

Abstract
The normal and a mutant (S248F) human neuronal α4β2 nicotinic receptors, and their interaction with the channel blocker carbamazepine (CBZ) have been modelled. The mutant, responsible for the autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), has an enhanced sensitivity to and a slower recovery from desensitization, a lower conductance, short open times, reduced calcium permeability, and is 3 fold more sensitive to CBZ, a drug used in the treatment of partial epilepsies.
Mutant channel properties are explained by the physicochemical properties of the two Phe248 side chains, including size and cation-π interaction, and their dynamic behaviour. A defective mechanism of dehydration might be responsible for the reduced calcium influx.
Phe248 residues are the main component of CBZ binding sites in the mutant, while this is not true for Ser248 in the normal receptor.
A higher number of blocking binding sites and a predicted higher affinity found for CBZ in the mutant account for its differential sensitivity to CBZ.
Aromatic–aromatic interactions between CBZ and the two Phe248 account for the difference in affinity, which is at least 12 times higher for the mutant, depending on the method used for calculating Ki.
Normal vs mutant differences in Ki, enhanced by the higher number of blocking binding sites in the mutant, seem excessive compared to the differential sensitivities to CBZ experimentally found. The negative cooperativity suggested by a predicted overlapping of blocking and non-blocking binding sites gives an explanation, as overlapping is higher in the mutant.
For both types of receptors we found that the carbamyl group of the best blocking conformers of CBZ forms hydrogen bonds with serine residues, which may explain the fundamental role of that moiety for this molecule to act as antiepileptic drug.
Keywords: Molecular modelling, nicotinic receptor, epilepsy, carbamazepine
Introduction
The nicotinic acetylcholine receptor (nAChR) is one of the best known signalling proteins. It belongs to the Ligand-Gated Ion Channel (LGIC) superfamily of receptors (Ortells & Lunt, 1995) which also comprises the 5HT3, GABAA and glycine receptors. The nAChR and the 5HT3 receptors are selective for cations (and hence excitatory) whereas GABA and glycine receptors are selective for anions (and are thus inhibitory). They are all oligomers, most probably pentamers, and sequence information also reveals that for any given member of the family, the subunits (around 500 residues long) are themselves homologous. For example, muscle nACh receptors (including Torpedo electroplax nAChR) are composed of four different subunits named α1, β1, γ1, and δ1, with an α1(2)β1γ1δ1 stoichiometry. Neuronal variants can be either heteromeric receptors, composed most probably of two α and three β subunits, or homomeric composed of five α subunits. Each of these subunits has an N-terminal extracellular domain which contains the ligand binding site, four putative transmembrane regions (M1–M4), and a short extracellular C-terminus (Karlin, 1993). The second of the transmembrane regions (M2) is the main contributor to the ion channel (Bertrand et al., 1993).
Nicotinic receptors are pharmacologically important as they are targets of many different types of drugs such as local (Arias, 1999) and general (Forman et al., 1995) anaesthetics, barbiturates (Dilger et al., 1997), toxins (Choi et al., 1995), drugs of abuse (Fryer & Lukas, 1999a), antidepressants (Fryer & Lukas, 1999b), neuroleptics (Picard et al., 1999) and other important drugs. The knowledge of the structure of these receptors would be extremely important to understand the mechanisms of action of many drugs and in the development of new or enhanced ones. However, the full tertiary structure of these receptors has not been obtained as yet; their membrane-bound nature and size have so far frustrated all efforts either to obtain suitable crystals for high resolution X-ray analysis, or to carry out NMR studies. Electron microscope images of the nAChR have been obtained at 4.6 Å resolution (Miyazawa et al., 1999), but they only show the general shape of the receptor. In the ion-channel, well defined regions of α-helices can be seen, but not individual amino acids. Actually, until very recently, most of the structural information available was derived from biochemical and mutational experiments. A new perspective came with the elucidation of the structure of a novel acetylcholine binding protein (ABP) by Brejc et al. (2001), which is involved in the regulation of synaptic transmission in snails, and is secreted through glial cells (Smit et al., 2001). This is a soluble protein homologous to the ligand binding domain of the LGIC superfamily. On the basis of the ABP tertiary structure, almost every biochemical and mutational information available for LGICs can be rationalized. Unfortunately, this ABP lacks the transmembrane region present in LGICs.
For these reasons, we have been building molecular models of the transmembrane region of these receptors to have an approximation to their three-dimensional structure at an atomic resolution (Ortells & Lunt, 1996; Ortells et al., 1997). These models were in agreement with all the structural information available (used or not used in the construction of the models), and gave also structural explanation to several unexpected experimental results. Another important feature of the models is that they were able to explain with relative accuracy the interactions between several and structurally different channel blockers and the ion channel (Ortells & Barrantes, 2001).
In the present work we used molecular modelling techniques to study, on a structural basis, the involvement of a nicotinic receptor in a genetic form of epilepsy and the pharmacology of one of the drugs used in the treatment of this disease. It was found that a mutation in the neuronal α4 subunit of the nicotinic receptor is responsible for the autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE). This was the first gene shown to be involved in a human idiopathic disease (Steinlein et al., 1995; 1997). After this finding, two other mutations in LGICs were discovered to be involved in epilepsy. The first was found in the nicotinic β2 subunit (de fusco et al., 2000) and is also involved in a nocturnal front lobe epilepsy. More recently, a mutation in a GABAA γ2 subunit (Baulac et al., 2001) was found to be directly involved in another type of human idiopathic epilepsy (generalized epilepsy with febrile seizures plus). The mutation of interest for the present work, the first discovered α4 S248F, is in the second transmembrane segment M2 of this subunit, which, as stated before, is believed to be the main domain that shapes the pore. The mutation results in an enhanced desensitization sensitivity and a slower recovery from desensitization (Bertrand et al., 1998; Kuryatov et al., 1997; Weiland et al., 1996), a lower conductance and short open times and a reduced permeability to calcium (Kuryatov et al., 1997), that might account for the ADNFLE phenotype. Among the common drugs used in the treatment of patients with partial epilepsies, carbamazepine (5H-Dibenz[b,f]lazepine-5-carboxamide, CBZ) is one of the most effective. α4β2 receptors bearing the ADNFLE mutation are 3 fold more sensitive to the blocking properties of CBZ than the normal (Picard et al., 1999). These authors also demonstrated that CBZ behaves as an open channel blocker (OCB), like the neuroleptic chlorpromazine.
S248 of the α4 subunit is part of a ring of polar amino acids, separated by three residues towards the extracellular side, from the so called central polar ring (Imoto et al., 1991). The latter ring, in conjunction with the intermediate ring of negatively charged residues (Imoto et al., 1988), are supposed to constitute the narrowest part of the channel, and to be involved in cation dehydration and selectivity. As phenylalanines are much larger and hydrophobic than serine residues, some observed modifications of the channel properties in the mutant receptor such as a reduced conductance or a different cation selectivity might be expected.
Using a model of the open α4β2 receptor transmembrane region, we try here to give a deeper structural explanation to the altered channel properties and to the higher affinity to CBZ in this ADNFLE mutant receptor. It is believed that the general mechanism of action of CBZ as an antiepileptic drug is by slowing the rate of recovery of voltage-activated Na+ channels from inactivation. In the case of the α4β2 receptor, and as stated before, CBZ acts as an open channel blocker. If the therapeutic properties of CBZ in this type of epilepsy are related to this blocking capacity, then there are chances to find better treatments if we understand the molecular basis of the enhanced sensitivity of the mutant. In ADNFLE patients, normal α4β2 receptors must be present, and hence, more potent mutant blockers will interfere less with the normal function of the former.
Methods
Molecular models of both normal and mutant α4(2)β2(3) receptors in their open state were constructed on the basis of our previous modelled structures (Ortells et al., 1997), which proved to be successful in predicting docking sites for OCBs (Ortells & Barrantes, 2001). A simple molecular dynamics simulation was performed for the mutant receptor to analyse possible variations in the channel structure. An automatic docking procedure was employed to investigate the binding modes of CBZ in the normal and mutant receptors. Subsequently, theoretical estimations of CBZ affinity to both receptors were carried out. A detailed description of the methods employed follows.
