

Abstract
Oncostatin M (OSM), a member of the interleukin-6 (IL-6) cytokine family, acts on a variety of cells and elicits diversified biological responses, suggesting potential roles in the regulation of cell survival, differentiation and proliferation.
We have examined the effect of OSM on the regulation of human lung fibroblast proliferation, collagen production and spontaneous apoptosis. The proliferative effects of OSM (0.5 – 100 ng ml−1) were assessed using a MTS assay as well as [3H]-thymidine incorporation and cell counts at 24 and 48 h. Hydroxyproline was measured as an index of procollagen production by high pressure liquid chromotography (HPLC). Apoptosis was determined by annexin staining.
OSM enhanced the mitotic activity of lung fibroblasts in a time and dose dependent manner. Maximum proliferation of 57% above control was observed after incubation for 48 h with 2 ng ml−1 OSM (P<0.05).
Incubation with the mitogen activated protein kinase (MAPK) kinase inhibitor, PD98059 or the tyrosine kinase inhibitor, genestein both significantly reduced the mitogenic effect of OSM (P<0.05).
In contrast, proliferation in response to OSM was not regulated by induction of cyclo-oxygenase and subsequent prostaglandin E2 (PGE2) release or by IL-6.
OSM also stimulated fibroblasts to synthesize pro-collagen by a maximum of 35% above control levels after 48 h (P<0.05).
OSM significantly inhibited the spontaneous apoptosis of fibroblasts at 24 and 48 h.
These results provide evidence that OSM has pro-fibrotic properties and suggest that it may play a role in normal lung wound repair and fibrosis.
Keywords: Cytokine, asthma, lung, tissue repair, fibrosis
Introduction
Chronic inflammatory airway diseases such as asthma and pulmonary fibrosis are characterized by an increased deposition of extracellular matrix (ECM), concomitant with proliferation and activation of sub-epithelial fibroblasts (Jeffery, 1991; Brewster et al., 1990; Gizycki et al., 1997). Fibroblasts are important sources of cytokines, growth factors and mediators that can influence the behaviour of adjacent cell types and thereby contribute to the initiation and resolution of airway inflammation. These include pro-inflammatory cytokines such as interleukin-1 beta (IL-1β), tumour necrosis factor alpha and anti-inflammatory cytokines such as macrophage inhibitory protein (Chung & Barnes, 1999). Fibroblasts also release multiple growth factors such as transforming growth factor beta (TGFβ), which directly influence the deposition and composition of the ECM (Chung & Barnes, 1999) and therefore may control multiple processes in wound repair (Border & Noble, 1994). However the regulatory role and relative importance of other more recently identified cytokines is far from clear and needs to be assessed.
OSM is a pleiotropic cytokine that was first described as a product of inflammatory cells such as T-lymphocytes, monocyte/macrophages and more recently neutrophils (Rose & Bruce, 1991; Zarling et al., 1986). OSM is a member of the IL-6 family of cytokines that also includes leukaemia inhibitory factor (LIF), interleukin-11, ciliary neurotrophic factor and cardiotrophin-1 (Taga, 1996; 1997). These cytokines produce a variety of overlapping and specific biological effects related to the regulation of inflammation. Recent studies have examined the localization of these cytokines and their role in airway inflammation (Knight et al., 1999a; 1999b). To date OSM has been shown to upregulate the production of lung epithelial cell anti-proteases (Cichy et al., 1998) and PGE2 (Knight et al., 2000) as well as the production of TIMP-1 in human lung fibroblasts (Richards et al., 1993). In dermal fibroblasts, OSM is an activator of the collagen α2 (1) promoter (Ihn et al., 1997) and potently induces collagen and glycosaminoglycan production (Duncan et al., 1995). Furthermore, mice with targeted overexpression of OSM to the pancreas, developed severe localized fibrotic lesions (Bamber et al., 1998). These studies suggest that this cytokine may have wound healing or profibrotic properties.
In the current study we have examined the effects of OSM on human lung fibroblast proliferation, pro-collagen synthesis and spontaneous apoptosis. The results demonstrate that OSM is a mitogen for these cells, induces procollagen production and is anti-apoptotic. Taken together, these results suggest that OSM may play an important role in the wound healing response, asthma and fibrosis.
Methods
Cytokines and chemicals
Recombinant human OSM and anti-gp130 antibody were purchased from R&D systems (Minneapolis, MN, U.S.A.). TGFβ1, indomethacin, DAPI, Propidium Iodide (PI) and rat-tail collagen were purchased from Sigma (Castle Hill, NSW, Australia). IL-1β and Annexin V-FITC were purchased from Boehringer Mannheim (Melbourne, Vic, Australia). Genestein and PD98059 were purchased from Calbiochem (La Jolla, CA, U.S.A.). Nimesulide and monoclonal antibodies recognizing COX-1 and COX-2 were purchased from Cayman Chemical Co (Ann Arbor, MI, U.S.A.). Peroxidase-conjugated secondary antibodies were purchased from Dako (Carpintaria, NSW, Australia). Alexa-fluor conjugated secondary antibodies were purchased from Molecular Probes (Brisbane, QLD, Australia). Monoclonal antibodies recognizing cyclin DI and IL-6 were purchased from Santa Cruz and Pharmingen (La Jolla, CA, U.S.A.). MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2H-tetrazolium, inner salt)] was purchased from Promega (Madison, WI, U.S.A.). [3H]-thymidine was purchased from Amersham (Sydney, NSW, Australia). The polyclonal antibody-OSMR antibody was a kind gift from Immunex Corp. (Seattle, WA, U.S.A.).
Cell culture
The normal diploid human foetal lung fibroblast cell line was obtained from American Type Culture Collection (ATCC, Rockville, MD, U.S.A) and was used between passages 11 and 24. Primary human lung fibroblasts were established from explant cultures (Keerthisingam et al., 2001). Cells were cultured in T-75 tissue culture flasks with DMEM medium (Life Technologies, Melbourne, VIC, Australia) supplemented with 10% foetal calf serum (FCS), 2 mM L-glutamine, 100 μg ml−1 penicillin and gentamycin. Cells were kept in a humidified atmosphere of 5% CO2 in air at 37°C. Medium was routinely changed every 3 to 4 days.
