

Abstract
Novel analogues of antisauvagine-30 (aSvg-30), a selective antagonist for CRF2 receptors, have been synthesized and characterized in vitro and in vivo.
The analogues were tested for their ability to compete for [125I-Tyr0]Svg binding and to inhibit Svg-stimulated adenylate cyclase activity in human embryonic kidney (HEK) 293 cells, permanently transfected with cDNA coding for the human CRF1 (hCRF1), hCRF2α and hCRF2β receptor. One analogue [D-Phe11, His12, Nle17]Svg(11-40), named K41498, showed high affinity binding to hCRF2α (Ki=0.66±0.03 nM) and hCRF2β (Ki=0.62±0.01 nM) but not the hCRF1 receptor (ki=425+50 nM) and decreased Svg-stimulated cAMP accumulation in hCRF2 expressing cells. In conscious Wistar-Kyoto rats, K41498 (1.84 μg, i.v.) antagonized the hypotensive response to systemic urocortin (1.4 μg, i.v.), but did not block the pressor response to centrally administered urocortin (2.35 μg, i.c.v.).
K41498 was subsequently radio-iodinated, and in autoradiographic studies, specific (sensitive to rat urocortin, astressin and aSvg30, but insensitive to antalarmin) binding of 125I-K41498 (100 pM) was detected in the heart and in selected brain regions including the nucleus tractus solitarius (NTS), spinal trigeminal nucleus, lateral septum and around the anterior and middle cerebral arteries.
Following unilateral nodose ganglionectomy, binding of 125I-K41498 was reduced by 65% in the ipsilateral NTS, indicative of presynaptic CRF2 receptors on vagal afferent terminals.
These data demonstrate that K41498 is a useful tool to study native CRF2 receptors in the brain and periphery.
Keywords: CRF2 receptors, K41498, nucleus tractus solitarius
Introduction
Corticotropin-releasing factor (CRF) is a primary regulator of the stress response (reviewed in Vale et al., 1997) and is implicated in the pathophysiology of several psychiatric disorders including anxiety, major depression and anorexia nervosa (Behan et al., 1996; Arborelius et al., 1999; Holsboer, 1999). The actions of CRF, and its structurally related analogues mammalian urocortin (Ucn), amphibian sauvagine (Svg) and fish urotensin I, are elicited through binding to at least two different G protein-coupled receptors: CRF receptor, type 1 (CRF1) and type 2 (CRF2) (reviewed in Dautzenberg et al., 2001a). Several alternatively spliced forms of CRF2 receptors, which are 70% identical in amino acid sequence, have been described including CRF2α, CRF2β and CRF2γ (reviewed in Kilpatrick et al., 1999). The recent discovery of further members of the CRF peptide family, namely urocortin II in the mouse and stresscopin (urocortin III) and stresscopin-related peptide in man (Reyes et al., 2001; Hsu & Hsueh, 2001; Lewis et al., 2001), appear to have identified selective, endogenous agonists for the CRF2 receptor.
Although there are a number of CRF1 receptor antagonists (reviewed in McCarthy et al., 1999), there is a relative lack of selective antagonists for the CRF2 receptor. We recently designed, synthesized and characterized for the first time a CRF2-specific peptide antagonist, antisauvagine-30 (aSvg-30) (Rühmann et al., 1998). This compound showed high selectivity in binding to CRF2α and CRF2β but not to the CRF1 receptor, in both in vitro and in vivo studies (Rühmann et al., 1998; Radulovic et al., 1999; Higelin et al., 2001). Mice lacking CRF2 receptors have been produced in a number of laboratories, and behavioural phenotyping of such mice has resulted in a degree of controversy. Thus, mice that are homozygous null for the CRF2 either display an anxious phenotype in both males and females (Bale et al., 2000), just in males (Kishimoto et al., 2000), or not at all (Coste et al., 2000). Furthermore, the anxious phenotype of male CRF2 receptor deficient mice could be mimicked by intracerebroventricular injection of aSvg-30 in wild type mice (Kishimoto et al., 2000). It is currently unclear as to why the different phenotypes prevail. In addition, homozygous null mice of either sex have normal feeding behaviour, but completely lack the hypotensive response to systemic Ucn injection (Bale et al., 2000). Given the lack of cohesion between the phenotypes of the different CRF2 deficient mice, there is a clear need for improved antagonist tools to further probe the nature of native CRF2 receptors in the brain and periphery.
A recent study has reported the synthesis of a stable radioligand for CRF receptors (Assil et al., 2001); however, this sauvagine analogue is non-selective and labels both CRF1 and CRF2 receptors. While aSvg-30 has recently been radiolabelled for use in membrane binding assays (Higelin et al., 2001), the aim of this study was to develop a chemically more stable analogue of aSvg-30 with similar selectivity for CRF2 receptors that could be radioactively labelled to high specific activity, and therefore be useful not only as a functional CRF2 receptor antagonist, but also as a radiotracer to study CRF2 receptors in the brain and periphery in autoradiographic studies.
Methods
All of the experiments described here were performed in accordance with the Prevention of Cruelty to Animals Act 1986 under the guidelines of the NH & MRC Code of Practice for the Care and Use of Animals for Experimental Purposes in Australia.
Synthesis and purification of peptides
The aSvg-30 analogue K41498 (Figure 1) was custom-synthesised by AusPep (Melbourne, Victoria). The CRF-like peptides rUcn, Svg and astressin were purchased from Bachem (Bubendorf, Switzerland). The CRF1-selective antagonist, antalarmin, was a gift from Dr G.P. Chrousos, NIH Bethesda, MD, U.S.A.
Figure 1.

Comparison of the amino acid sequence of human/rat corticotropin-releasing factor (h/rCRF), rat urocortin (rUcn), sauvagine (Svg), aSvg-30 and astressin with the modified aSvg-30 analogue K41498. Z, pyroglutamic acid; f, D-phenylalanine; B, norleucine; lactam bridge is indicated by a bracket. Identical amino acids are shaded.
