

Abstract
Organic cation transporters (OCTs) are involved in the elimination of monoamines and cationic xenobiotics. To examine whether some cell lines express several different OCTs, we investigated seven human cell lines for the mRNA expression pattern of the human (h) transporters hOCT1, hOCT2 and hOCT3. hOCT1 mRNA was found in all cell lines, six additionally expressed hOCT3 and only two cell lines contained all three hOCTs.
Among the three OCTs only for the OCT3 (also designated as ‘uptake2' or ‘extraneuronal monoamine transporter') ‘selective' inhibitors are described in the literature. The affinities of the OCT3 inhibitors for the other two OCTs are largely unknown. Therefore, we compared the potencies of eight compounds as inhibitors of hOCT-mediated uptake of the organic cation [3H]-1-methyl-4-phenylpyridinium ([3H]-MPP+) in human embryonic kidney 293 (HEK293) cells stably expressing hOCT1, hOCT2 or hOCT3. Decynium-22 inhibited hOCT3 with 10 fold higher potency than hOCT1 and hOCT2. Corticosterone was about 100 fold more potent as inhibitor of hOCT3 than of hOCT1 or hOCT2, and O-methylisoprenaline (OMI) inhibited almost exclusively hOCT3. Progesterone and β-Oestradiol preferentially inhibited hOCT3 and hOCT1, whereas prazosin was a potent inhibitor of hOCT1 and hOCT3. Phenoxybenzamine (PbA) inhibited with about equal apparent potency all three hOCTs, whereas the PbA derivative SKF550 ((9-fluorenyl)-N-methyl-β-chloroethylamine) preferentially inhibited hOCT3 and hOCT2.
PbA reversibly inhibited hOCT1 and irreversibly hOCT2 and hOCT3; SKF550 also irreversibly inhibited hOCT3 but hOCT2 in a reversible manner.
These compounds enable a functional discrimination of the three hOCTs: hOCT1 is selectively inhibited by prazosin, reversibly inhibited by PbA and it is not sensitive to inhibition by SKF550 and OMI; hOCT2 is reversibly inhibited by SKF550, irreversibly by PbA and not by prazosin, β-oestradiol and OMI, whereas hOCT3 is selectively inhibited by corticosterone, OMI and decynium22.
Keywords: Organic cation transporters, expression pattern, OCT inhibitors, irreversible inhibition
Introduction
Catecholamines are removed from the circulation by uptake into non-neuronal tissues through the action of the Na+- and Cl−-independent, corticosterone-sensitive extraneuronal monoamine transport system ‘uptake2' whereas synaptically released noradrenaline is mainly transported back into noradrenergic neurones via the Na+- and Cl−-dependent, desipramine-sensitive neuronal noradrenaline transport system ‘uptake1' (Iversen, 1965; 1973; Bönisch, 1980; Graefe & Bönisch, 1988; Eisenhofer, 2001). Molecular cloning of both transporter cDNAs has shown that they belong to two different transporter families. While the neuronal noradrenaline transporter is a member of the Na+- and Cl−-dependent monoamine neurotransmitter transporters, the ‘uptake2' transporter is represented by OCT3 and thus belongs to the family of organic cation transporters (OCTs) which are involved in the absorption, distribution and elimination of endogenous compounds (e.g. amines) as well as of drugs, toxins and other xenobiotics that are positively charged at physiological pH (Eisenhofer, 2001; Burckhardt & Wolff, 2000).
The first cloned OCT has been the rat OCT1 (rOCT1) (Gründemann et al., 1994). Meanwhile also the mouse (Schweifer & Barlow, 1996), rabbit (Terashita et al., 1998) and human OCT1 transporters have been cloned. The human OCT1 (hOCT1) (Zhang et al., 1997; Gorboulev et al., 1997) shows about 80% amino acid (aa) identity to the rOCT1. From the human glioma cell line SK-MG-1 which also expresses an ‘uptake2' transport system (Streich et al., 1996) we recently isolated four hOCT1 isoforms, the full-length hOCT1 and splice variants (Hayer et al., 1999).
The rat, human and porcine OCT2 have been cloned from kidney (Okuda et al., 1996; Gorboulev et al., 1997; Gründemann et al., 1997), and the human and porcine transporters show about 90% amino acid identity to the rat OCT2.
The rat OCT3 which has been isolated from placenta (Kekuda et al., 1998) represents the ‘uptake2' transport system (Wu et al., 1998a). The human OCT3 cDNA has been amplified from the human kidney carcinoma cell line Caki-1 (Gründemann et al., 1998), which previously had been shown to express the ‘uptake2' transport system (Schömig & Schönfeld, 1990). All three hOCT genes have been shown to be clustered on the long arm of chromosome 6 (Verhaagh et al., 1999). The close physical linkage of the three OCT genes and their structural similarities (Gründemann & Schömig, 2000; Wieland et al., 2000) indicate that they may have evolved from a common anchestor gene through duplication. Interestingly, the human colon adenocarcinoma cell line Caco-2 expresses the mRNAs of all three human OCTs (Bleasby et al., 2000; Martel et al., 2001). Therefore, one aim of the present study was to explore by examining a series of human cell lines whether expression of more than one OCT mRNA is a frequent phenomenon in tumour cells.
In spite of the similiarities between the three OCTs little is known about similarities in their pharmacological properties. Gründemann et al. (1999) have shown some differences between the three OCTs concerning the transport efficiencies of various substrates; however, some of the observed differences might reflect species differences since the pharmacological properties were examined in rat OCT1, rat OCT2 but human OCT3. Recently, Dresser et al. (2000) have demonstrated that some properties of the human OCT1 differ from those of the mouse, rat and rabbit OCT1 homologues. Species differences also have been observed between the rat and human OCT2 transporters concerning inhibition of 14C-TEA uptake by some compounds such as procainamide and MPP+ (Koepsell et al., 1999). Finally, species differences have also been described for the OCT3: while the rat OCT3 transports MPP+ and the prototypic organic cation TEA (Kekuda et al., 1998), virtually no transport of TEA could be demonstrated for the human homologue (Gründemann et al., 1998).
The main aim of the present study was to examine within one species (human) differences in the pharmacology of the three OCTs. Thus, we cloned the hOCT2- and hOCT3-cDNAs and stably transfected HEK293 cells with hOCT1-, hOCT2- or hOCT3-cDNA. Using these cells we examined inhibition of hOCT-mediated uptake of the organic cation [3H]-MPP+ by a series of compounds, which all have previously been shown to inhibit ‘uptake2' at low micromolar concentrations.