Analysis of OSMR expression
Flow cytometry was used to confirm the presence of OSMR on the cell surface. Fibroblasts (1×106 ml−1) were cultured in six well culture plates in 0.4% FCS/DMEM and allowed to adhere for 16 h as described above. Cells were detached from the well with either Trypsin or EDTA and resuspended in serum free DMEM supplemented with 1% BSA. Cells were washed three times in PBS (phosphate-buffered saline) containing 0.1% Tween-20 incubated with rabbit anti-human OSMR (1 : 1000) before incubation with FITC-conjugated goat α rabbit IgG (1 : 500) at 4°C for 30 min. Cells were washed, resuspended in serum free DMEM and analysed using a FACScan flow cytometer.
Cell proliferation assays
Fibroblasts were seeded in DMEM/10% FCS at a density of 3×104 cells per well in 24 well tissue culture plates and allowed to adhere for 24 h. The cells were the quiesced by replacing the medium with serum free DMEM for a further 24 h before being treated with either OSM at 0.5 – 100 ng ml−1 at 37°C for 24 or 48 h. Experiments were also performed in the presence of pharmacological inhibitors. These inhibitors were added 30 min prior to the addition of OSM (2 ng ml−1). At the end of each incubation period, proliferation was assessed using an MTS assay. This assay measures mitochondrial activity through the formation of an insoluble formazan salt and has been shown to correlate to cell density (Martinez et al., 1999). MTS was diluted 1 : 20 with serum free DMEM and 300 μl was added to each well. After 1 h incubation, cell proliferation was assessed at 450 nm in a Spectramax spectrophotometer (Molecular Devices Co., CA, U.S.A.). Results were expressed as absorbance or as a percentage change in absorbance compared to control (i.e. cells with medium alone). Direct cell counts and tritiated thymidine ([3H]-TdR) incorporation were also performed to confirm the results of the MTS assay. [3H]-TdR incorporation assays were performed by adding [3H]-TdR (1 μCi ml−1) to each well 6 h after the addition of treatments. The cells were incubated at 37°C for 18 h. Cells were then harvested using a cell harvester and radioactivity incorporation assessed using a Packard direct β counter (Canberra, NSW, Australia). For cell counts, cells were stained with Trypan blue and the number of viable cells counted using a Neubauer Haemocytometer.
IL-6 production
Following exposure of fibroblasts to OSM, supernatants were analysed for IL-6 using a specific enzyme-linked immunosorbent assay (ELISA). Briefly, 96 well plates (Maxisorp, Nunc) were coated with mouse anti-human-IL-6 (500 ng ml−1 in 0.1 M NaHCO3/NaCO3 buffer, pH 9.6) and incubated overnight at 4°C. After rinsing in PBS containing 0.5% v v−1 Tween-20, wells were blocked by addition of 1% (w v−1) BSA in PBS/Tween-20. After multiple washings, 100 μl of sample were added and the plate allowed to incubate at 4°C overnight. After washing, a biotinylated anti-mouse IgG antibody was added and the plate allowed to incubate at room temp for 1 h. Peroxidase-labelled streptavidin (S-HRP) was then added for a further 30 min and the plates washed three times in PBS/Tween-20. Peroxidase substrate (K-blue ELISA substrate) was added to each well and the reactions terminated by addition of 1 M phosphoric acid. Absorbances were read at 450 nm on an ELISA plate reader Spectramax 250 (Molecular Devices Co., CA, U.S.A.) at 450 nm. The detection limit of the assay was 15 pg ml−1.
PGE2 enzyme immunoassay
PGE2 was measured using a competitive enzyme immunoassay according to the manufacturer's instructions (Cayman Chemical, Ann Arbor, MI, U.S.A.). Plates were read at 450 nm using an ELISA plate reader Spectramax 250 (Molecular Devices Co., CA, U.S.A.). The manufacturer's specifications for this assay include an intra-assay coefficient of variation of P<10%, cross reactivity with PGD2 and PGF2α of less than 1% and linearity over the range of 10 – 1000 pg ml1
Western blotting
The effect of OSM on the expression of COX-1 or COX-2 in fibroblasts was assessed by Western analysis. Trypsinized cells (2×106) were incubated with lysis-buffer (50 mM Tris, 0.5 mM EGTA, 150 mM NaCl, 1% Triton X-100, pH 7.5) containing a cocktail of protease inhibitors (Sigma), passed through a fine gauge needle and centrifuged at 104×g for 20 min. The protein content of the resultant supernatant was determined using the Bradford method (BioRad). Equal amounts of protein (40 μg) were added to SDS-sample buffer (0.5 M Tris-HCl, 1% glycerol, 0.5% bromophenol blue, 0.5% β-mercaptoethanol), boiled for 5 min and electrophoresed through 12% polyacrylamide gel. Proteins were electroblotted onto PVDF membranes (BioRad, Sydney, NSW, Australia), which were blocked overnight in TTBS (tris-buffered saline/Tween-20) containing 5% skim milk. Membranes were probed with a mouse monoclonal antibody to human COX-1 or COX-2 for 1 h at a dilution of 1 : 1000. After repeated washes in TTBS, horse radish-peroxidase (HRP)-conjugated anti-mouse IgG antibody (1 : 1000) was added for 1 h. After further washing in TTBS, blots were developed with the ECL detection system and exposed to ECL-Hyperfilm.
Analysis of apoptosis
Following 24 or 48 h incubation (37°C, 5% CO2 in air) with and without OSM (2 ng ml−1), fibroblasts were harvested and stained for the differential analysis of apoptotic and necrotic cells according to the manufacturer's protocol (Boehringer Mannheim). Briefly, 20 μl each of Annexin V-FITC and PI (50 μg ml−1) was added per 1 ml of labelling buffer (10 mmol l−1 HEPES, 140 mmol l−1 NaCl, 5 mmol l−1 CaCl2, pH 7.4). Labelling solution (100 μl) was added to fibroblasts (2×105 per tube) and incubated in the dark (room temp, 50 min). Fibroblasts were washed in PBS and immediately analysed on a FACScan flow cytometer using CellQuest software (Becton Dickinson, San Jose, CA, U.S.A.). Ten thousand cells were acquired. Apoptotic cells stained positively for Annexin V (AV) but excluded PI (AV+PI−), whilst necrotic cells were double positive (AV+PI+).