Radioligand binding assays
Binding of the aSvg-30 analogue to the hCRF1, hCRF2α or hCRF2β receptor was performed essentially as described previously (Dautzenberg et al., 2000, 2001b). Briefly, membranes isolated from HEK293 cells stably expressing the hCRF1 (30 μg), hCRF2α (5 μg) or hCRF2β (5 μg) receptor were combined with 100 pM 125I-Tyr0-Svg (Amersham, Little Chalfont, U.K., 2000 Ci/mmol). Binding was performed in 96-well plates (Beckmann Instruments, Fullertown, CA, U.S.A.) with 0.5 mg wheat germ agglutinin beads using a scintillation proximity assay as described previously (Dautzenberg et al., 2000; Higelin et al., 2001). The inhibition constant (Ki) was calculated using the interactive curve-fitting program Xlfit (Dautzenberg et al., 2001b).
cAMP stimulation
HEK293 cells, stably expressing hCRF1, hCRF2α or hCRF2β receptors were plated at 104 cells per well in 96-well dishes. Cells were incubated for 10 min at 37°C with a submaximal (1 nM) concentration of Svg with or without 100 nM (hCRF2α and hCRF2β) to 10 μM (hCRF1) antagonist. The 1 nM Svg concentration, which was slightly above the EC50 value at the hCRF1, hCRF2α and hCRF2β receptors was chosen to detect even minimal antagonist activities. The data were analysed by one-way analysis of variance (ANOVA) and significance between groups was determined by post hoc analysis using Dunnett's test.
Cardiovascular studies
Male Wistar-Kyoto rats (WKY), obtained from the Biological Research Laboratories, Austin Hospital, Victoria. Rats (340–360 g) were anaesthetized with ketamine/xylazine (60/7.8 mg kg−1, i.p.) and placed in a stereotaxic frame. A stainless steel intra-cerebroventricular (i.c.v.) guide cannula (23 ga) was implanted, such that a 31 ga injector would protrude 2 mm beyond the tip of the guide cannula and enter the lateral ventricle (AP–0.7, ML–1.4, DV–2.2 mm from bregma), according to a stereotaxic atlas (Paxinos & Watson, 1986). Verification of implant locations were performed 4–6 days after surgery whereby angiotensin II (0.1 μg in 5 μl) was injected and the time taken for rats to drink recorded. In all cases, rats drank within 30 s of injection. At least one week after intial cannula implantation, rats were re-anaesthetized with ketamine/xylazine (60/7.8 mg kg−1, i.p.) and the tail artery cannulated and exteriorized between the scapulae for measurement of blood pressure and heart rate. The following day, the arterial cannula was attached to a pressure transducer (Gould) and cardiovascular parameters were recorded on a Grass Polygraph (79D).
A separate group of rats were anaesthetized with ketamine/xylazine (60/7.8 mg kg−1, i.p.) and the jugular vein and tail artery cannulated and exteriorized between the scapulae for drug administration and measurement of blood pressure and heart rate respectively. The following day, the arterial cannula was attached to a pressure transducer (Gould) and cardiovascular parameters were recorded on a Grass Polygraph (79D).
All cardiovascular data were analysed by a Paired t-test, P<0.05 was considered significant.
Iodination of K41498
K41498 was labelled with Na125I at histidine-12 essentially as described (Rühmann et al., 1996; Bonk & Rühmann, 2000). The peptides were analysed and purified by reverse-phase HPLC (RPHPLC) on a Vydac C18 silica gel column (0.46×25 cm, 5-μm particle size, 30-nm pore size) with solvents A (0.1% TFA in water) and B (80% MeCN in 0.1% TFA in water) at a flow rate of 0.5 ml min−1. The samples were eluted with 30% B for 5 min and then with a linear gradient of 30–80% B in 30 min (K41498: Rt=21.0 min, 125I-K41498: Rt=22.5 min). The 125I-iodinated product was obtained in 7–21% radiochemical yield with at least 99% radiochemical purity. The specific activity of the radioactively labelled peptide was 2000 Ci/mmol.
Unilateral vagal deafferentation
Nodose ganglionectomies were performed as previously described (Lawrence et al., 1995). In brief, male WKY rats (365–395 g) were anaesthetized with ketamine/xylazine (60/7.8 mg kg−1, i.p.) and placed on their back. A midline incision was made in the neck and the left nodose ganglion was exposed and excised (n=5) including trunks of vagus, superior laryngeal and inferior pharyngeal nerves at the respective points of contact with the nodose ganglion. In a second group (n=5), the left nodose ganglion was exposed but not removed (sham control). After a 14 day recovery period, the animals were re-anaesthetized with sodium pentobarbitone (60 mg kg−1, i.p.), immediately killed by decapitation and their brains and hearts removed. All tissue was frozen over liquid nitrogen at the time of removal and stored at −80°C until further processed.
Autoradiographic studies with 125I-K41498
Cryostat-cut coronal sections (14 μm) of brain were taken through the caudal half of the rat nucleus tractus solitarius (NTS) and through the septum, while hearts (14 μm) and nodose ganglia (10 μm) were sectioned in the horizontal plane. All tissue sections were thaw-mounted onto gelatine/chrome-alum-coated microscope slides and stored at −80°C prior to use.
The general procedure for receptor autoradiography with 125I-K41498 (an analogue of aSvg-30) was modified from an established protocol utilizing the structurally-related ligand, [125I-Tyr0]-Svg (Mar Sanchez et al., 1999). The concentration of radioligand employed (100 pM) was chosen from competition studies indicating a Ki for K41498 at CRF2 receptors of ∼0.6 nM, versus a Ki of ∼425 nM at CRF1 receptors (see Table 1). Autoradiographic conditions, including buffer composition, radioligand concentration, incubation and wash times were all established in preliminary experiments (not shown), and the following method represents the optimised conditions for the use of 125I-K41498. On the day of experimentation, tissue sections were equilibrated to room temperature and pre-incubated in Tris.HCl (50 mM, pH 7.4) containing MgCl2 (10 mM) and EGTA (2 mM) for 15 min at room temperature (∼22°C). Tissue was then transferred into buffer supplemented with bovine serum albumin (BSA, 0.8 %), aprotinin (0.04 T.I.U. ml−1), bacitracin (0.1 mM) and 125I-K41498 (100 pM) and incubated for 2 h at room temperature. Non-specific binding was defined with K41498 (1 μM), while adjacent sections were incubated in the presence of either rat Ucn, astressin or antalarmin (1 μM). Following this, sections were washed in ice-cold phosphate-buffered saline (0.1 M, pH 7.4) containing 0.8 % BSA (3×3 min) and rinsed twice in ice-cold distilled water. Finally, tissue sections were dried under a gentle stream of cool air, dessicated overnight and apposed to Kodak X-omat film in the presence of standard microscales (American Radiolabelled Chemicals Inc.).
