, followed by Scheffe's post hoc analysis. Changes in airway function (sGaw and breaths min−1) were compared using analysis of variance followed by the appropriate paired or unpaired Student's t-test. Differences were considered statistically significant when P<0.05 (Motulsky, 1995).
Results
Lung function
Exposure of guinea-pigs to ozone caused an immediate fall in sGaw, indicating bronchoconstriction (−52.2±6.5 peak per cent decrease from baseline sGaw, P<0.001), compared with medi-air (−13.4±3.3%) (Figure 1). This early-phase bronchoconstriction (EPB) recovered 2 h later. However, at 5 h after ozone exposure, a late-phase bronchoconstriction (LPB) commenced (5 h: −19.0±3.1% decrease from baseline sGaw, P<0.01). During the EPB, the respiratory rate was unaltered from baseline values (94±13 breaths min−1) (Figure 2). However, at 5 h after exposure to ozone, an increase in respiratory rate (120±18 breaths min−1, P>0.05) commenced that coincided with the development of a LPB. Both the increase in respiratory rate (186±32 breaths min−1, P<0.01) and LPB (−29.9±5.7 peak per cent decrease from baseline sGaw, P<0.01) peaked at 24 h after ozone exposure and recovered to baseline values at 72 h (Figures 1, 2). Interestingly, cyanosis was observed in the ears, paws and nose of the guinea-pigs, especially during the LBP, that also recovered at 72 h. Exposure to medi-air failed to significantly (P>0.05) alter lung function from baseline values of sGaw or respiratory rate (Figures 1, 2).
Figure 1.
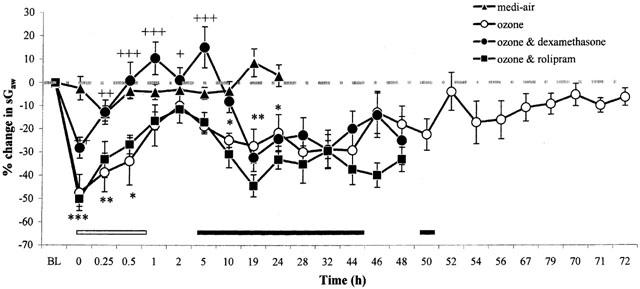
The effect of exposure (90 min) to medi-air (0.35 l min−1, 18.8 – 25% oxygen in nitrogen), or medi-air via an ozone generator (2.15±0.05 p.p.m.) with and without dexamethasone (20 mg kg−1) or rolipram (1 mg kg−1) treatment on lung function, in conscious guinea-pigs. Treatment was administered 24 and 0.5 h before exposure to ozone and 24 h afterwards. Each point represents the mean±s.e.mean (n=6) change in sGaw expressed as a percentage of the baseline sGaw values (sGaw (sec−1 cmH2O−1): medi-air=0.29±0.01; ozone=0.34±0.01). Negative values represent bronchoconstriction. The EPB is depicted from 0 – 1 h (white bar) and the LPB is shown from 5 – 50 h (black bar) after exposure to ozone. Significance of differences in changes of sGaw after ozone exposure: from baseline values (black and white bars: P<0.05); or between medi-air exposure (*P<0.05, **P<0.01 and ***P<0.001); or with and without dexamethasone treatment (+P<0.05, ++P<0.01 and +++P<0.001), were determined by analysis of variance (single factor), followed by a Student's t-test (paired, unpaired and unpaired, respectively). There was no significant (P>0.05) difference between the lung function in ozone exposed guinea-pigs with and without vehicle treatment (data not shown).
Figure 2.
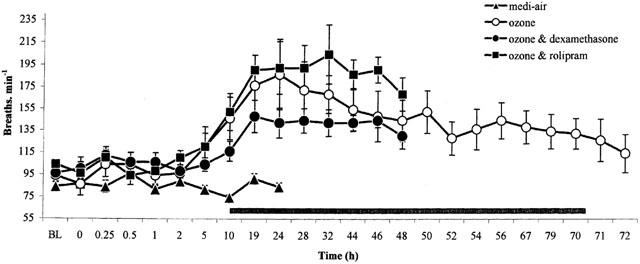
The effect of exposure (90 min) to medi-air (0.35 l min−1), or medi-air via an ozone generator (2.15±0.05 p.p.m.) with and without dexamethasone (20 mg kg−1) or rolipram (1 mg kg−1) treatment on respiratory rate, in conscious guinea-pigs. Treatment was administered 24 and 0.5 h before exposure to ozone and 24 h afterwards. Each point represents the mean±s.e.mean (n=6) change in respiratory rate (breaths min−1). Black bar denotes the significance of differences (P<0.05) between respiratory rate before and after exposure to ozone as determined by analysis of variance (single factor), followed by Student's unpaired t-test. There was no significant (P>0.05) difference between the respiratory rate in ozone exposed guinea-pigs with and without rolipram, dexamethasone or vehicle (data not shown) treatment.
In ozone-exposed animals treated with vehicle there was a similar profile of EPB followed by a LPB accompanied by increased respiratory rate (data not shown). Rolipram treatment did not affect this profile (Figures 1, 2). However, dexamethasone treatment attenuated (P<0.01) the EPB after ozone exposure, but failed to significantly alter the LPB (Figure 1). Animals treated with dexamethasone also displayed a slight (P>0.05) reduction in the increased respiratory rate associated with the LPB (24 h: 142±14 breaths min−1), compared with ozone exposed animals (24 h: 186±32 breaths min−1) (Figure 2). In vehicle, rolipram or dexamethasone treated animals, the LPB and associated increase in respiratory rate recovered to baseline levels at 72 h after ozone exposure (Figures 1, 2). There was no statistically significant difference between starting baseline values of sGaw or respiratory rate for any of the groups studied.
Airway responsiveness to inhaled histamine
Inhalation of histamine (1 mM, 20 s) before ozone exposure failed to produce a significant bronchoconstriction (Figure 3a,b). However, at 0.5 h after a single exposure to ozone, there was a significant (P<0.05) bronchoconstriction to inhaled histamine, compared to before exposure, indicating AHR (Figure 3a,b). There was also AHR to inhaled histamine at 2 h (P<0.01), 12 h (P<0.05), 24 h (P<0.05) and 48 h (P<0.05) after ozone exposure, a lack of bronchoconstriction before ozone exposure being converted to a significant bronchoconstriction afterwards (Figure 3b). At 72 h after exposure to ozone, the increased reactivity towards histamine (i.e. AHR) subsided (Figure 3b). Exposure to medi-air failed to alter airway responsiveness to histamine at 2 h after exposure (Figure 3b).
