

Abstract
Soluble guanylyl cyclase (sGC) is the only proven receptor for the ubiquitous biological messenger nitric oxide (NO) and is intimately involved in many signal transduction pathways, most notably in regulating vascular tone and platelet function. sGC is a heterodimeric (α/ß) protein that converts GTP to cyclic GMP; NO binds to its prosthetic haem group. Here, we report the discovery of a novel sGC activating compound, its interaction with a previously unrecognized regulatory site and its therapeutic implications.
Through a high-throughput screen we identified BAY 58-2667, an amino dicarboxylic acid which potently activates sGC in an NO-independent manner. In contrast to NO, YC-1 and BAY 41-2272, the sGC stimulators described recently, BAY 58-2667 activates the enzyme even after it has been oxidized by the sGC inhibitor ODQ or rendered haem deficient.
Binding studies with radiolabelled BAY 58-2667 show a high affinity site on the enzyme.
Using photoaffinity labelling studies we identified the amino acids 371 (α-subunit) and 231 – 310 (ß-subunit) as target regions for BAY 58-2667.
sGC activation by BAY 58-2667 results in an antiplatelet activity both in vitro and in vivo and a potent vasorelaxation which is not influenced by nitrate tolerance.
BAY 58-2667 shows a potent antihypertensive effect in conscious spontaneously hypertensive rats. In anaesthetized dogs the hemodynamic effects of BAY 58-2667 and GTN are very similar on the arterial and venous system.
This novel type of sGC activator is a valuable research tool and may offer a new approach for treating cardiovascular diseases.
Keywords: BAY 41-2272, BAY 58-2667, blood pressure, cyclic GMP, haemodynamics, nitrate tolerance, nitric oxide, photoaffinity labelling, soluble guanylate cyclase, YC-1
Introduction
Soluble guanylyl cyclase (sGC), the intracellular receptor for the ubiquitous biological messenger nitricoxide (NO), is a heterodimer consisting of α- and ß-subunits with haem as prosthetic group. Activation of the enzyme facilitates conversion of GTP to the intracellular second messenger cyclic GMP, which regulates various cyclic GMP effector systems such as phosphodiesterases, ion channels and protein kinases. Thus, the NO/cyclic GMP pathway is important in many physiological processes including vasodilatation, neurotransmission and platelet aggregation (Furchgott, 1999; Murad, 1999; Ignarro, 1999; Moncada et al., 1991). Due to its ubiquitous nature, the pathogenesis of various disease states, especially of the cardiovascular system, has been linked to inappropriate activation of sGC (Hobbs, 2000). Activators of sGC are therefore very desirable as both pharmacological tools to probe the NO-cGMP pathway and as potential therapeutics.
The best studied classes of sGC activators are organic nitrates which mimic the action of endogenous NO by bioconversion to NO and NO-related compounds which nitrosylate the haem of sGC. Organic nitrates have been used for decades as treatment for angina pectoris; however, this therapy suffers from the development of tolerance upon prolonged use and the absence of clinically relevant antiplatelet activity (Feelisch, 1998). Therefore, there is a need for novel activators of sGC. An indazole derivative, YC-1 has been described which stimulates sGC directly and sensitizes the enzyme towards its native activators NO and CO (Ko et al., 1994; Wu et al., 1995; Friebe et al., 1996; Mülsch et al., 1997; Hoenicka et al., 1999). The exact mechanism of YC-1 dependent stimulation is not fully understood and is currently a subject of intense investigation (Sharma et al., 1999, Koesling, 2000; Zhao et al., 2000; Martin et al., 2001).
Recently we discovered the new sGC stimulator BAY 41-2272, similar to YC-1 but about two orders of magnitude more potent (Stasch et al., 2001; Straub et al., 2001). Using a photoaffinity label approach we identified the cysteine 238 and cysteine 243 spanning region in the α-subunit of sGC as part of the target site for this new type of NO-independent, but haem-dependent sGC stimulator (Stasch et al., 2001; Becker et al., 2001). BAY 41-2272 is a potent vasodilator of aortic rings in vitro and reduces the mean arterial blood pressure in normal and hypertensive rats (Straub et al., 2001). These effects are probably mediated both by NO-independent stimulation of the enzyme and by sensitization of sGC towards endogenous NO. Very recently, the pharmacological in vitro and in vivo profile of a novel NO-independent sGC stimulator BAY 41-8543 which is structurally related to BAY 41-2272 has been described (Stasch et al., 2002a, 2002b).
Through a high-throughput screening of around 250,000 compounds using a read-out system consisting of a CHO cell line expressing sGC (Becker et al., 1999), a cyclic GMP-sensitive cation channel and aequorin, we unexpectedly identified a class of aminodicarboxylic acids as a new type of sGC activators (Alonso-Alija et al., 2001). Following chemical optimization, we identified BAY 58-2667 as the most potent member of series of compounds. The results presented here show that BAY 58-2667 potently activates sGC in an NO-independent manner; but in contrast to NO, YC-1 and BAY 41-2272, it activates the enzyme even after it has been oxidized by the sGC inhibitor ODQ or rendered haem deficient. Because this novel type of sGC activation is not influenced by nitrate tolerance and because it exhibits potent vasorelaxation, antiplatelet activities, antithrombotic effects and haemodynamic effects like organic nitrates, it may offer a new approach for treating cardiovascular diseases.
Methods
Substances
BAY 58-2667 (4-[((4-carboxybutyl){2-[(4-phenethylbenzyl)oxy] phenethyl}amino) methyl[benzoic]acid) and BAY 41-2272 (5-cyclopropyl - 2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine) were synthesized as described (Alonso-Alija et al., 2001; Straub et al., 2001). Tritium labellings of BAY 58-2667 and its analogue BAY 61-2573 were performed according to a method previously described (Shu & Heys, 2000). The exchange reaction was carried out with the appropriate diesters of BAY 61-2573 and BAY 58-2667. Specific activities of 14.5 Ci mmol−1 (BAY 61-2573) and 5.4 Ci mmol−1 (BAY 58-2667) were obtained. The labelling positions were confirmed by 3H-NMR spectroscopy.
sGC assay
We purified rat sGC by using a baculovirus/Sf9 expression system and measured enzyme activity as described by us (Hoenicka et al., 1999). Briefly, sGC (0.16 μg protein ml−1) was incubated for 10 min at 37°C in a final volume of incubation buffer ((mM): TEA/HCl 50, EGTA 100, IBMX 1, dithiothreitol 1, cyclic GMP 1, creatine phosphate 5, 12.5 U ml−1 creatine phosphokinase, MgCl2 3, 1 mg ml−1 BSA and 0.1 μCi [α-32P] GTP; pH 7.5) in the presence and absence of sGC activator. Incubations were stopped by coprecipitation of 5′-nucleotides with a 400 μl zinc acetate (100 mM) and 500 μl sodium carbonate (120 mM). Following centrifugation (5 min, 2800 g, 4°C), [α-32P]cGMP was isolated from the supernatant by chromatography on neutral aluminia colums. The amount of [α-32P]cGMP was determined by liquid scintillation counting. All measurements were performed in duplicate and were repeated at least three times. Haem-free sGC was prepared by low concentrations (0.5%) of the non-ionic detergent Tween-20 without destruction of basal activity (Hoenicka et al., 1999; Friebe et al., 1996).