Model construction
First, the model of the wild type α4(2)β2(3) receptor was built and energy minimized. For this purpose, the starting and not energy minimized structure of our previous homomeric α7 receptor (Ortells et al., 1997) was computationally mutated to the α4(2)β2(3) using the Biopolymer module in the Insight II program (InsightII, 2000). The two α4 subunits in the receptor were modelled with one β2 subunit in between. Prior to energy minimization, a search for the best side-chain conformers was made using a database of known protein structures employing the module Biopolymer. Energy minimization was performed using molecular mechanics (Keserü & Kolossváry, 1999) in three steps using the programme Discover and the CVFF force field (Discover, 2000; Doucet & Weber, 1996). First, an energy minimization was carried out forcing the main chain atoms to remain to their original positions with a forcing constant of 4500 kcal Å−1. This was done to avoid distorting the secondary structure and general shape of the model. During this minimization, no Morse functions and no cross terms were used and the steepest descents method was employed. The structure was minimized until the maximum derivative was less than 2.00 kcal Å−1. In a second step, the model was further minimized until the maximum derivative was less than 0.050 kcal Å−1, with the same forcing constant for the main chain atoms, but using the full model, with Morse functions and cross terms, and employing the conjugate gradients method. Finally, all the atoms were tethered to their positions, except those belonging to the ion channel, that were fully minimized until the maximum derivative was less than 0.050 kcal Å−1 without any restriction. This model was subsequently utilized to simulate the S248F mutation and was further energy minimized, as in the last described step, to relax the structure and to eliminate possible clashes caused by the mutation.
CBZ was modelled using the Insight II programme (InsightII, 2000). Minimization and partial charge calculations were done using the MOPAC programme as implemented in Insight II and employing the semiempirical AM1 method. Parameters for the electronic state were lowest electronic state, Hamiltonian spin restricted, and null total charge. All other parameters were left with default values.
Molecular dynamics
Starting from the final minimized structure of the mutant receptor, a molecular dynamics simulation was performed. The Verlet leapfrog integration method, as implemented in the Discover (2000) programme and the CVFF force field were employed. Only those atoms belonging to the ion channel were allowed to move. The system was equilibrated at 300 K by 10 ps of heating, followed by a production run of 100 ps. The time step was set to 1 fs. A cutoff distance of 8 Å was used for the electrostatic interactions and a distance-independent dielectric constant of 1 was employed.
Docking procedures
CBZ was docked on both receptors using a molecular mechanics/grid (Luty et al., 1995) method employing the Affinity programme (MSI, San Diego, CA, U.S.A.). The main molecular mechanics programme employed in Affinity is also Discover and the CVFF force field including a desolvation term (Stouten et al., 1993).
The region corresponding to the pore of the ion channel was defined as the binding site region. In order to make the docking procedure faster, the system was partitioned into ‘bulk' (represented by grids) and ‘movable' atoms. Bulk atoms were defined as atoms of the receptor that were not in the defined binding site. These atoms were held rigid during the course of the docking search and represented by a grid. The potential energies due to the rigid atoms were calculated only once. Movable atoms consisted of atoms in the binding site of the receptor (the ion-channel) and ligand atoms (CBZ) and were simulated explicitly. These atoms could move freely except for binding site atoms close to bulk atoms, which were restrained. These restrained atoms conformed a buffer which separated the freely movable atoms from bulk atoms. The grid method works only if the bulk atoms and movable atoms are fairly well separated, and hence this buffer was necessary to avoid inconsistencies in the calculations.
Initially, CBZ was placed arbitrarily in the ion-channel. This complex was then energy minimized to obtain the starting structure. This step removed bad contacts in the initial structure and obtained a reasonable starting point for subsequent search. The programme next moved CBZ by random combination of translation, rotation, and torsional changes. The random move of a ligand samples both the orientational and conformational spaces of the ligand with respect to the receptor. It can be considered as a first step to roughly dock the ligand. It has the advantage that it can get over any energy barrier on the potential energy surface. Affinity subsequently checked the energy of the resulting randomly moved structure. If it was within a specified energy tolerance parameter of the previous minimized structure, it was considered to have passed the first step and the structure was then subjected to energy minimization, the second step for fine-tuning the docking. The final minimized structure was accepted or rejected based on the energy criterion and its similarity to structures found before. The energy criterion can be either Metropolis or energy range, and we used both in different runs. With the Metropolis criterion, structures whose energy was lower than that of the last accepted structure or whose Boltzmann factor (computed using specified Monte Carlo temperature) was greater than a random number between 0 and 1, were accepted. If energy range is used, structures whose energy was within the specified energy range of the lowest energy found so far were accepted. The Metropolis criterion is best suited for finding a very small number of docked structures with very low energies, while the energy range criterion is designed for finding more and diverse structures. In checking structure similarity, the root mean square (RMS) distances between the current structure and structures found so far were computed for ligand atoms. In contrast to RMS deviation, in RMS distance the ligand molecule is not translated or rotated (i.e., no superimposition is done) because ligand/receptor structures with the ligand being translated or rotated are treated as different docked structures.
The parameters we used in the docking procedures were as follows. The grid spacing was 0.66 Å, and the restrain buffer size was of 0.5 Å. The energy range was set to 10 kcal mol−1, that is, the programme was asked to accept structures within 10 kcal mol−1 of the lowest energy structure found so far. We search for a maximum of 10 different structures using the energy range method, and for all found after 4 days of an R10000 based SGI O2 CPU time using Metropolis.
CBZ affinity estimations
The affinity of CBZ conformers for both types of receptors were based on two different types of estimations. The first calculation is a molecular mechanics based energy difference. The second is a regression based score function.
Molecular mechanics based calculation: In the first method, free energies of binding for each conformer were calculated as the difference between the energy of the minimized ligand-receptor complex and the energies of the minimized ligand and receptor alone. These energies were calculated using a molecular mechanics approximation employing the programme Discover and the VCFF force field. Estimations of the dissociation constants Ki for the different conformers were based on the equation:
which relates Ki to the free energy change of the formation of the ligand-receptor complex (ΔG).
There are several well known drawbacks to this sort of approach. First we can mention the inaccuracies of the parameters in the force fields employed. Second, these are static calculations, and hence, do not account for entropic factors such as those related to conformational changes in the ligand and receptor. Hence, these calculations are not actually referring to changes in free energy but mainly to its enthalpic component. Third, desolvation effects are also not considered. However, a desolvation term was used during the docking procedure. Fourth, the energy of binding is calculated from two very similar numbers, the energy of the complex and the energies of the receptor and ligand separately. Thus, small errors in these quantities can lead to large errors in the calculated binding energies. Moreover, if we translate these errors in terms of the dissociation constants, a ΔG of binding with an error of 2 kcal mol−1 results in an error similar to 30 fold in Ki.
Having described its negative aspects, we must say that molecular mechanics calculations have been used with some degree of success in analysing relative differences in binding affinities in related systems. Although estimations of the absolute binding affinities are usually incorrect, these are seldom necessary. As stated, molecular mechanics calculations do not refer to true free energies. However, parameters developed for force fields are based on the fitting of free energies or enthalpies. Hence, to some degree, these parameters must be implicitly taking into account these effects. For a detailed analysis of these kind of methods refer to Koehler et al. (1996), and Ajay et al. (1997).
Regression based score function: the second method employed is the described by Böhm (1992a, b; 1994a, b; 1995; 1998). This author developed an empirical scoring function for his de novo ligand design program LUDI, that predicts the binding free energy ΔG. In this function ΔG is partitioned into six terms:
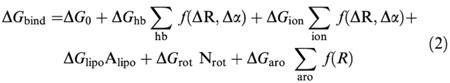 |
where ΔG0 represents loss of overall rotational and translational entropy of the ligand. ΔGhb is the free energy of ideal hydrogen bonded interactions and Σhb (fΔR,Δα) is a penalty function that accounts for large deviations of the hydrogen bond geometry from ideality. ΔGion and Σion f(ΔR,Δα) are similar functions to those for hydrogen bonding but for ionic interactions. ΔGlipo is the free energy of lipophilic interactions, that is proportional to Alipo, the lipophilic contact surface between the receptor and the ligand. ΔGrot describes the loss of binding energy for freezing the rotatable bonds in the ligand, being Nrot the number of rotatable bonds. The last term, ΔGaro accounts for specific interaction between aromatic rings; the angular dependence of aromatic–aromatic interactions is ignored and a simple distance cutoff is used (f(R)=1 for R<4.5 Å and f(R)=0 for R⩾4.5 Å). The programme returns score values for each analysed ligand, which are related to Ki values in the form: score=100 log Ki. The main problem with this approach is that it performs well with the training data set, but its behaviour with very different ligand-receptor complexes is unclear.
