

Abstract
The phorbol ester TPA, an activator of protein kinase C (PKC), inhibits cholinergic stimulation of gastric acid secretion but increases basal H+ secretion.
Since these contradictory findings suggest the action of different PKC isozymes we analysed the role of calcium-dependent PKC-α, and calcium-independent PKC-ε in gastric acid secretion.
Inhibition of PKC-α by the indolocarbazole Gö 6976 revealed that about 28% of carbachol-induced acid secretion was inhibited by PKC-α. In the presence of Gö 6976 approximately 64% of the carbachol-induced signal transduction is mediated by Ca2+/calmodulin-dependent protein kinase II (CaMKII), and 14% is conveyed by PKC-ε as deduced from the inhibition with the bisindolylmaleimide Ro 31-8220.
Inhibition of carbachol-induced acid secretion by TPA was accompanied by a decrease in CaMKII activity.
The stimulation of basal acid secretion by TPA was biphasic with a peak at a very low concentration (10 pM), resulting in an activation of the calcium-sensor CaMKII. The activation was determined with a phosphospecific polyclonal antibody against active CaMKII. The TPA-induced increase of H+ secretion was sensitive to the cell-permeable Ca2+-chelator BAPTA/AM, Ro 31-8220, and the CaMKII-inhibitor KN-62, but not to Gö 6976.
Since TPA induced the translocation of PKC-ε but not of PKC-α in resting parietal cells, PKC-ε seems to be at least responsible for an initial elevation of free intracellular calcium to initiate TPA-induced acid secretion.
Our data indicate the different roles of two PKC isoforms: PKC-ε activation appears to facilitate cholinergic stimulation of H+-secretion likely by increasing intracellular calcium. In contrast, PKC-α activation attenuates acid secretion accompanied by a down-regulation of CaMKII activity.
Keywords: Bisindolylmaleimide, Ca2+/calmodulin-dependent protein kinase, calcium, carbachol, gastric acid secretion, muscarinic acetylcholine receptor, phorbol ester, protein kinase C, protein kinase inhibition, signal transduction
Introduction
Protein kinase C (PKC) comprises a family of multifunctional serine/threonine protein kinases which are ubiquitously distributed in a variety of eukaryotic cells. The PKC family is subdivided into three groups. The calcium- and diacylglycerol-dependent or ‘conventional' PKC-isoforms (cPKC) are activated by Ca2+ and diacylglycerol or the phorbol ester TPA (12-O-tetradecanoyl phorbol-13-acetate), a structural analogue of diacylglycerol. The ‘novel' PKC's (nPKC) are activated by diacylglycerol or TPA but not by Ca2+. Finally, there are the ‘atypical' PKC's (aPKC) which are not regulated by Ca2+ or diacylglycerol or TPA (for reviews, see Nishizuka, 1992; Newton, 1997). In the case of rabbit gastric parietal cells, PKC-α is the only member of cPKC which could be detected by immunoblot analysis, and PKC-ε the only member of nPKC, respectively (Nandi et al., 1996; Chew et al., 1997). There also appears to be at least two atypical PKC isoforms (ι, λ) expressed in parietal cells. The expression of PKC-ζ is uncertain, as reports are contradictory (Nandi et al., 1996; Chew et al., 1997). A related enzyme, PKC-μ or PKD, which is also expressed in rabbit parietal cells (Chew et al., 1997), displays multiple unique properties that makes it a distant relative of the PKC isozymes (Johannes et al., 1994).
cPKC is capable of differentiating between different time-courses of [Ca2+]I (concentration of free intracellular Ca(II)-ions) and diacylglycerol. The rapid activation of cPKC by calcium is followed by the binding of diacylglycerol which potentiates the binding affinity of cPKC with the plasma membrane, and prolongs the kinase activity even after a decrease of [Ca2+]i (Oancea & Meyer, 1998). Upon stimulation of non-excitable secretory cells, PKC translocates from the cytosol to the cell membrane to be activated and targeted to the proximity of its substrate (Akita et al., 1994; Bastani et al., 1995; Hong et al., 1997; Yedovitzky et al., 1997).
The involvement of PKC during H+ secretion has been extensively investigated especially by the use of the phorbol ester TPA to directly activate PKC (Anderson & Hanson, 1985; Muallem et al., 1986; Brown & Chew, 1987; Beil et al., 1987; Chiba et al., 1989; Nandi et al., 1996; Chew et al., 1997; Kopp & Pfeiffer, 2000). PKC inhibitors were also utilized to analyse the function of PKC in parietal cells (McKenna & Hanson, 1993; Chew et al., 1997).
However, the data with regard to the role of PKC are conflicting, as either a stimulatory or inhibitory function of PKC in cholinergic stimulation of acid secretion was observed (reviewed in Fährmann, 2000). Although some of those contradictory results may derive from the action of different PKC isoforms, the specific function of these isoforms in acid secretion has been rarely investigated. To show that different PKC-isoforms are effectively involved in gastric acid secretion we address the action of PKC-α and PKC-ε in response to the muscarinic agent carbachol (2-hydroxyethyl)trimethylammonium chloride carbamate) in gastric parietal cells. The M3 acetylcholine muscarinic receptor is coupled to heterotrimeric Gq and phospholipase C. Cholinergic stimulation increases both intracellular calcium by action of inositol 1,4,5-trisphosphate, and diacylglycerol (Brown & Chew, 1989; Pfeiffer et al., 1989; 1990; Seidler & Pfeiffer, 1991; Kajimura et al., 1992) as well as acid secretion. As yet, the understanding of the molecular mechanism how [Ca2+]i and diacylglycerol mediate H+ secretion is incomplete.