Spectroscopic studies
U.v.-visual spectra were recorded from 300 to 600 nm on a DU 640 spectrophotometer (Beckman, Munich, Germany). NO was introduced via an aqueous solution of DEA/NO. A 100 mM stock solution of BAY 58-2667 in DMSO was prepared and added in a final concentration of 10 μM, resulting in a final DMSO concentration of 0.1% (v v−1), not interfering with properties of the enzyme (Hoenicka et al., 1999).
Labelling of purified sGC and sequence analysis
Fifteen μg sGC (100 pmol) were dissolved in PAL buffer ((mM): final TEA/HCl 50, EGTA 0.1, cyclic GMP 1, MgCl2 3, 200 μM GTP, pH 7.4) and incubated with 5, 15 or 50 μCi 3H-PAL (0.3, 0.9 and 3 nmol) in the presence or absence of a 50 fold excess of PAL in a volume of 200 μl (5 min, 37°C). Samples were irradiated at 254 nm (distance 3 cm, 4°C, 30 min), and the reaction was stopped by adding TCA to a final concentration of 10%. Protein was precipitated (4°C, 30 min), centrifuged (14,000 r.p.m., 4°C, 30 min) and washed twice with ice-cold ethanol/ether (1/1=v v−1). The pellets were dissolved in 60 μl Laemmli sample buffer and heated (80°C, 5 min). Separation was performed on a 10% SDS – PAGE (PROTEAN II xi cell, Bio-Rad, München, Germany). After electrophoresis, proteins were fixed for 20 min in methanol/acetic acid/MilliQ water (30/10/60), dried and exposed to BAS-TR 20/25 imaging plates (Fuji Photo Film, Tokyo, Japan) for 15 days as described (Ahr & Steinke, 1994). After exposure, the imaging plates were scanned (BAS 5000, Fuji Photo Film). Evaluation was performed by visual classification of radiographic pseudo-colours intensities. One hundred and fifty μg sGC (1 nmol) were labelled with 485 μCi 3H-PAL (30 nmol), and the CNBr-fragmentation was performed as previously described (Becker et al., 2001). Lyophilized and labelled CNBr fragments were dissolved in 100 μl O'Farrel lysis buffer (O'farrel, 1975). After prefocusing (15 min 200 V, 30 min 300 V, 30 min 400 V) the samples were loaded on tube gels (9.8 M urea/4% (v v−1) acrylamide/2% (v v−1) Nonidet® P-40/4% (v v−1) carrier ampholines 5 – 7/1% (v v−1) carrier ampholines 3 – 10) and focused according to their isoelectric point for 9600 Vh. The tube gels were extruded and equilibrated for 20 min in equilibration buffer (60 mM Tris/HCl pH 6.8/30% (v v−1) glyceryl/3% (w v−1) SDS). The second dimension was run on a 16% Tris/glycine gel (PROTEAN II xi cell, Bio-Rad). Two tube gels without probes were extruded, cut into 10 mm pieces and equilibrated in 1 ml MilliQ water for pH measurement. Finally CNBr fragments were transferred to a PVDF membrane (Trans-Blot® Electrophoretic Transfer Cell, Bio-Rad), stained with 0.025% (w v−1) Coomassie-blue-R/40% (v v−1) MeOH and destained in 50% (v v−1) MeOH (Towbin et al., 1979). Dried membranes were exposed to BAS-TR 20/25 imaging plates (Fuji Photo Film) for 6 h (Ahr & Steinke, 1994; Kolbe & Dietzel, 2000). After exposure, the imaging plates were scanned (BAS 5000, Fuji Photo Film Ltd., Tokyo, Japan) and evaluated as described above. N-terminal sequence analyses were performed using the gas-liquid-solid-phase protein sequencer Procise® with an RP-18-PTH-column from Applied Biosystems (Forster City, CA, U.S.A.). Labelled spots from the 2D-PAGE were cut off and washed twice with 100 μl 50% (v v−1) MeOH before sequencing. Fragment 8 (α) and 5 (β) were sequenced over 82 and 66 cycles, respectively. The single PTH amino acids were collected, lyophilized and radioactivity was counted after combustion (Oxidizer model 387, Oximate 80, Packard, IL, U.S.A.) in a liquid scintillation counter (LS-6500, Beckmann, München, Germany).
Receptor binding study
Ninety-six well filter plates (Millipore FC/B glasfibre, 1 μm) were coated with a solution of 0.5% polyvinylpyrrolidone (PVP, 360 kDa) and 0.1% Tween 20 (3 h, 25°C). Filter plates were washed six times with ice-cold buffer (10 mM Tris, 100 mM NaCl, pH 7.2). sGC (0.8 μg) was incubated in incubation buffer (mM: TEA 50, EDTA 0.1, DTT 1, MgCl2 3, pH 7.5; 5 min, 37°C). BAY 58-2667 and 3H-BAY 58-2667 dissolved in acetonitrile/H2O (60/40=v v−1) were added simultaneously and incubated (10 min, 37°C). Afterwards the samples were cooled (10 min, 4°C), IgG was added to a final concentration of 0.75 μg μl−1 and precipitation was performed with PEG 8000 (final 212 μg μl−1, 30 min, 4°C). Bound and free ligand were separated by filtration and washed twice (10% PEG in incubation buffer).
Rabbit saphenous artery
Chinchilla rabbits (2.0 – 3.3 kg) were sacrificed by an overdose of thiopental. Saphenous artery rings (3 mm width) were suspended under an initial tension of approximately 4 g in 5 ml organ baths containing Krebs – Henseleit solution (containing 0.001% BSA) at 37°C. Contractions were measured isometrically with Statham UC2 strain gauges connected to a DAS1802HC data acquisition board (Keithley Instruments, Germering, Germany). Rings were precontracted by 3×10−8 g ml−1 phenylephrine (submaximal contraction) four times. Each contraction was followed by a series of 16 washing cycles and a resting period of 28 min. The test compounds were added to the organ baths at the beginning of the last resting period. The concentration of the test compounds was increased by a factor of 10. Rings were subsequently contracted by phenylephrine (3×10−8 g ml−1).
Rat heart Langendorff preparation
The hearts of Wistar rats (200 – 250 g) were perfused according to Langendorff at 37°C with a non-recirculating system. The perfusion medium was a filtered Krebs – Henseleit solution containing 11 mmol l−1 glucose and 1.2 mmol l−1 CaCl2, equilibrated with O2+CO2 (95+5%), to give a pH of 7.4 and a pO2 of 650 to 700 mmHg. Perfusion was performed at a constant rate (10 ml min−1). A latex balloon filled with saline and connected to a pressure transducer (Gould Statham, Oxnard, CA, U.S.A.) via a metal cannula was inserted into the left ventricular cavity to measure the isovolumetric contractions of the left ventricle. A second pressure transducer was connected to the aortic cannula in order to record the perfusion pressure. Drug solutions were infused into the aortic cannula at a rate of 1% of the total flow rate.