Results
Modelling of ADNFLE mutation
Our results show that replacement of Ser 248 by a Phe in the α4 subunits greatly and directly obstructs the ion channel (Figures 1a,b and 2a). The side chain of one of the expressed mutant Phe residues (named here α4′ Phe 248) is pointing to the centre of the channel lumen, whilst the other (named here α4′′ Phe 248) is blocking the channel more laterally. In this way, the channel pore is nearly filled with hydrophobic moieties. The narrowest ion channel diameter in the normal receptor is between 6.5 Å and 5 Å, at the level of the central and intermediate rings (Brovtsyna et al., 1996a, b; Imoto et al., 1988; 1991), as shown in Figure 1a. In the mutant the narrowest part is moved towards the extracellular region to the level of the mutant Phe 248 and is around 2.5 Å (Figures 1b and 2a). Both phenyl ring planes of the mutated residues are parallel to the major ion-channel axis. Most of the remaining side chains remain in positions similar to the normal receptor. A serine residue (β2′′ Ser 271) in the β subunit sandwiched by the two α is also displayed in Figure 1. This residue has, on average, the highest interaction with CBZ in the normal receptor (see below). In the mutated receptor β2′′ Ser 271 changes its side chain orientation due to the interactions with both α4′ Phe 248 and α4′′ Phe 248. Molecular dynamics of the channel region showed that the movement of the mutant phenylalanines can occlude even more the lumen but never makes it wider. Moreover, in some conformation the channel is totally closed (Figure 2b).
Figure 1.
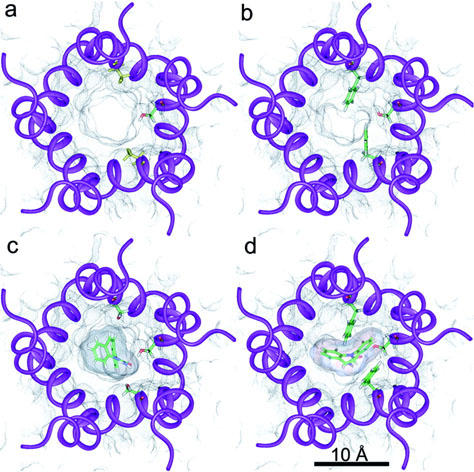
α4β2 ion channel models. Views of the normal (a, c) and mutant (b, d) receptors are extracellular and perpendicular to the membrane plane. Transmembrane segments M2 are schematically represented by violet ribbons. Black dots represent the solvent accessible surface within the ion-channel. (a, c) Side chains of the two α4 Ser 248 (upper and lower) and β2′′ Ser 271 (right) residues are shown in stick representation. Colour coding is atom based (C: green; H: white; N: blue; O: red). (b, d) Side chains of the two α4 Phe 248 (upper and lower) and β2′′ Ser 271 (right) residues are shown in stick representation. Colour coding as before. (c, d) The best ranked CBZ conformers in their docked conformations are displayed in these panels in stick representation. Colour coding as before. Their solvent accessible surface is also represented by dense black dots.
Figure 2.
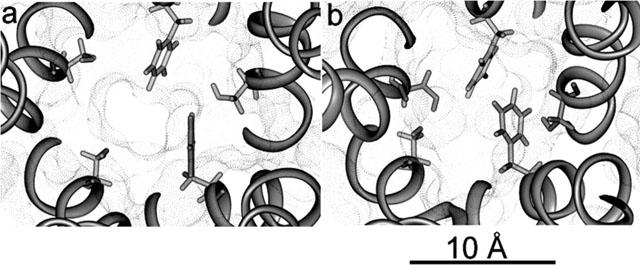
Mutant ion channel sizes. Views of the mutant α4β2 ion channel in the minimized conformation (a) and in a transient closed conformation observed during a molecular dynamics simulation (b). Both are extracellular views perpendicular to the membrane plane, where all residues below the level of the mutant ring are not displayed to show the actual size of the pore. Black dots represent the solvent accessible surface within the ion-channel. Displayed side chains in stick representation are α4 Phe 248 and β2 Ser 271.
Docking of CBZ
Visual inspection of the docked CBZ molecules in both the normal and mutant receptors (13 and 16 conformers respectively) showed that actually only six block the channel in the former compared to 10 in the latter. Given the docking methodology employed, we may assume that the search of possible and relevant binding sites was exhaustive for the model.
Energetics and binding constants
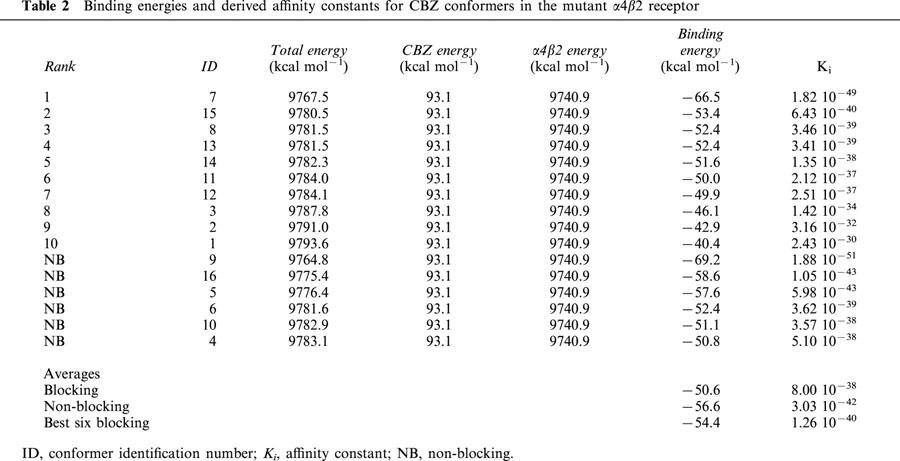
The binding affinity values obtained from the two different methods, for both the normal and mutant receptors, are not directly comparable. As stated in Methods, the absolute values of the molecular mechanics approximation are not expected to be reliable, and hence we are only interested in the relative values found for the normal and mutant receptors. For the same reasons, LUDI based affinities are not comparable to those from molecular mechanics, and although these absolute values can be considered more realistic, we are again only interested in their relative values. Tables 1 and 2 show the calculated binding energies and the derived Ki for the different conformers of CBZ in the normal and mutant receptors respectively. These binding energies are based on the difference between the total energy of the receptor-ligand complex and the energies of the ligand and the receptor alone, which are also shown in these tables. The blocking conformers are ranked according to binding energies, followed by the non-blocking conformers, ordered by their binding energies. An identification number ID for each conformer is also given for comparison purposes. At the end of these tables are the energy and Ki averages for blocking and non-blocking conformers. For the mutant (Table 2), we also show the average for the best six scored blocking conformers, to have another comparison with all (six) blocking conformers in the normal receptor. Comparing blocking and non-blocking conformers, the binding energies of the latter are more negative, and their Ki values are two orders of magnitude higher in the normal and four orders of magnitude higher in the mutant receptor. However, if compared to the mutant best six blocking conformers the differences in energies and Ki are also of two orders of magnitude.
Table 1.
Binding energies and derived affinity constants for CBZ conformers in the normal α4β2 receptor
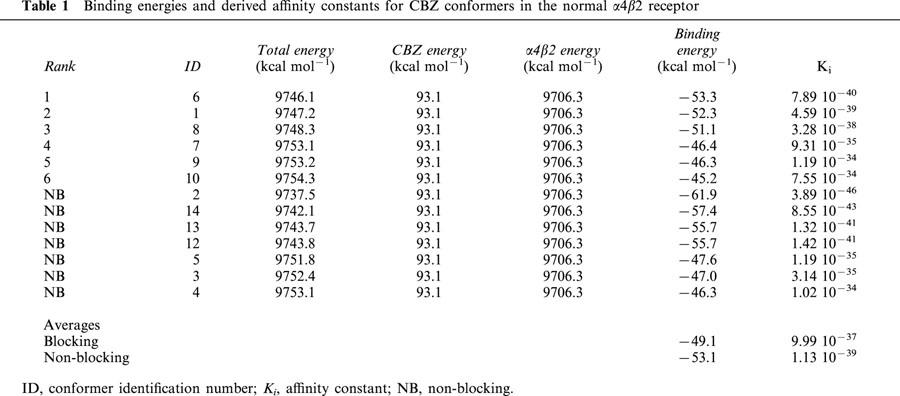
Table 2.