Methods
Chemicals
BAPTA/AM, (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl) ester), calmodulin, calyculin A, microcystin LR, Gö 6976 (12-(2-cyanoethyl)-6,7,12,13-tetrahydro - 13 - methyl - 5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole), KN-62 (1-[N, O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenyl-piperazine), Ro 31-8220, (3-[1-[3-(amidinothio)propyl - 1H - indol -3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide, bisindolylmaleimide IX), and TPA (12-O-tetradecanoyl phorbol-13-acetate) were obtained from Calbiochem-Novabiochem (Bad Soden, Germany). ATP containing 370 GBq mol−1 γ-[32P]-ATP was purchased from ICN (Meckenheim, Germany). [Dimethylamine-14C]-aminopyrine of 3.92 GBq mol−1 was from Amersham-Pharmacia Biotech (Freiburg, Germany). Leupeptin and pepstatin A were from Bachem (Heidelberg, Germany). Pefabloc SC was obtained from Roche (Strasbourg, France). All other fine chemicals were from Sigma (Deisenhofen, Germany) if not mentioned otherwise.
Cell isolation and culture of rabbit gastric parietal cells
Gastric parietal cells of male New Zealand white rabbits weighing 2–3 kg were isolated as recently described (Fährmann et al., 2002). High pressure perfusion of the stomach in situ was followed by scraping off the gastric mucosa. Parietal cells were isolated by pronase and collagenase digestion. Subsequently, parietal cells were enriched by centrifugal elutriation in a Beckman JM6-C centrifuge with a JE-5.0 elutriator rotor (Beckman, München, Germany). Final enrichment to >95% purity of parietal cells was achieved by centrifugation in a Nycodenz (Nycomed, Oslo, Norway) step gradient. Highly enriched parietal cells were suspended in culture medium (DMEM-Ham's F-12 medium containing 2 mg ml−1 BSA (bovine serum albumine), 800 nM insulin, 5 μg ml−1 transferrin, 5 ng ml−1 sodium selenite, 10 nM hydrocortisone, 8 nM EGF, 5 μg ml−1 geneticin, 50 μg ml−1 novobiocin, 100 μg ml−1 gentamicin, 10 μg ml−1 phenol red) to 5×106 cells ml−1, and seeded in 24-well plates coated with Matrigel (Becton Dickinson, Bedford, MA, U.S.A.). Parietal cells were allowed to attach and grow in a humidified atmosphere containing 5% (v v−1) CO2 at 37°C.
Estimation of acid secretion by [14C]-aminopyrine uptake
The secretion of H+ of cultured rabbit parietal cells was indirectly determined by accumulation of aminopyrine utilizing dimethylamine [14C]-aminopyrine as previously described (Fährmann et al., 2002). Parietal cells were incubated in oxygen-saturated buffer A (10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES, pH 7.4), plus (mM): NaCl 114.4, KCl 5.4, Na2HPO4 5, NaH2PO4 1, MgSO4 1.2, CaCl2 2, BSA 2 mg ml−1, glucose 10, DTT (dithiothreitol) 0.5, pyruvate 1) in the absence or presence of various concentrations of divers protein kinase inhibitors with subsequent stimulation by carbachol or TPA for 45 min. Incorporation of [14C] was measured in a β-scintillation counter after two-times washing, and detergent lysis of the cells. The basal acid secretion was determined in the presence of the H2-receptor blocker ranitidine (0.1 mM). The non-specific [14C]-aminopyrine accumulation was obtained by blocking acid-secretion with omeprazole (5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulphinyl]-1H-benzimidazole) in control assays (10 μM; AstraZeneca, Wedel, Germany). The nonspecific background was subtract from each specific value.
Preparation of apical membrane, tubulovesicles,and SA-vesicles
Preparation of the postnuclear supernatant, apical membrane, tubulovesicles, and SA (stimulus-associated)-vesicles of rabbit gastric mucosal cells was performed as described (Fährmann et al., 2002). For preparation of membranes of the resting state (apical membrane, tubulovesicles), male ‘New Zealand' white rabbits weighing 2–3 kg were fasted for 24 h, and had free access to drinking water containing ranitidine (1 mg l−1) offered for 24 h before killing, and intravenously (50 mg kg−1 h−1) 30 min before killing. For the isolation of SA-vesicles (secretory canaliculi) male ‘New Zealand' white rabbits weighing 2–3 kg received carbachol (30 nmol kg−1 h−1) for 30 min before killing.
Preparation of membrane fraction of parietal cells
Parietal cells (5×106 cells ml−1) were lysed by short-pulse sonication in buffer B (mM): HEPES 40, with 10 mM EGTA pH 7.4., EDTA 2, DTT 1, leupeptin 0.1, pepstatir A 1 μg ml−1, pefabloc SC 0.1, aprotinin 0.25. The lysate was separated in the cytosolic and the microsomal fraction by centrifugation at 100,000×g for 1 h at 4°C. The microsomal fraction was resuspended in buffer B containing 0.5% (v v−1) triton X-100. After incubation on ice for 30 min, the suspension was centrifuged at 100,000×g for 1 h at 4°C. The supernatant was used as the membrane fraction.
CaMKII phosphotransferase assay
Membrane preparations of gastric mucosal cells were monitored for CaMKII (Ca2+/calmodulin-dependent protein kinase II) phosphotransferase activity by using buffer C (mM): HEPES/NaOH 25 pH 7.4, MgCl2 10, DTT 1, leupeptin 0.1, aprotinin 0.1, pepstatin A 1 μg ml−1, EGTA 0.1, CaCl2 0.2, 10–100 μM adenosine-5′-triphosphate, containing 370 MBq mmol−1 adenosine-5′-[γ-32P]-triphosphate (ICN, Meckenheim, Germany). Calmodulin (1 μM) was added to the phosphorylation buffer to activate CaMKII activity. Autocamtide-II (KKALRRQETVDAL; Bachem, Heidelberg, Germany), a specific CaMKII substrate peptide, was used with a concentration of 60 μM. Phosphoprotein-phosphatases were inhibited in the presence of 0.05 μM calyculin A, and 0.1 μM microcystin LR.