Methods
RT–PCR
Total RNA from about 107 cells each of the human cell lines SK-MG-1 (glioma), Ski-1 (glioma), Caki-1 (kidney carcinoma), HepG-2 (hepatocellular carcinoma), FL (amnion epithelium), Caco-2 (colon adenocarcinoma), and JAR (choriocarcinoma) was isolated (RNeasy Mini Kit, Qiagen, Hilden) and primed with an Oligo(dT)18 primer to synthesize the first strand cDNA using SuperScriptII Reverse Transcriptase (Gibco, Life Technologies) according to the manufacturer's protocol. Primers for the amplification of full-length hOCT3 were deduced from the 5′- and 3′-nontranslated region of the hOCT3 cDNA (accession number AJ001417) published by Gründemann et al. (1998) (sense, position: bp −12–+12, 5′- GGCGGGCGCACCATGCCCTCCTTC-3′; antisense, position: bp 1711-1685; 5′- GCTCCTGGATAGCTCCTTCTTTCTGTC-3′). PCR was performed with cDNA from the human glioma cell line SK-MG-1 with the ‘Fail Safe PCR System' (Biozym) using buffer G according to the manufacturer's protocol and the following temperature program: inital denaturation (94°C) for 3 min, 94°C for 1 min, 68°C for 2 min, for 40 cycles, followed by a final 10 min extension at 68°C. For the amplification of full length hOCT2, human Marathon kidney cDNA (Clontech, Palo Alto, CA, U.S.A.) was used as template for PCR amplification. Primers were deduced from the 5′- and 3′-nontranslated region of the hOCT2 cDNA (accession number X98333; Gorboulev et al., 1997) (sense, position: −125-−105, 5′- GTCACTTGCAGAGGTAAACTC-3′; antisense, position: 1700–1680, 5′- CTAGGTCATGACAGCAGCAAC-3′). PCR was performed using Expand Long Template PCR System (Roche, Germany) and the following temperature program: 94°C for 1 min, 62°C for 1 min and 68°C for 2 min for 40 cycles, followed by a final 10 min extension at 68°C. PCR products were separated by electrophoresis on 0.8% agarose gels and the fragments were isolated and subcloned into the pCR2.1 vector (TA Cloning Kit, Invitrogen, Netherlands) using T4 DNA Ligase (Roche, Mannheim, Germany) followed by transformation into E. coli INVαF′ competent cells. Plasmid DNA was isolated by means of the Spin Miniprep Kit (Qiagen, Hilden, Germany). The cDNAs were sequenced with an automated sequencer (Li-COR 4200, MWG Biotech, Ebersberg, Germany) and the Thermo Sequenase fluorescent labelled primer cycle sequencing kit with 7-deaza-dGTP (Amersham, Freiburg, Germany). Sequence alignment was performed by the PC/GENE software (IntelliGenetics).
Amplification of hOCT1, hOCT2 and hOCT3 fragments from the human cell lines was performed using primers (0.2 μM each) deduced from the the published hOCT1, hOCT2 and hOCT3 cDNA sequences: hOCT1 (Zhang et al., 1997; accession number U77086): sense bp 1579–1596, 5′-GACGCCGAGAACCTTGGG-3′ and antisense bp 1776–1759, 5′-GGGTAGGCAAGTATGAGG-3′; hOCT2 (Gorboulev et al., 1997): sense bp 691–712, 5′-CGGAGATATCGGAGAACAGT-3′ and antisense bp 890–870, 5′-GCATTCTTATTCTGGGAGATC-3′; hOCT3 (Gründemann et al., 1998): sense bp 1408–1431, 5′-GTTTCGCTCTGTTCAGGTCTGTGT-3′ and antisense bp 1881–1861, 5′-TTATGTGTTCCCAGAAACTTC-3′. PCR was performed using 2 U Taq DNA polymerase (Gibco–BRL), dNTPs (200 μM each), MgCl2 (2 mM), 1×PCR buffer, and the following temperature program: initial denaturation (94°C) for 2 min, 94°C for 30 s, 58°C (hOCT1), 54°C (hOCT2), 64°C (hOCT3) for 30 s, 72°C for 30 s for 35 cycles. Beta-actin amplification served as internal standard; specific primers (sense, bp 840–862, 5′-CACTCTTCCAGCCTTCCTTCCTG-3′ and antisense, bp 1197–1175, 5′-TAGTCCGCCTAGAAGCATTTGCG-3′) were used with the following temperature program: 94°C for 1 min, 64°C for 30 s, 72°C for 1 min, for 25 cycles. The PCR products were separated on 1% agarose gels. The predicted sizes of the PCR products were (in bp): 197 (hOCT1), 199 (hOCT2), 473 (hOCT3) and 357 (β-actin).
Cell lines
For RT–PCR we used seven established human tumour cell lines: SK-MG-1 (human glioma; a gift from Dr Cairncross, London, Ontario), Ski-1 (human glioma; a gift from Dr Panasci, Montreal, Quebec, Canada), Caki-1 (human kidney carcinoma; ATCC HTB 46), HepG-2 (human hepatoma; ATCC HB-8065), FL (human amnion; ATCC CCL-62), Caco-2 (human colon adenocarcinoma; ATCC HTB-37) and JAR (human placenta choriocarcinoma; ATCC HTB-144). HEK293 (human embryonic kidney; ATCC CRL-1573) cells were used for stable transfection of hOCT1, hOCT2 or hOCT3.
Cell culture
HEK293 cells stably transfected with the cDNAs of hOCT1, hOCT2 or hOCT3 were maintained at 37°C in a humified atmosphere (5% CO2) on plastic culture flasks (Gibco, Life Technologies, Eggenstein, Germany). The medium was DMEM/HAMS F12 (Sigma-Aldrich, Deisenhofen, Germany) supplemented with 10% foetal calf serum (FKS; Biochrom, Berlin Germany), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. The culture medium was changed every 2–3 days. SK-MG-1, Ski-1 and Caki-1 cells were grown in McCoy's 5A, HepG-2 and FL cells were grown in MEM, JAR cells were grown in RPMI medium (all supplemented with 10% FKS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin) and Caco-2 cells were grown in DMEM medium supplemented with 20% FKS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin.
Stable transfection of hOCT1, hOCT2 and hOCT3
Full-length hOCT1 cDNA was subcloned into the mammalian expression vector pcDNA3 (Invitrogen, Netherlands), full-length hOCT2 and hOCT3 were subcloned into the mammalian expression vector pIRESneo (Invitrogen, Netherlands) and HEK293 cells were transfected with these vectors by the modified calcium phosphate precipitation method (Chen & Okayama, 1987). Selection of stably transfected cells was carried out with geneticin (G418, 800 μg ml−1, Life Technologies, Karlsruhe, Germany) for 3 weeks. Expression of hOCT1, hOCT2 and hOCT3 was verified by RT–PCR (data not shown) and functional characterization.