Determination of fibroblast procollagen production
Fibroblast procollagen production was assessed in vitro by quantitating hydroxyproline (hyp) using reverse-phase high pressure liquid chromatography (HPLC) as previously described (Campa et al., 1990; Chambers et al., 1998). Briefly, cells were grown to confluence in 2.4 cm diameter wells in DMEM supplemented with 10% FCS and preincubated in DMEM supplemented with 1% FCS, 50 g ml−1 ascorbic acid (BDH, Poole, U.K.) and 0.2 mM proline (Sigma) at 37°C for 24 h. The cells were then exposed to 10 and 100 ng ml−1 OSM or 1 ng ml−1 TGF-β1 in similar 1% FCS supplemented culture medium (n=10 for each dose: four for cell counts and six for hyp measurement) for a further 48 h. For hyp measurement, the cell layer was scraped into the culture medium and the contents of each well collected. Wells were washed with 1 ml PBS and combined with the original medium. Proteins were precipitated by the addition of ethanol to a final concentration of 67% (v v−1) at 4°C overnight. Precipitated proteins were separated from free amino acids by filtration through a 0.45 mm pore-size filter (Millipore, Watford, Herts, U.K.) and hydrolyzed in 6 N HCl at 110°C for 16 h. Hydrolysates were mixed with activated charcoal, filtered and hyp quantified using reverse-phase-HPLC (Beckman, System Gold, High Wycombe, Bucks., U.K.) after derivatization with 7-chloro-4-nitrobenzofuran (Sigma) as previously described (Chambers et al., 1998). The hyp content in each sample was determined by comparing peak areas of samples from the chromatogram to those generated from standard solutions. All values obtained were corrected for the amount of hyp measured in the cell layer and culture medium at the onset of the incubation period and for cell number. Procollagen production is expressed as nM of hyp 106 cells−1.
Statistical analysis
Data were expressed as mean±s.e.mean. Statistical comparisons of mean data were performed using a statistical t-test or a one-way ANOVA with post-hoc Bonferroni correction for multiple comparisons. A P value of <0.05 was considered significant.
Results
Detection of OSMR on lung fibroblasts
FACS analysis with specific OSMR antibodies shows that fibroblasts express specific OSMR on their cell surface (Figure 1).
Figure 1.
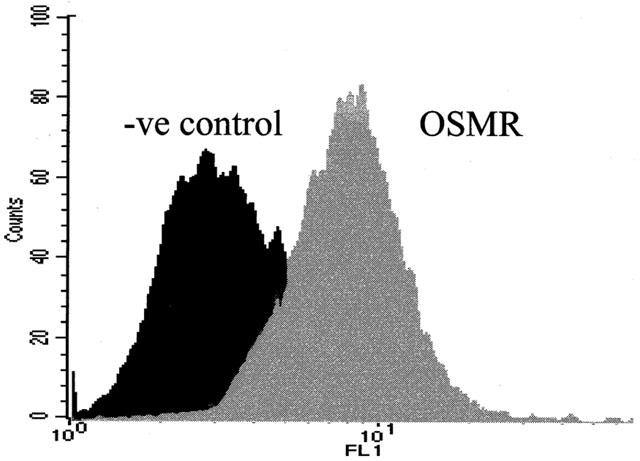
Detection of the OSMR on lung fibroblasts. Cells were stained with no antibody or with anti-OSMR followed by FITC-labelled goat anti-rabbit IgG. Immunostaining was then analysed by flow cytometry.
Effect of OSM on the proliferation of lung fibroblasts
OSM increased the mitotic activity of fibroblasts in a time and dose dependent manner with a maximal response of 57% above control at 2 ng ml−1 after 48 h (Figure 2A) (P<0.05). These findings were consistent with those obtained either by [3H]-TdR incorporation (Figure 2B) and direct cell counting (Table 1). Proliferative responses to OSM were completely abrogated by pre-incubating the cells with specific neutralizing antibodies against OSMR or gp 130 (Figure 2C). Fibroblasts were also stained with antibodies to cyclin D1 (Figure 3). Incubation of fibroblasts with OSM (2 ng ml−1) induced an increase in the number of cyclin D1 positive cells (Figure 3E) compared to unstimulated fibroblasts (Figure 3D).
Figure 2.
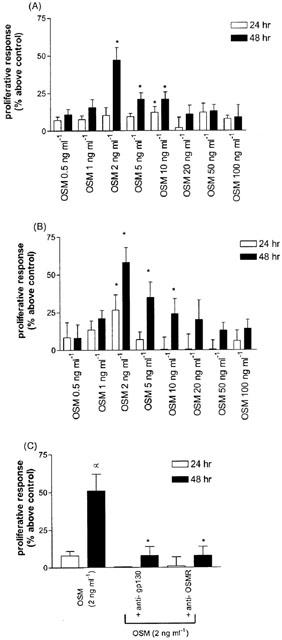
Proliferation of human lung fibroblasts in response to OSM. Cells were exposed to OSM at concentrations ranging from 0.5 – 100 ng ml−1 for 24, 48 or 72 h. Proliferative effect was examined using an MTS assay (A) and confirmed by [3H]-TdR uptake (B). The proliferative effect of OSM was completely abrogated by incubation of cells with neutralizing antibodies against OSMR and gp 130. Results are means of four separate experiments performed in triplicate. *P<0.05 compared to cells in serum free conditions.
Table 1.
Cell counts of human lung fibroblasts after incubation with OSM

Figure 3.