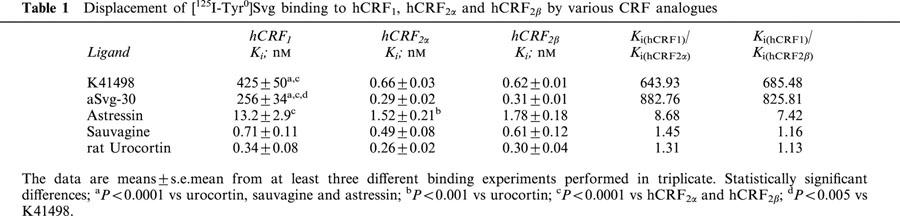
Table 1.
Displacement of [125I-Tyr0]Svg binding to hCRF1, hCRF2α and hCRF2β by various CRF analogues
Developed autoradiograms were quantified under constant illumination using the SCION image analysis system (P.C. version of NIH Image), by comparison of the optical densities of autoradiographic images with those of standard microscales. A paired t-test was employed to compare binding of 125I-K41498 between the denervated and intact sides of the NTS. Brain regions were identified following microscopic examination of adjacent tissue sections stained with 0.1% thionin with reference to a stereotaxic atlas (Paxinos & Watson, 1986).
Results
Binding of CRF analogues to hCRF1, hCRF2α and hCRF2β receptors
The CRF peptide agonists rUcn and Svg showed similar high binding affinities (0.26–0.71 nM) to all three receptors (Table 1). In contrast to astressin, K41498 showed high affinity binding to hCRF2α and hCRF2β (sub-nanomolar Ki's at both receptors, Table 1) but not the hCRF1 receptor (Ki=425±50 nM) resulting in ∼700 fold greater affinity for the hCRF2α and hCRF2β receptor than for the hCRF1 receptor (Table 1, Figure 2A). As was the case for aSvg-30, K41498 did not differ in binding to the CRF2 receptor splice variants (Table 2A).
Figure 2.
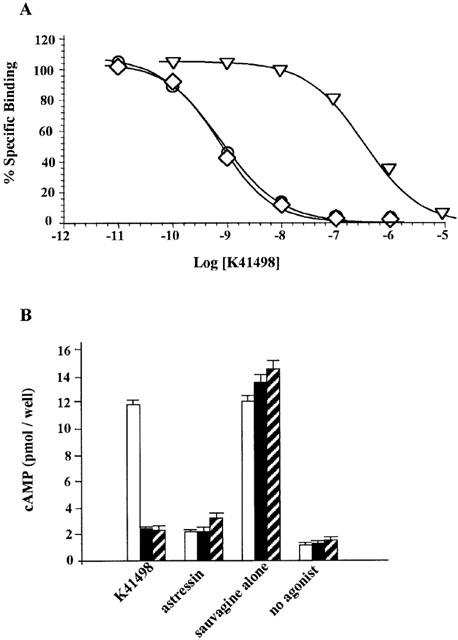
Pharmacological properties of K41498 at human CRF receptors. (A) Competitive binding of K41498 was performed using [125ITyr0]Svg (∼100 pM) and membrane proteins from HEK293 cells expressing hCRF1 (30 μg, open triangles), hCRF2α (open diamonds) or hCRF2β (open circles) receptors (5 μg each). Results are triplicates from three independent experiments. (B) Inhibition of Svg-stimulated cAMP inhibition in HEK293 cells expressing hCRF1 (open columns), hCRF2α (closed columns), or hCRF2β (hatched columns) receptors by K41498. Cells were stimulated as described in Methods with 1 nM Svg in the absence or presence of 100 nM astressin or 100 nM (hCRF2α and hCRF2β) or 10 μM (hCRF1) K41498. For comparison, the basal cAMP values (no agonist) are listed. The data are the mean of one experiment performed in quadruplicates and repeated at least twice.
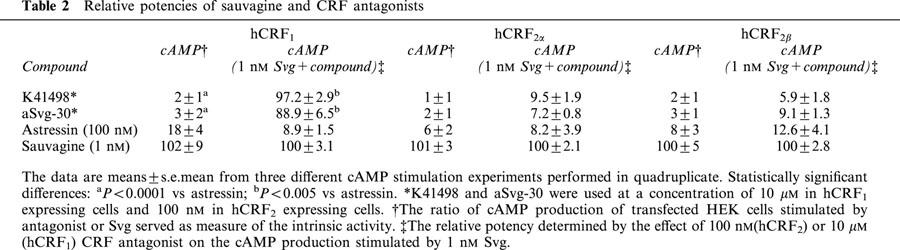
Table 2.
Relative potencies of sauvagine and CRF antagonists
cAMP accumulation
The peptide agonist Svg exhibited high potency to increase cAMP concentration in HEK293 cells stably expressing any of the three receptors investigated; EC50 values: 0.42±0.03 nM (hCRF1), 0.34±0.03 nM (hCRF2α), and 0.49±0.08 nM (hCRF2β). The antagonists showed minimal intrinsic activity ranging from a low of 1–3% for K41498 at CRF1 and CRF2 receptors to a high of 6% (hCRF2α), 8% (hCRF2β), and 18% (hCRF1) for astressin at the three receptor-expressing lines (Table 2).
The ability of the CRF antagonists to inhibit Svg-stimulated cAMP accumulation in hCRF1 cells was high in the presence of astressin but low in the presence of K41498 (Table 2, Figure 2B). In contrast, K41498 and astressin were equipotent in inhibiting Svg-mediated cAMP accumulation in hCRF2α- and hCRF2β-expressing cells (Table 2, Figure 2B).