Measurement of platelet aggregation
Platelet aggregation was measured with an aggregometer (Carat, IDC, Langewiesen) as previously described by us (Becker et al., 2000; Stasch et al., 2002a). In the cuvette, platelet rich plasma or washed platelets were pre-incubated for 10 min at 37°C after the addition of BAY 58-2667 or the vehicle. Aggregation was induced by the addition of collagen, U 46619, thrombin, TRAP-6, or ADP. The concentration of the agonists was individually adjusted to achieve maximal aggregation response. In platelet rich plasma the final concentrations of collagen, U 46619, ADP and TRAP-6 were 1 – 2, 1, 2 – 5 and 30 – 50 μg ml−1, respectively, and in washed platelets the final concentration of thrombin was 5 μg ml−1. In order to quantify the inhibitory effect, the maximal increase in light transmission was determined from the aggregation curve 5 min after the addition of the agonist. The effect of BAY 58-2667 was expressed as percentage inhibition of agonist-induced platelet aggregation compared to vehicle of six independent experiments.
FeCl3 arterial thrombosis model in rats
Male Wistar rats (HSD CPB:WU; Harlan Winkelman, Borchen, Germany) weighing 180 – 220 g were anaesthetized with xylazine (12 mg kg−1 i.p.) followed by ketamine hydrochloride (50 mg kg−1 i.p.). After exposure of the left common carotid artery vascular damage was produced by placing a piece of filter paper (8×6 mm) placed on a strip of parafilm under the vessel and adding 20 μl of 10% FeCl3 (in 1N HCl) onto the filter paper according to a method described previously (Stasch et al., 2002a). The filter paper was removed after 3 min and the vessel was rinsed with 0.9% NaCl. The carotid artery was removed 15 min after the application of the filter paper. The thrombus was withdrawn and weighed immediately. Bay 58-2667 was given 75 min before damage of the vessel as a solution in ETOH/ Solutol®/H2O (10/40/50=v/v/v). The animals of the control group received the vehicle. Ten animals were used for each group.
Rat tail bleeding time
Male Wistar rats weighing 280 – 320 g (Harlan Winkelmann, Borchen, Germany), were orally treated with BAY 58-2667, acetylsalicylic acid or vehicle (Transcutol®/Cremophor® EL/H2O; 10/20/70=v/v/v). Fifty minutes after the oral dosing rats were anaesthetized with thiopental (Nycomed®, Munich, Germany) 100 mg kg−1 i.p. and placed in a tube holder with the tail allowed to protrude. After additional 20 min, the terminal 2 mm tip of the tail was removed with a sterile razor blade, and the tail was vertically immersed into normal saline at 37°C. The bleeding time was then measured as described (Stasch et al., 2002b).
Haemodynamics in conscious SHR
Female conscious SHR (Moellegaard, Denmark, 220 – 290 g) were equipped with implantable radiotelemetry, and a data acquisition system (Data Sciences, St. Paul, MN, U.S.A.), comprising a chronically implantable transducer/transmitter unit equipped with a fluid-filled catheter was used. The transmitter was implanted into the peritoneal cavity and the sensing catheter was inserted into the descending aorta.
Single administration of BAY 58-2667 was performed asa solution in Transcutol®/Cremophor®/H2O (10/20/70=v/v/v) given orally by gavage. The animals of control groups only received the vehicle.
Haemodynamics in anaesthetized dogs
Studies were performed on anaesthetized dogs of either sex and a body weight between 20 – 30 kg as described previously (Stasch et al., 2002b). Arterial blood pressure, electrocardiogram (lead II), left ventricular pressure, first derivative of left pressure (dP dt−1), heart rate, coronary blood flow, and oxygen saturation in the coronary sinus were continuously recorded on a pen recorder after implantation of the respective catheters. After completion of surgery an interval of 60 min was allowed for stabilization before BAY 58-2667 was intravenously applied. Care was taken that all measured cardiovascular parameters had returned to control level before injection of the next dose. BAY 58-2667 was dissolved in a solution of glycerol/water/polyethylenglycol 400 (60/100/949=g/g/g).
Induction of in vivo nitrate tolerance
Female chinchilla rabbits (2 – 3 kg) were used. The skin was shaved between both scapulae. To induce nitrate tolerance, isosorbide dinitrate (ISDN, TD Spray Iso Mack®, Heinrich Mack, Illertissen, Germany) was applied percutaneously with a dose of approximately 75 mg kg−1 three times a day over a period of 3 days (Mülsch et al., 2001).
Statistics
Differences were checked for significance by Student's t-test (one-way ANOVA) for unpaired data. All values in the tables and figures are given in the form of means±s.e.mean if not otherwise indicated. IC50 values were expressed as means with 95% confidential intervals in parenthesis. * P<0.05, ** P<0.01, *** P<0.001 compared to values in untreated controls.
Results
sGC stimulation
We studied the effects of BAY 58-2667 (Figure 1A) on sGC activity, alone and in the presence of NO or the sGC inhibitor ODQ. BAY 58-2667 from 0.001 to 10 μM showed a concentration-dependent stimulation of sGC from 2 to 37 fold (maximum 4136 nmol min−1 mg−1; Figure 1B). In combination, BAY 58-2667 (0.001 to 10 μM) and DEA/NO (10 and 100 nM) over the whole range of concentrations showed an additive effect on sGC activity. These observations have been repeated with higher and lower concentrations of DEA/NO, and it remained additive. Even in the presence of ODQ (10 μM) the activity of sGC was increased over a wide range of concentrations. At the highest concentration of BAY 58-2667 in combination with 10 μM ODQ, the specific sGC activity increased 19707 nmol min−1 mg−1 resulting in a 187 fold increase above the baseline (Figure 1B). Moreover, BAY 58-2667 activates the haem-free enzyme concentration-dependently up to 31974 nmol min−1 mg−1 or 190 fold at 10 μM (Figure 1C). In this experimental setting, the addition of the sGC inhibitor ODQ (10 μM) did not influence haem-free sGC activation induced by BAY 58-2667 (Figure 1C).
Figure 1.
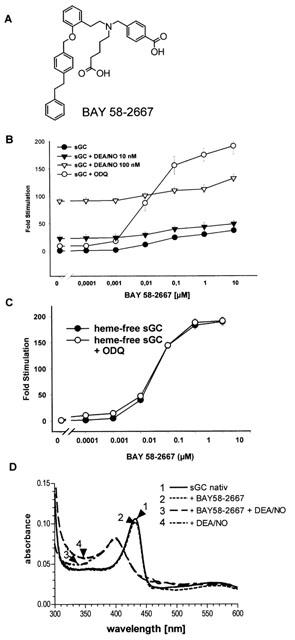
Effects of BAY 58-2667 on sGC activity. (A) Structure of BAY 58-2667. (B) Stimulation of purified sGC by BAY 58-2667 in the absence and presence of DEA/NO (10 and 100 nM) and ODQ (10 μM). The specificity is expressed as fold stimulation versus basal activity in the presence of Mg2+: 111 nmol min−1 mg−1 for the purified sGC. (C) Stimulation of haem free sGC by BAY 58-2667 in the absence and presence of ODQ. The specificity of sGC is expressed as fold stimulation versus basal activity in the presence of Mg2+: 169 nmol min−1 mg−1 for the haem-free sGC. Each value represents the mean±s.e.mean from three (B) and five (C) independent experiments performed in duplicate. (D) Haem spectra of sGC in the presence of BAY 58-2667 and DEA/NO. These data are representative of three independent determinations.