Transport assays
For measurement of [3H]-MPP+ uptake, HEK293 cells stably expressing the hOCT1, hOCT2 or hOCT3 were grown in 12 well cell culture plates (Falcon, Becton Dickinson, Heidelberg, Germany) precoated with 0.1 g l−1 poly-L-ornithine in 0.15 M sodium borate (pH 8.4). After 2 days in culture, the cells were used for uptake experiments. Since all three OCTs (Gründemann et al., 1999) transport the neurotoxin 1-methyl-4-phenylpyridinium (MPP+), 10 min rates of uptake of 25 nM [3H]-MPP+ (which represent initial rates of uptake; data not shown) were measured at 37°C using a Krebs–Ringer–HEPES (KRH) buffer of the following composition: (mM) HEPES 25 (pH 7.4), NaCl 125, KCl 4.8, D-glucose 5.6, CaCl2 1.2, KH2PO4 1.2 and MgSO4 1.2. If not stated otherwise, inhibitors were also present during 15 min preincubation of the cells with KRH. Uptake was stopped by rinsing the cells four times with 1 ml of ice-cold KRH buffer. Cells were then solubilized in 0.1% (v v−1) Triton X-100, and 0.1 ml and 0.8 ml samples were assayed for protein content (by the method of Lowry et al., 1951) and for [3H] content (by scintillation counting), respectively.
HEK293 cells stably expressing hOCT1, hOCT2 or hOCT3 were cultured as described above. Two days after subculturing, cells were used for uptake and wash-out experiments. For controls, cells were preincubated for 15 min in the absence of an uptake inhibitor and then uptake of [3H]-MPP+ (25 nM, 10 min at 37°C) was measured using KRH buffer again without an uptake inhibitor. For determination of reversibility of hOCT inhibition, cells were preincubated for 15 min with 10 μM PbA, 0.3 μM SKF550 or 100 μM OMI. After preincubation, cells were incubated for 10 min with 25 nM [3H]-MPP+ either without previous washing of the cells (zero min wash-out, i.e., in the continued presence of an uptake inhibitor) or after washing of the cells for 2, 30 or 60 min with inhibitor-free solution. Uptake of [3H]-MPP+ was stopped and cells were then solubilized as described above.
Calculations and statistics
Nonlinear regression analysis (GraphPad Prism software, San Diego, CA, U.S.A.) was used for calculation of IC50 values. Since Km values for MPP+ uptake by the three OCTs are between 10 and 100 μM (Burckhardt & Wolff, 2000) and since nonsaturating concentrations of [3H]-MPP+ (25 nM) were used, the IC50 values reflect Ki values at least for those compounds which are not irreversible inhibitors. Shown are arithmetic means±s.e.mean of n experiments. Statistical significance was analysed using Student's t-test or ANOVA, as appropriate. Differences were considered to be significant when P<0.05.
Drugs
[3H]-MPP+ iodide (N-[methyl-3H]-4-phenylpyridinium iodide; specific activity 80 Ci mmol−1) was obtained from Biotrend (Cologne, Germany), decynium-22 [D22] (1,1′-diethyl-2,2′cyanine iodide), corticosterone, prazosin hydrochloride, phenoxybenzamine hydrochloride [PbA] and progesterone were from Sigma (Munich, Germany), β-oestradiol (17-β-oestradiol) was from ICN Biomedicals (Eschwege, Germany), O-methylisoprenaline hydrochloride [OMI] and SKF 550 ((9-fluorenyl)-N-methyl-β-chloroethylamine hydrochloride) were gifts from Boehringer (Ingelheim, Germany) and SmithKline Beecham (King of Prussia, PA, U.S.A.), respectively.
Results
Cloning of full-length hOCT2 and hOCT3
The full-length hOCT2 and hOCT3 cDNAs were cloned by RT–PCR from SK-MG-1 cells and human kidney, respectively (Figure 1). Sequence analysis of the isolated cDNAs demonstrated that the deduced amino acid sequences were identical to the published hOCT2 and hOCT3 sequences (Gorboulev et al., 1997, Gründemann et al., 1998). Due to a very high GC content of the hOCT3 cDNA, Gründemann et al. (1998) failed to amplify the full-length cDNA; using the fail safe PCR system (see Methods) we were able to clone for the first time the full-length hOCT3 cDNA. The cloning of the hOCT1 cDNA has been reported previously (Hayer et al., 1999).
Figure 1.
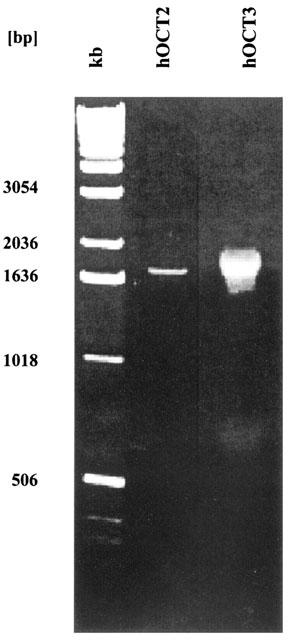
Agarose gel electrophoresis (0.8% agarose gel) of the PCR products from human kidney cDNA (hOCT2) and cDNA from the human glioma cell line SK-MG-1 (hOCT3). kb: 1 kb DNA ladder (Gibco, Life Technologies, Karlsruhe, Germany).
Expression of hOCT1, hOCT2 and hOCT3 in various human tumour cell lines
RT–PCR analysis of mRNA expression of hOCT1, hOCT2 and hOCT3 was performed with RNA from seven human cell lines which previously had been shown to express at least one human OCT (see Discussion). Figure 2 shows that the placenta cell line JAR expresses only the hOCT1 while all other examined cell lines express the hOCT1 and another hOCT. Thus, expression of hOCT1 and hOCT3 was observed in the glioma cell line Ski-1, the kidney carcinoma line Caki-1, the hepatoma line HepG-2 and the amnion line FL (Figure 2). Expression of all three hOCTs was shown for the intestinal cell line Caco-2 and the glioma line SK-MG-1 (Figure 2).
Figure 2.
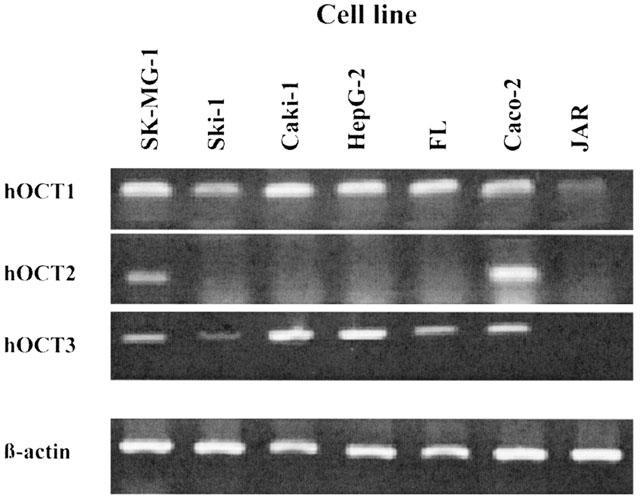
Expression pattern of hOCT1, hOCT2 and hOCT3 in seven human cell lines. The 1% agarose gel shows the 197 bp hOCT1, 199 bp hOCT2, 473 bp hOCT3 and 357 bp β-actin PCR product.