Binding energies and derived affinity constants for CBZ conformers in the mutant α4β2 receptor
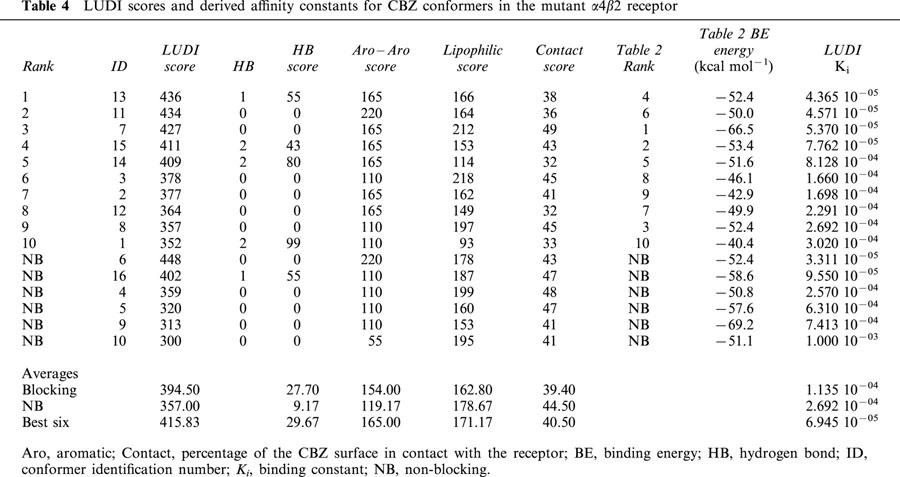
Tables 3 and 4 show the LUDI scores and derived Ki for CBZ in the normal and mutant receptors respectively. For reference purposes, the conformer ID, their corresponding binding energies and the binding energy based rank order (taken from Tables 1 and 2) are also given. As with Tables 1 and 2, the blocking conformers are ranked according to their LUDI scores, followed by the non-blocking conformers, ordered by their scores. Another information given in Tables 3 and 4 is the partitioning of the scores as described in Methods. The absent terms (e.g. ionic interactions) have a null score for all conformers. Contact column refers to the percentage of the CBZ conformer surface in contact with the ion-channel. For the hydrogen bonding scores, the number of hydrogen bonds present for each conformer is also given. The scores of non-blocking conformers for the normal and mutant receptors are very similar, and their corresponding Ki of the same order. In the normal receptor, and in a similar way with what was observed with binding energies estimated using molecular mechanics, the average LUDI score for the blocking conformers are lower than the average of non-blocking conformers, with their corresponding Ki one order of magnitude lower. However, in the mutant the blocking and non-blocking averages are similar, with Ki of the same order. Moreover, the best six blocking conformers have a higher average score, with a Ki one order of magnitude higher with respect to the non-blocking conformers.
Table 3.
LUDI scores and derived affinity constants for CBZ conformers in the normal α4β2 receptor
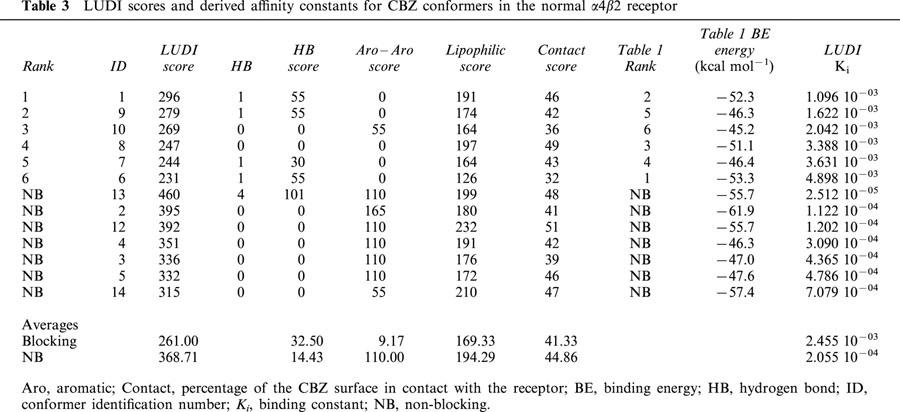
Table 4.
LUDI scores and derived affinity constants for CBZ conformers in the mutant α4β2 receptor
Comparison of CBZ blocking conformers in the normal and mutant receptors
The average binding energy of the blocking conformers is more negative in the mutant receptor (Tables 1 and 2). The average difference is similar to 1.5 kcal mol−1 (Table 5). This value corresponds to a 12 fold change in the value of the binding constant, i.e. the affinity of CBZ for the mutant receptor is 12 times higher. Much higher differences arise (Table 5) if the top ranked conformers in the normal and mutant receptor are compared (nine orders of magnitude) or when comparing the average of the best six conformers of each receptor (similar to an 8000 fold change).
Table 5.
Comparison of CBZ binding energies, LUDI scores and binding constants between the normal and mutant α4β2 receptors
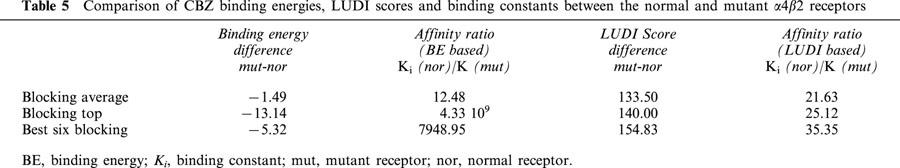
When LUDI scores for the normal and mutant receptor are compared, a similar result is found, that is, CBZ has a higher affinity for the mutant. Table 5 shows that the score differences are 133, 140 and 154 for the total average, top ranked and best six scored conformers respectively. These values correspond to a 21, 25 and 35 fold change in binding constants in that order. The lowest change (for the total average) is much higher than predicted on the basis of binding energies (almost 2 fold). However, for the top ranked and the best six, the values are much lower (Table 5). Tables 3 and 4 show that the main contributors to these differences in affinities are the aromatic–aromatic interactions. CBZ is mainly an aromatic molecule, and the presence of the two Phe residues in the mutant is responsible for this difference (see below).
CBZ–ion channel interactions
A view of the top scored CBZ conformations docked in the normal and mutant receptors are given in Figure 1c and d respectively (top scored according to LUDI scores). In the normal receptor (Figure 1c) β2′′ Ser 271 is hydrogen bonded to the CBZ amide oxygen (see below). In the mutant, docking of CBZ forces α4′′ Phe 248 side chain to move towards the channel wall, while β2′′ Ser 271 adopts a conformation similar to the present in the normal receptor. β2′′ Ser 271 is also hydrogen bonded to the oxygen of the amide moiety of CBZ, although the conformations of CBZ docked to the normal and mutant receptors are different. The interactions of this conformer of CBZ with α4′ Phe 248 and α4′′ Phe 248 are quite strong (see details below). One phenyl group of CBZ interacts with the α4′ Phe 248 aromatic side chain and the other does it with the corresponding of α4′′ Phe 248. What is interesting is that these aromatic–aromatic interactions between each of the phenyl groups of CBZ and α4 Phe 248 residues are of a different type. Described, there are two most favourable, isoenergetic, types of aromatic–aromatic interactions, named off-centred parallel orientation and the T-shaped packing geometry (Brocchieri & Karlin, 1994; Kannan & Vishveshwara, 2000; Klebe & Diederich, 1993; McGaughey et al., 1998). In the first type, also denoted as π-stacking, the ring planes are parallel (with ring centroids not centred). The second type is characterized by a T-shaped geometry, where two aromatic rings adopt an edge to face conformation, with their ring planes orthogonal. α4′′ Phe 248 is involved in the first type, with its aromatic ring plane nearly parallel to one of the phenyl ring planes of CBZ. On the contrary, the aromatic ring of α4′ Phe 248 is perpendicular to the other phenyl ring of CBZ. In this case, one of the C-H bond dipoles of α4′ Phe 248 is making a hydrogen bond with a negative carbon atom of the phenyl ring of CBZ.
To assess the interactions of CBZ conformers with the ion channel, all the protein residues at a distance equal or lower to 4 Å to all blocking CBZ conformers are listed in Table 6 for the normal receptor, and in Table 7 for the mutant. For each amino acid residue, we calculated their interaction energy with all blocking conformers of CBZ, and these values were subsequently added. The residues in Tables 6 and 7 are ranked according to these sums, which are proportional to both, the energy contribution and the times an amino acid is implicated in CBZ binding.
Table 6.