To determine whether BAY 58-2667 interacts directly with the prosthetic haem group, we recorded the ultraviolet-visual spectra of the purified sGC under unstimulated and stimulated conditions. NO caused the characteristic shift of the Soret peak to lower wavelength, and the addition of BAY 58-2667 resulted in no change of the Soret band of either the non-stimulated (431 nM) or NO-stimulated (398 nM) enzyme.
sGC binding studies
For binding studies, 3H-BAY 58-2667 was used in a range from 0.1 to 300 nM (Figure 2A). A nonlinear regression for one site saturation binding yields a Kd of 3.2 nM and a Bmax of 470 pmol mg−1 protein. Preincubation of the enzyme with ODQ led to a Bmax of 1048 pmol mg−1 protein. The best regression calculated a curve fitting two binding sites: a high affinity binding site with KD of 1.2 nM and a Bmax of 548 pmol bound mg−1 protein and a second binding site with a KD of 53.4 nM and a Bmax of 500 pmol mg−1 protein (Figure 2B). Competition binding performed in the absence and presence of ODQ resulted in Ki values of 6.3 and 6.5 nM respectively (Figure 2C).
Figure 2.

Receptor binding studies. (A) Structure of 3H-BAY 58-2667. (B) Saturation binding of 3H-BAY 58-2667 on sGC in the absence and presence of ODQ (10 μM). (C) Competition binding of 3H-BAY 58-2667 on sGC in the absence and presence of ODQ (10 μM) by BAY 58-2667. Unspecific binding was maximum 20% of total binding. Each value represents the mean±s.e.mean from four independent experiments performed in duplicate.
Photoaffinity labelling studies
To localize the binding region of BAY 58-2667 a photoaffinity labelling (PAL) study was performed. In the chemical core structure of a close analogue of BAY 58-2667, a photolabile azido group and a tritium label was introduced (3H-PAL, BAY 61-2573, Figure 3A). Unlabelled PAL activates isolated sGC with similar potency as BAY 58-2667 (data not shown) and displaced 3H-BAY 58-2667 from the enzyme (Ki=1.3 nM). sGC and 3H-PAL (5, 15, 50 μCi) were irradiated and processed as described. The autoradiogram of the SDS – PAGE (Figure 3B) shows labeling of the α- and β-subunit which could be diminished up to 81% by a 50 fold excess of PAL. The control sample incubated with 50 μCi 3H-PAL but without irradiation showed only 10% labelling compared to the irradiated sample. Irradiation of 3H-PAL and sGC in the presence of BAY 41-2272 (50 fold excess) or DEA/NO did not show any distinct alteration of the labelling pattern (Figure 3C). In addition, irradiation of 3H-PAL with the haem depleted or oxidized form of sGC results in a shift of radioactivity from the α- to the β-subunit (Figure 3C,D). For localization of the binding region of 3H-PAL, labelled sGC was cleaved by CNBr. The fragments were separated by high resolution 2D-PAGE and blotted onto a PVDF-membrane for sequencing. The autoradiogram on imaging plates shows numerous labelled spots which all include either fragment 8 of the α-subunit or fragment 5 of the β-subunit (Figure 3E). After sequencing and determination of radioactivity, fragment 8 of the α-subunit shows a 70 fold increase in radioactivity over background at the second position on tyrosine 371. Fragment 5 of the β-subunit (amino acids 165 – 310) was sequenced over 66 cycles without detection of radioactivity over basal activity. Therefore, it is concluded that the radiolabel is localized in the region of the amino acids 231 – 310 of the β-subunit of sGC.
Figure 3.
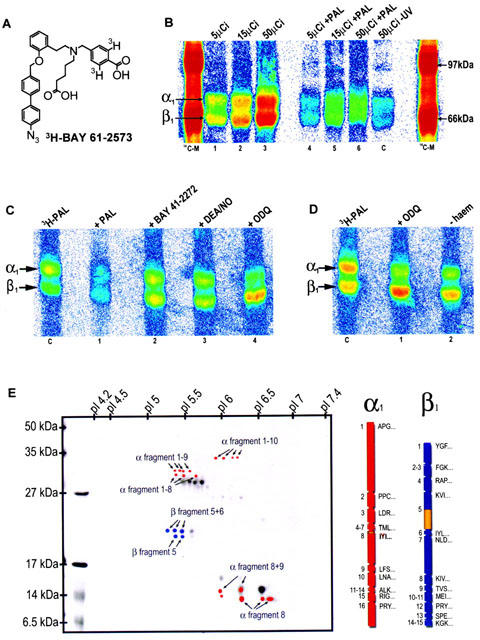
Photoaffinity labelling. (A) Structure of the 3H-photoaffinity label (3H-PAL). (B) Autoradiogram of photoaffinity-labelled sGC after separation by SDS – PAGE (10%). lane 1: 15 μg (100 pmol) sGC with 5 μCi 3H-PAL after irradiation; lane 2: as lane 1 with 15 μCi 3H-PAL; lane 3: as lane 1 with 50 μCi 3H-PAL; lanes 4 – 6: as lanes 1 – 3, but in the presence of a 50 fold excess of PAL. (C) control, as lane 3 but without irradiation. (C) Autoradiogram of photoaffinity-labelled sGC after separation by SDS – PAGE (10%). Control: 10 μg sGC with 15 μCi 3H-PAL after irradiation: lane 1: as control with a 50 fold surplus PAL; lane 2: as control with BAY 41-2272 (250 μM), lane 3 as control with DEA/NO (2.5 μM); lane 4: as control with ODQ (250 μM). (D) Autoradiogram of photoaffinity-labelled sGC after separation by SDS – PAGE (10%). Control: 10 μg sGC with 15 μCi 3H-PAL after irradiation; lane 1: as control with a 50 fold surplus ODQ; lane 2: as control with haem-free sGC (final concentration 0.5% Tween 20). (E) 2D-PAGE of CNBr fragments. The first dimension was performed in a tube gel and the second dimension was run on a 16% Tris/glycine gel. The gel was blotted to a PVDF membrane, Coomassie stained, and autoradiography was performed. Labelled spots were highlighted in red for α-subunit fragments and blue for β-subunit fragments. On the right side the theoretical cleavage fragments of both subunits with the first amino acids are shown. The labelled positions are highlighted in yellow.