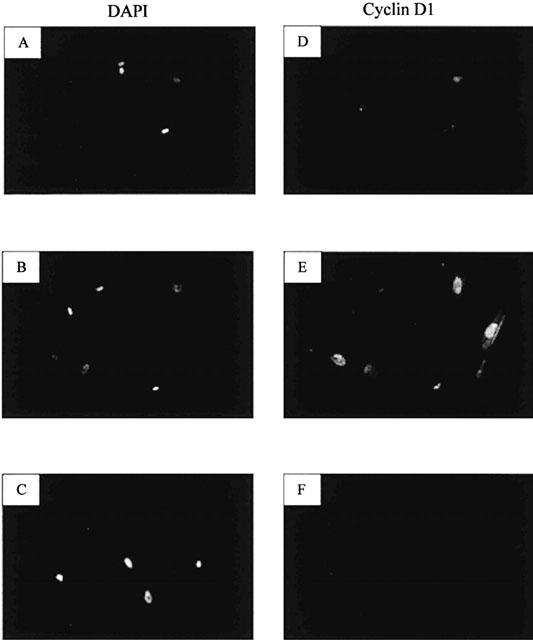
Detection of cyclin D1 expression in lung fibroblasts. Immunohistochemical staining for cyclin D1 was performed on unstimulated cells (D) or cells exposed to OSM (2 ng ml−1) for 24 h (E). The specificity of the cyclin D1 antibody was confirmed by substituting the primary antibody with non-immune mouse IgG1 (F). Cells were also stained with DAPI in order to enumerate cell nuclei (A – C). Immunofluorescent images from 1 μm sections were obtained by scanning laser confocal microscopy at 40×magnification.
Effects of p42/44 MAPK and tyrosine kinase inhibition on OSM-induced proliferation
OSM induces cellular responses via tyrosine phosphorylation of multiple intracellular proteins (Heinrich et al., 1998). Therefore the effect of the tyrosine kinase inhibitor genestein (10 μM) on OSM-induced mitogenesis was investigated. Incubation of fibroblasts with genestein completely abolished the proliferative effects of OSM (P<0.05) (Figure 4). In order to examine the role of p42/p44 MAPK in OSM-induced proliferation, fibroblasts were treated with the MEK inhibitor PD98059 (50 μM) for 1 h before the addition of OSM for 24 and 48 h. This concentration is consistent with the IC50 values of PD98059 for MEK1 (4 μM) and MEK2 (50 μM) in other cell systems (Xiao et al., 2001). The proliferative responses of these cells were reduced compared to OSM alone at both time points (Figure 4). However the proliferative effects of OSM was not reduced to the same degree when the tyrosine kinase pathway was abolished.
Figure 4.
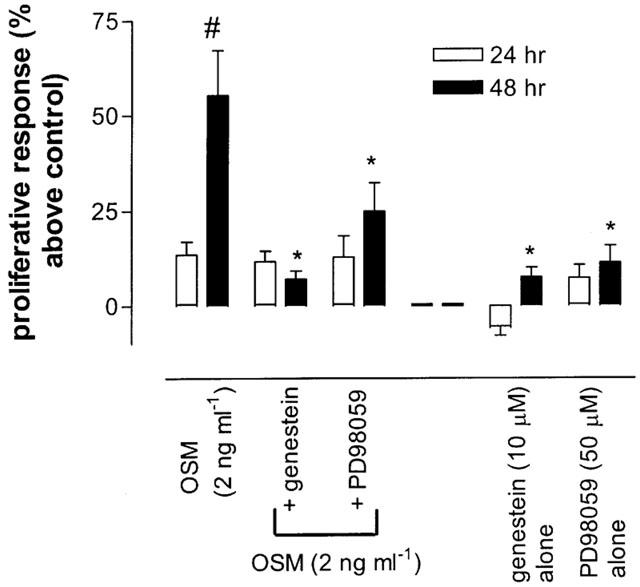
Effects of the tyrosine kinase inhibitor (genestein) and p42/44 MAPK inhibitor (PD98059) on OSM-induced proliferation. Fibroblasts were seeded in 24 well plates and incubated with genestein (10 μM) or PD98059 (50 μM) in the presence or in the absence of 2 ng ml−1 OSM. At 24 and 48 h cell numbers were assessed using an MTS assay. Results are means of four separate experiments performed in triplicate. *P<0.05 versus cells exposed to OSM 2 ng ml−1. #P<0.05 compared to 24 h time point.
Effect of inhibition of COX-2 and PGE2 release on OSM-induced proliferation
OSM has been shown to be a COX-2 dependent mitogen for vascular smooth muscle cells (Bernard et al., 1999), while in other cells, PGE2 is a negative regulator of cell growth (Belvisi et al., 1998). Thus, COX-2 expression and PGE2 release in response to OSM were investigated by EIA and Western blotting. The non-selective COX inhibitor indomethacin (5 μM) and the COX-2 selective inhibitor, nimesulide (5 μM) had no effect on fibroblast proliferation alone and did not alter OSM-induced proliferation (Figure 5a). Furthermore, OSM did not appear to induce the expression of COX-1 or COX-2 (Figure 5b) or the production of PGE2 (Figure 5c).
Figure 5.
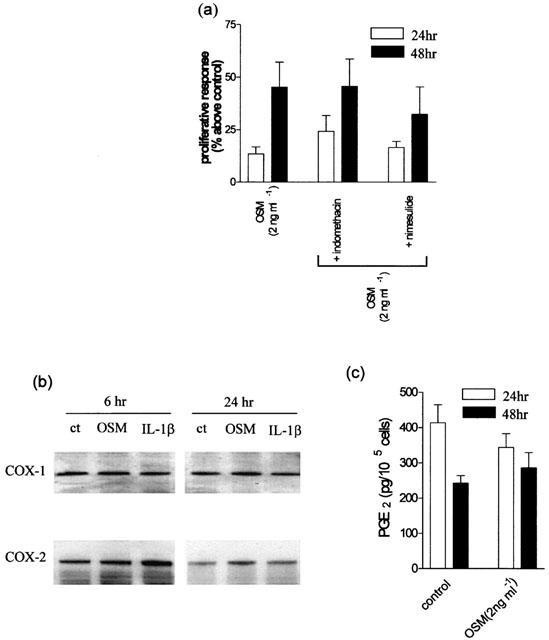
Effects of the non-selective COX inhibitor indomethacin and the selective COX-2 inhibitor nimesulide on OSM-induced proliferation, COX expression and PGE2 release. (a) Cells were treated with indomethacin (5 μM) or the selective COX-2 inhibitor nimesulide (5 μM) prior to incubation with OSM (2 ng ml−1). Proliferative responses to OSM were not influenced by either drug at 24 or 48 h. Detection of COX-1 and COX-2 protein. Fibroblasts were incubated for either 6 or 24 h with OSM (2 ng ml−1) or IL-1β (2 ng ml−1) and expression of both COX-1 and COX-2 was determined by Western analysis (b). PGE2 release in response to OSM. PGE2 release was measured by EIA in supernatants taken from fibroblasts treated with OSM (2 ng ml−1) for 24 and 48 h (c). Results are shown as means of four separate experiments. *P<0.05 compared to cells in serum free medium.