Energy contribution of α4β2 normal receptor residues at a distance equal or lower to 4 Å
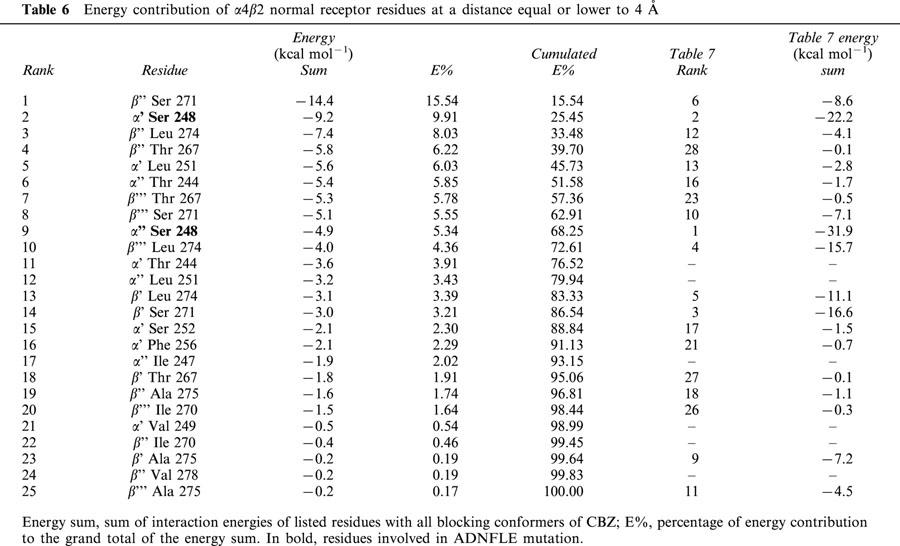
Table 7.
Energy contribution of α4β2 mutant receptor residues at a distance equal or lower to 4 Å
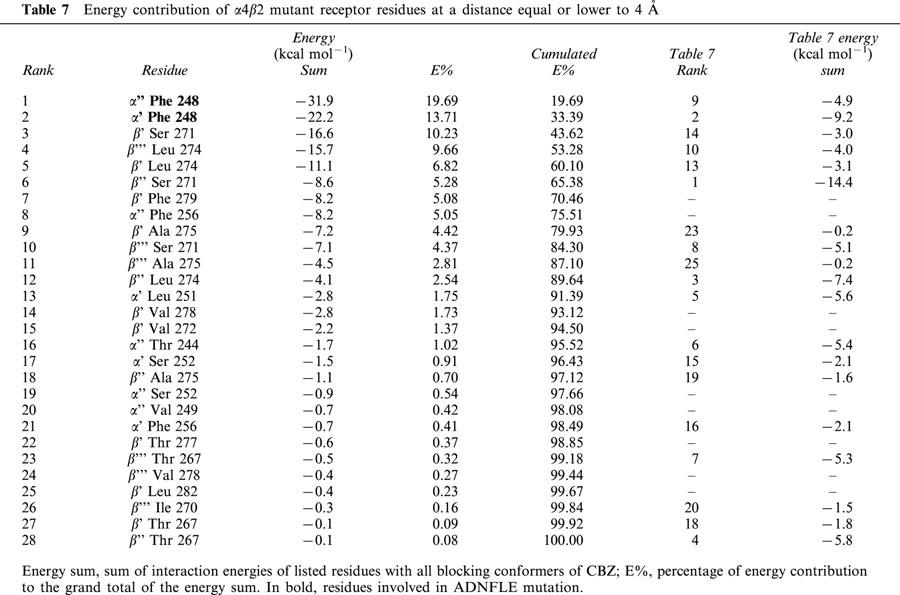
β2′′ Ser 271, also shown in Figure 1d, is on average the residue that most contributes to the binding of CBZ conformers to the normal receptor. Second in place is α4′ Ser 248, one of the α4 expressed residues, mutated in ADNFLE. The other residue, α4′′ Ser 248, is well below in the ninth place. When these two residues are mutated to Phe, they become the main contributors to the binding energy, with an accumulated 33% of the total contribution of those residues at a distance of 4 Å or less. A 33% is achieved in the normal receptor with three residues. The corresponding mutated residue of α4′ Ser 248, α4′ Phe 248, remains in the second place, and the other escalates from the ninth to the first position. The main contributor in the normal receptor is still important, but is now in the sixth place (Tables 6 and 7). In addition to these movements in their importance as participants in CBZ binding, many residues are involved only in either the normal or the mutant receptor.
Blocking–non blocking CBZ interactions
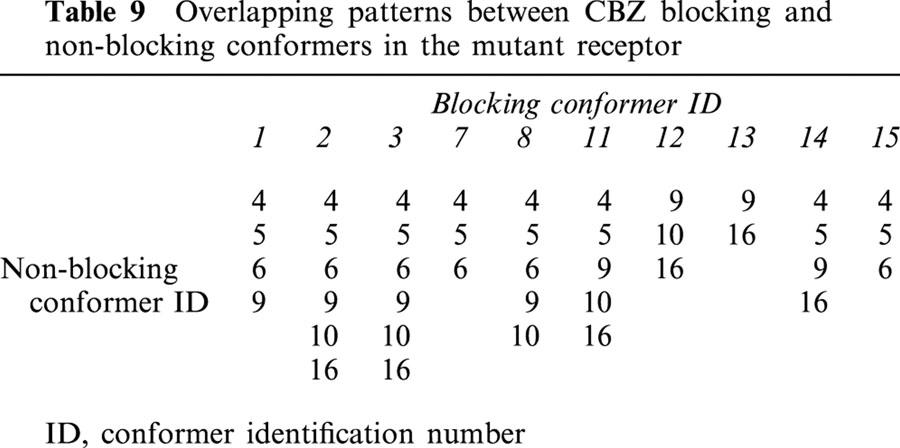
In a real system, many CBZ molecules must bind with high or low affinity to many non-blocking sites outside the ion-channel, and these have no effect on neither IC50 measurements nor on Ki calculations. However, the situation is different with those that may bind within the ion-channel. The reason for this is that the binding sites of non-blocking conformers superimpose partially with those with blocking properties, and hence, they have an interfering effect on channel blocking. Tables 8 and 9 give the patterns of overlapping between blocking and non-blocking conformers in the normal and mutant receptors respectively. In the normal receptor (Table 8), the degree of overlapping is quite low. Only conformers 6 and 7 overlap with one and two non-blocking conformers respectively. The situation is quite different in the mutant (Table 9), where all the blocking conformers overlap from two to all non-blocking conformers.
Table 8.
Overlapping patterns between CBZ blocking and non-blocking conformers in the normal receptor

Table 9.
Overlapping patterns between CBZ blocking and non-blocking conformers in the mutant receptor
Discussion
The replacement of a single residue in the α4β2 neuronal nicotinic receptor derives in a major neurological disorder as is ADNFLE. This mutation has profound consequences in the ion channel properties and in the sensitivity to the open channel blocker carbamazepine. The aim of the present work was to give structural explanations to both phenomena.
Ion channel properties
Figures 1b and 2a show that the mutant phenylalanine residues are almost blocking the ion channel. Thus, the narrowest region of the ion channel is changed from the very cytoplasmic ‘central' polar ring to the mutant ring composed of three serines and two phenylalanines. Under these conditions the channel, upon agonist activation, will open allowing only a reduced flow of cations because of the size of the pore. Perhaps most of the ion flow through the channel is produced before Phe248 side chains reach the minimized conformations shown in Figures 1b and 2a. At this level of the channel, only fully dehydrated ions would be allowed to passage as the diameter size of this asymmetrical pore is around 2.5 Å. In the normal receptor, an alleged function for this polar ring of serine residues is to partially dehydrate incoming cations before they reach the last ‘central' polar ring. Crystal radius for Na+ and Ca2+ are 0.98 Å and 0.94 Å respectively. However, the interaction of water with Ca2+ is much stronger than with Na+, thus, dehydration of the former is more difficult. Although highly hydrophobic, phenylalanines are also polar moieties that can strongly bind electrostatically to cations (Dougherty, 1996; Dunbar, 2000; Felder et al., 2001; Gallivan & Dougherty, 1999; Miklis et al., 1998; Wouters, 2000; Zhu et al., 1999). Thus, this noncovalent force, termed ‘cation-π interaction' can help in dehydration. As we stated, Na+ ions are more easily dehydrated and hence would be allowed to flow, albeit at a lower rate than in the normal receptor. For calcium ions it has been shown that they react quickly with aromatic rings when hydrated with four or five water molecules, but not with six or seven (Rodriguez & Wilson, 2001). Therefore, if calcium ions reach the mutated polar ring with a shell of six or more water molecules, dehydration at this point would be more difficult. Considering that calcium flow is highly reduced, this might be the case.
On the other hand, if cation-π interactions are too strong, cations can be trapped between the aromatic rings of the two phenylalanines adding a steric hindrance that further blocks the channel. According to our molecular dynamics analysis, Phe248 side chains are highly mobile and their movement causes the channel pore to periodically change its shape and size, making it more narrow or even totally closed (Figure 2b). Although not directly tested by molecular dynamics because cation-π interactions are not explicitly parametrized in the force field employed, it is expected that if any of these conformations favours a stronger interaction with either sodium or calcium ions, side chains will be stabilized, and the channel blocked.