Isolated organs
The effect of BAY 58-2667 has been examined on the contraction of rabbit saphenous artery rings preconstricted with phenylephrine. BAY 58-2667 elicited a potent concentration-dependent relaxation with an IC50 of 0.4 (0.3 – 0.5) nM, whereas BAY 41-2272, SNP and SIN-1 used as controls exhibited IC50's of 64 (50 – 80) nM, 635 (335 – 1270) nM and 1100 (480 – 1910) nM, respectively (Figure 4A). To determine whether the vasorelaxant effect of BAY 58-2667 is preserved under nitrate tolerant conditions, the relaxant effect of BAY 58-2667 was examined on isolated artery rings taken from normal and nitrate tolerant rabbits. GTN inhibited the phenylephrine-induced contraction with IC50=0.3 (0.2 – 0.4) μM in control vessels and with IC50=2.8 (1.4 – 5.0) μM in tolerant vessels, confirming the presence of nitrate tolerance. In contrast, BAY 58-2667 relaxed saphenous arteries taken from normal and tolerant rabbits with similar IC50 values of 0.16 (0.09 – 0.24) nM and 0.22 (0.16 – 0.29) nM (Figure 4B). In the rat heart Langendorff preparation, BAY 58-2667 decreased the coronary perfusion pressure in a concentration-dependent manner as shown in Figure 4C (controls, 110±4.3; 0.1 ng ml−1, 98.4±4.8; 1 ng ml−1, 60.0±5.8, P<0.001; 10 ng ml−1, 40.0±2.5, P<0.001; 100 ng ml−1, 39.2±5.2, P<0.001; values are means mmHg±s.e.mean, n=5). No effect on left ventricular pressure (controls, 38.0±4.5; 0.1 ng ml−1, 36.8±4.3; 1 ng ml−1, 36.0±4.2; 10 ng ml−1, 35.2±3.3; 100 ng ml−1, 36.0±4.2; values are means mmHg±s.e.mean, n=5) and heart rate (controls, 267±10; 0.1 ng ml−1, 260±10; 1 ng ml−1, 264±10; 10 ng ml−1, 263±8; 100 ng ml−1, 262±8; values are means b.p.m.±s.e.mean, n=5) was observed.
Figure 4.

Pharmacological effects of BAY 58-2667 in vitro. (A) Inhibition of phenylephrine (30 μg ml−1) induced contractions of isolated rabbit saphenous arteries by BAY 58-2667, BAY 41-2272, SNP and SIN-1. Values are means±s.e.mean of six, 16, eight and seven experiments. (B) Effects of BAY 58-2667 and GTN on phenylephrine-induced contractions of isolated rabbit saphenous arteries from normal and tolerant rabbits. Values are means±s.e.mean of eight and 11 vessels. *P<0.05 and ** P<0.01 vs normal saphenous artery rings. (C) Effect of BAY 58-2667 and GTN on coronary perfusion pressure at the rat heart Langendorff preparation. (D) The inhibitory effects of BAY 58-2667 on U 46619, collagen, ADP, TRAP-6-induced platelet aggregation in human platelet rich plasma and on thrombin-induced platelet aggregation in washed human platelets. The final concentrations of U 46619, collagen, ADP, TRAP-6 and thrombin were 1, 1 – 2, 30 – 50, and 5 μg ml−1, respectively. The effects were expressed as percentage inhibition of platelet aggregation compared to vehicle control. Each value represents the mean±s.e.mean from eight (U46619), 14 (collagen), five (ADP), five (TRAP-6), four (thrombin) experiments.
Antiplatelet activity
BAY 58-2667 produced a concentration-dependent inhibition of platelet aggregation induced by the thromboxane A2 mimic U 46619 (IC50=0.046 μM), collagen (IC50=1.1 μM) and ADP (IC50=7.5 μM) in human platelet rich plasma. Aggregation mediated by TRAP-6, a synthetic thrombin receptor agonist, was markedly resistant to inhibition by BAY 58-2667 (Figure 4D). In addition, thrombin-induced aggregation was not affected (IC50 > 16 μM) using washed platelets to avoid coagulation (Figure 4D). In the FeCl3 arterial thrombosis rat model, administration of BAY 58-2667 (0.3 to 10 mg kg−1 p.o.) inhibited thrombus formation in the carotid artery dose-dependently with ED50=0.9 mg kg−1 p.o. (Figure 5A). In this study clopidogrel (3 mg kg−1 p.o.) was used as positive control. Moreover, BAY 58-2667 prolonged rat tail bleeding time significantly from 0.3 to 10 mg kg−1 p.o. (Figure 5B). In this study acetylsalicylic acid (30 mg kg−1 p.o.) was used as positive control. Consistent with these results, BAY 58-2667 induced a significant and dose-dependent increase in cyclic GMP content in washed platelets of rats 1 h after oral administration (controls, 29±4.6 (5); 0.3 mg kg−1 p.o., 104±30 (6), P<0.05; 1 mg kg−1 p.o., 223±34 (4), P<0.001; 3 mg kg−1 p.o., 354±36 (6), P<0.001; values are means fmol cyclic GMP/2×10−8 platelets±s.e.mean (n)).
Figure 5.
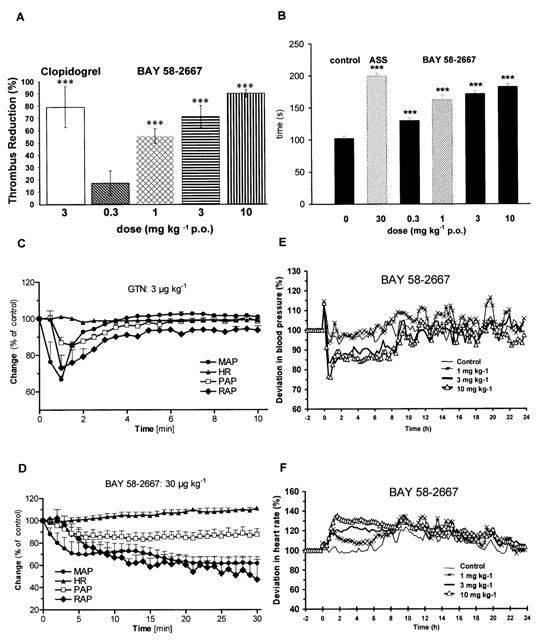
Pharmacological effects of BAY 58-2667 in vivo. (A) Effect of BAY 58-2667 (0.3, 1.0, 3.0 and 10 mg kg−1 p.o.) and clopidrogel (3.0 mg kg−1 p.o.) on the thrombus formation in the FeCl3 arterial thrombosis rat model (n=10 per group). *** P<0.001 compared with the values in the untreated controls. (B) Rat tail bleeding time after oral administration of BAY 58-2667 (0.3, 1.0, 3.0, and 10 mg kg−1), acetylsalicylic acid (30 mg kg−1) or vehicle (n=10 per group). *** P<0.001 compared with the values in the untreated controls. Haemodynamic effects of intravenous bolus injections of (C) GTN (3 μg kg−1) and (D) BAY 58-2667 (30 μg kg−1). The relative effects of each compound on mean arterial blood pressure (MAP), heart rate (HR), diastolic pulmonary artery pressure (PAP), and on mean right atrial pressure (RAP) are shown. Mean arterial blood pressure before administration (at time 0): GTN 90±1.7; BAY 58-2667 89.3±3.6 mmHg; heart rate: GTN 103±5.9; BAY 58-2667 97.3±6.8 beats min−1, diastolic pulmonary artery pressure: GTN 7.7±1.1; BAY 58-2667 7.5±1.1 mmHg; mean right atrial pressure: GTN 2.8±0.4; BAY 58-2667 2.6±0.5 mmHg (n=4 per group). (E) Effect of orally administered BAY 58-2667 (1.0, 3.0 and 10 mg kg−1) on mean arterial blood pressure and (F) on heart rate of spontaneously hypertensive rats. Values depicted represent changes in heart rate and mean arterial blood pressure (n=6 per group). The heart rate at baseline was 318±16, 288±9, 297±7 and 302±6 b.p.m., respectively, and the mean arterial blood pressure at baseline was 122±11, 130±10, 130±5 and 139±7 mmHg, respectively. Values are mean±s.e.mean.