Cardiovascular studies
Figures 3 and 4 show the temporal and group data respectively for the effect of pre-treatment with K41498 on urocortin-evoked changes in blood pressure. In WKY rats, urocortin caused reproducible depressor responses after intra-venous (i.v.) injection (1.4 μg i.v., not shown). As shown (Figures 3A and 4A), this dose of urocortin resulted in a substantial (>30 mm Hg) and long-lasting hypotension. Systemic administration of K41498 itself caused a small (∼10–15 mm Hg) pressor response in three out of the four rats; however, this was not statistically significant. In all cases, pre-treatment with K41498 (1.84 μg i.v., n=4) completely abolished the hypotensive response to urocortin (Figures 3B and 4A). In contrast, central administration of urocortin (2.35 μg i.c.v., n=4) caused a pressor response (Figures 3C and 4B), that was not affected by pre-treatment with K41498 (1.84 μg i.c.v., Figures 3D and 4B). Interestingly, this dose of K41498 caused a reproducible, statistically significant, pressor response by itself (∼25 mm Hg, P<0.05 Paired t-test) when centrally administered (Figure 3D). While pre-treatment with K41498 had no effect on the pressor response to centrally administered urocortin, pre-treatment with antalarmin (2.1 μg i.c.v., n=3, Figure 4C) effectively abolished urocortin evoked hypertension. Antalarmin itself caused no change in blood pressure.
Figure 3.
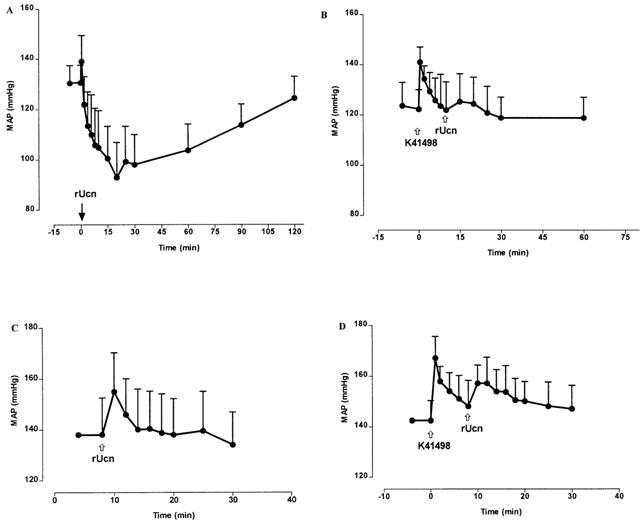
Temporal data for the effects of K41498 on urocortin-mediated changes in blood pressure. (A) The hypotensive response following intra-venous administration of urocortin (1.4 μg, n=4). (B) Pre-treatment with K41498 (1.84 μg i.v., n=4) 10 min prior to urocortin prevents the hypotensive response. (C) Intracerebroventricular administration of urocortin (2.35 μg, n=4) results in a pressor response. (D) Pre-treatment with K41498 (1.84 μg i.c.v., n=4) 10 min prior to urocortin, causes a pressor response by itself, but has no impact upon the pressor response to urocortin.
Figure 4.
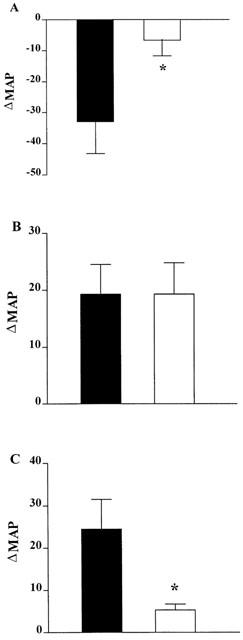
Group data for the effects of K41498 on urocortin-mediated changes in blood pressure. (A) The hypotensive response to urocortin (1.4 μg, i.v.) before (closed symbols) or 10 min after (open symbols) systemic injection of K41498 (1.84 μg i.v., n=4). (B) The hypertensive response to urocortin (2.35 μg, i.c.v.) before (closed symbols) or 10 min after (open symbols) central injection of K41498 (1.84 μg i.c.v., n=4). (C) The hypertensive response to urocortin (2.35 μg, i.c.v.) before (closed symbols) or 10 min after (open symbols) central injection of antalarmin (2.1 μg i.c.v., n=3). *P<0.05 response to urocortin significantly different after pretreatment with antagonist, Paired t-test.
Autoradiographic studies
125I-K41498 (100 pM) bound specifically and topographically to slices of rat heart, brain and nodose ganglia. Particularly dense binding was observed around coronary vessels and also the middle and anterior cerebral arteries. In rat heart, rUcn, astressin and K41498 (1 μM each) all completely displaced the binding of 125I-K41498 to both cardiac tissue and coronary vessels, whereas antalarmin caused only 12% inhibition (Figure 5). Similarly, in the nodose ganglia, astressin and K41498 (1 μM each) displaced the binding of 125I-K41498 by 77–87% whereas antalarmin (1 μM) caused only 18% inhibition of binding (not shown).
Figure 5.
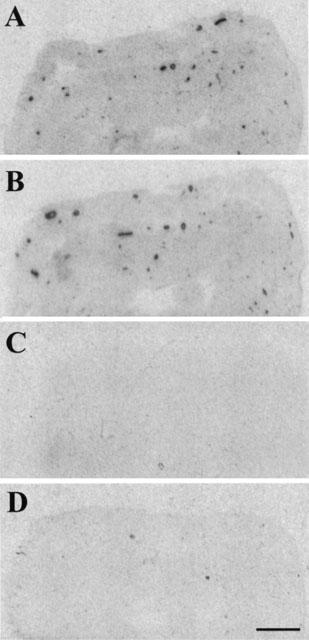
Autoradiograms demonstrating the distribution of 125I- K41498 binding in sections of rat heart (scale bar=1.4 mm). (A) Total binding. Note the particularly dense binding around coronary vessels. (B) Binding of 125I- K41498 in the presence of antalarmin (1 μM). (C) Binding of 125I- K41498 in the presence of astressin (1 μM). (D) Binding of 125I- K41498 in the presence of K41498 (1 μM).
The pharmacological profile of 125I-K41498 binding to rat brain slices resembled that found in heart and nodose ganglia. Thus, binding in forebrain (lateral septum and choroid plexus) and brain stem was sensitive to rUcn, astressin and K41498 (1 μM each) and insensitive to antalarmin (1 μM). In medulla oblongata, specific 125I-K41498 binding was restricted to the NTS, area postrema and the lateral border of the interpolar sub-region of the spinal trigeminal nucleus (Figure 6). Unilateral vagaldeafferentation resulted in 65±4% reduction in the binding of 125I-K41498 in the ipsilateral NTS compared to the contralateral (intact) NTS (n=5 rats, Figure 6).
Figure 6.