Inhibition of hOCT1, hOCT2 and hOCT3 expressed in stably transfected HEK293 cells
Compounds known to inhibit the ‘uptake2' transport system (Bönisch, 1980) were examined for inhibition of hOCT-mediated uptake of [3H]-MPP+ in HEK293 cells stably expressing either hOCT1, hOCT2 or hOCT3. The inhibition curves are shown in Figure 3 and the calculated IC50 values are summarized in Table 1.
Figure 3.
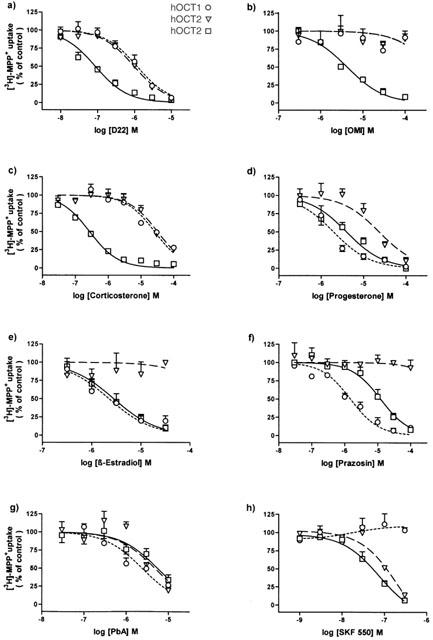
Inhibition of 10 min uptake of 25 nM [3H]-MPP+ in HEK293 cells stably expressing hOCT1, hOCT2 or hOCT3. Initial rates of [3H]-MPP+ transport were determined in the presence of various concentrations of (a) Decynium22 (D22), (b) O-Methylisoprenaline (OMI), (c) Corticosterone, (d) Progesterone, (e) β-Oestradiol, (f) Prazosin, (g) Phenoxybenzamine (PbA), (h) SKF550 and expressed in per cent of control. Shown are arithmetic means±s.e.mean of n=4–8 independent experiments carried out in triplicates. Uptake of [3H]-MPP+ under control conditions was: 247±14 fmol mg−1 protein for hOCT1, 737±39 fmol mg−1 protein for hOCT2 and 1106±72 fmol mg−1 protein for hOCT3.
Table 1.
IC50 values of various compounds for inhibition of [3H]-MPP+ uptake (25 nM, 10 min) by hOCT1, hOCT2 or hOCT3 expressed in stably transfected HEK293 cells
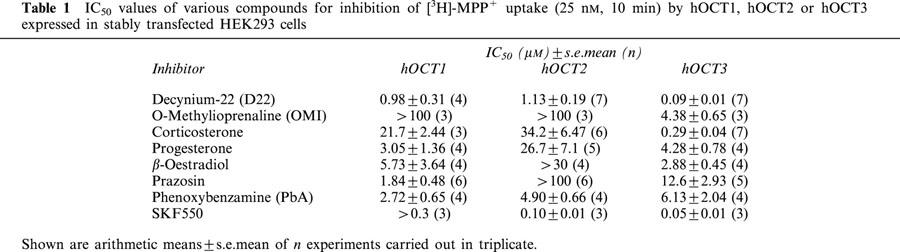
Decynium-22 (D22) inhibited [3H]-MPP+ uptake by all three hOCTs (Figure 3a). However, D22 was most potent at hOCT3 (IC50: 0.09 μM) and was about 10 fold less potent as inhibitor of hOCT1 and hOCT2 (IC50: about 1 μM).
O-methylisoprenaline (OMI) exhibited a selective hOCT3 inhibition (IC50: 4.4 μM), and it caused in concentrations up to 100 μM no significant inhibition of hOCT1 or hOCT2 (Figure 3b).
Corticosterone was found to be a potent, selective inhibitor of hOCT3 (IC50: 0.29 μM). With IC50 values of 22 and 34 μM corticosterone was about 100 fold less potent as inhibitor of hOCT1 and hOCT2 (Table 1).
As shown in Figure 3d and Table 1, progesterone inhibited with about the same potency hOCT3 (IC50: 4.3 μM) and hOCT1 (IC50: 3.0 μM), and with an about 6 fold lower potency hOCT2. A similar preferential inhibition of hOCT3 and hOCT1 was also observed for β-oestradiol (IC50: 3–6 μM; Figure 3e; Table 1). However, in contrast to progesterone, β-oestradiol caused virtually no inhibition of hOCT2, at least not in concentrations up to 30 μM which almost completely inhibited hOCT3 and hOCT1 (Figure 3e).
The α1-adrenoceptor antagonist prazosin most potently inhibited hOCT1 (IC50: 1.8 μM); it was 7 fold less potent at hOCT3, and concentrations up to 100 μM had no effect on hOCT2 (Figure 3f; Table 1).
The irreversible α-adrenoceptor antagonist phenoxybenzamine (PbA) inhibited with about the same potency (IC50 values between 2.7 and 6.1 μM) all three hOCTs, and the PbA analog SKF550 was highly potent at both, hOCT3 (IC50: 50 nM) and hOCT2 (IC50: 100 nM). However, SKF550 concentration up to 0.3 μM which almost completely inhibited hOCT3 and hOCT2 had no effect on hOCT1 (Figure 3g, h; Table 1).
Since there are conflicting reports concerning the reversibility of PbA as inhibitor of ‘uptake2' (see Discussion), we examined whether PbA (10 μM) and its analogue SKF550 (0.3 μM) are reversible or irreversible hOCT inhibitors. The reversibility of inhibition of [3H]-MPP+ uptake was studied after wash-out of the inhibitors for 2, 30 and 60 min. Wash-out of the inhibitory effect at hOCT3 of the competitive ‘uptake2' inhibitor OMI (100 μM) was examined in parallel experiments. The presence of OMI (100 μM) during pre-incubation and incubation with [3H]-MPP+ caused about 90% inhibition of [3H]-MPP+ uptake, and a 2 min wash of the cells (after preincubation with OMI) with inhibitor-free solution was enough to restore [3H]-MPP+ uptake to values not significantly different from controls (Figure 4b).
Figure 4.