Haemodynamics
The effects of BAY 58-2667 were compared with those of glyceryl trinitrate (GTN) in anaesthetized dogs under autonomic blockade (Figure 5C, D). Both compounds caused a pronounced decrease in mean arterial blood pressure. The dose for bolus injections of BAY 58-2667 (30 μg kg−1) and GTN (3 μg kg−1) was adjusted to give a comparable decrease in mean arterial blood pressure (BAY 58-2667: −39% and GTN: −31%). There was no major change in heart rate (BAY 58-2667: +9% and GTN: −1%). The haemodynamic profile of BAY 58-2667 and GTN was very similar: in addition to the decrease in arterial blood pressure both compounds caused a decrease in diastolic pulmonary artery pressure (BAY 58-2667: −18%, GTN: −13%) and in mean right atrial pressure (BAY 58-2667: −48%, GTN: −24%). The duration of action was much longer for BAY 58-2667 compared to GTN. In conscious spontaneously hypertensive rats equipped with a telemetric device, BAY 58-2667 is also a potent and long-lasting antihypertensive. After oral administration of 3 and 10 mg kg−1 maximal blood pressure lowering effects of −15% and −22% were achieved in combination with an increase in heart rate (Figure 5E, F).
Discussion
Here we report the discovery of a novel sGC activating compound, its interaction with a previously unrecognized regulatory site and its therapeutic implications. Through a high throughput screening we identified BAY 58-2667, an amino dicarboxylic acid which potently stimulates sGC in an NO-independent manner. Although BAY 58-2667 does not activate sGC as strongly as does NO, concentrations as low as 1 nM activate sGC sufficiently to yield biologically important increases in cyclic GMP (Stasch et al., 2001). An additive effect with NO is observed over a wide range of concentrations. In contrast, BAY 41-2272 and YC-1 synergize with NO (Friebe et al., 1996; Hoenicka et al., 1999; Stasch et al., 2001).
NO stimulates sGC by forming a nitrosyl-haem complex. Removal of the haem group by the non-ionic detergent Tween-20 yields an NO-insensitive sGC without destruction of basal enzyme activity as judged by spectroscopic analysis (Friebe et al., 1996; Hoenicka et al., 1999). YC-1 and BAY 41-2272 did not significantly activate the haem-free enzyme (Zhao et al., 1998; Hoenicka et al., 1999; Stasch et al., 2001). In contrast, BAY 58-2667 activates the haem-free enzyme concentration-dependently up to 190 fold.
The binding of NO to the haem group can be visualized in haem spectra, which show a characteristic shift of the absorption maximum to lower wavelengths. To determine whether BAY 58-2667 directly interacts with the prosthetic haem group, we recorded the u.v.-visual spectra of the purified sGC under unstimulated and stimulated conditions (Hoenicka et al., 1999). NO elicited the characteristic shift of the Soret peak to lower wavelength, while the addition of BAY 58-2667 resulted in no change of the Soret band of either the non-stimulated or NO-stimulated enzyme. Therefore, unlike NO, BAY 58-2667 does not bind to the haem moiety of sGC. Because BAY 58-2667 showed activity at the haem-free enzyme, we conclude that BAY 58-2667 activates sGC by an NO- and haem-independent mechanism in contrast to YC-1 or BAY 41-2272 (Hoenicka et al., 1999; Stasch et al., 2001).
Studies with purified sGC revealed that the sGC inhibitor ODQ binds in an NO-competitive manner, oxidizes the haem iron, and leads to an apparently irreversible inhibition of the stimulated enzyme (Garthwaite et al., 1995; Schrammel et al., 1996). As expected by the haem-independence of BAY 58-2667, the activity of sGC was markedly increased by BAY 58-2667 even in the presence of ODQ. In contrast to these findings the activation of sGC by other known stimulators (such as NO, YC-1 and BAY 41-2272) is prevented by ODQ (Hoenicka et al., 1999; Stasch et al., 2001). Because BAY 58-2667 activates sGC by an NO- and haem-independent mechanism, it may be a useful tool for understanding of the mechanisms of sGC activation and for the detection of sGC activity in biological material independent of the presence or oxidation status of the haem group.
For the first time classical receptor binding studies at the sGC could be performed. In the presence of ODQ saturation of the binding of 3H-BAY 58-2667 could be reached at submicromolar concentrations. Concomitant with these results a very potent and concentration dependent activation of sGC was observed by BAY 58-2667 in the presence of ODQ. However, in the absence of ODQ stimulation was less at the native enzyme with BAY 58-2667 even at micromolar concentrations and did not reach a plateau. In receptor binding studies at the native enzyme the maximum saturation observed with BAY 58-2667 was about half of the value determined in the presence of ODQ. These findings could be explained by postulating two binding sites for BAY 58-2667. Both sites show low KD values in the presence of ODQ resulting in maximal binding and saturably sGC activation by BAY 58-2667. One might speculate that at the native enzyme the ferrous state of the haeme moiety distorts one binding site of BAY58-2667 and thereby shifts its KD value to a distinct higher concentration. This could explain the missing saturation in the activation profile of BAY 58-2667 at native sGC. In the enzyme assay the high affinity site could be saturated at similar concentrations as known from the receptor binding studies, however, this saturation could be masked by increased enzyme activity due to ligand binding to the putative ultra-low affinity site at higher concentrations. Due to the limited range of concentrations which can be applied in the receptor binding study, a putative ultra-low affinity site might not be detectable. Interestingly, in the photoaffinity labelling study we observed a shift of the labelling of 3H-BAY 58-2667 to the β-subunit induced by ODQ. This might also be a hint for conformational changes in the enzyme structure by oxidation of its haem moiety.
The photoaffinity labelling studies suggest that both the α- and ß-subunit of rat sGC are involved in the binding of BAY 58-2667. After sequencing, we found that the label was associated with the amino acids 371 on the α-subunit and 231 – 310 in the ß-subunit. Alignment analysis show that the identified residues are within a highly conserved region between different species. However, the mechanism of sGC activation is rather more complicated than simple interaction with one or two amino acids. Since there is a distance between the photolabile azido group and the pharmacophore of the compound, a covalent binding of 3H-PAL to reactive amino acids slightly outside the binding pocket would not be surprising. Nevertheless, we assume that the labelled amino acids are in the direct region of access to 3H-PAL because no other labelled amino acids have been detected and a diffusion over a wide distance can be excluded. One might speculate that BAY 58-2667 influences the three-dimensional structure of both subunits of sGC via its carboxylic functions and thereby stabilizes a transition state complex and increases its catalytic rate. This hypothesis is supported by the fact that both carboxylic functions in the molecule are essential for the activity of BAY 58-2667. Whether these amino acids are in the binding pocket(s) or merely adjacent to them and whether they represent one or two pockets remains to be determined once cocrystallization studies as shown for adenylyl cyclase with forskolin become available (Tesmer et al., 1997).