Figure 3.
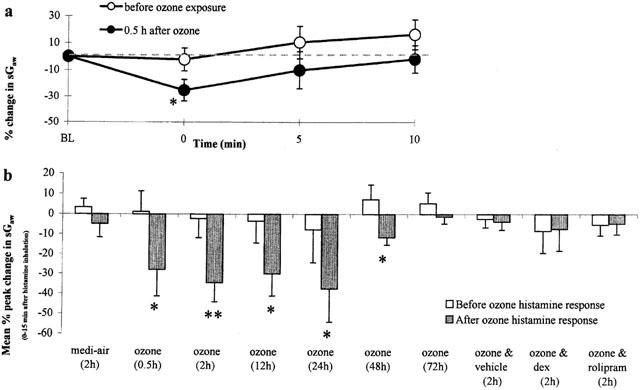
The airways responsiveness of a nose-only exposure (20 s) to histamine (1 mM) before and (a) 0.5 h after exposure (90 min) to ozone (2.15±0.05 p.p.m.); (b) 0.5, 2, 12, 24, 48 and 72 h after exposure (90 min) to ozone, 2 h after exposure (90 min) to medi-air, and 2 h after ozone with vehicle, dexamethasone (dex, 20 mg kg−1), or rolipram (1 mg kg−1) treatment, in conscious guinea-pigs. Treatment was administered (i.p.) 24 and 0.5 h before exposure to ozone. Each point (n=6) represents the (a) mean±s.e.mean change in sGaw expressed as a percentage of the baseline sGaw values (sGaw (sec−1 cmH2O−1) before=0.37±0.02 and after=0.40±0.02); (b) mean±s.e.mean of individual peak change in sGaw from baseline sGaw values, 0 – 10 min following inhalation of histamine expressed as a percentage of the baseline, 24 h before and: 2 h after medi-air (before=0.27±0.01 and after=0.28±0.01); 0.5 h (before=0.37±0.02 and after=0.40±0.02), 2 h (before=0.31±0.01 and after=0.33±0.02), 12 h (before=0.33±0.02 and after=0.31±0.03), 24 h (before=0.34±0.03 and after=0.35±0.02), 48 h (before=0.32±0.01 and after=0.28±0.01), 72 h (before=0.31±0.01 and after=0.30±0.01) after ozone, and 2 h after ozone and vehicle (before=0.33±0.01 and after=0.32±0.01), dex (before=0.34±0.02 and after=0.33±0.03), or rolipram (before=0.33±0.02 and after=0.27±0.02) treatment. Negative values represent bronchoconstriction. Differences between before and after exposure to histamine were determined by analysis of variance (single factor), followed by a Student's (paired) t-test and denoted as *P<0.05 and **P<0.01.
Compared with before ozone (−2.2±8.4 peak per cent decrease from baseline sGaw), the peak increase in airway responsiveness to histamine (−34.3±9.6%, P<0.01) occurred at 2 h after exposure to ozone (Figure 3b). The airway responsiveness to histamine was not significantly (P>0.05) increased at 2 h after ozone exposure in guinea-pigs treated with rolipram (−4.7±5.3%) or dexamethasone (−7.4±10.9%), compared with before exposure (−5.2±5.7% and −8.4±4.9%, respectively), indicating an inhibition of the acute ozone-induced AHR (Figure 3b). Treating animals with the rolipram and dexamethasone vehicle failed (P>0.05) to alter the airway responsiveness to histamine (Figure 3b). Treating control animals with dexamethasone or rolipram fails to affect the responsiveness to histamine as shown in our previous studies (Toward & Broadley, 2001a).
Leukocyte infiltration
In animals exposed to ozone, there was no significant (P>0.05) increase in the population of leukocytes in BALF removed at 0.5 and 2 h later, compared with naïve animals (Figure 4). However, there was a marked increase in the number of macrophages (P<0.05), eosinophils (P<0.05) and neutrophils (P<0.01) retrieved from the BALF at 12 h after ozone exposure, compared to that removed from naïve animals. Macrophages and eosinophils further increased at 24 h and remained elevated at 48 h, which coinciding with the peak in LPB and peak increase in respiratory rate. However, the neutrophil influx that peaked at 12 h began to wane at 24 h. At 48 h, the eosinophil and macrophage influx peaked. At 72 h, the eosinophils began to subside and along with the neutrophils were not significantly different from naïve levels. Macrophages were also significantly reduced, but remained significantly (P<0.01) elevated compared with naïve animals. At 48 h after exposure to media-air, the levels of leukocytes in BALF were not significantly (P>0.05) different from those observed in naïve guinea-pigs (Figure 4).
Figure 4.
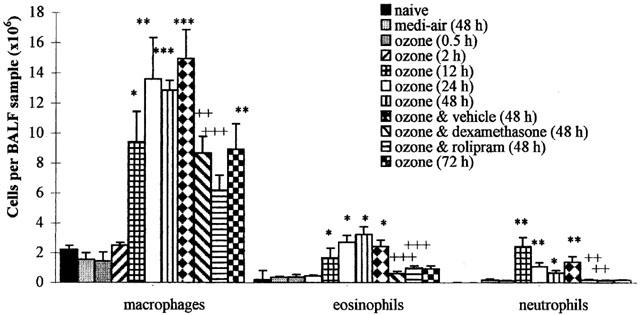
Differential cell (macrophage, eosinophil and neutrophil) counts of bronchoalveolar lavage fluid (BALF) removed before (naïve) and after exposure (90 min) to medi-air (48 h), ozone (2.15±0.05 p.p.m.) (0.5, 2, 12, 24, 48 and 72 h), or ozone with vehicle, dexamethasone (20 mg kg−1), or rolipram (1 mg kg−1) treatment (48 h). Treatment was administered (i.p.) at 24 and 0.5 h before exposure to ozone and at 24 h afterwards. Each point represents the mean±s.e.mean (n=6) of the differential cells per BALF sample (×106) removed from the left lung lobes, via the left bronchi. The significance of differences in airway cell influx compared with naïve guinea-pigs (*P<0.05, **P<0.01 and ***P<0.001), and ozone exposure and vehicle vs dexamethasone or rolipram treatment (++P<0.01 and +++P<0.001), as determined by analysis of variance (single factor), followed by Scheffe's post hoc analysis.