Western blot analysis
All immunoblotting steps followed the protocol according to Fährmann et al. (1999). Membrane proteins were quantified by the method of Stoschek (1990). Equal amounts of membrane proteins (15 μg) of rabbit parietal cells were separated in 12.5% acrylamide/2.67% bisacrylamide (w v−1) SDS-gels, followed by a transfer onto a polyvinylidene difluoride membrane (Pierce, Rockford, IL, U.S.A.). PKC-α was probed with a monoclonal anti-(mouse/rat/human) PKC-α (code sc 8393), and PKC-ε by monoclonal anti-(mouse/rat/human) PKC-ε (code sc 1681). Both antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, U.S.A.). Rabbit polyclonal anti-active (rat) CaMKII against phosphorylated threonine of the autonomy site was from Promega, (Mannheim, Germany). Each antibody was incubated for 1 h. Antibody-binding was visualized by the ECL (enhanced chemiluminescence) detection plus system (Amersham Pharmacia Biotech). The immunolabelled CaMKII bands on ECL films were quantified by densitometry as described (Fährmann et al., 2002).
Data analysis
Data values are shown as means±standard deviance (s.d.) as σn. The statistical significance of differences was estimated by analysis of variance (ANOVA) followed by a post hoc Dunnett test. Differences were taken to be significant at P<0.05. Quantitation of the relative importance of signalling pathways was performed according to Krauss & Brand (2000).
Results
Carbachol-stimulated acid secretion is enhanced by the PKC inhibitors Gö 6976 or Ro 31-8220
The fact that TPA activates basal acid secretion but inhibits carbachol-induced H+ secretion, suggests that different PKC isoforms have a differential effect on acid secretion. To discriminate between the action of PKC-α and -ε, we compared the effects of two PKC inhibitors, the indolocarbazole Gö 6976 (Martiny-Baron et al., 1993) being selective for cPKC or in the case of rabbit parietal cells, for PKC-α, and the bisindolylmaleimide Ro 31-8220 (David et al., 1989) which inhibits both PKC-α and PKC-ε being 4.8-times more efficient to inhibit PKC-α.
Gö 6976 (10 μM) potentiated the carbachol stimulated acid secretion 1.4±0.1 fold (Figure 1, lanes 2 and 6). The inhibition of carbachol-induced aminopyrine uptake by TPA (1 μM; Figure 1, lane 4) was completely abolished by Gö 6976 (1 μM) (data not shown). Remarkably, in the presence of 10 μM Gö 6976 and TPA (1 μM) carbachol-induced secretory response was 1.8±0.2 fold stimulated (Figure 1, bar 8) compared to control with carbachol alone (Figure 1, bar 2). This is surprising, since stimulation of parietal cells with higher concentrations of carbachol than applied (0.1 mM) did not result in a significant rise of acid secretion (Kopp & Pfeiffer, 2000; Fährmann et al., 2002). The stimulatory effect of Gö 6976 was reversed by KN-62 (Hidaka & Yokokura, 1996), an inhibitor of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) (20 μM) (Tsunoda et al., 1992; Fährmann et al., 2002) (Figure 1, lanes 7 and 9). In sum, the enhanced stimulation of carbachol-evoked acid secretion by Gö 6976 or a combination of TPA and Gö 6976 reveals, first, a suppressant effect of PKC-α on carbachol-evoked H+ formation, and second, the involvement of another, H+ stimulatory PKC isoform.
Figure 1.
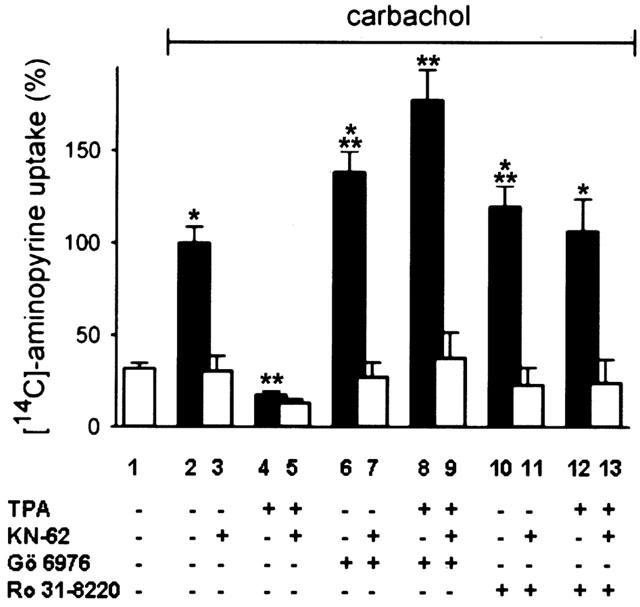
Effects of the PKC inhibitors Gö 6976 and Ro 31-8220 on carbachol-induced acid secretion. Parietal cells were pretreated with Gö 6976 (10 μM) or Ro 31–8220 (10 μM) for 15 min, then with TPA (1 μM) or the CaMKII-inhibitor KN-62 (20 μM, grey bars) or both for 15 min with subsequent stimulation of aminopyrine uptake by carbachol (0.1 mM). Basal aminopyrine-uptake which means in the absence of any secretagogue is represented by the white-filled bar. Results represent means±s.d. from 4–6 experiments. *Significantly different from the corresponding control group (+KN-62); **significantly different from carbachol alone, P<0.05.
Therefore, we tested the potent inhibitor of PKC-α and PKC-ε, Ro 31-8220 (David et al., 1989). Carbachol (0.1 mM)-induced aminopyrine uptake (Figure 1, lane 10) was 1.2±0.1 fold enhanced if parietal cells were pretreated with Ro 31-8220 (10 μM).
Furthermore, Ro 31-8220 (10 μM) antagonized the inhibitory effect of TPA (1 μM) on carbachol-stimulated acid formation (Figure 1, lane 12). Both stimulatory effects of Ro 31-8220 were reversed by KN-62 (20 μM) (Figure 1, lane 11 and 13). As control, the inhibition of carbachol-evoked aminopyrine uptake with KN-62 (20 μM) (Figure 1, lane 3) was in the range of basal acid secretion (Figure 1, bar 1). With TPA (1 μM) (Figure 1, bar 4), or both TPA and KN-62 (Figure 1, lane 5) the inhibition was below basal H+-secretion (Figure 1, bar 1).