In previous photoaffinity studies using an analogue of BAY 41-2272, amino acids 238 and 243 of the α-subunit were labelled (Becker et al., 2001; Stasch et al., 2001). In the current studies the addition of BAY 41-2272 did not alter the 3H-PAL labelling pattern. Further BAY 41-2272 does not compete with 3H-BAY 58-2667 at sGC. The results clearly indicate that the two compounds occupy different independent sites.
BAY 58-2667 possesses a unique pharmacological profile in vitro and in vivo. BAY 58-2667 is a very potent relaxing agent on isolated artery rings with an IC50 value in the subnanomolar range and thereby about 160 fold more potent than BAY 41-2272 (Stasch et al., 2001; Straub et al., 2001). Moreover, in the rat heart Langendorff preparation, BAY 58-2667 reduces the coronary perfusion pressure in a concentration-dependent manner about two orders of magnitude more potently than GTN.
Despite the widespread use of organic nitrates in the treatment of angina, the development of tolerance limits the therapeutic value of this class of compounds for chronic treatment (Elkayam, 1991; Münzel et al., 1996; Feelisch, 1998). Therefore, compounds which can stimulate sGC in a NO-independent manner may show a significant advantage over existing nitrovasodilator therapy. We used artery rings from normal and nitrate-tolerant rabbits to test the hypothesis that sGC is not involved in nitrate tolerance and that the vasorelaxing effect of the sGC stimulator is the same under normal and tolerant conditions. Most notably, the vasorelaxation caused by BAY 58-2667 in aortic rings taken from normal and tolerant rabbits was almost the same. These observations indicate that BAY 58-2667 represents a new class of therapeutics which may be useful in overcoming the tolerance developed during sustained GTN therapy.
sGC stimulation in platelets correlates with inhibition of aggregation, platelet cyclic GMP increase, prolongation of bleeding time, and antithrombotic effects in vitro (Hobbs, 2000; Stasch et al., 2002a, 2002b; Becker et al., 1999; Teng et al., 1997). BAY 58-2667 potently inhibited platelet aggregation induced by the thromboxane mimic U46619 and collagen, whereas TRAP-6- and thrombin-mediated aggregation was not affected. The significance of the antiplatelet effect was confirmed by the in vivo results. We observed a significant prolongation in rat tail bleeding time. More importantly, BAY 58-2667 reduces thrombus formation in the FeCl3 thrombosis rat model. Consistent with these results, BAY 58-2667 induced a dose-dependent increase in platelet cyclic GMP content. However, it should be mentioned that the blood pressure lowering effect of BAY 58-2667 at higher doses could contribute to its antiplatelet effect observed in vivo. These results reveal the potential of BAY 58-2667 as a representative of a new class of antiaggregatory drugs.
The oral administration of BAY 58-2667 to hypertensive rats equipped with a radiotelemetric device for the continuous recording of haemodynamic parameters induced a dose-dependent and long-lasting decrease in blood pressure and a reflex increase in heart rate. The effects of the BAY 58-2667 on the high (arterial) and low (venous) pressure system were compared with those of GTN in anaesthetized dogs. The haemodynamic profile of BAY 58-2667 and GTN was very similar: in addition to the decrease in arterial blood pressure both compounds caused a decrease in diastolic pulmonary artery pressure and in mean right atrial pressure. Thus, BAY 58-2667 is the first compound which has the same haemodynamic profile as organic nitrates causing in vivo relaxation of both arterial and venous blood vessels. However, the pharmacokinetic profile of BAY 58-2667 and GTN are markedly different. BAY 58-2667 has a lower clearance, longer half-life and a much lower free fraction in plasma compared to GTN (unpublished data). Therefore, higher total plasma concentrations are needed to lead to similar effects than GTN. The lower clearance and longer half-life of BAY 58-2667 led to a longer lasting effect than observed after administration of GTN.
In summary, characteristics of BAY 58-2667 on sGC activity, the binding studies, and the photoaffinity labelling studies suggest the existence of a new NO- and haem-independent regulatory site on sGC in the region of the amino acids 371 (α-subunit) and 231 – 310 (ß-subunit), which modulates the catalytic rate. Our data offer a new approach in the understanding of sGC regulation and manipulation of this signal transduction pathway. A very potent new activator of sGC, BAY 58-2667, is presented with a potent vasorelaxating effect on isolated vessels, an antiplatelet activity in vitro and in vivo, a strong blood pressure lowering effect and a haemodynamic profile comparable to that of organic nitrates. This novel pharmacological principle may be clinically useful for the treatment of cardiovascular diseases.
Acknowledgments
We thank Dr U. Janssen-Bienhold for support on 2D-PAGE and Dr D. Wood for critical comments on the manuscript.
Abbreviations
- b.p.m.
beats per min
- DEA/NO
diethylamine NONOate
- GTN
glyceryl trinitrate
- NO
nitric oxide
- ODQ
(1H-(1,2,4)-Oxadiazolo-(4,3a)-quinoxazin-1-one)
- PAL
photoaffinity label
- sGC
soluble guanylyl cyclase
- SHR
spontaneously hypertensive rats
- YC-1
3-(5′-hydroxymethyl-2′furyl)-1-benzyl-indazole
References
- AHR H.J., STEINKE W. Imaging plate: validation and first experience with quantitative studies in whole-body autoradiography during drug development. Xenobiotic Metab. Dispos. 1994;9:371–378. [Google Scholar]
- ALONSO-ALIJA C., HEIL M., FLUBACHER D., NAAB P., STASCH J.-P., WUNDER F., DEMBOWSKY K., PERZBORN E., STAHL E.Novel derivatives of dicarboxylic acid having pharmaceutical properties 2001. WO-119780-A 2001.03.22
- BECKER E.M., ALONSO-ALIJA C., APELER H., GERZER R., MINUTH T., PLEISS U., SCHMIDT P., SCHRAMM M., SCHRÖDER H., SCHROEDER W., STEINKE W., STRAUB A., STASCH J.-P. NO-independent regulatory site of direct sGC stimulators like YC-1 and BAY 41-2272. BMC Pharmacol. 2001;1:13. doi: 10.1186/1471-2210-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BECKER E.M., SCHMIDT P., SCHRAMM M., SCHRÖDER H., WALTER U., HOENICKA M., GERZER R., STASCH J.-P. The vasodilator-stimulated phosphoprotein VASP: mediator of YC-1 and nitric oxide effects in human and rat platelets. J. Cardiovasc. Pharmacol. 2000;35:390–397. doi: 10.1097/00005344-200003000-00007. [DOI] [PubMed] [Google Scholar]
- BECKER E.M., WUNDER F., KAST R., ROBYR C., HOENICKA M., GERZER R., SCHRÖDER H., STASCH J.-P. Generation and characterization of a stable soluble guanylate cyclase overexpressing CHO cell line. Nitric Oxide. 1999;3:55–66. doi: 10.1006/niox.1999.0207. [DOI] [PubMed] [Google Scholar]
- ELKAYAM U. Tolerance to organic nitrates: evidence, mechanisms, clinical relevance, and strategies for prevention. Ann. Int. Med. 1991;114:667–677. doi: 10.7326/0003-4819-114-8-667. [DOI] [PubMed] [Google Scholar]