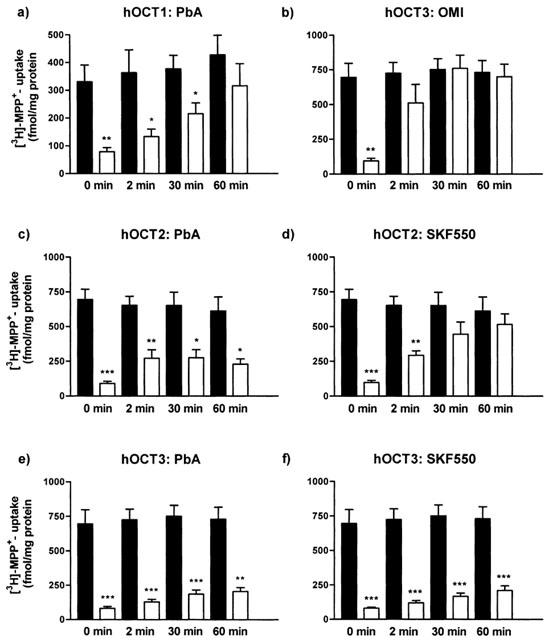
Reversibility of inhibition by OMI, PbA or SKF550 of 10 min uptake of 25 nM [3H]-MPP+ in HEK293 cells stably expressing hOCT1, hOCT2 or hOCT3. Cells were preincubated with 100 μM OMI, 10 μM PbA or 0.3 μM SKF550. Control cells (filled columns) were preincubated in the absence, experimental cells (open columns) in the presence of a transport inhibitor. After preincubation, cells were washed for different times (0, 2, 30 or 60 min) and subsequently [3H]-MPP+ uptake was determined. Shown are arithmetic means±s.e.mean of n=4 independent experiments carried out in triplicates. *P<0.05, **P<0.01, ***P<0.001 for differences between neighbouring columns, i.e., control cells and cells exposed to an inhibitor, and both washed for a defined time.
The reversibility of inhibition by PbA (100 μM) of the three hOCTs was different. At hOCT1, the PbA-effect was slowly reversed by washing of the cells, and after 60 min of wash-out no significant inhibition was further detectable (Figure 4a). However, at hOCT2 and hOCT3 even wash-out for 60 min caused only a very small recovery of [3H]-MPP+ uptake (Figure 4c and e), indicating irreversible inhibition by PbA.
Since SKF550 (0.3 μM) had no effect at hOCT1, the reversibility of action of 0.3 μM SKF550 was examined only at cells expressing hOCT2 or hOCT3. At hOCT2 transport was re-established after 30 min of wash out (Figure 4d), indicating reversible inhibition, whereas inhibition of hOCT3 by SKF550 was irreversible (Figure 4d).
Discussion
We investigated the expression pattern of the three human organic cation transporters hOCT1, hOCT2 and hOCT3 in the human tumour cell lines Caki-1, SK-MG-1, Ski-1, HepG-2, FL, Caco-2 and the human choriocarcinoma line JAR. From these cells, expression of a functional ‘uptake2' transporter and/or of OCT3 mRNA has been described for the kidney carcinoma cell line Caki-1 (Schömig & Schönfeld, 1990; Gründemann et al., 1998), the glioma cell lines SK-MG-1 (Streich et al., 1996) and Ski-1 (Chen et al., 1999), the amnion epithelial cell line FL (Marino et al., 1993), and the colon adenocarcinoma cell line Caco-2 (Martel et al., 2001). The hepatocellular carcinoma line HepG-2 is known to express the hOCT1 (Sinclair et al., 2000), and the human placenta (choriocarcinoma) line JAR has been shown to express the organic cation/carnitine transporter hOCTN2 (Wu et al., 1998b). Only Caco-2 cells were known to express the mRNAs of all three hOCTs (Zhang et al., 1999; Bleasby et al., 2000; Martel et al., 2001).
Using RT–PCR we now demonstrate that all examined cell lines, except JAR, express at least two hOCTs, namely hOCT3 and hOCT1. OCT2 could only be detected in SK-MG-1 and Caco-2 cells, which expressed all three hOCTs. Although expression of hOCT2 had been described in placenta and kidney (Gorboulev et al., 1997), we could not amplify hOCT2 fragments from the placenta cell line JAR or the kidney cell line Caki-1.
Although SK-MG-1 cells express all three hOCT mRNAs, uptake of [3H]-MPP+ in these cells had previously been shown to be nearly completely sensitive to inhibition by OMI and thus almost exclusively mediated through ‘uptake2', i.e., hOCT3 (Streich et al., 1996). The small OMI-resistant component may represent uptake through hOCT1. Thus, whether cell lines which express two or three hOCT mRNAs also exhibit the corresponding transport activity remains to be shown. The here presented data on selective hOCT inhibitors should strongly facilitate such an effort.
In our search for selective inhibitors of hOCT1, hOCT2 or hOCT3 we examined eight compounds known from the literature to inhibit ‘uptake2'.
Decynium-22 (D22) has first been described as a potent inhibitor of ‘uptake2' (hOCT3) in the human cell line Caki-1 (Ki: 16 nM; Russ et al., 1992), and it was later shown to inhibit with about 20–30 fold lower potency rat OCT1 (IC50 of 0.36 μM; Gründemann et al., 1994) and rat OCT2 (IC50 of 0.58 μM; Koepsell et al., 1999). In the present study D22 exhibited highest potency (IC50: 90 nM) at hOCT3 and 10 fold lower potency at hOCT1 and hOCT2. The IC50 value at hOCT1 (1 μM) agrees with literature values whereas the IC50 value at hOCT2 is 10 fold lower (Zhang et al., 1998). The reason for this discrepancy is unclear. At the porcine OCT2 D22 was a very potent inhibitor (IC50: 51 nM; Gründemann et al., 1997), indicating species differences in the D22 sensitivity of OCT2.
O-Methylisoprenaline (OMI) is known as reversible and competitive ‘uptake2' inhibitor which does not inhibit ‘uptake1' (Salt, 1972; Bönisch & Trendelenburg, 1974). In the present study OMI revealed to be the most selective hOCT3 inhibitor (IC50: 4.4 μM) with no effect at hOCT1 or hOCT2 at concentrations up to 100 μM. Inhibition by OMI of hOCT3 was rapidly washed-out and thus reversible. The IC50 value at hOCT3 in HEK293 cells corresponds to that reported in SK-MG-1 cells which express ‘uptake2' (Streich et al., 1996), i.e., hOCT3. The low potency of OMI at hOCT2 agrees with previous reports at hOCT2 (IC50: 570 μM; Gorboulev et al., 1997), rat OCT2 (IC50: 2474 μM; Koepsell et al., 1999) and at porcine OCT2 (IC50: 880 μM; Gründemann et al., 1997). However, while 100 μM OMI caused no inhibition of the human OCT1, half maximal inhibition of the rat OCT1 by 43 μM OMI was reported by Gründemann et al. (1994) indicating species-specific differences in the OMI sensitivity of the two species homologues of OCT1.