The translocation of both PKC-α (Figure 2a) or PKC-ε (Figure 2b) to the membrane of parietal cells during carbachol-stimulation was detected with specific monoclonal antibodies for each PKC isoenzyme. The translocation of each PKC isoform was potentiated by TPA (1 μM). In the absence of carbachol TPA induced the translocation of PKC-ε (Figure 2b) but not of PKC-α (Figure 2a). The translocation of PKC-α appears to depend on the presence of free intracellular Ca2+ elicited by carbachol-stimulation, since we observed no significant translocation of PKC-α in the absence of carbachol even in the presence of TPA. This is consistent with current knowledge about PKC-α: It was shown that the translocation and, hence, activation of PKC-α depends on phospholipids, diacylglycerol, and calcium(II)ions (Nishizuka, 1992; Newton, 1997; Oancea & Meyer, 1998). TPA or other diacylglycerol analogues such as DiC8 alone efficiently induce translocation of cPKC only either in combination with Ca2+ or phospholipids, or at extremely high concentrations (Sakai et al., 1997; Shirai et al., 1998).
Figure 2.
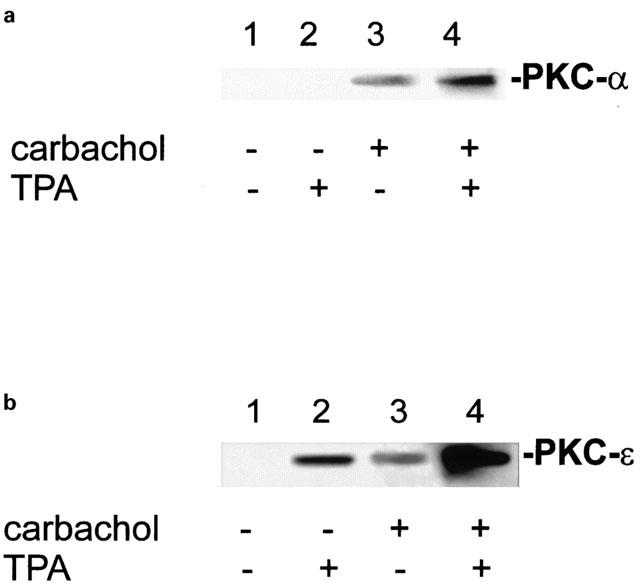
PKC-α and PKC-ε differ in translocation to the parietal cell membrane. Gastric parietal cells were incubated with TPA (1 μM) or carbachol (0.1 mM) or both for 15 min. After subfractionization of parietal cells equal amounts of protein (15 μg) of membrane fraction were subjected to SDS–PAGE and Western-blotting. PKC-α (a) and PKC-ε (b) were immunolabeled with each a specific monoclonal antibody. PKC-ε was translocated to the cell membrane even in the absence of carbachol. PKC-α showed no translocation to the parietal cell membrane in the absence of carbachol. Detection was performed with the ECL system.
Besides the qualitative identification of different protein kinases (PKC-α, PKC-ε, CaMKII), which are involved in carbachol-induced H+-secretion, we deduced the quantitative importance of these protein kinases in the signal transduction from the effects of the utilized protein kinase inhibitors according to Krauss & Brand (2000) (Figure 3). Quantitation of signal transduction of each kinase was determined at maximally achievable stimulation by carbachol (0.1 mM). The most important effect in carbachol-evoked acid secretion has CaMKII which contributes with ∼36% shown by the inhibition with KN-62. PKC-ε activates with ∼14% which was deduced from the effects of Ro 31-8220 and Gö 6976, respectively. About ∼22% of stimulation are unidentified. PKC-α suppresses about 28% of carbachol-induced acid secretion. Supposed the attenuation of PKC-α in acid secretion affects only CaMKII, its share in the signal transduction would be ∼64% if PKC-α activity is totally blocked. In our analyses we have only focussed on three different protein kinases but using other inhibitors may reveal unidentified signalling components in acid secretion. The future may provide more specific inhibitors to discriminate more precisely the action of protein kinases.
Figure 3.
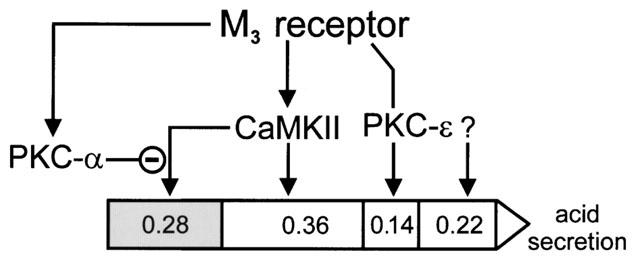
Quantitation of relative importance of signal transduction intermediates in acid secretion. Gastric acid secretion via the M3 muscarinic receptor is illustrated as arrow. The quantity of three different protein kinases (PKC-α, PKC-ε, CaMKII) in carbachol-stimulated H+ secretion and in the presence of Gö 6976 (10 μM) is deduced from inhibitor studies presented in Figure 1. About 36% of the signal is transmitted through CaMKII, and about 14% through PKC-ε in carbachol-induced acid secretion. About 22% are mediated by unidentified signalling components (question mark). PKC-α activity suppresses CaMKII activity, and attenuates about 28% of carbachol-stimulated acid secretion (grey section). Thus, if PKC-α activity is inhibited, CaMKII transduces about 64% (36+28%) of total signalling.