In ozone-exposed guinea-pigs treated with the vehicle there was no significant (P>0.05) difference in the peak increase in BALF macrophages (14.9±1.9×106 cells sample−1), eosinophils (2.4±0.4×106) and neutrophils (1.4±0.4×106) at 24 h, from that observed in ozone-only exposed animals (Figure 4). Treatment with rolipram or dexamethasone significantly attenuated the 48 h peak airway influx of macrophages, eosinophils and neutrophils by 59, 59 and 42% and 42, 75 and 56% respectively, compared with ozone-exposed animals treated with vehicle (Figure 4). Treatment of control guinea-pigs with dexamethasone and rolipram enhance the levels of macrophages and eosinophils to a small extent, as shown in our previous studies (Toward & Broadley, 2001a).
Nitric oxide
The variation in concentrations of the individual NO metabolites (nitrate and nitrite) in BALF were synchronous. However, lower levels of nitrate were analysed, due to the rate limiting nitrate reductase conversion of nitrate to measurable nitrite (data not shown). Therefore, to describe changes in the NO metabolites assayed, the combined nitrate and nitrite values are shown (Figure 5) and referred to.
Figure 5.
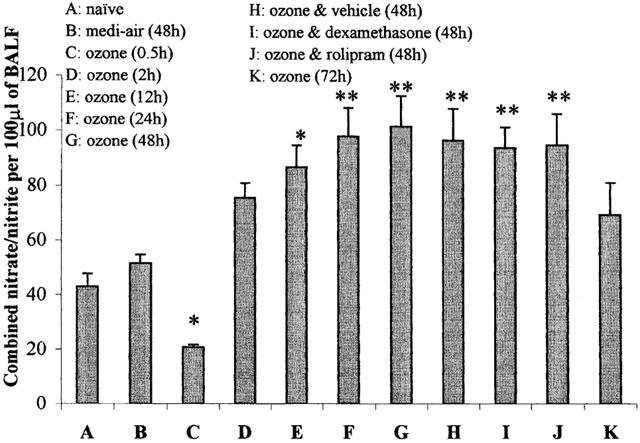
Airways levels of combined nitric oxide (NO) metabolites (nitrate and nitrite) recovered before (naïve) and after exposure (90 min) to medi-air (48 h), ozone (2.15±0.05 p.p.m.) (0.5, 2, 12, 24, 48 and 72 h), or ozone with vehicle, dexamethasone (20 mg kg−1), or rolipram (1 mg kg−1) treatment (48 h). Treatment was administered (i.p.) at 24 and 0.5 h before exposure to ozone and at 24 h afterwards. Each point represents the mean±s.e.mean (n=6) of the concentration of combined NO metabolites (μM) per 100 μl of BALF removed from the left lung lobes, via the left bronchi. *P<0.05, and **P<0.01 denotes the significance of differences in airway NO levels compared with naïve guinea-pigs, as determined by analysis of variance (single factor), followed by Scheffe's post hoc analysis.
There was a basal level of combined NO metabolites in BALF removed from naïve animals (Figure 5). At 0.5 h after ozone exposure, the concentration of combined NO metabolites in the BALF were significantly (P<0.05) decreased, by 52%, compared to that found in naïve animals (Figure 5). At 2 h after ozone, the NO-deficiency recovered and was increased by 75% (P>0.05), compared to that observed in naïve animals. At 12 h (P<0.05) and 24 h (P<0.01), the levels of combined NO metabolites in BALF were further increased (101 and 127% increase from naïve levels, respectively). This also coincided with the LPB, increase in respiratory rate and cell influx. At 48 h after ozone exposure, the NO levels remained significantly (131%, P<0.01) elevated compared with naïve animals, but like the LPB, increase in respiratory rate and leukocyte influx, began to subside at 72 h. Exposure to medi-air failed to affect the basal production of NO at 48 h.
The significantly (P<0.01) increased NO metabolites at 48 h after ozone exposure, were not affected by treatment with vehicle (96.7± 11.2 μM 100 μl BALF−1, P<0.01) (Figure 5). Treating the animals with either rolipram (93.8±7.7 μM, P<0.01) or dexamethasone (96.6±11.5 μM, P<0.01) failed to alter the significantly elevated levels of BALF NO metabolites, compared with vehicle treated animals at 48 h after ozone exposure. Dexamethasone or rolipram have also been shown in our previous studies to not affect BALF NO metabolites in control animals not exposed to ozone (Toward & Broadley, 2001a).
Lung wet/dry weight
At 0.5 and 2 h after exposure to ozone there was no significant (P>0.05) increase in the lung wet/dry weight difference, compared with naïve animals (Figure 6). However, at 12 and 24 h there was a significant increase (P<0.05) in the lung wet/dry weight difference. At 48 h, the lung wet/dry weight difference further significantly (P<0.001) increased, but at 72 h, it decreased and was not significantly (P>0.05) different from that observed in naïve animals.
Figure 6.
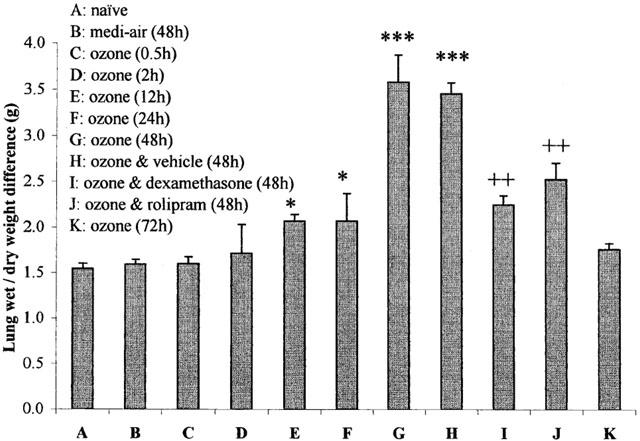
Difference in wet and oven-dried lung weights before (naïve) and after exposure (90 min) to medi-air (48 h), ozone (2.15±0.05 p.p.m.) (0.5, 2, 12, 24, 48 and 72 h), or ozone with vehicle, dexamethasone (20 mg kg−1), or rolipram (1 mg kg−1) treatment (48 h). Treatment was administered (i.p.) at 24 and 0.5 h before exposure to ozone and at 24 h afterwards. Each point represents the mean±s.e.mean (n=6) of the difference in right lung lobe weights (grams), removed below the right bronchi, before (wet) and after oven drying. The significance of differences in wet/dry lung weights compared with naïve guinea-pigs (*P<0.05 and ***P<0.001), and ozone exposure and vehicle versus dexamethasone or rolipram treatment (++P<0.01), was determined by analysis of variance (single factor), followed by Scheffe's post hoc analysis.