Under these hypotheses, an explanation of the differential channel properties of the mutant ADNFLE receptor compared to the wild type can be given. The lower conductance might be due mainly to the reduced size of the pore, as stated elsewhere (Kuryatov et al., 1997), but also as a consequence of the less efficient mechanism of dehydration. The short open times seen using patch clamp techniques (Kuryatov et al., 1997) can be viewed as a direct consequent of early channel closing mechanisms, either because of the movement of phenylalanine side chains alone, or due to cation trapping.
We believe that cation blocking is an important mechanism in the mutant channel which may explain its desensitization properties. Once blocked, any cation released can be replaced by another from the pool, resulting in an early and long lasting blockade. Hence, what is seen as an enhanced desensitization sensitivity might be due to the stabilization of a blocked, uncharacteristic open conformation. Eventually, receptors will gradually change their conformations to the true desensitized state. The channel gate in this conformation is above (more extracellular) the level of the mutant residues (Bertrand et al., 1992; Revah et al., 1991), thus new cations are not allowed to replace those released from the blocking site, giving rise to the observed slow recovery from desensitization.
The common factor among all mutations causing ADNFLE seems to be a reduced influx of calcium ions (Cohen et al., 2001), but other channel properties are different. For example, receptors bearing the leucine insertion (776ins3) in the M2 transmembrane segment of the α4 subunit, do have a reduced calcium influx as in the S248F mutation (Bertrand et al., 1998). However, a critical reduction in conductance is neither expected nor observed, as the inserted leucine is well above the level of the S248F mutation and a reduction in the diameter of the narrowest part of the pore is not predicted. Aromatic residues which, as described above, might be causative of the enhanced desensitization sensitivity and slower recovery from desensitization observed in the S248F mutation, are not present. In accordance, 776ins3 containing receptors lack these desensitization properties.
If the reduced calcium influx is the common factor among ADNFLE mutations, all the other channel properties are probably of secondary importance. We explained the reduced calcium influx in the S248F mutant on the basis of the weaker propensity to dehydration of calcium ions because in this mutant one of the polar rings involved in this process is altered. Hence, it is probable that all ADNFLE mutations have an altered dehydration mechanism, which mainly affects the strongly solvated calcium ions.
CBZ-ion channel interactions
The higher sensitivity of the mutant receptor to CBZ (Picard et al., 1999) is in agreement with our results. The mutation creates a new and more favourable environment for CBZ binding, where the Phe248 side chains themselves are directly involved and are the main contributors to the binding of this channel blocker.
First, one factor that can be taken into consideration to account for the differences in sensitivity to CBZ, is the difference in the number of binding modes for this drug in both receptors. The ion channel is a large entity compared, for example, to the agonist binding pocket, and thus channel blockers might be expected to have many ways to exert their action (Ortells & Barrantes, 2001), albeit one most favoured. As described, we found six binding modes for the normal and 10 for the mutant receptor. In this way, after entering the channel, more CBZ orientations have chances to find a binding site where to block the channel. In addition, after being released from the first site, the probability to be trapped in another one is higher.
Second, our estimations of binding affinities for CBZ are also in agreement with the experimental results. Both types of estimations, energy and LUDI score based, show that CBZ has a higher affinity for the mutant. This seems to be mainly due to the aromatic–aromatic interaction between CBZ and the mutant Phe248 side chains. The difference in LUDI scores between the normal and mutant receptors are considerable, 133 units more favourable to the mutant, and represents a 21 fold difference in affinity. As we stated, we are assuming that we actually found all possible binding modes for CBZ in the ion channel model, and hence we are not dealing with a sample but with the whole population. As a consequence, no statistical analysis of this difference should be necessary. The same applies to the binding energy estimations. As we stated in Methods, there is an intrinsic possible error in the estimations of binding energies based on molecular mechanics calculations. However, we must remember again that the observed energy difference, even quite low (1.5 kcal mol−1) compared to the possible error is based not on one, but on the average of six and 10 ligand-receptor complexes, for the normal and mutant receptors respectively. If we consider that this binding energy difference is enhanced (to 5 kcal mol−1) when considering only the top scored or the best six CBZ conformations on each receptor (which represents all blocking conformations for the normal), and also that the same trend is found using a different approach to calculate binding affinities, it seems that the energy difference observed is valid, at least for the model.
On the other hand, the estimated differences in binding affinities in favour of the mutant receptor seem to be, however, too high (in spite that IC50 and Ki values are not directly comparable). If we also consider that there are more CBZ blocking binding sites in the mutant receptor, this appreciation is reinforced. Picard et al. (1999), showed that the mutant receptor is three times more sensitive to the blocking properties of CBZ than the normal according to IC50 measurements. However, our binding affinity estimations suggest that the ADNFLE receptor should be at least 12 times more sensitive to CBZ when using molecular mechanics calculations and at least 21 times when using LUDI scores (Table 5). Hence, it is possible that other factors, apart from the affinity of CBZ to both receptors, are contributing to the overall blocking capacity of CBZ in these receptors, as measured by the IC50. One of these factors appears to be the interactions between blocking and non-blocking conformers of CBZ observed in our simulations. In effect, the chances that CBZ blocks the channel are reduced if another CBZ molecule was already bound to a non-blocking site. As shown in Tables 8 and 9, the probability of interactions between blocking and non-blocking conformers are higher for the mutant, as all the blocking sites overlap with two or more non-blocking. On the other hand, in the normal receptor only two conformer sites overlap with one or two non-blocking sites. This negative cooperativity in both types of receptors may be reflected in the Hill coefficient values obtained by Picard et al. (1999) when fitting the dose-response inhibition profiles of the normal and mutant receptors. For the normal receptor they found a Hill coefficient value of 0.85±0.09 for the peak and plateau responses. For the mutant they found values of 0.75 and 0.8 for the peak and plateau responses respectively (no standard errors were given). According to our results, we expect Hill coefficients for the mutant to be lower than those of the normal receptor. Hill values found by Picard et al. (1999) are lower for the mutant as we expected, but they might not be significantly different. Nevertheless, we think that the experimental evidence supports our idea that there are interactions between blocking and non-blocking conformers that reduce the potential capacity of CBZ to block the channel, and that these are stronger in the mutant.
As stated before, the average binding affinity calculated with molecular mechanics for non-blocking conformers is higher in the mutant. Although we expected that both methods should give similar results, this difference is not reflected in the average binding affinities calculated with LUDI, which are almost the same. This discrepancy may be attributed to a higher sensitivity of molecular mechanics calculations to the interactions between aromatic moieties. As described in Methods, these interactions are measured with LUDI using a simple distance cutoff, i.e. only those interactions within 4.5 Å are considered. Blocking conformers in the mutant are very close to the mutant residues. In this way both methods account for these aromatic interactions. As shown in Table 9, non-blocking conformers interact to some degree with the mutant residues, as they superimpose with blocking conformers. However, most of these interactions with mutant residues are at greater distance than 4.5 Å, and hence are not accounted by LUDI. Tables 3 and 4 show that the average aromatic–aromatic scores for the normal and mutant receptors are almost the same, which means that the interactions with the mutant aromatic residues are not taken into account.
CBZ as an antiepileptic drug
CBZ is used as a primary drug for the treatment of partial and tonic-clonic seizures. The presence of the carbamyl group is essential for its potent antiseizure activity (McNamara, 2001). We found that in both the normal and mutant receptors, this moiety is involved in hydrogen bonding with serine residues in the best blocking conformers and that the interactions between serines and CBZ contribute substantially to the energy of binding. This may explain the importance of the carbamyl moiety, not only in the interaction of CBZ with the nicotinic receptor, but in a general way. Most of the structure of CBZ is composed of aromatic rings, and these are responsible for obstructing the channel path (as occurs probably with other channel blockers like chlorpromazine). However, in the mutant receptor the aromatic moieties are, most probably, also responsible for the increased affinity of CBZ. It was not the aim of this work to analyse possible ways of improving the affinity of CBZ or other molecules for the mutant, but most likely enhancing the aromatic–aromatic interactions between the blocker and the receptor might be a way.