The inhibition of carbachol-stimulated acid secretion by TPA results in the inhibition of CaMKII activity
The key signalling protein kinase in cholinergic stimulation of H+ secretion is CaMKII (Tsunoda et al., 1992; Mayer et al., 1994; Fährmann et al., 1998; 1999; 2002; Fährmann & Pfeiffer, 1999; 2000). Carbachol-stimulation causes a doubling of CaMKII activity (Figure 4a), as well as a strong increase of autoactivated CaMKII in the secretory apical membrane of rabbit gastric mucosal cells (Figure 4b). Since carbachol-stimulated acid secretion is accompanied by an activation of CaMKII phosphotransferase activity, we examined the possibility that during inhibition of carbachol-induced H+ secretion by TPA, CaMKII was less or not activated. Cultured parietal cells were preincubated with different kinase modulators (TPA, Gö 6976, KN-62, Ro 31-8220), stimulated by carbachol (0.1 mM), lysed and subfractionized. Parietal cell membrane proteins were separated by SDS–PAGE and blotted. A phosphospecific anti-active CaMKII antibody directed against the phosphorylated autonomy site of CaMKII was used to detect activated CaMKII. Phosphorylation of the autonomy site indicates activated CaMKII (for review, Soderling et al., 2001). In parietal cells, carbachol induced the activation of CaMKII (Figure 5a, lane 2), an effect that was antagonized by the CaMKII inhibitor KN-62 (20 μM) (Figure 5a, lane 7), or TPA (1 μM) (Figure 5a, lane 8). Notably, TPA (1 μM) exerted a moderate activation of CaMKII in basal secreting cells (Figure 5b, lane 2) which was reversed by KN-62 (20 μM) (Figure 5b, lane 8). The inhibition of carbachol-induced CaMKII activity by TPA was antagonized by either Gö 6976 (Figure 5a, lane 4) or by Ro 31-8220 (Figure 5b, lane 6). Ro 31-8220 (10 μM) had no significant effect on CaMKII activity in the presence of carbachol (Figure 5b, lane 5 compared to lane 3) but inhibited the TPA-induced activation of CaMKII (Figure 5b, lane 7 compared to lane 2). Unstimulated parietal cells contained no activated CaMKII (Figure 5a,b, each lane 1).
Figure 4.
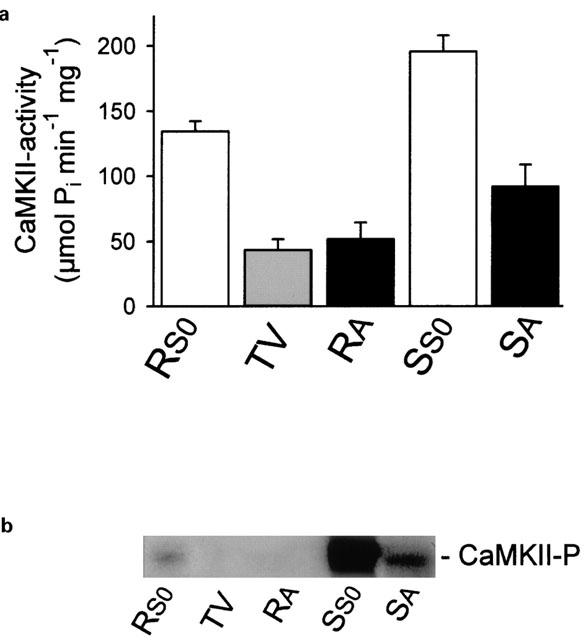
The abundance of CaMKII in SA vesicles doubled after carbachol-stimulation whereas the amount of autoactivated CaMKII was strongly increased. (a) The specific CaMKII transphosphorylating activity of SA vesicles was 2 fold increased after carbachol-stimulation compared to the apical membrane of the resting state. Apical membranes of resting or carbachol-stimulated rabbit gastric mucosal cells were incubated under CaMKII phosphorylation conditions to phosphorylate the CaMKII-specific substrate autocamtide-II. (b) A strong increase of autoactivated CaMKII (CaMKII-P) was detected for the postnuclear supernatant as well as for the SA vesicles after carbachol-stimulation compared to each corresponding membrane fraction of the resting state. Autoactivated CaMKII was probed with a phospho-specific, anti-autoactivated CaMKII. The post nuclear supernatant (S0) represents almost the total of cellular CaMKII activity; (RS0: postnuclear supernatant of the resting state, TV: tubulovesicles, RA: apical membrane of the resting state, SS0: postnuclear supernatant of the carbachol-stimulated state, SA: SA vesicles).
Figure 5.
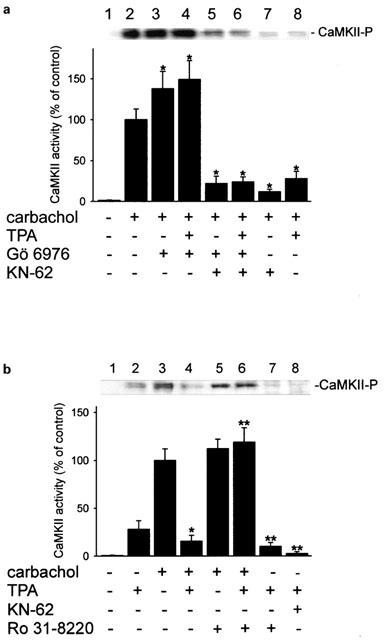
Effects of Gö 6976 and Ro 31-8220 on the activity of CaMKII in carbachol-stimulated parietal cells. Active CaMKII (CaMKII-P) was detected in membrane fraction with a phospho-specific antibody by immunoblot analysis (each upper panel), and quantified by densitometry (each lower panel). Parietal cells were pretreated with either Gö 6976 (10 μM) (a), or Ro 31-8220 (10 μM) (b), for 15 min. Cells were then incubated with TPA (1 μM) or KN-62 (20 μM) or both for 15 min, and optionally stimulated with carbachol (0.1 mM). Equal amounts of cell membrane proteins (15 μg) per lane were subjected to SDS-gel electrophoresis with subsequent Western blotting. Results represent means±s.d. from three experiments. *Significantly different from carbachol alone; **significantly different from TPA alone, P<0.05.