In guinea-pigs treated with vehicle there was no significant (P>0.05) difference in the peak increase in oedematous fluid (3.5±0.3 g) at 48 h after ozone exposure, to that observed in ozone-only exposed animals (Figure 6). Treatment with rolipram or dexamethasone significantly attenuated the peak increase in lung wet/dry weight difference by 27 and 35% respectively, compared with ozone-exposed animals treated with vehicle at 48 h (Figure 6).
Discussion
In COPD and severe asthma, chronic inflammatory-derived ROS in the airways are thought to contribute to pulmonary oxidative stress (Repine et al., 1997; Barnes et al., 1999). The aim of this study was to induce oxidative stress in conscious guinea-pig airways by exposure to ozone and to establish a model that displays the features of COPD. To test the validity of this model, the modification of these features by two standard anti-inflammatory agents, dexamethasone and rolipram were examined.
Exposure of guinea-pigs to ozone caused an immediate EPB that recovered at 2 h. This was, followed at 5 h, by a LPB that peaked at 24 h and recovered at 72 h. These two distinct phases of bronchoconstriction have not been reported before following ozone exposure, probably because a prolonged time-course, as used here, has not previously been examined in conscious guinea-pigs. Early- and late-phase bronchoconstrictions are commonly associated features of the asthmatic response to inhaled allergen and are thought to reflect two distinct pathological events (Bousquet et al., 2000). In asthmatics and atopic animals, allergen exposure causes an immediate mast cell-derived release of histamine and other bronchoconstrictor mediators into the airways that contribute to the EPB (Bousquet et al., 2000). The allergen-induced LPB, however, is often associated with an eosinophilic infiltration of activated leukocytes and a further increase in airways pro-inflammatory mediators that contribute to bronchoconstriction, AHR and oedema (Barnes et al., 1999; Bousquet et al., 2000).
In this study, the EPB after ozone exposure was not associated with a change in respiratory rate, oedema, or an increase in BALF leukocyte levels, although an increase in peripheral airway leukocytes cannot be ruled out. There was, however, a deficiency in airway NO and AHR to histamine at 0.5 h after ozone. In human macrophages, Cardile et al. (1995) have also shown that NO synthesis was attenuated after exposure to ozone. The authors suggested that oxidative damage to NOS was an unlikely cause of the NO-deficiency, but that enzymatic synthesis of NO was antagonized by ozone-derived ROS. An increase in mucosal mast cells and increased histamine in guinea-pig airways has been reported immediately after ozone exposure (Murlas & Roum, 1985; Igarashi et al., 1998). Other bronchoconstrictor metabolites may also occur from the generation of ROS-induced lipid peroxidation of arachidonic acid to form isoprostanes. The most prevalent of which, 8-epi-prostaglandin F2, causes potent constriction of human airways (Barnes et al., 1999). In human airway smooth muscle, NO is the sole inhibitory nonadrenergic noncholinergic (NANC) bronchodilator (Nijkamp & Folkerts, 1995). Thus, in this study, an absence of opposing bronchodilatory NO coinciding with a mast cell-derived release of histamine or ROS-induced peroxidation of arachidonic acid to yield isoprostanes, may contribute to the development of the ozone-induced EPB. Another explanation for the bronchoconstriction is that ozone inhibits cholinesterase activity in guinea-pig lungs (Gordon et al., 1981) and also causes a loss of pre-junctional inhibitory M2 muscarinic receptor function either directly or via inflammatory cell-derived mediators (Schultheis et al., 1994). Both of these effects would increase the release and longevity of acetylcholine in the synapse, and subsequently enhance post-synaptic vagal nerve stimulation and explain the bronchoconstrictor activity.
In this study, AHR to histamine occurred as early as 0.5 h after ozone exposure. Previously, we have shown in LPS-exposed guinea-pigs (Toward & Broadley, 2001b) that AHR to inhaled histamine coincided with NO-deficiency, at 1 h after exposure. Furthermore, we demonstrated that NO might aid recovery from LPS-induced AHR, as inhibiting NO generation with the non-selective NOS inhibitor, L-NAME, prolonged the duration of AHR (Toward & Broadley, 2000b). The AHR to inhaled histamine at 0.5 h after ozone, might also be a consequence of NO-deficiency failing to oppose an inhaled histamine-induced bronchoconstriction or to the reduced airway calibre due to the EPB. At 2 h after ozone exposure, however, there was no residual bronchoconstriction or NO-deficiency, but there was a maximal increase in airways responsiveness to histamine. One explanation for the AHR at 2 h is that ozone inactives neutral endopeptidase (NEP) in the airway (Holbrook et al., 1996), which is a peptidase involved in the degradation of pro-inflammatory peptides, such as tachykinins released from sensory nerve endings in guinea-pig lung (Spina et al., 1998; Barnes et al., 1999). Tachykinins cause hypersensitive vagal stimulation and bronchoconstriction (Spina et al., 1998; Barnes et al., 1999) and an ozone-induced inactivation of NEP would elevate their levels and cause neuronal hypersensitivity and an increased bronchoconstrictor response to inhaled histamine (i.e. AHR).