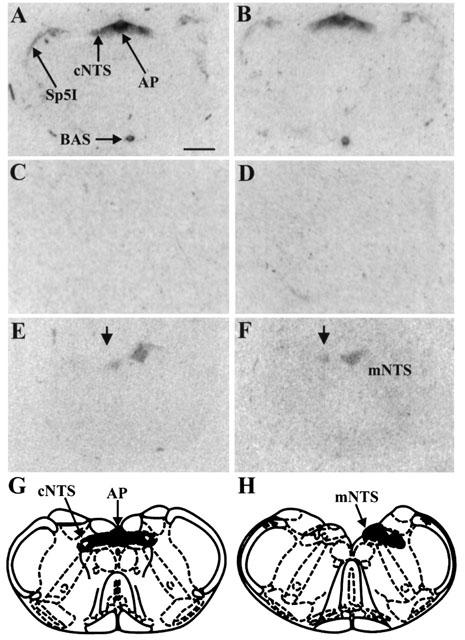
Autoradiograms demonstrating the distribution of 125I-K41498 binding in sections of rat brainstem, at the level of the nucleus tractus solitarius (NTS)/area postrema (scale bar=1 mm). (A) Total binding. (B) Binding of 125I- K41498 in the presence of antalarmin (1 μM). (C) Binding of 125I- K41498 in the presence of astressin (1 μM). (D) Binding of 125I- K41498 in the presence of K41498 (1 μM). (E) Binding of 125I- K41498 following unilateral nodose ganglionectomy. The arrow indicates the denervated side of the rat NTS, demonstrating a dramatic reduction in binding following vagal deafferentation. (F) Binding of 125I- K41498, in the presence of antalarmin (1 μM) following unilateral nodose ganglionectomy. The arrow indicates the denervated side of the rat NTS, and demonstrates that the remaining binding following deafferentation is insensitive to antalarmin. (G) Schematic diagram demonstrating the level of the rat brain stem depicted in A–D. (H) Schematic diagram demonstrating the level of the rat brain stem depicted in G and H. Abbreviations as follows: AP, area postrema; BAS, basilar artery, cNTS, commissural nucleus tractus solitarius; mNTS, medial nucleus tractus solitarius; Sp5I, interpolar subdivision of the spinal trigeminal nucleus.
Discussion
The present study details the characterization of a high affinity CRF2 receptor antagonist, K41498, that is not only highly selective for CRF2 over CRF1 receptors, but is also amenable to radio-iodination without loss of properties. K41498 therefore represents a milestone compound for neurochemical, functional and autoradiographic studies of CRF2 receptors and thus will help shed more light on the physiological role of this CRF receptor subtype. Replacement of the methionine residue of aSvg30 with norleucine was to prevent sulphoxidation and therefore increase chemical and metabolic stability of the peptide. However, it is not guaranteed that the pharmacological properties of such a peptide would be the same as aSvg30. Hence we investigated the binding affinity of K41498 and found the selectivity of K41498 for CRF2 receptors to be essentially the same as aSvg30.
The CRF2 receptor is the predominant CRF receptor in the periphery and the mRNA encoding CRF2 receptors has been found in the skeletal muscle, heart, vasculature, testis, ovaries and the gastrointestinal tract (Chalmers et al., 1995; Lovenberg et al., 1995; Perrin et al., 1995; Palchaudhuri et al., 1999; Muramatsu et al., 2000). The recent discovery of further members of the CRF peptide family, namely urocortin II and III in the mouse and stresscopin and stresscopin-related peptide in man (Reyes et al., 2001; Hsu & Hsueh, 2001; Lewis et al., 2001), appears to have identified selective, endogenous agonists for the CRF2 receptor. Until then Ucn had been considered as the endogenous ligand for this receptor, although some debate surrounded this issue (Bittencourt et al., 1999).
We recently developed aSvg-30, a specific antagonist for the CRF2 receptor (Rühmann et al., 1998; Higelin et al., 2001). This peptide ligand to date is the only CRF2 receptor-specific antagonist. Pharmacological studies have confirmed that aSvg-30 does act as a competitive antagonist at CRF2 receptors (Brauns et al., 2001). We have now advanced the study of CRF2 receptors by designing a more metabolically stable analogue of aSvg-30, K41498 that can also be radiolabelled to high specific activity. In membranes isolated from HEK293 cells stably expressing the hCRF1, hCRF2α or hCRF2β receptor, K41498 displayed sub-nanomolar affinity for CRF2 receptors with ∼700 fold selectivity over CRF1 receptors. The antagonist properties of K41498 were confirmed in assays measuring agonist-stimulated accumulation of cAMP.
In addition to the in vitro characterization, we have also demonstrated that K41498 can be used in vivo. K41498 blocked hypotension following systemic administration of urocortin, an established CRF2 receptor-mediated response; however, it had no impact upon the pressor response to centrally administered urocortin, an established CRF1 receptor-mediated response (Richter & Mulvany, 1995), confirmed by the ability of antalarmin to antagonize the central pressor response to urocortin. Interestingly, K41498 alone caused a pressor response following central administration. It is unlikely that the pressor effect of central K41498 represents an agonist action at CRF1 receptors for a number of reasons.
Firstly, as demonstrated in the present study, K41498 is devoid of agonist properties at CRF1 or CRF2 receptors transfected into HEK293 cells. Secondly, recent Schild analysis studies have confirmed that aSvg-30 acts as a competitive antagonist at CRF2 receptors (Brauns et al., 2001). Thirdly, aSvg-30 when administered centrally in conscious rats results in anxiolysis (Takahashi et al., 2001), whereas CRF1 receptor agonists are anxiogenic. It would therefore appear that the pressor response to i.c.v. K41498 is due to blockade of central CRF2 receptors that consequently enables endogenously released CRF to act upon CRF1 receptors and elevate pressure.
Previous studies of CRF2 receptor distribution have essentially been confined to the use of non-selective agonist radioligands in the presence of suppressing concentrations of CRF1 ligands (Rominger et al., 1998; Mar Sanchez et al., 1999). K41498 was therefore radio-iodinated for autoradiographic studies of CRF2 receptors in selected tissues. Dense binding of 125I-K41498 was observed over slices of rat heart and also over the smooth muscle of coronary vessels, in accordance with the expression of the mRNA encoding CRF2 receptors in these tissues (Kageyama et al., 2000). In all cases, binding was totally insensitive to displacement by antalarmin, a CRF1 receptor selective antagonist (Webster et al., 1996). The same profile of 125I-K41498 binding was observed over rat nodose ganglia, suggestive of the presence of CRF2 receptors on vagal perikarya. Consistent with this was the visualization of antalarmin-insensitive 125I-K41498 binding in rat NTS and area postrema, the terminal fields of vagal afferent neurons (Lawrence & Jarrott, 1996).