In conclusion, we propose that the S248F mutation involved in the ADNFLE has a lower conductance due to both a reduced pore size and a less efficient mechanism of dehydration. However, perhaps some or most of the observed current is transiently produced before the mutant phenylalanine side chains reach their most stable conformation. Also, the altered dehydration mechanism is unable to properly desolvate calcium ions, and as a consequence they are not capable of crossing through the narrow channel pore. The enhanced sensitivity and the slower recovery from desensitization is explained as the result of an early channel blocking by cations trapped by cation-π interactions with the aromatic side chains of the mutant phenylalanines. The observed short open times might be the outcome of either the movement of phenylalanine side chains that periodically adopt a conformation that totally closes the channel, or the blocking of the channel by cations, as described.
The presence of Phe248 residues also directly modifies the way CBZ binds to the ion channel. In the normal and mutant receptors we found that several blocking and non-blocking binding modes for CBZ can exist. The affinities of non-blocking conformers are almost the same in both receptors, but those of the blocking conformers are higher in the mutant (actually much higher that what would be expected considering the correspondingly observed IC50 values). This higher affinity is also a direct consequence of the presence of the two mutant residues, as it is based on the aromatic–aromatic interactions between the phenyl rings of CBZ and Phe248 side chains. However, we observed overlapping between blocking and non-blocking binding sites, and this is much higher in the mutant. This could explain both the possible disagreement between the differences in experimental IC50 values and the predicted binding affinities between the normal and mutant receptors, and the negative cooperativity experimentally observed and based on the Hill coefficient values in the dose-response curves of Picard et al. (1999).
We propose that the importance of CBZ carbamyl moiety related to the antiepileptic activity might be due to its capability of forming hydrogen bonds with serine (and probably threonine) residues frequently present in ion-channels.
A word must be stated with respect to the ion-channel model used. We are far from claiming that it represents the actual structure of the receptor ion-channel. However, it seems that its structural features are accurate enough to model not only experimental structural data, but also to be used as a working hypothesis to study some of its functional properties.
Acknowledgments
This work was supported by grants of Fundación Antorchas, CONICET PIP No 721/98 and ANPCyT BID 1201/OC-AR PICT 98 No 05-04048 to M.O. Ortells and CONICET PEI No 313/99 to G.E. Barrantes.
Abbreviations
- ABP
acetylcholine binding protein
- ADNFLE
autosomal dominant nocturnal frontal lobe epilepsy
- CBZ
5H-Dibenz[b,f]lazepine-5-carboxamide, carbamazepine
- LGIC
Ligand-gated ion channel
- OCB
open channel blocker
- RMS
root mean square
References
- AJAY MURCKO M.A., STOUTEN P.F.W.Recent advances in the prediction of binding free energy Practical application of computer-aided drug design. 1997New York: Marcel Dekker, Inc; 355–410.ed. Charifson, P.S. pp [Google Scholar]
- ARIAS H.R. Role of local anesthetics on both cholinergic and serotonergic ionotropic receptors. Neurosci. Biobehav. Rev. 1999;23:817–843. doi: 10.1016/s0149-7634(99)00020-2. [DOI] [PubMed] [Google Scholar]
- BAULAC S., HUBERFELD G., GOURFINKEL-AN I., MITROPOULOU G., BERANGER A., PRUD'HOMME J.F., BAULAC M., BRICE A., BRUZZONE R., LEGUERN E. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the γ2-subunit gene. Nature Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- BERTRAND D., DEVILLERSTHIERY A., REVAH F., GALZI J.L., HUSSY N., MULLE C., BERTAND S., BALLIVET M., CHANGEUX J.P. Unconventional pharmacology of a neuronal nicotinic receptor mutated in the channel domain. Proc. Natl. Acad. Sci. U. S. A. 1992;89:1261–1265. doi: 10.1073/pnas.89.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERTRAND D., GALZI J.-L., DEVILLERS-THIÉRY A., BERTRAND S., CHANGEUX J.-P. Stratification of the channel domain in neurotransmitter receptors. Curr. Opin. Cell. Biol. 1993;5:688–693. doi: 10.1016/0955-0674(93)90141-c. [DOI] [PubMed] [Google Scholar]
- BERTRAND S., WEILAND S., BERKOVIC S.F., STEINLEIN O.K., BERTRAND D. Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br. J. Pharmacol. 1998;125:751–760. doi: 10.1038/sj.bjp.0702154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BÖHM H.J. LUDI – rule-based automatic design of new substituents for enzyme-inhibitor leads. J. Computer-Aided Mol. Design. 1992a;6:593–606. doi: 10.1007/BF00126217. [DOI] [PubMed] [Google Scholar]
- BÖHM H.J. The computer-program LUDI – a new method for the de novo design of enzyme-inhibitors. J. Computer-Aided Mol. Design. 1992b;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- BÖHM H.J. On the use of LUDI to search the fine chemicals directory for ligands of proteins of known 3-dimensional structure. J. Computer-Aided Mol. Design. 1994a;8:623–632. doi: 10.1007/BF00123669. [DOI] [PubMed] [Google Scholar]
- BÖHM H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein ligand complex of known 3-dimensional structure. J. Computer-Aided Mol. Design. 1994b;8:243–256. doi: 10.1007/BF00126743. [DOI] [PubMed] [Google Scholar]
- BÖHM H.J. Site-directed structure generation by fragment-joining. Perspect. Drug Discov. Design. 1995;3:21–33. [Google Scholar]
- BÖHM H.J. Prediction of binding constants of protein ligands: A fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J. Computer-Aided Mol. Design. 1998;12:309–323. doi: 10.1023/a:1007999920146. [DOI] [PubMed] [Google Scholar]
- BREJC K., VAN DIJK W.J., KLAASSEN R.V., SCHUURMANS M., VAN DER OOST J., SMIT A.B., SIXMA T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- BROCCHIERI L., KARLIN S. Geometry of interplanar residue contacts in protein structures. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9297–9301. doi: 10.1073/pnas.91.20.9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROVTSYNA N.B., GMIRO V.E., GORBOUNOVA O.B., SERDYUK S.E., LUKOMSKAYA N.Y. The structure of the neuronal nicotinic acetylcholine-receptor ion-channel as estimated on the basis of the structure-activity-relationships in a series of open-channel blocking-drugs. Biologicheskie Membrany. 1996a;13:57–70. [Google Scholar]
- BROVTSYNA N.B., TIKHONOV D.B., GORBUNOVA O.B., GMIRO V.E., SERDUK S.E., LUKOMSKAYA N.Y., MAGAZANIK L.G., ZHOROV B.S. Architecture of the neuronal nicotinic acetylcholine-receptor ion-channel at the binding-site of bis-ammonium blockers. J. Membr. Biol. 1996b;152:77–87. doi: 10.1007/s002329900087. [DOI] [PubMed] [Google Scholar]
- CHOI S.K., KALIVRETENOS A.G., USHERWOOD P.N.R., NAKANISHI K. Labeling studies of photolabile philanthotoxins with nicotinic acetylcholine-receptors – mode of interaction between toxin and receptor. Chem. Biol. 1995;2:23–32. doi: 10.1016/1074-5521(95)90077-2. [DOI] [PubMed] [Google Scholar]
- COHEN B.N., PINQUET N., LI M., FIGL A., JIA L., TRUONG A. All ADNFLE mutations reduce Ca2+ potentiation of the ACh response. Biophys. J. 2001;80:1979. [Google Scholar]
- DE FUSCO M., BECCHETTI A., PATRIGNANI A., ANNESI G., GAMBARDELLA A., QUATTRONE A., BALLABIO A., WANKE E., CASARI G. The nicotinic receptor β2 subunit is mutant in nocturnal frontal lobe epilepsy. Nature Genet. 2000;26:275–276. doi: 10.1038/81566. [DOI] [PubMed] [Google Scholar]
- DILGER J.P., BOGUSLAVSKY R., BARANN M., KATZ T., VIDAL A.M. Mechanisms of barbiturate inhibition of acetylcholine receptor channels. J. Gen. Physiol. 1997;109:401–414. doi: 10.1085/jgp.109.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DISCOVER . Molecular Simulations Inc.: San Diego; 2000. [Google Scholar]