In contrast to the EPB, the LBP was associated with an increase in leukocytes. Initially, neutrophils transiently infiltrated the airways and peaked at 12 h. As neutrophils subsided, macrophage and eosinophil levels peaked between 24 and 48 h, and at 72 h, the levels of BALF granulocytes recovered to those in naïve animals. However, macrophage levels remained significantly elevated. Ozone and other ROS activate nuclear transcription factor-κB (NF-κB), which orchestrates the expression of multiple inflammatory genes that are up-regulated in both COPD and asthma (Stockley, 1995; Barnes & Adcock, 1997; Barnes et al., 1999). Ozone-induced activation of NF-κB would cause resident macrophages, lymphocytes and epithelial cells in the airway to express the neutrophil chemotactic factors, interleukin (IL)-8 and tumour necrosis factor (TNF)-α, which may explain the initial neutrophilia in this study (Barnes & Adcock, 1997). NF-κB and TNF-α also induce a complex cascade of pro-inflammatory mediators responsible for macrophage and eosinophil recruitment (macrophage inflammatory protein-1α, monocyte chemotactic protein-3 and eotaxin), adhesion molecule expression and promoting leukocyte survival (granulocyte-macrophage colony stimulating factor) (Barnes & Adcock, 1997; Barnes et al., 1999). As in asthma and COPD, in this study during the LBP, AHR to histamine correlated with a decline in airway function and the development of underlying inflammation (Postma & Kerstjens, 1998). Furthermore, the recovery in AHR also coincided with a resolution in leukocyte influx and the LPB. This may suggest that during the ozone-induced LBP, leukocyte influx and airway function are related to AHR.
The inducible inflammatory enzymes, iNOS and cyclo-oxygenase-2 (iCOX2) are also up-regulated by NF-κB and TNF-α. In this study, the elevated levels of NO during the LBP could be due to NF-κB-mediated iNOS induction. Haddad et al. (1995) demonstrated an increase in iNOS mRNA expression at 2 and 8 h after ozone exposure of Brown Norway rats. Interestingly, Colasanti & Suzuki (2000) suggest that basal levels of NO suppress NF-κB dissociation from the inhibitory nuclear transcription factor (I-κB) complex and also prevent NF-κB binding to the promoter region of the iNOS gene. Thus, in the present study, the initial NO-deficiency during the ozone-induced EPB, could facilitate activation of the machinery to up-regulate iNOS expression during the LBP. The high levels of bronchodilatory NO at 24 and 48 h, may be a rebound response to attenuate the ozone-induced LBP (Barnes et al., 1999). However, excessive NO also causes leukocyte adhesion and microvascular leakage that promotes leukocyte influx and oedema (Barnes et al., 1999). These effects might be further compounded due to underlying oxidative stress from exposure to ozone and ROS derived from activated inflammatory cells, that combine with NO to form oxidative nitrogen species (e.g. peroxynitrite). These oxidative nitrogen species would exacerbate airway damage and oedema and also cause the AHR associated with the LBP in this study (Nijkamp & Folkerts, 1995).
In the present study, the peak leukocyte influx and generation of NO (metabolites) was also associated with a doubling in lung wet/dry weight difference at 48 h after ozone. The increase in wet lung weight is likely to arise from oedema (Hwang et al., 2001). An increase in lung oedematous fluid may be a result of an increased capillary blood pressure or an increase in permeability of the capillary wall. Ozone and pro-inflammatory-derived arachidonic acid metabolites, synthesised by iCOX2 (e.g. thromboxane) or 5-lipoxygenase (e.g. leukotriene: C4, D4, E4), induce vasoconstriction to elevate trans-capillary blood pressure. Tachykinins, ROS, mast cell-derived histamine, excess NO and ROS-activated proteolytic enzymes may cause increased permeability of the capillary wall (Barnes et al., 1999). Oedematous extravasation from the capillaries into the airway and oedematous airway wall swelling, would reduce airway calibre and contribute to the LBP (Pare & Hogg, 1989). However, as ozone and ROS are potent mucus secretagogues, the secretion of mucus from goblet cells and submucosa glands could also increase the lung wet weight and reduce airway calibre, thus causing bronchoconstriction (Barnes et al., 1999; Bousquet et al., 2000). A reduced airway calibre is also a likely cause of the AHR during the LBP, since it parallels the increase in lung wet weight (Hwang et al., 2001), and a reduced geometric airway calibre would potentiate spasmogenic contractility to inhaled histamine (Pare & Hogg, 1989). However, tachykinin-induced neuronal hypersensitivity, peroxynitrite, or epithelial damage exposing airway sensory nerves are also likely contributors of the AHR associated with the LBP in this study (Nijkamp & Folkerts, 1995; Spina et al., 1998).
The profile of LBP, unlike the EBP, was also paralleled with an increase in respiratory rate. As far as the authors are aware, this is the first report of distinct phases of bronchoconstriction and an increase in respiratory rate associated with only the LPB, in conscious animals exposed to ozone. One explanation for the increased respiratory rate is that ROS compete with oxygen, for haemoglobin in erythrocytes (Repine et al., 1997; Barnes et al., 1999). This would increase carbon dioxide tension in the blood, causing acidosis. Chemoreceptors located on the ventrolateral medulla region of the brainstem and peripherally in the carotid bodies, in response to acidosis, would vagally stimulate an increase in respiratory rate (Staub, 1993). Levels of acidosis in the blood and vagal activity were not examined. However, cyanosis in the animals after ozone exposure, would indicate a reduced haemoglobin oxygen capacity, a possible trigger of increased respiratory rate.
Treatment of the ozone-exposed animals with dexamethasone or rolipram, inhibited the AHR at 2 h and attenuated the leukocyte influx and oedema at 48 h. Interestingly, dexamethasone but not rolipram also attenuated the EPB. However, neither treatment significantly affected the LPB or the associated increase in respiratory rate and excess NO generation at 48 h. The inhibitory effects of dexamethasone can be attributed to inhibition of NF-κB activation and subsequent expression of pro-inflammatory mediators responsible for leukocyte influx into the airways (e.g. iCOX2 and iNOS), through an increased transcription of I-κB (Barnes & Adcock, 1997). Rolipram is a PDE4-inhibitor and elevates intracellular levels of cAMP that suppress cell activation and inflammatory processes (Torphy et al., 1999), which would also explain the attenuated leukocyte influx at 48 h after ozone. That both treatments attenuated the oedema was probably a consequence of a reduced leukocyte influx into the airways, inflammatory cell activity and a subsequent release of pro-inflammatory mediators capable of inducing oedema (e.g. eicosanoids, leukotrienes and proteolytic enzymes) (Barnes et al., 1999; Torphy et al., 1999). However, neither dexamethasone nor rolipram treatment attenuated the LPB, nor the excess generation of airways NO. This is contrary to our previous findings in parainfluenza 3-infected guinea-pigs (Toward & Broadley, 2001b) and LPS-exposed guinea-pigs (Toward & Broadley, 2001a), whereby both excess airways NO and leukocyte influx were inhibited using this dosing regime. Similarly, Haddad et al. (1995) showed that dexamethasone inhibited iNOS mRNA expression in ozone-exposed rats. It was unlikely that ozone itself artificial generated the free radical NO, since passing ozone through BALF removed from naïve animals did not affect the level of NO metabolites (unpublished observations). Furthermore, the increase in NO occurred 48 h after ozone exposure.