To establish whether or not vagal afferent terminals house CRF2 receptors, a series of rats were subjected to unilateral vagal deafferentation (nodose ganglionectomy) prior to autoradiography. In these rats, the binding of 125I-K41498 was dramatically reduced in the denervated side of the NTS, indicating that the majority of CRF2 receptors in the rat NTS are located on vagal afferents. The validity of this technique for the determination of the anatomical location of neurotransmitter receptor populations is well established; being previously used to demonstrate the presynaptic localization of opioid receptors (Atweh et al., 1978), dopamine D2 receptors (Lawrence et al., 1995), GABAA receptors (Ashworth-Preece et al., 1997), neuropeptide Y receptors (McLean et al., 1996) and nicotinic receptors (Ashworth-Preece et al., 1998) on rat vagal afferent terminals. While it was originally thought that the NTS and area postrema lacked the mRNA encoding the CRF2 receptor (Chalmers et al., 1995; Mar Sanchez et al., 1999), more recent studies have demonstrated CRF2 receptor mRNA (van pett et al., 2000) in both of these structures. Taken together, this would suggest that the non-vagal component of 125I-K41498 binding largely represents binding to postsynaptic receptors on intrinsic NTS neurons.
Interestingly, previous studies using [125I]Tyr0-sauvagine have suggested the presence of CRF1, but not CRF2 receptors in NTS (Mar Sanchez et al., 1999). It is clear therefore that previous studies of CRF2 receptor distribution using non-selective radioligands must be treated with caution, and furthermore that 125I-K41498 represents a significant development for the study of CRF2 receptors. Consistent with the presence of presynaptic CRF2 receptors in the rat NTS is the presence of urocortin in fibres, but not cells, in this nucleus (Bittencourt et al., 1999). The presence of CRF2 receptors on vagal afferents is a significant observation, and provides an anatomical correlate for the potential modulation of vagal activity by CRF. For example, it is possible that part of the hypotension caused by systemic injections of urocortin and other CRF analogues (Richter & Mulvany, 1995) is mediated by CRF2 receptors modulating the activity of vagal baroreceptor afferent input to the NTS, and/or modulating the release of glutamate, a transmitter of arterial baroreceptor afferents, within the NTS (Lawrence & Jarrott, 1994; 1996). Similarly, CRF effects on feeding may have a vagal component. In agreement with this hypothesis, both central and systemic administration of CRF and urocortin induce the expression of Fos protein in the NTS and area postrema of rats (Benoit et al., 2000; Wang et al., 2000).
In addition to the medulla oblongata, 125I-K41498 binding was also examined in rat forebrain at the level of the lateral septum, an area enriched in CRF2 receptors. Our data indicate dense populations of CRF2 receptors in the lateral septum, choroid plexus and also around cerebral vessels, in complete agreement with previous studies (Rominger et al., 1998). Future studies will undoubtedly provide a detailed account of the distribution and regulation of 125I-K41498 binding throughout the entire neuraxis.
In summary we have designed, synthesized and characterized a high affinity, metabolically stable CRF2-specific antagonist, K41498. Contrary to previous suggestions that central CRF2 receptors are post-synaptic (Rominger et al., 1998), the present study has demonstrated the presence of presynaptic CRF2 receptors on vagal afferent terminals within the rat NTS. 125I-K41498 is thus a useful tool to further study the distribution and regulation of central and peripheral CRF2 receptors.
Acknowledgments
We are grateful to Dr G.P. Chrousos (NIH, Bethesda, MD, U.S.A.) for the generous gift of antalarmin. A.J. Lawrence is a Senior Research Fellow of the NHMRC, Australia. This work was funded in part by the Australian Institute of Nuclear Science & Engineering.
Abbreviations
- cAMP
adenosine 3′, 5′-cyclic monophosphate
- ANOVA
analysis of variance
- AP
area postrema
- aSvg-30
antisauvagine-30
- BAS
basilar artery
- BSA
bovine serum albumin
- cNTS
commissural nucleus tractus solitarius
- CRF
corticotropin-releasing factor
- HEK
human embryonic kidney
- i.c.v.
intra-cerebroventricular
- i.v.
intra-venous
- mNTS
medial nucleus tractus solitarius
- NTS
nucleus tractus solitarius
- RPHPLC
reverse-phase HPLC
- Sp5I
spinal trigeminal nucleus, interpolar
- Svg
sauvagine
- Ucn
urocortin
References
- ARBORELIUS L., OWENS M.J., PLOTSKY P.M., NEMEROFF C.B. The role of corticotropin-releasing factor in depression and anxiety disorders. J. Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- ASHWORTH-PREECE M.A., KRSTEW E., JARROTT B., LAWRENCE A.J. Functional GABAA receptors on rat vagal afferent neurons. Br. J. Pharmacol. 1997;120:469–475. doi: 10.1038/sj.bjp.0700909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASHWORTH-PREECE M.A., JARROTT B., LAWRENCE A.J. Nicotinic acetylcholine receptors in the rat and primate nucleus tractus solitarius and on rat and human inferior vagal nodose ganglia: evidence from in vivo microdialysis and [125I]α-bungarotoxin autoradiography. Neuroscience. 1998;83:1113–1122. doi: 10.1016/s0306-4522(97)00476-4. [DOI] [PubMed] [Google Scholar]
- ASSIL I.Q., SHOMALI M.E., ABOU-SAMRA A.B. An oxidation resistant radioligand for corticotropin-releasing factor receptors. Peptides. 2001;22:1055–1061. doi: 10.1016/s0196-9781(01)00424-7. [DOI] [PubMed] [Google Scholar]