Effect of endogenous IL-6 production on OSM-induced proliferation
In other cell types OSM is a potent regulator of IL-6 production (Bernard et al., 1999; Brown et al., 1991) and in turn IL-6 has been shown to be a mitogen for human fibroblasts (Roth et al., 1995). Exposure of fibroblasts to OSM for either 24 or 48 h did not evoke the release of measurable quantities of IL-6 (data not shown).
Effect of OSM on spontaneous apoptosis
Human lung fibroblasts were incubated with OSM (20 ng ml−1) for 24 or 48 h. Using Annexin V, we confirmed that at these time points OSM significantly decreased the percentage of spontaneous apoptotic cells (Table 1). At 24 h spontaneous apoptosis was 9.3±1.2% in untreated cells and 7.4±0.6% in the OSM treated cells. At 48 h, spontaneous apoptosis had increased to 16.3±3.6% in control cells, compared to 9.9±1.79% in the OSM treated cells (P<0.05). The number of necrotic fibroblasts tended to increase with time in culture, but represented less than 5% of the total cell number in all experiments (data not shown).
The effect of OSM on procollagen production
The effect of OSM on fibroblast procollagen production was assessed by measuring hyp levels using HPLC. The concentrations of OSM used were those previously shown to be optimal for procollagen production by mesenchymal cells (Duncan et al., 1995; Levy et al., 2000). OSM increased procollagen production at 10 and 100 ng ml−1 by 35±2% and 22±2% respectively above control although this increase was only significant at 10 ng ml−1 (P<0.05; Figure 6). For comparison, an optimal stimulatory dose of 1 ng ml−1 TGFβ1 was used as a positive control and stimulated fibroblast procollagen production by 224±10% (P<0.05).
Figure 6.
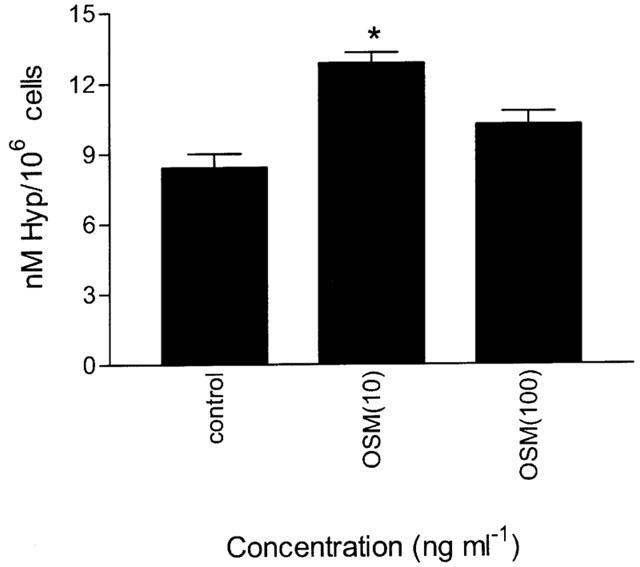
Effect of OSM on human lung fibroblast pro-collagen production. Fibroblasts were grown to confluence and incubated with 10 or 100 ng ml−1 OSM for 48 h and the production of hyp measured by HPLC. Data are means of six independent experiments. *P<0.05 compared to cells in serum free medium.
Discussion
OSM is a growth regulatory cytokine and is synthesized by a number of circulating inflammatory cells, including T-lymphocytes, macrophages and neutrophils (Rose & Bruce, 1991; Grenier et al., 1999). Although OSM has been shown to inhibit the proliferation of cells derived from solid tumours as well as normal and malignant mammary epithelium (Liu et al., 1998), it is also a potent mitogen for dermal fibroblasts (Ihn & Tamaki, 2000), endothelial cells (Pourteau et al., 1999) and vascular smooth muscle cells (Grove et al., 1993). Furthermore, OSM stimulates extracellular matrix production in dermal fibroblasts, hepatic stellate cells and in the pancreas (Duncan et al., 1995, Ihn et al., 1997; Bamber et al., 1998; Levy et al., 2000) suggesting that it may be involved in a variety of fibroproliferative diseases. Whether OSM has similar effects on human lung fibroblasts is unknown. In the current study, we have demonstrated that OSM, acting through its specific type II receptor (1) is a mitogen, (2) is anti-apoptotic (3) stimulates collagen production from human lung fibroblasts.
OSM has profound effects on the behaviour of dermal fibroblasts (Ihn & Tamaki, 2000) as well as synovial and lung fibroblasts (Richards et al., 1997), suggesting that OSMR are present on fibroblasts. However, OSM has also been shown to induce cellular responses by engaging and activating the LIFR, which has also been demonstrated on human lung fibroblasts (Knight et al., 1999b; Mosley et al., 1996). It was therefore important to confirm the presence of OSMR on lung fibroblasts. In the current study we have identified specific OSMR on the cell surface by flow cytometry and immunohistochemistry.
Proliferation of human lung fibroblasts in response to OSM were completely abrogated by specific antibodies against OSMR and gp130, which confirmed that OSM preferentially engages and activates its specific type II receptor. The maximal effect observed was at a concentration of 2 ng ml−1. This mitogenic effect is consistent with a recent study, which showed that OSM induced the proliferation of dermal fibroblasts (Ihn & Tamaki, 2000).