The AHR at 2 h after ozone exposure was sensitive to both dexamethasone and rolipram. In previous studies, we have demonstrated using this dosing regime that neither dexamethasone nor rolipram caused an anti-histamine or bronchodilator effect that would functionally antagonise a histamine-induced bronchoconstriction (Toward & Broadley, 2001a). Thus, the inhibition of AHR by these compounds may be related to their anti-inflammatory activity. It is plausible that ozone or ROS derived from inflammatory cells inactivate NEP and thus elevate airway tachykinin levels that cause neuronal hypersensitivity and contribute to the AHR at 2 h after ozone. Murlas et al. (1993) have reported in ozone-exposed guinea-pigs a reduction in tachykinin levels after dexamethasone treatment. Furthermore, Jacoby et al. (2001) have shown that dexamethasone reduced stimulation of tachykinin release from sensory nerves by activated leukocytes in rats exposed to ozone. Rolipram has also been reported to prevent tachykinin release via inhibiting vagal stimulation of sensory nerves (Holbrook et al., 1996). Thus, dexamethasone or rolipram treatment may inhibit AHR at 2 h after ozone by prevention of tachykinin-dependent AHR.
In summary, this study demonstrates, that oxidative stress induced by ozone exposure of conscious guinea-pigs causes an EPB and LPB. The EPB was associated with a deficiency in NO metabolites, whereas the LPB, as in COPD, was associated with dyspnoea, elevated NO levels, leukocyte influx and an increase in lung oedema. In this model, as with COPD and asthma, AHR also correlated with the airways leukocyte influx, oedema, excess NO generation and dyspnoea associated with the LPB. Both dexamethasone and rolipram inhibited the ozone-induced AHR, leukocyte influx and oedema. Although dexamethasone attenuated the EPB, neither dexamethasone nor rolipram affected the excess NO, dyspnoea or associated LPB. Thus, this model provides many of the features of COPD and the results support the development of PDE4 inhibitors for the treatment of oxidant stress-induced inflammation and AHR in COPD or severe asthma.
Acknowledgments
This work was financially supported through a Glaxo-Wellcome studentship to T.J. Toward. The authors gratefully acknowledge Dr A.T. Nials at GlaxoSmithKline for his assistance and helpful discussions.
Abbreviations
- AHR
airway hyperreactivity
- BALF
bronchoalveolar lavage fluid
- breaths min−1
respiratory rate
- cAMP
adenosine 3′, 5′ cyclic monophosphate
- COPD
chronic obstructive pulmonary disease
- DMSO
dimethylsulphoxide
- EPB
early-phase bronchoconstriction
- FAD
flavin adenine dinucleotide
- FEV1
Forced expiratory volume in 1 s
- HEPES
N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid]
- I-κB
inhibitory nuclear transcription factor-κB
- iCOX2
inducible cyclo-oxygenase2
- iNOS
inducible nitric oxide synthase
- L-NAME
Nω-nitro-L-arginine methyl ester
- LPB
late-phase bronchoconstriction
- LPS
lipopolysaccharide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NANC
nonadrenergic noncholinergic
- NED
naphthyleneethylenediamine
- NEP
neutral endopeptidase
- NF-κB
nuclear transcription factor-κB
- NO
nitric oxide
- NOS
nitric oxide synthase
- PDE4
phosphodiesterase isoenzyme 4
- ROS
reactive oxygen species
- sGaw
specific airways conductance
- TNF-α
tumour necrosis factor-α
References
- AGUSTI A.G.N., VILAVERDE J.M., TOGORES B., BOSCH M. Serial measurements of exhaled nitric oxide during exacerbations of chronic obstructive pulmonary disease. Eur. Respir. J. 1999;14:523–528. doi: 10.1034/j.1399-3003.1999.14c08.x. [DOI] [PubMed] [Google Scholar]
- ALVING K., WEITZBERG E., LUNDBERG J.M. Increased amount of nitric oxide in exhaled air of asthmatics. Eur. Respir. J. 1993;6:1368–1370. [PubMed] [Google Scholar]
- ARAKIDA Y., OHGA K., SUWA K., OKADA Y., MORIO H., YOKOTA M., MIYATA K., YAMADA T., HONDA K. Effect of YM158, a dual lipid mediator antagonist, on immediate and late asthmatic responses, and on airway hyper-responsiveness in guinea pigs. Jpn. J. Pharmacol. 2000;82:287–294. doi: 10.1254/jjp.82.287. [DOI] [PubMed] [Google Scholar]
- ARIS R.M., CHRISTIAN D., HEARNE P.Q., KERR K., FINKBEINER W.E., BALMES J.R. Ozone-induced airway inflammation in human subjects as determined by airway lavage and biopsy. Am. Rev. Respir. Dis. 1993;148:1363–1372. doi: 10.1164/ajrccm/148.5.1363. [DOI] [PubMed] [Google Scholar]
- BARNES P.J, , CHUNG K.F, PAGE C.P. Inflammatory mediators of asthma: an update. Pharmacol Rev. 1999;50:515–575. [PubMed] [Google Scholar]
- BARNES P.J., ADCOCK I.M. NF-κB: a pivotal role in asthma and a new target for therapy. Trends Pharmacol Sci. 1997;18:46–51. doi: 10.1016/s0165-6147(97)89796-9. [DOI] [PubMed] [Google Scholar]
- BHOWMIK A., SEEUNGAL T., ROLAND M., WEDZICHA J.A. Exhaled nitric oxide in patients with COPD. Proceedings from the British Thoracic Society winter meeting. Thorax. 1998;53:158P. [Google Scholar]