Sensitivity to corticosteroids has often been used to specify monoamine transport as transport by the extraneuronal ‘uptake2' transporter (Salt, 1972; Bönisch, 1980). Corticosteroids inhibit ‘uptake2' through a non-genomic, competitive interaction (Bryan & O'Donnell, 1981). Here we show that corticosterone is a potent (IC50: 0.29 μM) and selective inhibitor of hOCT3. Corticosterone was 100 fold less potent at hOCT1 (IC50: 22 μM) and hOCT2 (IC50: 34 μM). The high potency at hOCT3 corresponds to data obtained at Caki-1 and SK-MG-1 cells which express hOCT3 (Schömig & Schönfeld, 1990; Streich et al., 1996; Gründemann et al., 1998). At the rat OCT3 corticosterone is by a factor of 10 less potent (IC50 about 5 μM; Kekuda et al., 1998; Wu et al., 1998a). At hOCT2 Koepsell et al. (1999) observed a similar potency of corticosterone (IC50: 29 μM) as we (see above). However, corticosterone exhibited higher potencies at OCT2 of the rat (IC50: 5.8 and 4.2 μM; Koepsell et al., 1999; Wu et al., 1998a) and the pig (IC50: 0.67 μM; Gründemann et al., 1997). The IC50 value of 22 μM for corticosterone at hOCT1 obtained in the present study is in the same order of magnitude as that reported at hOCT1 by Koepsell et al. (1999). However, at the rat OCT1 corticosterone exhibits a more than 20 fold lower potency (IC50: 160 μM; Gründemann et al., 1994) than at the hOCT1 (see above). Thus, OCTs show also pronounced species differences in their sensitivity to corticosterone.
The sex steroids progesterone and β-oestradiol are known to inhibit competitively at micromolar concentrations the ‘uptake2' transport system (Salt, 1972; Bönisch, 1980; Grohmann & Trendelenburg, 1984). In the present study progesterone inhibited with the same potency (IC50: 3–4 μM) hOCT3 and hOCT1 and with an about 7 fold lower potency (IC50: 26 μM) hOCT2. However, progesterone was shown to be about 7 fold more potent at rOCT2 than at rOCT3 (Wu et al., 1998a). At the human OCTs β-oestradiol was a preferential inhibitor of hOCT3 and hOCT1 (IC50: 3–6 μM) and concentrations up to 30 μM caused no inhibition of hOCT2. The inhibition data of β-oestradiol at hOCT3 and hOCT2 agree with those reported by Wu et al. (1998a) at the corresponding rat OCTs.
The α1-adrenoceptor antagonist prazosin had previously been shown to competitively inhibit rat ‘uptake2' with a potency similar to that of OMI (Grohmann & Trendelenburg, 1984; Akimoto et al., 1989). We now demonstrate that prazosin is not only an inhibitor of hOCT3 (IC50: 12.5 μM) but an even more selective and more potent inhibitor of hOCT1 (IC50: 1.8 μM) which in concentrations up to 100 μM causes no inhibition of hOCT2.
Lightman & Iversen (1969) showed that the irreversible α-adrenoceptor antagonist phenoxybenzamine (PbA) is a potent and competitive inhibitor of ‘uptake2' in the isolated perfused rat heart (IC50: 2.5 μM). However, according to Bryan & O'donnell (1981) PbA is a non-competitive and irreversible ‘uptake2' inhibitor. Since nothing was known about PbA effects at human OCTs we studied the potency and reversibility of its interaction at the three hOCTs. We could show that PbA inhibits with about the same apparent potency (IC50: 3–6 μM) all three human OCTs. The inhibition by PbA of hOCT2 and hOCT3 was found to be irreversible (see Figure 4). According to Iversen et al. (1972) the PbA derivative SKF550 is in the rat heart the most potent ‘uptake2' inhibitor (IC50: 0.08 μM). In the present study SKF550 revealed to be the most potent inhibitor of both, hOCT3 (IC50: 50 nM) and hOCT2 (IC50: 100 nM); however, SKF550 had no effect on hOCT1 in concentrations up to 0.3 μM. Haloalkylamines such as PbA and SKF550 form an unstable aziridinium ion which via the highly reactive intermediate ethyleneiminium ion forms covalent bonds with sulfhydryl, hydroxy, amino and carboxy groups of amino acids of target proteins (Jenkinson, 1996). Cysteines are known to be the most reactive among the amino acid residues (Shulman-Roskes et al., 1998) and a cysteine residue in transmembrane domain 3 of the α2-adrenoceptor has recently been identified as a possible site for the irreversible PbA interaction at this receptor (Frang et al., 2001). However, amino acid sequence alignment of the three hOCTs (data not shown) did not give a hint for a cysteine (or serine or threonine or tyrosine) as possible site of interaction of PbA and SKF550 at the hOCT3.
In summary, this is the first pharmacological comparison of the three OCTs of one species (human) performed under identical conditions in human cells stably expressing hOCT1, hOCT2, or hOCT3. Using various substances known to inhibit at least the ‘uptake2' transporter OCT3, we identified inhibitors that allow functional discrimination of the three human OCTs. Thus, hOCT1 is characterized by the potency order D22>prazosin>PbA>progesterone⩾β-oestradiol>corticosterone>>OMI, hOCT2 by the order SKF550>D22>PbA>progesterone⩾corticosterone>β-oestradiol>OMI=prazosin, and hOCT3 by the order SKF550>D22>corticosterone>β-oestradiol>progesterone=OMI⩾PbA>prazosin. The availability of some relatively selective OCT inhibitors should facilitate the investigation of the physiological functions of these transport systems in kidney, liver, intestine and brain.
Acknowledgments
We thank Gundula Hesse for skillful technical assistance. We also gratefully acknowledge the donations of the compounds OMI and SKF550 by Boehringer Ingelheim (Germany) and SmithKline Becham (U.S.A.), respectively. This study was supported by the Deutsche Forschungsgemeinschaft, BONFOR and the Caroline-Wenzel-Stiftung.