The signal transduction pathways utilized by OSM have traditionally been thought to involve tyrosine phosphorylation of multiple proteins including the Janus kinase (JAK)-STAT pathway (Heinrich et al., 1998). To examine the role of the tyrosine kinase activation in OSM-induced mitogenesis, we treated fibroblasts with the specific tyrosine kinase inhibitor genestein. In keeping with previous studies (Li & Zafarullah, 1998; Heinrich et al., 1998), our results demonstrate that OSM-induced mitogenesis of these cells was completely inhibited by genestein. Phosphorylation of tyrosine residues on the OSMR also provides binding sites for a variety of signalling molecules containing SH2 domains, including those that activate the ras-MAPK pathway. In mammalian cells, three distinct MAPK families exist, p42/p44 MAPK, p38 MAPK and JNK (Gutkind, 1998). However, it is the p42/p44 MAPK that plays a pivotal role in stimulating fibroblasts to re-enter the cell cycle (Pages et al., 1993). Activation of p42/p44 MAPK occurs through phosphorylation of threonine and tyrosine residues under the control of the enzyme MAP kinase kinase (MEK) 1/2. In this study we used the MEK-1/2 inhibitor PD98059, which prevents down-stream activation of p42/p44 MAPK, but does not inhibit p38 MAPK or JNK. Incubation of fibroblasts with PD98059 markedly diminished OSM-induced mitogenesis, strongly suggesting that OSM stimulates the proliferation of fibroblasts via a MAPK-dependent pathway. Similar findings have recently been reported for dermal fibroblasts (Ihn & Tamaki, 2000).
Bornfeldt et al. (1997) recently demonstrated that the MAPK pathway can either mediate or inhibit cell proliferation, depending on the induction of down-stream events, most notably COX-2 meditated production of PGE2. Indeed, PGE2 has been demonstrated to be a potent inhibitor of fibroblast (Mcanulty et al., 1997) and airway smooth muscle proliferation (Belvisi et al., 1998). This is even more pertinent since OSM induces COX-2 expression in vascular endothelial cells (Pourteau et al., 1999; Bernard et al., 1999). However, in the current study, incubation of fibroblasts with the selective COX-2 inhibitor nimesulide or the non-selective inhibitor indomethacin did not influence OSM-induced proliferation, suggesting that the mitogenic effect of OSM was not mediated by COX activation and PGE2 production. This was confirmed by Western analysis of COX-1 and COX-2 expression and by measurement of PGE2 release following OSM exposure.
OSM is also a potent regulator of IL-6 release from a variety of cell types (Brown et al., 1991). In turn, IL-6 has been shown to be a mitogen for human lung fibroblasts (Roth et al., 1995). However, the results of this study clearly show that OSM does not induce IL-6 production in fibroblasts, suggesting that fibroblast production of IL-6 is not directly involved is OSM-induced proliferation of lung fibroblasts.
Alterations in the proliferation and activation of fibroblasts may influence the progression of wound healing. As such, an increase in viable fibroblast numbers through inhibition of apoptosis may enhance the fibrotic response. However, the role of OSM or related cytokines in apoptosis has received little attention. The results of the current study support this hypothesis since OSM inhibited the development of spontaneous apoptosis in human lung fibroblasts over a 48 h period. We have previously shown that while LIF has no effect on either spontaneous or Fas-induced eosinophil apoptosis, OSM significantly inhibits spontaneous apoptosis in these cells (Zheng et al., 1999). OSM has also been shown to inhibit osteoblast apoptosis by preventing an increase in the expression of the pro-apoptotic protein Bax (Jilka et al., 1998). Taken together, these data suggest that OSM may be anti-apoptotic.
Further evidence of a profibrotic role for OSM is its ability to increase collagen in mesenchymal cells (Duncan et al., 1995; Ihn et al., 1997; Bamber et al., 1998, Levy et al., 2000) although the mechanisms involved may differ between cell types. Duncan et al. (1995) demonstrated an increase in both types I and III procollagens and their mRNA transcripts in human dermal fibroblasts and more recently a response element has been characterized in this cell type that mediates OSM stimulation of the human α2(I) collagen promoter (Ihn et al., 1997). OSM also increased collagen protein levels in human hepatic stellate cells (Levy et al., 2000) although an increase in collagen α2(I) mRNA was not detected. As OSM stimulated TIMP-1 production in these cells it was suggested that TIMP-1 was responsible for or contributed to the increased collagen protein by the inhibition of collagen degradation. In the current study OSM also stimulated collagen production in human lung fibroblasts although the increase observed was only moderate when compared to the TGF-β control. The mechanism for this increase is currently being investigated.
In summary we have demonstrated that OSM has a number of effects on fibroblast behaviour, including mitogenesis, inhibition of apoptosis and the capacity to stimulate collagen deposition. These actions of OSM suggest an involvement in limiting the tissue damage of an inflammatory response as well as regulating tissue repair. Coupled with previously demonstrated effects on TIMP-1, plasminogen activator and acute phase proteins and the inhibition of pro-inflammatory cytokines these data suggest that OSM may play an important role in normal wound healing. However, dysregulated production of OSM or its receptors may also play a role in a variety of pathological conditions leading to excessive collagen deposition such as pulmonary fibrosis and asthma.
Table 2.
Spontaneous apoptosis of human lung fibroblasts after incubation with OSM

Acknowledgments
The authors would like to acknowledge the financial support of the National Health and Medical Research Council of Australia and the Asthma Foundation of Western Australia. We also thank Immunex Corp. for the provision of the OSMR antibody and Dr. Neil Misso for his review of the manuscript. Yuben Moodley is the inaugural recipient of the South African Pulmonology Society Post-doctoral Research Fellowship to Australia and New Zealand.