- FEELISCH M. The use of nitric oxide donors in pharmacological studies. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358:113–122. doi: 10.1007/pl00005231. [DOI] [PubMed] [Google Scholar]
- FRIEBE A., SCHULTZ G., KOESLING D. Sensitizing soluble guanylate cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- FURCHGOTT R.F. Endothelium-derived relaxing factor: Discovery, early studies, and identification as nitric oxide (Nobel lecture) Angew. Chem. - Int. Ed. Engl. 1999;38:1870–1880. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1870::AID-ANIE1870>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- GARTHWAITE J., SOUTHAM E., BOULTON C.L., NIELSEN E.B., SCHMIDT K., MAYER B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- HOBBS A.J. Soluble guanylate cyclase. Emerg. Therap. Targets. 2000;4:735–749. [Google Scholar]
- HOENICKA M., BECKER E.M., APELER H., SHIRICHOKE T., SCHRÖDER H., GERZER R., STASCH J.-P. Purified soluble guanylyl cyclase expressed in a baculovirus Sf9 system: stimulation by YC-1, nitric oxide, and carbon monoxide. J. Mol. Med. 1999;77:14–23. doi: 10.1007/s001090050292. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J. Nitric oxide: A unique endogenous signaling molecule in vascular biology (Nobel lecture) Angew. Chem. - Int. Ed. Engl. 1999;38:1882–1892. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1882::AID-ANIE1882>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- KO F.N., WU C.C., KUO S.C., LEE F.Y., TENG C.M. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. [PubMed] [Google Scholar]
- KOESLING D.Enzymology of soluble guanylyl cyclase Nitric Oxide 2000San Diego, USA: Academic Press; 485–493.ed. Ignarro, L.J. pp [Google Scholar]
- KOLBE H., DIETZEL G. Technical validation of radioluminography systems. Regul. Toxicol. Pharmacol. 2000;31:S5–S14. doi: 10.1006/rtph.2000.1380. [DOI] [PubMed] [Google Scholar]
- MARTIN E., LEE Y.C., MURAD F. YC-1 activation of human soluble guanylyl cyclase has both heme-dependent and heme-independent components. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12938–12942. doi: 10.1073/pnas.231486198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M., HIGGS E.A. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- MÜLSCH A., BAUERSACHS J., SCHÄFER A., STASCH J.-P., KAST R., BUSSE R. Effect of YC-1, an NO-independent, superoxide-sensitive stimulator of soluble guanylyl cyclase, on smooth muscle responsiveness to nitrovasodilators. Br. J. Pharmacol. 1997;120:681–689. doi: 10.1038/sj.bjp.0700982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MÜLSCH A., OELZE M., KLOSS S., MOLLNAU H., TOPFER A., SMOLENSKI A., WALTER U., STASCH J.-P., WARNHOLTZ A., HINK U., MEINERTZ T., MÜNZEL T. Effects of in vivo nitroglycerine treatment on activity and expression of guanylyl cyclase and cGMP-dependent protein kinase and their down-stream target vasodilator-stimulated phosphoprotein in aorta. Circulation. 2001;103:2188–2194. doi: 10.1161/01.cir.103.17.2188. [DOI] [PubMed] [Google Scholar]
- MÜNZEL T., KURZ S., RAJAGOPALAN S., THOENES M., BERRINGTON W.R., THOMPSON J.A., FREEMAN B.A., HARRISON D.G. Hydralazine prevents nitroglycerin tolerance by inhibiting activation of a membrane-bound NADH oxidase. A new action for an old drug. J. Clin. Invest. 1996;98:1465–1470. doi: 10.1172/JCI118935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAD F. Discovery of some of the biological effects of nitric oxide and its role in cell signaling (Nobel lecture) Angew. Chem. - Int. Ed. Engl. 1999;38:1857–1868. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1856::AID-ANIE1856>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- O'FARREL P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- SCHRAMMEL A., BEHRENDS S., SCHMIDT K., KOESLING D., MAYER B. Characterization of 1H[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol. Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- SHARMA V.S., MAGDE D., KHARITONOV V.G., KOESLING D. Soluble guanylate cyclase: effect of YC-1 on ligation kinetics with carbon monoxide. Biochem. Biophys. Res. Commun. 1999;254:188–191. doi: 10.1006/bbrc.1998.9812. [DOI] [PubMed] [Google Scholar]
- SHU A.Y.L., HEYS J.R.Extension of organoiridium catalyzed hydrogen isotope exchange: photoaffinity labels and paclitaxel Proceedings of the 7th International Symposium 2000Vol. 7Dresden, Germany: John Wiley & Sons; 68–70.(eds) Pleiss U., Voges R. [Google Scholar]
- STASCH J.-P., ALONSO-ALIJA C., APELER H., DEMBOWSKY K., FEURER A., MINUTH T., PERZBORN E., SCHRAMM M., STRAUB A. Cardiovascular actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41-8543: in vitro studies. Br. J. Pharmacol. 2002a;135:333–343. doi: 10.1038/sj.bjp.0704484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STASCH J.-P., BECKER E.M., ALONSO-ALIJA C., APELER H., DEMBOWSKY K., FEURER A., GERZER R., MINUTH T., PERZBORN E., PLEIB U., SCHROEDER H., SCHROEDER W., STAHL E., STEINKE W., STRAUB A., SCHRAMM M. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- STASCH J.-P., DEMBOWSKY K., PERZBORN E., STAHL E., SCHRAMM M. Cardiovascular actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41-8543: in vivo studies. Br. J. Pharmacol. 2002b;135:344–355. doi: 10.1038/sj.bjp.0704483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRAUB A., STASCH J.P., ALONSO-ALIJA C., BENET-BUCHHOLZ J., DUCKE B., FEURER A., FÜRSTNER C. NO-independent stimulators of soluble guanylate cyclase. Bioorg. Med. Lett. 2001;11:781–784. doi: 10.1016/s0960-894x(01)00073-7. [DOI] [PubMed] [Google Scholar]
- TENG C.M., WU C.C., KO F.N., LEE F.Y., KUO S.C. YC-1, a nitric oxide-independent activator of soluble guanylate cyclase, inhibits platelet-rich thrombosis in mice. Eur. J. Pharmacol. 1997;320:161–166. doi: 10.1016/s0014-2999(96)00911-9. [DOI] [PubMed] [Google Scholar]
- TESMER J.J.G., SUNAHARA R.K., GILMAN A.G., SPRANG S.R. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha. GTPgammaS. Science. 1997;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- TOWBIN H., STAEHELIN T., GORDON J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU C.C., KO F.N., KUO S.C., LEE F.Y., TENG C.M. YC-1 inhibited human platelet aggregation through NO-independent activation of soluble guanylate cyclase. Br. J. Pharmacol. 1995;116:1973–1978. doi: 10.1111/j.1476-5381.1995.tb16400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO Y., BRANDISH P.E., DIVALENTIN M., SCHELVIS J.P.M., BABCOCK G.T., MARLETTA M.A. Inhibition of soluble guanylate cyclase by ODQ. Biochemistry. 2000;39:10848–10854. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]
- ZHAO Y., SCHELVIS J.P.M., BABCOCK G.T., MARLETTA M.A. Identification of histidine 105 in the ß1 subunit of soluble guanylate cyclase as the heme proximal ligand. Biochemistry. 1998;37:4502–4509. doi: 10.1021/bi972686m. [DOI] [PubMed] [Google Scholar]