- ATWEH S.F., MURRIN L.C., KUHAR M.J. Presynaptic localization of opiate receptors in the vagal and accessory optic systems: an autoradiographic study. Neuropharmacology. 1978;17:65–71. doi: 10.1016/0028-3908(78)90175-2. [DOI] [PubMed] [Google Scholar]
- BALE T.L., CONTARINO A., SMITH G.W., CHAN R., GOLD L.H., SAWCHENKO P.E., KOOB G.F., VALE W.W., LEE K.F. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nature Genetics. 2000;24:410–414. doi: 10.1038/74263. [DOI] [PubMed] [Google Scholar]
- BEHAN D.P., GRIGORIADIS D.E., LOVENBERG T., CHALMERS D., HEINRICHS S., LIAW C., DE SOUZA E.B. Neurobiology of corticotropin releasing factor (CRF) receptors and CRF-binding protein: implications for the treatment of CNS disorders. Mol. Psych. 1996;1:265–277. [PubMed] [Google Scholar]
- BENOIT S.C., THIELE T.E., HEINRICHS S.C., RUSHING P.A., BLAKE K.A., STEELEY R.J. Comparison of central administration of corticotropin-releasing hormone and urocortin on food intake, conditioned taste aversion, and c-Fos expression. Peptides. 2000;21:345–351. doi: 10.1016/s0196-9781(00)00153-4. [DOI] [PubMed] [Google Scholar]
- BITTENCOURT J.C., VAUGHAN J., ARIAS C., RISSMAN R.A., VALE W.W., SAWCHENKO P.E. Urocortin expression in rat brain: evidence against a pervasive relationship of urocortin-containing projections with targets bearing type 2 CRF receptors. J. Comp. Neurol. 1999;415:285–312. [PubMed] [Google Scholar]
- BONK I., RÜHMANN A. Novel high-affinity photoactivatable antagonists of corticotropin-releasing factor (CRF) photoaffinity labeling studies on CRF receptor, type 1 (CRFR1) Eur. J.Biochem. 2000;267:3017–3024. doi: 10.1046/j.1432-1033.2000.01321.x. [DOI] [PubMed] [Google Scholar]
- BRAUNS O., LIEPOLD T., RADULOVIC J., SPIESS J. Pharmacological and chemical properties of astressin, antisauvagine-30 and α-helCRF: significance for behavioral experiments. Neuropharmacology. 2001;41:507–516. doi: 10.1016/s0028-3908(01)00094-6. [DOI] [PubMed] [Google Scholar]
- CHALMERS D.T., LOVENBERG T.W., DE SOUZA E.B. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J. Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSTE S.C., KESTERSON R.A., HELDWEIN K.A., STEVENS S.L., HEARD A.D., HOLLIS J.H., MURRAY S.E., HILL J.K., PANTELY G.A., HOHIMER A.R., HATTON D.C., PHILLIPS T.J., FINN D.A., LOW M.J., RITTENBERG M.B., STENZEL P., STENZEL-POORE M.P. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nature Genetics. 2000;24:403–409. doi: 10.1038/74255. [DOI] [PubMed] [Google Scholar]
- DAUTZENBERG F.M., HUBER G., HIGELIN J., PY-LANG G., KILPATRICK G.J. Evidence for the abundant expression of arginine 185 containing human CRF2 receptors and the role of position 185 for receptor-ligand selectivity. Neuropharmacology. 2000;39:1368–1376. doi: 10.1016/s0028-3908(00)00044-7. [DOI] [PubMed] [Google Scholar]
- DAUTZENBERG F.M., KILPATRICK G.J., HAUGER R.L., MOREAU J. Molecular biology of the CRH receptors– in the mood. Peptides. 2001a;22:753–760. doi: 10.1016/s0196-9781(01)00388-6. [DOI] [PubMed] [Google Scholar]
- DAUTZENBERG F.M., PY-LANG G., HIGELIN J., FISCHER C., WRIGHT M.B., HUBER G. Different binding modes of amphibian and human corticotropin-releasing factor type 1 and type 2 receptors: evidence for evolutionary differences. J. Pharmacol. Exp. Ther. 2001b;296:113–120. [PubMed] [Google Scholar]
- HIGELIN J., PY-LANG G., PATERNOSTER C., ELLIS G.J., PATEL A., DAUTZENBERG F.M. 125I-Antisauvagine-30: a novel and specific high-affinity radioligand for the characterization of corticotropin-releasing factor type 2 receptors. Neuropharmacology. 2001;40:114–122. doi: 10.1016/s0028-3908(00)00105-2. [DOI] [PubMed] [Google Scholar]
- HOLSBOER F. The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. J. Psychiat. Res. 1999;33:181–214. doi: 10.1016/s0022-3956(98)90056-5. [DOI] [PubMed] [Google Scholar]
- HSU S.Y., HSUEH A.J. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nature Medicine. 2001;7:605–611. doi: 10.1038/87936. [DOI] [PubMed] [Google Scholar]
- KAGEYAMA K, GAUDRIAULT G.E., BRADBURY M.J., VALE W.W. Regulation of corticotropin-releasing factor receptro type 2β messenger ribonucleic acid in the rat cardiovascular system by urocortin, glucocorticoids and cytokines. Endocrinology. 2000;141:2285–2293. doi: 10.1210/endo.141.7.7572. [DOI] [PubMed] [Google Scholar]
- KILPATRICK G.J., DAUTZENBERG F.M., MARTIN G.R., EGLEN R.M. 7TM receptors: the splicing on the cake. Trends Pharmacol. Sci. 1999;20:294–301. doi: 10.1016/s0165-6147(99)01355-3. [DOI] [PubMed] [Google Scholar]
- KISHIMOTO T., RADULOVIC J., RADULOVIC M., LIN C.R., SCHRICK C., HOOSHMAND F., HERMANSON O., ROSENFELD M.G., SPIESS J. Deletion of CRHr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nature Genetics. 2000;24:415–419. doi: 10.1038/74271. [DOI] [PubMed] [Google Scholar]
- LAWRENCE A.J., JARROTT B. L-Glutamate as a neurotransmitter at baroreceptor afferents: evidence from in vivo microdialysis. Neuroscience. 1994;58:585–591. doi: 10.1016/0306-4522(94)90083-3. [DOI] [PubMed] [Google Scholar]
- LAWRENCE A.J., JARROTT B. Neurochemical modulation of cardiovascular control in the nucleus tractus solitarius. Prog. Neurobiol. 1996;48:21–53. doi: 10.1016/0301-0082(95)00034-8. [DOI] [PubMed] [Google Scholar]