Low concentrations of TPA induces acid secretion in resting parietal cells which was blocked by Ro 31-8220 but not by Gö 6976
The phorbol ester TPA is known to inhibit carbachol-evoked acid secretion. However, a low concentration of TPA stimulated basal acid secretion up to 2.4 fold at 10 pM (Figure 6a,b). Higher concentrations than 10 pM TPA were less efficient to stimulate acid formation. An inhibition of basal acid secretion was observed at 0.1 μM TPA but not at 1 μM (Figure 6a,b). No significant effect of Gö 6976 (10 μM) was observed for TPA-induced acid secretion except at a concentration of 0.1 μM TPA at which the basal H+ secretion was antagonized (Figure 6a). In contrast, the TPA-dependent increase of H+ secretion was totally abolished by Ro 31-8220 (Figure 6a). These effects of Gö 6976 and Ro 31-8220 suggest that the TPA-stimulated H+ secretion is conveyed by PKC-ε but not PKC-α. The TPA-evoked H+ secretion was also blocked when cells were preincubated with the cell-permeable Ca2+-chelator BAPTA/AM (0.1 mM) showing an involvement of [Ca2+]i (concentration of free intracellular Ca(II)-ions) (Figure 6b). KN-62 (20 μM) also efficiently blocked the TPA-stimulated H+ secretion, and indicates the involvement of CaMKII (Figure 6b). This suggestion was addressed by additional experiments.
Figure 6.
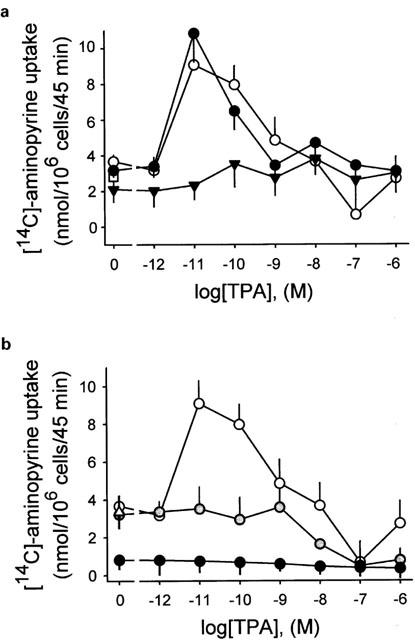
Effects of protein kinase inhibitors on TPA-induced acid secretion. Parietal cells were dose-dependently treated with TPA (0–0.1 μM) resulting in a biphasical course of [14C]-aminopyrine uptake (white-filled points). (a) TPA-stimulated acid secretion was abolished by Ro 31-8220 (10 μM; black triangles) but not by Gö 6976 (10 μM; black points). (b) TPA-induced acid secretion (white-filled points) was abolished by KN-62, a potent, cell-permeable inhibitor of the calcium-sensitive CaMKII at 20 μM (gray-filled points), and the cell-permeable calcium-chelator BAPTA/AM (0.1 mM; black points), respectively. Results are expressed means±s.d. from four experiments.
The calcium-sensor CaMKII is activated duringTPA-induced acid secretion
The above described results suggest a role for CaMKII in the TPA-evoked [14C]-aminopyrine accumulation. Therefore, experiments were carried out to determine if TPA-induced acid secretion is indeed mediated by activation of CaMKII. The most efficient activation of CaMKII in parietal cells was achieved in the presence of 10 pM TPA, and was about 60% of activity as in carbachol (0.1 mM)-induced acid secretion (Figure 7a). Remarkably, higher concentrations of TPA resulted in a reduced response of CaMKII activity. At 0.1 μM TPA, no CaMKII activity was detectable but at 1 μM TPA a moderate increase of CaMKII activity was obtained (Figure 7a). The moderate CaMKII activity at 1 μM TPA was 2.1±0.5 fold increased by Gö 6976 (10 μM), and 1.1±0.2 fold augmented by Ro 31-8220 (10 μM) (Figure 7b). The effect of Gö 6976 suggests that PKC-α suppressed TPA (1 μM)-stimulated acid formation by inhibition of CaMKII activity (Figure 7b, lane 4). The specificity of CaMKII reaction was shown by inhibition with KN-62 (20 μM) (Figure 7b, lane 6). The effect of Ro 31-220 implicates a more complex regulation involving both PKC-α and -ε. As we have recently demonstrated for submaximal stimulation of acid secretion by carbachol (<1 μM) the inhibition by KN-62 is relatively less efficient than at higher concentrations of carbachol (>1 μM) (Fährmann et al., 2002). In the case of TPA (1 μM)-treated parietal cells (Figure 6b, Figure 7b, lanes 3) inhibition of acid secretion and CaMKII, respectively, by KN-62 was not as efficient as after carbachol (0.1 mM)-stimulation (Figure 7c, lane 9; Fährmann et al., 2002).
Figure 7.
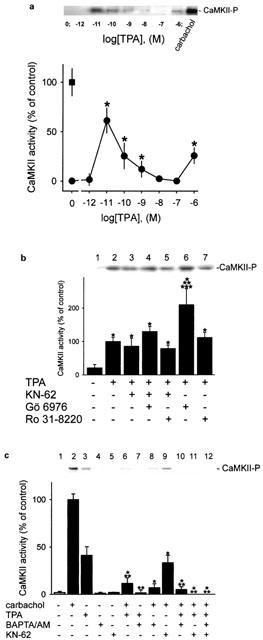
CaMKII is activated when resting parietal cells are treated with TPA. After treatment of parietal cells with TPA (0–1 μM), activated CaMKII (CaMKII-P) was detected in the membrane by immunoblot analysis (each upper panel). Bands of activated CaMKII were quantified by densitometry (each lower panel). Results are expressed as means±s.d. from 3–5 experiments. Absence of any chemical agent corresponds with basal acid secretion. (a) Resting parietal cells were treated with various concentrations of TPA (0–1 μM), and analysed for active CaMKII (black points). Controls were stimulated with carbachol (0.1 mM; black rectangle). *Significantly different from the corresponding control group (absence of any secretagogue or TPA), P<0.05. (b) Parietal cells were incubated with different protein kinase modulators (Gö 6976 (10 μM), KN-62 (20 μM), Ro 31-8220 (10 μM)) for 15 min with subsequent treatment by TPA (1 μM) for 30 min. *Significantly different from control (absence of TPA); **significantly different from TPA alone; ***significantly different from TPA+Gö 6976+KN-62, P<0.05. (c) Cells were pretreated with BAPTA/AM (0.1 mM) or KN-62 (20 μM) for 15 min, then incubated with carbachol (0.1 mM) or TPA (1 μM) or both for 30 min. *Significantly different from carbachol alone; **significantly different from TPA alone, P<0.01.