- BOUSQUET J., JEFFERY P., BUSSE W.W., JOHNSON M., VIGNOLA A.M. Asthma: from bronchoconstriction to airway inflammation and remodelling. Am. J. Respir. Crit. Care Med. 2000;161:1720–1745. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- BURGE P.S. Editorial: EUROSCOP, ISOLDE and the Copenhagen City Lung Study. Thorax. 1999;54:287–288. doi: 10.1136/thx.54.4.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALVERLEY P.M.A. Inhaled corticosteroids are beneficial in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2000;161:341–342. doi: 10.1164/ajrccm.161.2.16125_1. [DOI] [PubMed] [Google Scholar]
- CARDILE V., JIANG X., RUSSO A., CASELLA F., RENIS M., BINDONI M. Effects of ozone on some biological activities of cells in vitro. Cell Biol. Tox. 1995;11:11–21. doi: 10.1007/BF00769988. [DOI] [PubMed] [Google Scholar]
- COLASANTI M., SUZUKI H. The dual personality of NO. Trends Pharmacol. Sci. 2000;21:249–252. doi: 10.1016/s0165-6147(00)01499-1. [DOI] [PubMed] [Google Scholar]
- CORRADI M., MAGATI M., CACCIANI G.C., CONSIGLI G.F., DE MUNARI E., PESCI A. Increased exhaled nitric oxide in patients with stable chronic obstructive pulmonary disease. Thorax. 1999;54:572–575. doi: 10.1136/thx.54.7.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANAHAY H., BROADLEY K.J. Effects of inhibitors of phosphodiesterase, on antigen-induced bronchial hyperreactivity in conscious sensitised guinea-pigs and airway leukocyte infiltration. Br. J. Pharmacol. 1997;120:289–297. doi: 10.1038/sj.bjp.0700901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINNEY M.J., FORSBERG K.J. Quantification of nasal involvement in a guinea-pig plethysmography. J. App. Physiol. 1994;76:1432–1438. doi: 10.1152/jappl.1994.76.4.1432. [DOI] [PubMed] [Google Scholar]
- GIAID A., SALEH D., LIM S., BARNES P.J., ERNST P. Formation of peroxynitrite in asthmatic airways. FASEB. J. 1998;12:929–937. [PubMed] [Google Scholar]
- GORDON T., TAYLOR B.F., AMDUR M.O. Ozone inhibition of tissue cholinesterase in gunea-pigs. Arch. Envriron. Health. 1981;36:284–288. doi: 10.1080/00039896.1981.10667639. [DOI] [PubMed] [Google Scholar]
- GRIFFITHS-JOHNSON D.A., NICHOLLS P.J., MCDERMOTT M. Measurement of specific airways conductance in guinea-pigs: A non-invasive method. J. Pharmacol. Meth. 1988;19:233–242. doi: 10.1016/0160-5402(88)90025-3. [DOI] [PubMed] [Google Scholar]
- GRISHAM M.B., JOHNSON G.G., LANCASTER J.R. Quantification of nitrate and nitrite in extracellular fluids. Meth. Enzymology. 1996;268:237–246. doi: 10.1016/s0076-6879(96)68026-4. [DOI] [PubMed] [Google Scholar]
- HADDAD E.B., LIU S.F., SALMON M., ROBICHAUD A., BARNES P.J., CHUNG K.F. Expression of inducible nitric oxide synthase mRNA in Brown Norway rats exposed to ozone: effect of dexamethasone. Eur. J. Pharmacol. 1995;293:287–290. doi: 10.1016/0926-6917(95)00032-1. [DOI] [PubMed] [Google Scholar]
- HAZUCHA M.J. Relationship between ozone exposure and pulmonary function changes. J. Appl. Physiol. 1987;62:1671–1680. doi: 10.1152/jappl.1987.62.4.1671. [DOI] [PubMed] [Google Scholar]
- HOLBROOK M., GOZZARD N., JAMES T., HIGGS G., HUGHES B. Inhibition of bronchospasm and ozone-induced airway hyperresponsiveness in the guinea-pig by CDP840, a novel phosphodiesterase type 4 inhibitor. Br. J. Pharmacol. 1996;118:1192–2000. doi: 10.1111/j.1476-5381.1996.tb15523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HWANG Y.S., LEFFERTS P.L, SNAPPER J.R. Correlation between increased airway responsiveness and severity of pulmonary oedema. Pulmon. Pharmacol. Ther. 2001;14:47–53. doi: 10.1006/pupt.2000.0266. [DOI] [PubMed] [Google Scholar]
- ICHINOSE M., SUGIURA H., YAMAGATA S., KOARAI A., SHIRATO K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am. J. Respir. Crit. Care Med. 2000;162:701–706. doi: 10.1164/ajrccm.162.2.9908132. [DOI] [PubMed] [Google Scholar]
- IGARASHI A., IIJIMA H., TAMURA G., SHIRATO K. Tazanolast inhibits ozone-induced airway hyperresponsiveness in guinea pigs. Am. J. Respir. Crit. Care Med. 1998;157:1531–1535. doi: 10.1164/ajrccm.157.5.9707049. [DOI] [PubMed] [Google Scholar]
- INOUE H., AIZAWA H., NAKANO H., MATSUMOTO K., KUWANO K., NADEL J.A., HARA N. Nitric oxide synthase inhibitors attenuate ozone-induced airway inflammation in guinea pigs. Possible role of interleukin-8. Am. J. Respir. Crit. Care Med. 2000;161:249–258. doi: 10.1164/ajrccm.161.1.9804096. [DOI] [PubMed] [Google Scholar]
- JACOBY D.B., COSTELLO R.M., FRYER A.D. Eosinophil recruitment to the airway nerves. J. Allergy Clin. Immunol. 2001;107:211–218. doi: 10.1067/mai.2001.112940. [DOI] [PubMed] [Google Scholar]
- JOAD J.P., MCDONALD R.J. , GIRI S.N., BRIC J.M. Ozone effects on mechanics and arachadonic acid metabolite concentrations in isolated rat lungs. Environmental Research. 1994;66:186–197. doi: 10.1006/enrs.1994.1054. [DOI] [PubMed] [Google Scholar]