- DOUCET J.-P., WEBER J.Empirical force field methods and molecular mechanics Computer-aided molecular design. 1996San Diego: Academic Press; 124–170.ed. Doucet, J.-P. & Weber, J. pp [Google Scholar]
- DOUGHERTY D.A. Cation-π interactions in chemistry and biology – a new view of benzene, phe, tyr, and trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- DUNBAR R.C. Complexation of Na+ and K+ to aromatic amino acids: A density functional computational study of cation-π interactions. J. Phys. Chem. A. 2000;104:8067–8074. [Google Scholar]
- FELDER C., JIANG H.L., ZHU W.L., CHEN K.X., SILMAN I., BOTTI S.A., SUSSMAN J.L. Quantum/classical mechanical comparison of cation-π interactions between tetramethylammonium and benzene. J. Phys. Chem. A. 2001;105:1326–1333. [Google Scholar]
- FORMAN S.A., MILLER K.W., YELLEN G. A discrete site for general-anesthetics on a postsynaptic receptor. Mol. Pharmacol. 1995;48:574–581. [PubMed] [Google Scholar]
- FRYER J.D., LUKAS R.J. Noncompetitive functional inhibition at diverse, human nicotinic acetylcholine receptor subtypes by bupropion, phencyclidine, and ibogaine. J. Pharmacol. Exp. Ther. 1999a;288:88–92. [PubMed] [Google Scholar]
- FRYER J.D., LUKAS R.J. Antidepressants noncompetitively inhibit nicotinic acetylcholine receptor function. J. Neurochem. 1999b;72:1117–1124. doi: 10.1046/j.1471-4159.1999.0721117.x. [DOI] [PubMed] [Google Scholar]
- GALLIVAN J.P., DOUGHERTY D.A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. U.S.A. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IMOTO K., BUSCH C., SAKMANN B., MISHINA M., KONNO T., NAKAI J., BUJO H., MORI Y., FUKUDA K., NUMA S. Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature. 1988;335:645–648. doi: 10.1038/335645a0. [DOI] [PubMed] [Google Scholar]
- IMOTO K., KONNO T., NAKAI J., WANG F., MISHINA M., NUMA S. A ring of uncharged polar amino acids as a component of channel constriction in the nicotinic acetylcholine receptor. FEBS Lett. 1991;289:193–200. doi: 10.1016/0014-5793(91)81068-j. [DOI] [PubMed] [Google Scholar]
- INSIGHTII . Molecular Simulations Inc.: San Diego; 2000. [Google Scholar]
- KANNAN N., VISHVESHWARA S. Aromatic clusters: a determinant of thermal stability of thermophilic proteins. Prot. Engng. 2000;13:753–761. doi: 10.1093/protein/13.11.753. [DOI] [PubMed] [Google Scholar]
- KARLIN A. Structure of nicotinic acetylcholine receptors. Curr. Opin Neur. 1993;3:299–309. doi: 10.1016/0959-4388(93)90121-e. [DOI] [PubMed] [Google Scholar]
- KESERÜ G., KOLOSSVÁRY I. Molecular mechanics and conformational analysis in drug design. Oxford: Blackwell Science Ltd; 1999. p. 168. [Google Scholar]
- KLEBE G., DIEDERICH F. A comparison of the crystal packing in benzene with the geometry seen in crystalline cyclophane benzene complexes – guidelines for rational receptor design. Philosoph. Trans. Roy. Soc. Lond. Ser. A. 1993;345:37–48. [Google Scholar]
- KOEHLER K.F., RAO S.N., SNYDER J.P.Modeling drug-receptor interactions Guidebook on molecular modeling in drug design. 1996San Diego: Academic Press; 235–336.ed. Cohen, N.C. pp [Google Scholar]
- KURYATOV A., GERZANICH V., NELSON M., OLALE F., LINDSTROM J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca++ permeability, conductance, and gating of human alpha 4 beta 2 nicotinic acetylcholine receptors. J. Neurosci. 1997;17:9035–9047. doi: 10.1523/JNEUROSCI.17-23-09035.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUTY B.A., WASSERMAN Z.R., STOUTEN P.F.W., HODGE C.N., ZACHARIAS M., MCCAMMON J.A. A molecular mechanics grid method for evaluation of ligand-receptor interactions. J. Comput. Chem. 1995;16:451–464. [Google Scholar]
- MCGAUGHEY G.B., GAGNE M., RAPPE A.K. π-stacking interactions – alive and well in proteins. J. Biol. Chem. 1998;273:15458–15463. doi: 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- MCNAMARA J.O.Drugs effective in the therapy of the epilepsies The pharmacological basis of therapeutics. 2001New York: McGraw-Hill; 521–547.ed. Hardman, J.G. & Limbird, L.E. pp [Google Scholar]
- MIKLIS P.C., DITCHFIELD R., SPENCER T.A. Carbocation-π interaction: Computational study of complexation of methyl cation with benzene and comparisons with related systems. J. Am. Chem. Soc. 1998;120:10482–10489. [Google Scholar]
- MIYAZAWA A., FUJIYOSHI Y., STOWELL M., UNWIN N. Nicotinic acetylcholine receptor at 4.6 Å resolution: Transverse tunnels in the channel wall. J. Mol. Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- ORTELLS M.O., BARRANTES G.E. Understanding channel blocking in the nicotinic acetylcholine receptor. Receptors Channels. 2001;7:273–288. [PubMed] [Google Scholar]
- ORTELLS M.O., BARRANTES G.E., BARRANTES F.J., WOOD C., LUNT G.G. Molecular modelling of the nicotinic acetylcholine receptor transmembrane region in the open state. Prot. Engng. 1997;10:511–517. doi: 10.1093/protein/10.5.511. [DOI] [PubMed] [Google Scholar]
- ORTELLS M.O., LUNT G.G. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci. 1995;18:121–127. doi: 10.1016/0166-2236(95)93887-4. [DOI] [PubMed] [Google Scholar]
- ORTELLS M.O., LUNT G.G. A mixed helix-beta sheet model of the transmembrane region of the nicotinic acetylcholine receptor. Prot. Engng. 1996;9:51–59. doi: 10.1093/protein/9.1.51. [DOI] [PubMed] [Google Scholar]
- PICARD F., BERTRAND S., STEINLEIN O.K., BERTRAND D. Mutated nicotinic receptors responsible for autosomal dominant nocturnal frontal lobe epilepsy are more sensitive to carbamazepine. Epilepsia. 1999;40:1198–1209. doi: 10.1111/j.1528-1157.1999.tb00848.x. [DOI] [PubMed] [Google Scholar]
- REVAH F., BERTRAND D., GALZI J.-L., DEVILLERS-THIÉRY A., MULLE C., HUSSY N., BERTRAND S., BALLIVET M., CHANGEUX J.-P. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- RODRIGUEZ A.D., WILSON G.G. Gas-phase reactions of hydrated alkaline earth metal ions, M2+(H2O)n (M=Mg, Ca, Sr, Ba and n=4–7), with benzene. J. Am. Soc. Mass Spectrom. 2001;12:250–257. doi: 10.1016/S1044-0305(00)00224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMIT A.B., SYED N.I., SCHAPP D., VAN MINNEN J., KLUMPERMAN J., KITS K.S., LODDER H., VAN DER SCHORS R.C., VAN ELK R., SORGEDRAGER G., BREJC K., SIXMA T.K., GERAERTS W.P.M. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature. 2001;411:261–268. doi: 10.1038/35077000. [DOI] [PubMed] [Google Scholar]
- STEINLEIN O.K., MAGNUSSON A., STOODT J., BERTRAND S., WEILAND S., BERKOVIC S.F., NAKKEN K.O., PROPPING P., BERTRAND D. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum. Mol. Genet. 1997;6:943–947. doi: 10.1093/hmg/6.6.943. [DOI] [PubMed] [Google Scholar]
- STEINLEIN O.K., MULLEY J.C., PROPPING P., WALLACE R.H., PHILLIPS H.A., SUTHERLAND G.R., SCHEFFER I.E., BERKOVIC S.F. A missense mutation in the neuronal nicotinic acetylcholine-receptor alpha-4 subunit is associated with autosomal-dominant nocturnal frontal-lobe epilepsy. Nature Genet. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- STOUTEN P.F.W., FROMMEL C., NAKAMURA H., SANDER C. An effective solvation term based on atomic occupancies for use in protein simulations. Molecular Simulation. 1993;10:97–120. [Google Scholar]
- WEILAND S., WITZEMANN V., VILLARROEL A., PROPPING P., STEINLEIN O. An amino-acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett. 1996;398:91–96. doi: 10.1016/s0014-5793(96)01215-x. [DOI] [PubMed] [Google Scholar]
- WOUTERS J. Use of theoretical descriptors to characterize cation-π binding sites in (macro)molecules. J. Comput. Chem. 2000;21:847–855. [Google Scholar]
- ZHU W.L., JIANG H.L., TAN X.J., CHEN J.Z., ZHAI Y.F., GU J.D., LIN M.W., CHEN K.X., JI R.Y., CAO Y. Theoretical studies on cation-π interactions II. Density-functional theory investigation on the configurations of and interactions in ammonium cation-bibenzene complexes. Acta Chimica Sinica. 1999;57:852–859. [Google Scholar]