- LAWRENCE A.J., KRSTEW E., JARROTT B. Functional dopamine D2 receptors on rat vagal afferent neurones. Br. J. Pharmacol. 1995;114:1329–1334. doi: 10.1111/j.1476-5381.1995.tb13352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEWIS K., LI C., PERRIN M.H., BLOUNT A., KUNITAKE K., DONALDSON C., VAUGHAN J., REYES T.M., GULYAS J., FISCHER W., BILEZIKJIAN L., RIVIER J., SAWCHENKO P.E., VALE W.W. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc. Natl. Acad. Sci. U.S.A. 2001;98:7570–7575. doi: 10.1073/pnas.121165198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVENBERG T.W., CHALMERS D.T., LIU C., DE SOUZA E.B. CRF2 alpha and CRF2 beta receptor mRNAs are differentially distributed between the rat central nervous system and peripheral tissues. Endocrinology. 1995;136:4139–4142. doi: 10.1210/endo.136.9.7544278. [DOI] [PubMed] [Google Scholar]
- MAR SANCHEZ M., YOUNG L.J., PLOTSKY P.M., INSEL T.R. Autoradiographic and in situ hybridization localization of corticotropin-releasing factor 1 and 2 receptors in nonhuman primate brain. J. Comp. Neurol. 1999;408:365–377. [PubMed] [Google Scholar]
- MCCARTHY J.R., HEINRICHS S.C., GRIGORIADIS D.E. Recent advances with the CRF1 receptor: design of small molecule inhibitors, receptor subtypes and clinical indications. Curr. Pharm. Des. 1999;5:289–315. [PubMed] [Google Scholar]
- MCLEAN K.J., JARROTT B., LAWRENCE A.J. Neuropeptide Y gene expression and receptor autoradiography in hypertensive and normotensive rat brain. Mol. Brain Res. 1996;35:249–259. doi: 10.1016/0169-328x(95)00219-i. [DOI] [PubMed] [Google Scholar]
- MURAMATSU Y., FUKUSHIMA K., IINO K., TOTSUNE K., TAKAHASHI K., SUZUKI T., HIRASAWA G., TAKEYAMA J., ITO M., NOSE M., TASHIRO A., HONGO M., OKI Y., NAGURA H., SASANO H. Urocortin and corticotropin-releasing factor receptor expression in the human colonic mucosa. Peptides. 2000;21:1799–1809. doi: 10.1016/s0196-9781(00)00335-1. [DOI] [PubMed] [Google Scholar]
- PALCHAUDHURI M.R., HAUGER R.L., WILLE S., FUCHS E., DAUTZENBERG F.M. Isolation and pharmacological characterization of two functional splice variants of corticotropin-releasing factor type 2 receptor from Tupaia belangeri. J. Neuroendocrinol. 1999;11:419–428. doi: 10.1046/j.1365-2826.1999.00348.x. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. Australia: Academic Press; 1986. The rat brain in stereotaxic coordinates. [Google Scholar]
- PERRIN M., DONALDSON C., CHEN R., BLOUNT A., BERGGREN T., BILEZIKJIAN L., SAWCHENKO P., VALE W. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc. Natl. Acad. Sci. U.S.A. 1995;92:2969–2973. doi: 10.1073/pnas.92.7.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RADULOVIC J., RÜHMANN A., LIEPOLD T., SPIESS J. Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress: differential roles of CRF receptors 1 and 2. J. Neurosci. 1999;19:5016–5025. doi: 10.1523/JNEUROSCI.19-12-05016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REYES T.M., LEWIS K., PERRIN M.H., KUNITAKE K.S., VAUGHAN J., ARIAS C.A., HOGENESCH J.B., GULYAS J., RIVIER J., VALE W., SAWCHENKO P.E. Urocortin II: A member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc. Natl. Acad. Sci. U.S.A. 2001;98:2843–2848. doi: 10.1073/pnas.051626398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RICHTER R.M., MULVANY M.J. Comparison of hCRF and oCRF effects on cardiovascular responses after central, peripheral and in vitro application. Peptides. 1995;16:843–849. doi: 10.1016/0196-9781(95)00035-i. [DOI] [PubMed] [Google Scholar]
- ROMINGER D.H., ROMINGER C.M., FITZGERALD L.W., GRZANNA R., LARGENT B.L., ZACZEK R. Characterization of [125I]sauvagine binding to CRH2 receptors: membrane homogenate and autoradiographic studies. J. Pharmacol. Exp. Ther. 1998;286:459–468. [PubMed] [Google Scholar]
- RÜHMANN A., BONK I., LIN C.R., ROSENFELD M.G., SPIESS J. Structural requirements for peptidic antagonists of the corticotropin-releasing factor receptor (CRFR): development of CRFR2beta-selective antisauvagine-30. Proc. Natl. Acad. Sci. U.S.A. 1998;95:15264–15269. doi: 10.1073/pnas.95.26.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RÜHMANN A., KOPKE A.K., DAUTZENBERG F.M., SPIESS J. Synthesis and characterization of a photoactivatable analogue of corticotropin releasing factor for specific receptor labeling. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10609–10613. doi: 10.1073/pnas.93.20.10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKAHASHI L.K., HO S.P., LIVANOV V., GRACIANA N., ARNERIC S.P. Antagonism of CRF2 receptors produces anxiolytic behavior in animal models of anxiety. Brain Res. 2001;902:135–142. doi: 10.1016/s0006-8993(01)02405-2. [DOI] [PubMed] [Google Scholar]
- VALE W., VAUGHAN J., PERRIN M. Corticotropin-releasing factor (CRF) family of ligands and their receptors. Endocrinologist. 1997;7:3S–9S. [Google Scholar]
- VAN PETT K., VIAU V., BITTENCOURT J.C., CHAN R.K., LI H.Y., ARIAS C., PRINS G.S., PERRIN M., VALE W., SAWCHENKO P.E. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J. Comp. Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- WANG L., MARTINEZ V., VALE W., TACHE Y. Fos induction in selective hypothalamic neuroendocrine and medullary nuclei by intravenous injection of urocortin and corticotropin-releasing factor in rats. Brain. Res. 2000;855:47–57. doi: 10.1016/s0006-8993(99)02200-3. [DOI] [PubMed] [Google Scholar]
- WEBSTER E.L., LEWIS D.B., TORPY D.J., ZACHMAN E.K., RICE K.C., CHROUSOS G.P. In vivo and in vitro characterization of antalarmin, a nonpeptide corticotropin-releasing hormone (CRH) receptor antagonist: suppression of pituitary ACTH release and peripheral inflammation. Endocrinology. 1996;137:5747–5750. doi: 10.1210/endo.137.12.8940412. [DOI] [PubMed] [Google Scholar]