Pretreatment of parietal cells with BAPTA/AM (0.1 mM) effectively inhibited the activation of CaMKII by TPA (1 μM) (Figure 7c, lane 7) indicating that TPA induced the elevation of [Ca2+]i. The use of BAPTA/AM also showed that the carbachol-induced increase of CaMKII activity totally depends on [Ca2+]i (Figure 7c, lane 10). The inhibition of carbachol-stimulated acid by chelating intracellular Ca2+ with BAPTA/AM was recently demonstrated (Brown & Chew, 1989).
Discussion
The results in this study demonstrate that cholinergically-stimulated gastric acid secretion involves both PKC isozymes α and ε. PKC-α has an inhibitory, PKC-ε a stimulatory effect on carbachol-induced acid secretion. A number of studies have examined the role of PKC in gastric acid secretion with sometimes conflicting results. It was reported that TPA inhibits carbachol-stimulated acid secretion (Anderson & Hanson, 1985; Muallem et al., 1986; Brown & Chew, 1987; Beil et al., 1987; Chiba et al., 1989; Nandi et al., 1996). Brown & Chew (1987) described the effect of TPA on carbachol-evoked H+ secretion in rabbit gastric glands as biphasic with an early enhanced response and subsequent inhibition. Kopp & Pfeiffer (2000) showed a biphasic course of acid secretion for rat parietal cells with an increment at 5 nM TPA, and a decrease of acid secretion at 100 nM. In the same study it was shown that TPA inhibits the carbachol-evoked formation of inositol monophosphate which was used as an indicator of inositol trisphosphate production (Kopp & Pfeiffer, 2000). This effect was only observed at concentrations of TPA >5 nM. However, we determined a clear inhibition of carbachol-induced acid secretion even at concentrations below 1 nM (data not shown). Hence, the PKC-dependent down-regulation of inositol 1,4,5-trisphosphate seems to be only one mechanism in the PKC-induced attenuation of H+-secretion. The use of the diacylglycerol kinase inhibitor RHC 80267 causes an inhibition of carbachol-induced H+ secretion (Pfeiffer et al., 1989), and supports the assumption that the intracellular target of TPA is PKC. Additionally, an important property of PKC is its activation by translocation to the plasma membrane. Translocation of PKC on stimulation was observed for a variety of non-excitable secretory cells (Akita et al., 1994; Bastani et al., 1995; Hong et al., 1997; Yedovitzky et al., 1997). In fact, both PKC-α and PKC-ε translocated to the parietal cell membrane upon carbachol-stimulation, and, therefore, were activated during acid secretion.
We utilized a pharmacological approach to discriminate between the actions of PKC-α and -ε, respectively, on the H+ secretory response. Both Gö 6976 and Ro 31-8220 antagonized the inhibition by TPA of carbachol-induced acid secretion whereas only Gö 6976 enhanced carbachol-stimulated acid formation. Since Ro 31-8220 inhibits both PKC-α and -ε, and Gö 6976 only PKC-α in rabbit parietal cells, this likely reflects the attenuation of cholinergic acid secretion by PKC-α, and the augmentation by PKC-ε.
The main intracellular regulator of carbachol-induced H+ secretion appears to be CaMKII (Fährmann et al., 2002). CaMKII is abundant in gastric mucosa as holoenzyme (Fährmann et al., 1998; Fährmann & Pfeiffer, 2000). Carbachol-stimulation both induces translocation of CaMKII to the apical membrane of rat mucosal cells as well as a strong activation of CaMKII phosphotransferase activity (Fährmann et al., 2002). In this report we show similar findings for rabbit mucosal cells resulting in an about 10% higher activity of apically located CaMKII after carbachol-stimulation than in the case of rat mucosal cells (Fährmann et al., 2002). Recently, we suggested that PKC-α suppresses the activity of CaMKII by direct transphosphorylation (Fährmann & Pfeiffer, 1999). As carbachol induces both the localization of PKC-α and CaMKII to the parietal cell membrane the spatial, intracellular conditions for interaction of both kinases are given. In fact, the intracellular activity of CaMKII was increased in the same order of magnitude as the acid secretion when Gö 6976 and carbachol were used. The inhibition of carbachol-evoked acid release by TPA was accompanied by the inhibition CaMKII activity. Both inhibitory effects of TPA on carbachol-induced H+ secretion as well as on CaMKII activity were abolished by either Gö 6976 or Ro 31-8220. The use of Gö 6976 reveals that about two-third of carbachol-induced H+ secretion are conveyed by CaMKII. However, approximately half of the CaMKII activity is suppressed by PKC-α. In contrast, only one-eighth for carbachol-evoked acid release is mediated by PKC-ε.
Another study discussed that Gö 6976 had no effect on carbachol-induced acid secretion of gastric glands, but the potentitation of carbachol-induced acid secretion by Ro 31-8220 was observed (Chew et al., 1997). There are several differences in the morphology and the physiology between freshly isolated gastric glands and cultured parietal cells which makes it difficult to compare pharmacological effects. It was also reported that Ro 31-8220 inhibited carbachol-stimulated acid formation (McKenna & Hanson, 1993). Since a suspension of unenriched parietal cells was used there may have been paracrine effects of accompanying endocrine cells of the gastric gland which were also influenced by Ro 31-8220.