Abbreviations
- ANOVA
analysis of variance
- D22
Decynium-22
- MPP+
1-Methyl-4-phenylpyridinium iodide
- HEK
human embryonic kidney
- hOCT
human organic cation transporter
- OMI
O-Methylisoprenaline
- PbA
Phenoxybenzamine
- PCR
polymerase chain reaction
- SKF550
((9-fluorenyl)-N-methyl-β-chloroethylamine)
References
- AKIMOTO Y., SONO K., KURAHASHI K., FUJIWARA M. Effects of specific alpha-adrenoceptive agents on extraneuronal uptake (uptake2) of isoproterenol in perfused rat heart. Life Sci. 1989;44:945–950. doi: 10.1016/0024-3205(89)90493-1. [DOI] [PubMed] [Google Scholar]
- BLEASBY K., CHAUHAN S., BROWN C.-D. Characterization of MPP+ secretion across human intestinal Caco-2 cell monolayers: role of P-glycoprotein and a novel Na+-dependent organic cation transport mechanism. Br. J. Pharmacol. 2000;129:619–625. doi: 10.1038/sj.bjp.0703078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BÖNISCH H. Extraneuronal transport of catecholamines. Pharmacology. 1980;21:93–108. doi: 10.1159/000137422. [DOI] [PubMed] [Google Scholar]
- BÖNISCH H., TRENDELENBURG U. Extraneuronal removal, accumulation and O-methylation of isoprenaline in the perfused heart. Naunyn Schmiedebergs. Arch. Pharmacol. 1974;283:191–218. doi: 10.1007/BF00501145. [DOI] [PubMed] [Google Scholar]
- BRYAN L.-J., O'DONNELL S.-R. Kinetic analyses of the mechanisms of action of inhibitors of the extraneuronal uptake of adrenaline in smooth muscle cells of guinea-pig trachea. Naunyn Schmiedebergs. Arch. Pharmacol. 1981;315:249–254. doi: 10.1007/BF00499842. [DOI] [PubMed] [Google Scholar]
- BURCKHARDT G., WOLFF N.-A. Structure of renal organic anion and cation transporters. Am. J. Physiol. Renal Physiol. 2000;278:F853–F866. doi: 10.1152/ajprenal.2000.278.6.F853. [DOI] [PubMed] [Google Scholar]
- CHEN C., OKAYAMA H. High efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Z.-P., REMACK J., BRENT T.-P., MOHR G., PANASCI L.-C. Extraneuronal monoamine transporter expression and DNA repair vis-a-vis 2-chloroethyl-3-sarcosinamide-1-nitrosourea cytotoxicity in human tumor cell lines. Clin. Cancer Res. 1999;5:4186–4190. [PubMed] [Google Scholar]
- DRESSER M.-J., GRAY A.-T., GIACOMINI K.-M. Kinetic and selectivity differences between rodent, rabbit, and human organic cation transporters (OCT1) J. Pharmacol. Exp. Ther. 2000;292:1146–1152. [PubMed] [Google Scholar]
- EISENHOFER G. The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines. Pharmacol. Therapeut. 2001;91:35–62. doi: 10.1016/s0163-7258(01)00144-9. [DOI] [PubMed] [Google Scholar]
- FRANG H., COCKCROFT V., KARSKELA T., SCHEININ M., MARJAMAKI A. Phenoxybenzamine binding reveals the helical orientation of the third transmembrane domain of adrenergic receptors. J. Biol. Chem. 2001;276:31279–31284. doi: 10.1074/jbc.M104167200. [DOI] [PubMed] [Google Scholar]
- GORBOULEV V., ULZHEIMER J.-C., AKHOUNDOVA A., ULZHEIMER-TEUBER L., KARBACH U., QUESTER S., BAUMANN C., LANG F., BUSCH A.-E., KOEPSELL H. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16:871–881. doi: 10.1089/dna.1997.16.871. [DOI] [PubMed] [Google Scholar]
- GRAEFE K.-H., BÖNISCH H.The transport of amines across the axonal membranes of noradrenergic and dopaminergic neurones Handbook of Experimental Pharmacology 90/I, Catecholamines I 1988Berlin, Heidelberg, New York: Springer; 193–245.ed. Trendelenburg, U., Weiner, N. pp [Google Scholar]
- GROHMANN M., TRENDELENBURG U. The substrate specificity of uptake2 in the rat heart. Naunyn Schmiedebergs Arch. Pharmacol. 1984;328:164–173. doi: 10.1007/BF00512067. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., BABIN-EBELL J., MARTEL F., ÖRDING N., SCHMIDT A., SCHÖMIG E. Primary structure and functional expression of the apical organic cation transporter from kidney epithelial LLC-PK1 cells. J. Biol. Chem. 1997;272:10408–10413. doi: 10.1074/jbc.272.16.10408. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., GORBOULEV V., GAMBAEYAN S., VEHYL M., KOEPSELL H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature. 1994;372:549–552. doi: 10.1038/372549a0. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., LIEBICH G., KIEFER N., KÖSTER S., SCHÖMIG E. Selective substrates for non-neuronal monoamine transporters. Mol. Pharmacol. 1999;56:1–10. doi: 10.1124/mol.56.1.1. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., SCHECHINGER B., RAPPOLD G.-A., SCHÖMIG E. Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nat. Neurosci. 1998;1:349–351. doi: 10.1038/1557. [DOI] [PubMed] [Google Scholar]
- GRÜNDEMANN D., SCHÖMIG E. Gene structures of the human non-neuronal monoamine transporters EMT and OCT2. Hum. Genet. 2000;106:627–635. doi: 10.1007/s004390000309. [DOI] [PubMed] [Google Scholar]
- HAYER M., BÖNISCH H., BRÜSS M. Molecular cloning, functional characterization and genomic organization of four alternatively spliced isoforms of the human organic cation transporter 1 (hOCT1/SLC22A1) Ann. Hum. Genet. 1999;63:473–482. doi: 10.1017/S0003480099007770. [DOI] [PubMed] [Google Scholar]
- IVERSEN L.-L. The uptake of catecholamines at high perfusion concentrations in the rat isolated heart: a novel catecholamine uptake process. Br. J. Pharmacol. 1965;25:18–33. doi: 10.1111/j.1476-5381.1965.tb01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IVERSEN L.-L. Catecholamine uptake processes. Br. Med. Bull. 1973;29:130–135. doi: 10.1093/oxfordjournals.bmb.a070982. [DOI] [PubMed] [Google Scholar]
- IVERSEN L.-L., SALT P.-J., WILSON H.-A. Inhibition of catecholamine uptake in the isolated rat heart by haloalkylamines related to phenoxybenzamine. Br. J. Pharmacol. 1972;46:647–657. doi: 10.1111/j.1476-5381.1972.tb06890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENKINSON D. Textbook of Receptor Pharmacology. 1996Boca Raton: CRC Press; 3–62.ed. Foreman, J., Johansen, T. pp [Google Scholar]