Abbreviations
- BSA
bovine serum albumin
- COX
cyclo-oxygenase
- DAPI
4,6 diamidino-2-phenylindole
- EIA
enzyme immunoassay
- ELISA
enzyme-linked immunosorbent assay
- ECM
extracellular matrix
- FCS
foetal calf serum
- [3H]-TdR
tritiated thymidine
- HPLC
high pressure liquid chromatography
- hyp
hydroxyproline
- IL-6
interleukin-6
- LIF
leukaemia inhibitory factor
- MAPK
mitogen activated protein kinase
- MEK
mitogen activated protein kinase kinase
- MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2H-tetrazolium inner salt)
- OSM
Oncostatin M
- OSMR
OSM receptor
- PBS
phosphate-buffered saline
- PI
propidium iodide
- PGE2
prostaglandin E2
- TGFβ
transforming growth factor beta
- TIMP-1
tissue inhibitor of metalloprotease −1
- TTBS
tris-buffered saline/Tween-20
References
- BAMBER B., REIFE R.A., HAUGEN H.S., CLEGG C.H. Oncostatin M stimulates excessive extracellular matrix accumulation in a transgenic mouse model of connective tissue disease. J. Mol. Med. 1998;76:61–69. doi: 10.1007/s001090050191. [DOI] [PubMed] [Google Scholar]
- BELVISI M.G., SAUNDERS M., YACOUB M., MITCHELL J.A. Expression of cyclo-oxygenase-2 in human airway smooth muscle is associated with profound reductions in cell growth. Br. J. Pharmacol. 1998;125:1102–1108. doi: 10.1038/sj.bjp.0702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERNARD C., MERVAL R., LEBRET M., DELERIVE P., DUSANTER-FOURT I., LEHOUX S., CREMINON C., STAELS B., MACLOUF J., TEDGUI A. Oncostatin M induces interleukin-6 and cyclooxygenase-2 expression in human vascular smooth muscle cells: synergy with interleukin-1 beta. Circ. Res. 1999;85:1124–1131. doi: 10.1161/01.res.85.12.1124. [DOI] [PubMed] [Google Scholar]
- BORDER W.A., NOBLE N.A. Transforming growth factor beta in tissue fibrosis. New Engl. J. Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- BORNFELDT K.E., CAMPBELL J.S., KOYAMA H., ARGAST G.M., LESLIE C.C., RAINES E.W., KREBS E.G., ROSS R. The mitogen-activated protein kinase pathway can mediate growth inhibition and proliferation in smooth muscle cells. J. Clin. Invest. 1997;100:875–885. doi: 10.1172/JCI119603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BREWSTER C.E., HOWARTH P.H., DJUKANOVIC R., WILSON J., HOLGATE S.T., ROCHE W.R. Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 1990;3:507–511. doi: 10.1165/ajrcmb/3.5.507. [DOI] [PubMed] [Google Scholar]
- BROWN T.J., ROWE J.M., LIU J., SHOYAB M. Regulation of IL-6 expression by oncostatin M. J. Immunol. 1991;147:2175–2180. [PubMed] [Google Scholar]
- CAMPA J.S., MCANULTY R.J., LAURENT G.J. Application of high-pressure liquid chromatography to studies of collagen production by isolated cells in culture. Anal. Biochem. 1990;186:25–63. doi: 10.1016/0003-2697(90)90076-l. [DOI] [PubMed] [Google Scholar]
- CHAMBERS R.C., DABBAGH K., MCANULTY R.J., GRAY A.J., BLANC-BRUDE O.P., LAURENT G.J. Thrombin stimulates fibroblast procollagen production via proteolytic activation of protease-activated receptor 1. Biochem. J. 1998;333:121–127. doi: 10.1042/bj3330121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUNG K.F., BARNES P.J. Cytokines in asthma. Thorax. 1999;54:825–857. doi: 10.1136/thx.54.9.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CICHY J., ROSE-JOHN S., TRAVIS J. Oncostatin M, leukemia inhibitory factor and interleukin 6 trigger different effects on α1-proteinase inhibitor synthesis in human lung derived epithelial cells. Biochem. J. 1998;329:335–339. doi: 10.1042/bj3290335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNCAN M.R., HASAN A., BERMAN B. Oncostatin M stimulates collagen and glycosaminoglycan production by cultured normal dermal fibroblasts: insensitivity of sclerodermal and keloidal fibroblasts. J. Invest. Dermatol. 1995;104:128–133. doi: 10.1111/1523-1747.ep12613623. [DOI] [PubMed] [Google Scholar]
- GIZYCKI M.J., ADELROTH E., ROGERS A.V., O'BYRNE P.M., JEFFERY P.K. Myofibroblast involvement in the allergen-induced late response in mild atopic asthma. Am. J. Respir. Cell Mol. Biol. 1997;16:664–673. doi: 10.1165/ajrcmb.16.6.9191468. [DOI] [PubMed] [Google Scholar]
- GRENIER A., DEHOUX M., BOUTTEN A., ARCE-VICIOSO M., DURAND G., GOUGEROT-POCIDALO M.A., CHOLLET-MARTIN S. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood. 1999;93:1413–1421. [PubMed] [Google Scholar]
- GROVE R.I., EBERHARDT C., ABID S., MAZZUCCO C., LIU J., KIENER P., TODARO G., SHOYAB M. Oncostatin M is a mitogen for rabbit vascular smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 1993;90:823–827. doi: 10.1073/pnas.90.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUTKIND J.S. Cell growth control by G protein-coupled receptors: from signal transduction to signal integration. Oncogene. 1998;17:1331–1342. doi: 10.1038/sj.onc.1202186. [DOI] [PubMed] [Google Scholar]
- HEINRICH P.C., BEHRMANN I., MULLER-NEWEN G., SCHAPER F., GRAEVE L. Interleukin 6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IHN H., LEROY E.C., TROJANOWSKA M. Oncostatin M stimulates transcription of the human alpha2(I) collagen gene via the Sp1/Sp3-binding site. J. Biol. Chem. 1997;272:24666–24672. doi: 10.1074/jbc.272.39.24666. [DOI] [PubMed] [Google Scholar]
- IHN H., TAMAKI K. Oncostatin M stimulates the growth of dermal fibroblasts via a mitogen-activated protein kinase-dependent pathway. J. Immunol. 2000;165:2149–2155. doi: 10.4049/jimmunol.165.4.2149. [DOI] [PubMed] [Google Scholar]
- JEFFERY P.K. Morphology of the airway wall in asthma and in chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1991;143:1152–1158. doi: 10.1164/ajrccm/143.5_Pt_1.1152. [DOI] [PubMed] [Google Scholar]