- JOHNSON A., LEWIS C.A., BROADLEY K.J. Effects of low-level ozone exposure on reactivity and conductance in guinea pig airways. Inhalation Toxicol. 1998;10:101–118. [Google Scholar]
- MOTULSKY H. Intuitive Biostatistics. Oxford: Oxford University Press; 1995. Common and advanced statistical tests; pp. 207–284. [Google Scholar]
- MURLAS C.G., LANG Z., CHODIMELLA V. Dexamethasone reduces tachykinin but not ACh airway hyperreactivity after O3. Lung. 1993;171:109–121. doi: 10.1007/BF00542338. [DOI] [PubMed] [Google Scholar]
- MURLAS C.G., ROUM J.H. Sequence of pathologic changes in the airway mucosa of guinea pigs during ozone-induced bronchial hyperreactivity. Am. Rev. Respir. Dis. 1985;131:314–320. doi: 10.1164/arrd.1985.131.3.314. [DOI] [PubMed] [Google Scholar]
- NAKANO H., AIZAWA H., MATSUMOTO K., FUKUYAMA S., INOUE H., HARA N. Cyclooxygenase-2 participates in the late phase of airway hyperresponsiveness after ozone exposure in guinea pigs. Eur. J. Pharmacol. 2000;403:267–275. doi: 10.1016/s0014-2999(00)00524-0. [DOI] [PubMed] [Google Scholar]
- NIJKAMP F.P., FOLKERTS G. Nitric oxide and bronchial hyperresponsiveness. Arch. Int. Pharmacodyn. 1995;329:97–110. [PubMed] [Google Scholar]
- PARE P.D., HOGG J.C. Mechanics of airway narrowing. Am. Rev. Respir. Dis. 1989;139:242–246. doi: 10.1164/ajrccm/139.1.242. [DOI] [PubMed] [Google Scholar]
- POSTMA D.S., KERSTJENS H.A.M. Characteristics of airway hyperresponsiveness in asthma and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998;158:S187–S192. doi: 10.1164/ajrccm.158.supplement_2.13tac170. [DOI] [PubMed] [Google Scholar]
- REPINE J.E., BAST A, , LANKHORST I., THE OXIDATIVE STRESS STUDY GROUP Oxidative stress in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1997;156:341–357. doi: 10.1164/ajrccm.156.2.9611013. [DOI] [PubMed] [Google Scholar]
- SALEH D., ERNST P., LIM S., BARNES P., GIAID A. Increased formation of the potent oxidant peroxynitrite in the airways of asthmatic patients is associated with induction of nitric oxide synthase: effect of inhaled glucocorticosteroids. FASEB J. 1998;12:929–937. [PubMed] [Google Scholar]
- SCHULTHEIS A.H., BASSETT D.J.P., FRYER A.D. Ozone-induced airway hyperresponsiveness and loss of neuronal M2 muscarinic receptor function. J. Appl. Physiol. 1994;76:1088–1097. doi: 10.1152/jappl.1994.76.3.1088. [DOI] [PubMed] [Google Scholar]
- SELTZER J., BIGBY B.G., STULBARG M., HOLTZMAN M.J., NADEL J.A, , UEKI I.F., LEIKAUF G.D., GOETZL E.J, BOUSHEY H.A. O3-induced change in bronchial reactivity to methacholine and airway inflammation in humans. J. Appl. Physiol. 1986;60:1321–1326. doi: 10.1152/jappl.1986.60.4.1321. [DOI] [PubMed] [Google Scholar]
- SPINA D., SHAH S., HARRISON S. Modulation of sensory nerve function in the airways. Trends Pharmacol. Sci. 1998;19:460–465. doi: 10.1016/s0165-6147(98)01261-9. [DOI] [PubMed] [Google Scholar]
- STAUB N.C., SrThe respiratory System Physiology 1993London: Mosby Year Book Press; 547–599.3. ed. Berne, R.M., Levy, M.N., pp [Google Scholar]
- STOCKLEY R.A.Biochemical and cellular mechanisms Chronic Obstructive Pulmonary Disease 1995London: Chapman & Hall Medical Press; 93–135.1. ed. Calverley, P.M.A., Pride, N., pp [Google Scholar]
- TORPHY T.J., BARNETTE M.S., UNDERWOOD D.C., GRISWOLD D.E., CHRISTENSEN S.B., MURDOCH R.D., NIEMAN R.B., COMPTON C.H. Ariflo (SB 207499), a second generation phosphodiesterase 4 inhibitor for the treatment of asthma and COPD: from concept to clinic. Pulmonary Pharmacol. Ther. 1999;12:131–135. doi: 10.1006/pupt.1999.0181. [DOI] [PubMed] [Google Scholar]
- TOWARD T.J., BROADLEY K.J. Early and late phase bronchoconstrictions, airway hyperreactivity, cell influx and steroid- or rolipram-sensitivity, after inhaled ozone, in conscious guinea-pigs. Br. J. Pharmacol. 2000a;131:113P. [Google Scholar]
- TOWARD T.J., BROADLEY K.J. Airway reactivity, inflammatory cell influx and nitric oxide in guinea-pig airways after lipopolysaccharide inhalation. Br. J. Pharmacol. 2000b;131:271–281. doi: 10.1038/sj.bjp.0703589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOWARD T.J., BROADLEY K.J. Chronic lipopolysaccharide exposure on airway function, cell infiltration, and nitric oxide generation in conscious guinea-pigs: Effects of rolipram and dexamethasone. J.Pharmacol.Exp.Ther. 2001a;298:298–306. [PubMed] [Google Scholar]
- TOWARD T.J., BROADLEY K.J. Airway function, hyperreactivity, cell influx and nitric oxide, in conscious parainfluenza-3 virus infected guinea-pigs: Effects of rolipram and dexamethasone. Br. J. Pharmacol. 2001b;135:70P. [Google Scholar]
- VRUGT B., AALBERS R. Inflammation and bronchial hyperresponsiveness in allergic asthma and chronic obstructive pulmonary disease. Respir. Med. 1993;87:B3–B7. doi: 10.1016/0954-6111(93)90117-i. [DOI] [PubMed] [Google Scholar]