- KEKUDA R., PRASAD P.-D., WU X., WANG X., FEI Y.-J., LEIBACH F.-H., GANAPATHY V. Cloning and functional characterization of a potential-sensitive, polyspecific organic cation transporter (OCT3) most abundantly expressed in placenta. J. Biol. Chem. 1998;273:15971–15979. doi: 10.1074/jbc.273.26.15971. [DOI] [PubMed] [Google Scholar]
- KOEPSELL H., GORBOULEV V., ARNDT P. Molecular pharmacology of organic cation transporters in kidney. J. Membrane. Biol. 1999;167:103–117. doi: 10.1007/s002329900475. [DOI] [PubMed] [Google Scholar]
- LIGHTMAN S.L., IVERSEN L.L. The role of uptake2 in the extraneuronal metabolism of catecholamines in the isolated rat heart. Br. J. Pharmacol. 1969;37:638–649. doi: 10.1111/j.1476-5381.1969.tb08502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOWRY O.-H., ROSENBROUGH N.-J., FARR A.-L., RANDALL R.-J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MARTEL F., GRÜNDEMANN D., CALHAU C., SCHÖMIG E. Apical uptake of organic cations by human intestinal Caco-2 cells: putative involvement of ASF transporters. Naunyn Schmiedebergs Arch. Pharmacol. 2001;363:40–49. doi: 10.1007/s002100000335. [DOI] [PubMed] [Google Scholar]
- MARINO V., DE LA LANDE I.-S., NEWLYN M., PARKER D.-A. Evidence for uptake2-mediated O-methylation of noradrenaline in the human amnion FL cell-line. Naunyn Schmiedebergs Arch. Pharmacol. 1993;347:371–378. doi: 10.1007/BF00165386. [DOI] [PubMed] [Google Scholar]
- OKUDA M., SAITO H., URAKAMI Y., TAKANO M., INUI K. cDNA cloning and functional expression of a novel rat kidney organic cation transporter, OCT2. Biochem. Biophys. Res. Commun. 1996;224:500–507. doi: 10.1006/bbrc.1996.1056. [DOI] [PubMed] [Google Scholar]
- RUSS H., GLIESE M., SONNA J., SCHÖMIG E. The extraneuronal transport mechanism for noradrenaline (uptake2) avidly transports 1-methyl-4-phenylpyridinium (MPP+) Naunyn Schmiedebergs Arch. Pharmacol. 1992;346:158–165. doi: 10.1007/BF00165297. [DOI] [PubMed] [Google Scholar]
- SALT P.-J. Inhibition of noradrenaline uptake2 in the isolated rat heart by steroids, clonidine and methoxylated phenylethylamines. Eur. J. Pharmacol. 1972;20:329–340. doi: 10.1016/0014-2999(72)90194-x. [DOI] [PubMed] [Google Scholar]
- SCHÖMIG E., SCHÖNFELD C.-L. Extraneuronal noradrenaline transport (uptake2) in a human cell line (Caki-1 cells) Naunyn Schmiedebergs Arch. Pharmacol. 1990;341:404–410. doi: 10.1007/BF00176331. [DOI] [PubMed] [Google Scholar]
- SCHWEIFER N., BARLOW D.-P. The Lx1 gene maps to mouse chromosome 17 and codes for a protein that is homologous to glucose and polyspecific transmembrane transporters. Mammalian Genome. 1996;7:735–740. doi: 10.1007/s003359900223. [DOI] [PubMed] [Google Scholar]
- SHULMAN-ROSKES E.-M., NOE D.-A., GAMCSIK M.-P., MARLOW A.-L., HILTON J., HAUSHEER F.-H., COLVIN O.-M., LUDEMAN S.-M. The partitioning of phosphoramide mustard and its aziridinium ions among alkylation and P-N bond hydrolysis reactions. J. Med. Chem. 1998;41:515–529. doi: 10.1021/jm9704659. [DOI] [PubMed] [Google Scholar]
- SINCLAIR C.-J., CHI K.-D., SUBRAMANIAN V., WARD K.-L., GREEN R.-M. Functional expression of a high affinity mammalian hepatic choline/organic cation transporter. J. Lipid Res. 2000;41:1841–1848. [PubMed] [Google Scholar]
- STREICH S., BRÜSS M., BÖNISCH H. Expression of the extraneuronal monoamine transporter (uptake2) in human glioma cells. Naunyn Schmiedebergs Arch. Pharmacol. 1996;353:328–333. doi: 10.1007/BF00168636. [DOI] [PubMed] [Google Scholar]
- TERASHITA S., DRESSER M.-J., ZHANG L., GRAY A.-T., YOST S.-C., GIACOMINI K.-M. Molecular cloning and functional expression of a rabbit renal organic cation transporter. Biochim. Biophys. Acta. 1998;1369:1–6. doi: 10.1016/s0005-2736(97)00207-1. [DOI] [PubMed] [Google Scholar]
- VERHAAGH S., SCHWEIFER N., BARLOW D.-P., ZWART R. Cloning of the mouse and human solute carrier 22a3 (Slc22a3/SLC22A3) identifies a conserved cluster of three organic cation transporters on mouse chromosome 17 and human 6q26-q27. Genomics. 1999;55:209–218. doi: 10.1006/geno.1998.5639. [DOI] [PubMed] [Google Scholar]
- WIELAND A., HAYER-ZILLGEN M., BÖNISCH H., BRÜSS M. Analysis of the gene structure of the human (SLC22A3) and murine (Slc22a3) extraneuronal monoamine transporter. J. Neural Transm. 2000;107:1149–1157. doi: 10.1007/s007020070028. [DOI] [PubMed] [Google Scholar]
- WU X., KEKUDA R., HUANG W., FEI Y.-J., LEIBACH F.-H., CHEN J., CONWAY S.-J., GANAPATHY V. Identity of the organic cation transporter OCT3 as the extraneuronal monoamine transporter (uptake2) and evidence for the expression of the transporter in the brain. J. Biol. Chem. 1998a;273:32776–32786. doi: 10.1074/jbc.273.49.32776. [DOI] [PubMed] [Google Scholar]
- WU X., PRASAD P.-D., LEIBACH F.-H., GANAPATHY V. cDNA sequence, transport function, and genomic organization of human OCTN2, a new member of the organic cation transporter family. Biochem. Biophys. Res. Commun. 1998b;246:589–595. doi: 10.1006/bbrc.1998.8669. [DOI] [PubMed] [Google Scholar]
- ZHANG L., BRETT C.-M., GIACOMINI K.-M. Role of organic cation transporters in drug absorption and elimination. Annu. Rev. Pharmacol. Toxicol. 1998;38:431–460. doi: 10.1146/annurev.pharmtox.38.1.431. [DOI] [PubMed] [Google Scholar]
- ZHANG L., DRESSER M.-J., GRAY A.-T., YOST A.-C., TERASHITA S., GIACOMINI K.-M. Cloning and functional expression of a human liver organic cation transporter. Mol. Pharmacol. 1997;51:913–921. doi: 10.1124/mol.51.6.913. [DOI] [PubMed] [Google Scholar]
- ZHANG L., GORSET W., DRESSER M.-J., GIACOMINI K.-M. The interaction of n-tetraalkylammonium compounds with a human organic cation transporter, hOCT1. J. Pharmacol. Exp. Ther. 1999;288:1192–1198. [PubMed] [Google Scholar]