- JILKA R.L., WEINSTEIN R.S., BELLIDO T., PARFITT A.M., MANOLAGAS S.C. Osteoblast programmed cell death (apoptosis)–modulation by growth factors and cytokines. J. Bone Min. Res. 1998;13:793–802. doi: 10.1359/jbmr.1998.13.5.793. [DOI] [PubMed] [Google Scholar]
- KEERTHISINGAM C.B., JENKINS R.G., HARRISON N.K., HERNANDEZ-RODRIGUEZ N.A., BOOTH H., LAURENT G.J., HART S.L., FOSTER M.L., MCANULTY R.J. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am. J. Pathol. 2001;158:1411–1422. doi: 10.1016/s0002-9440(10)64092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNIGHT D.A., ASOKANANTHAN N., WATKINS D.N., MISSO N.L., THOMPSON P.J., STEWART G.A. Oncostatin M synergises with house dust mite proteases to induce the production of PGE(2) from cultured lung epithelial cells. Br. J. Pharmacol. 2000;131:465–472. doi: 10.1038/sj.bjp.0703612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNIGHT D.A., CARROLL N.G., LENZO J.C., MCKAY K.O., JAMES A.L., THOMPSON P.J. Immunohistochemical localisation of oncostatin M receptors in human airways: altered pattern of expression in asthma. Eur. Respir. J. 1999a;14:528s. [Google Scholar]
- KNIGHT D.A., LYDELL C.P., ZHOU D., WEIR T.D., SCHELLENBERG R., BAI T.R. Leukemia inhibitory factor (LIF) and LIF receptor in human lung. Distribution and regulation of LIF release. Am. J. Respir. Cell Mol. Biol. 1999b;20:834–841. doi: 10.1165/ajrcmb.20.4.3429. [DOI] [PubMed] [Google Scholar]
- LEVY M.T., TROJANOWSKA M., REUBEN A. Oncostatin M: a cytokine upregulated in human cirrhosis increases collagen production by human hepatic stellate cells. J. Hepatol. 2000;32:218–226. doi: 10.1016/s0168-8278(00)80066-5. [DOI] [PubMed] [Google Scholar]
- LI W.Q., ZAFARULLAH M. Oncostatin m up-regulates tissue inhibitor of metalloproteinases-3 gene expression in artricular chondrocytes via de novo transcription, protein synthesis, and tyrosine kinase-and mitogen-activated protein kinase-dependent mechanisms. J. Immunol. 1998;161:5000–5007. [PubMed] [Google Scholar]
- LIU J.W., HADJOKAS N., MOSLEY B., ESTROV Z., SPENCE M.J., VESTAL R.E. Oncostatin M-specific receptor expression and function in regulating cell proliferation of normal and malignant mammary epithelial cells. Cytokine. 1998;10:295–302. doi: 10.1006/cyto.1997.0283. [DOI] [PubMed] [Google Scholar]
- MARTINEZ E.J., OWA T., SCHREIBER S.L., COREY E.J. Pthalascidin, a synthetic anti-tmour agent with potency and mode of action comparable to ecteinascidin 743. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3496–3501. doi: 10.1073/pnas.96.7.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCANULTY R.J., HERNANDEZ-RODRIGUEZ N.A., MUTSAERS S.E., COKER R.K., LAURENT G.J. Indomethacin suppresses the anti-proliferative effects of transforming growth factor-beta isoforms on fibroblast cell cultures. Biochem. J. 1997;321:639–643. doi: 10.1042/bj3210639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOSLEY B., IMUS C.D., FRIEND D., BOIANI N., THOMA B., PARK L.S., COSMAN D. Dual oncostatin M receptors. J. Biol. Chem. 1996;50:32636–32643. doi: 10.1074/jbc.271.51.32635. [DOI] [PubMed] [Google Scholar]
- PAGES G., LENORMAND P., L'ALLEMAIN G., CHAMBARD J.C., MELOCHE S., POUYSSEGUR J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POURTEAU J., MIRSHAHI F., LI H., MURAINE M., VINCENT L., TEDGUI A., VANNIER J.-P., SORIA J., VASSE M., SORIA C. Cycloxygenase activity is necessary for the angiogenic properties of oncostatin M. FEBS Lett. 1999;459:1–5. doi: 10.1016/s0014-5793(99)01301-0. [DOI] [PubMed] [Google Scholar]
- RICHARDS C.D., KERR C., TANAKA M., HARA J., MIYAJIMA A., PENNICA D., BOTELHO F., LANGDON C.M. Regulation of tissue inhibitor of metalloproteinase-1 in fibroblast and acute phase proteins in hepatocytes in vitro by mouse oncostatin M, cardiotrophin-1, and IL-6. J. Immunol. 1997;159:2431–2436. [PubMed] [Google Scholar]
- RICHARDS C.D., SHOYAB M., BROWN T.J., GAULDIE J. Selective regulation of metalloproteinases inhibitor (TIMP-1) by Oncostatin M in fibroblasts in culture. J. Immunol. 1993;150:5596–5603. [PubMed] [Google Scholar]
- ROSE T.M., BRUCE A.G. Oncostatin M is a member of a cytokine family that includes leukemia-inhibitory factor, granulocyte colony-stimulating factor, and interleukin 6. Proc. Natl. Acad. Sci. U.S.A. 1991;88:8641–8645. doi: 10.1073/pnas.88.19.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTH M., NAUCK M., TAMM M., PERRUCHOUD A., ZIESCHE R., BLOCK L. Intracellular interleukin 6 mediates platelet-derived growth factor-induced proliferation of non-transformed cells. Proc. Natl. Acad. Sci. 1995;92:1312–1316. doi: 10.1073/pnas.92.5.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAGA T. gp130, a shared signal transducing receptor component for hematopoietic and neuropoietic cytokines. J. Neurochem. 1996;67:1–10. doi: 10.1046/j.1471-4159.1996.67010001.x. [DOI] [PubMed] [Google Scholar]
- TAGA T. gp130 and the interleukin-6 family of cytokines. Ann. Rev. Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- XIAO L., PIMENTAL D.R., AMIN J.K., SINGH K., SAWYER D.B., COLUCCI W.S. MEK 1/2-ERK mediates α1-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell. Cardiol. 2001;33:779–787. doi: 10.1006/jmcc.2001.1348. [DOI] [PubMed] [Google Scholar]
- ZARLING J.M., SHOYAB M., MARQUARDT H., HANSON B., LIOUBIN N., TODARO G.J. Oncostatin M: A growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 1986;83:9739–9743. doi: 10.1073/pnas.83.24.9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG X., KNIGHT D., ZHOU D., WEIR T., PEACOCK C., SCHELLENBERG R.R., BAI T. Leukemia inhibitory factor is synthesized and released by human eosinophils and modulates activation state and chemotaxis. J. Allergy Clin. Immunol. 1999;104:136–144. doi: 10.1016/s0091-6749(99)70125-9. [DOI] [PubMed] [Google Scholar]