To understand the role of PKC in acid secretion it is also valuable to study basal acid secretion, i.e. in the absence of any secretagogue. Previous studies have shown that TPA stimulates basal acid secretion (Brown & Chew, 1987; Hatt & Hanson, 1989), a finding which was confirmed in the present study. Remarkably, we observed that the TPA-induced acid secretion exhibited a biphasic concentration-dependence. The highest stimulation of acid secretion occurred at relatively low concentrations (10–100 pM TPA) and, interestingly, coincided with the highest TPA-dependent activation of CaMKII. Since CaMKII activity as well as acid secretion depend on free intracellular calcium, a PKC-dependent elevation of [Ca2+]i is implicated. The clear inhibition of TPA-potentiated basal acid secretion by KN-62 shows that the Ca2+-dependent form of CaMKII was activated since KN-62 cannot inhibit the Ca2+-independent form (Hidaka & Yokokura, 1996). A direct activation of CaMKII activity by TPA is neither known nor suggested by our data as CaMKII is not concentration-dependently activated by TPA. Rather, we assume that TPA activates PKC-ε to increase an at least initial increase of [Ca2+]i, resulting in an activation of CaMKII via calmodulin. This hypothesis was confirmed by abolishing both the activation of CaMKII and TPA-induced H+ secretion in the presence of the Ca2+-chelator BAPTA/AM. As in the case of carbachol-stimulated acid secretion, CaMKII appears also to be an important regulator in TPA-stimulated acid secretion.
TPA has been shown to induce an increase of [Ca2+]i and cell function in excitable (Redman et al., 1997), and non-excitable cells (Baranska et al., 1995). Although the precise mechanism of TPA action is unknown a direct regulation of calcium channels has been excluded (Vitale et al., 1995; Redman et al., 1997). Rather, a PKC-mediated change in the cytoskeleton architecture is assumed to regulate [Ca2+]i (Baranska et al., 1995). It is known that the actin cytoskeleton in non-excitable cells is implicated in vesicle trafficking, docking and fusion (Muallem et al., 1995; Vitale et al., 1995). The depolymerization of filamentous actin (F-actin) at the apical membrane triggers exocytosis (Muallem et al., 1995). Treatment of TPA disrupts the F-actin of the apical domain, presumably by activating PKC (Vitale et al., 1995; Muallem et al., 1995), and increased the secretion of pancreatic acinar cells (Muallem et al., 1995). It is speculated that PKC-ε is involved in the disassembly of F-actin for exocytosis in neuronal cells (Saitoh et al., 2001). Previously, it was demonstrated that PKC-ε is the only PKC-isoform which carries an actin-binding sequence (Prekeris et al., 1998). In fact, PKC-ε can directly bind to F-actin which retains PKC-ε in the catalytically active conformation (Prekeris et al., 1998). Gastric parietal cells are rich of actin: About 5% of total protein in parietal cells is presented by actin with about 90% as F-actin (Forte et al., 1998). Actin is found in the apical domain of parietal cells, and may be involved in the secretory exocytosis of acid (Yao et al., 1995). PKC-ε was observed to be colocalized with F-actin in rabbit parietal cells (Chew et al., 1997).
Higher concentrations of TPA than 0.1 nM reversed both stimulatory effects on acid secretion as well as on CaMKII activity. The stimulation of basal acid secretion by TPA was sensitive to Ro 31-8220 but not to Gö 6976. These data strongly corroborate the involvement of PKC-ε but not of PKC-α in TPA-evoked acid secretion as Ro 31-8220 would also inhibit activity of PKC-α. At excessive concentrations of TPA (>0.1 μM) an inhibitory function of PCK-α on both acid secretion and CaMKII activity is indicated. Further investigation of the PKC-dependent signalling in parietal cells is likely to enhance our understanding how acid secretion is regulated.
In summary, our results indicate that the activity of the PKC isozymes α and ε in acid secretion are coupled to the M3 muscarinic receptor, and that in the case of basal acid secretion at least PKC-ε is active. The ε isoform facilitates whereas the α isoform attenuates acid secretion. The consequences of this dual regulatory mechanism for parietal cell signalling is currently unknown. Moreover, we showed that CaMKII activity is essential for acid secretion which is either induced by carbachol or TPA.
Abbreviations
- BAPTA/AM
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl) ester
- BSA
bovine serum albumine
- [Ca2+]i
concentration of free intracellular Ca(II)-ions, CaMKII, Ca2+/calmodulin-dependent protein kinase II
- carbachol
(2-hydroxyethyl)trimethylammonium chloride carbamate
- DTT
dithiothreitol
- ECL
enhanced chemiluminescence
- Gö 6976
12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole
- KN-62
1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenyl-piperazine
- omeprazole
5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulphinyl-1H-benzimidazole
- PKC
protein kinase C (Ca2+-sensitive, phospholipid-dependent protein kinase)
- Ro 31-8220
3-[1-[3-(amidinothio)propyl-1H-indol-3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide
- SA
stimulus-associated
- TPA
12-O-tetradecanoyl phorbol-13-acetate
References
- AKITA Y., OHNO S., YAJIMA Y., KONNO Y., SAIDO T.C., MIZUNO K., CHIDA K., OSADA S., KUROKI T., KAWASHIMA S., SUZUKI K. Overproduction of a Ca(2+)-independent protein kinase C isozyme, nPKC epsilon, increases the secretion of prolactin from thyrotropin-releasing hormone-stimulated rat pituitary GH4C1 cells. J. Biol. Chem. 1994;269:4653–4660. [PubMed] [Google Scholar]
- ANDERSON N.G., HANSON P.J. Involvement of calcium-sensitive phospholipid-dependent protein kinase in control of acid secretion by isolated rat parietal cells. Biochem. J. 1985;232:609–611. doi: 10.1042/bj2320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARANSKA J., CHABAN V., CZARNY M., SABALA P. Changes in Ca2+ concentration in phorbolester and thapsigargin treated glioma C6 cells. The role of protein kinase C in regulation of Ca2+ entry. Cell Calcium. 1995;17:207–215. doi: 10.1016/0143-4160(95)90035-7. [DOI] [PubMed] [Google Scholar]
- BASTANI B., YANG L., BADASSASARE J.J., POLLO D.A., GARDNER J.D. Cellular distribution of isoforms of protein kinase C (PKC) in pancreatic acini. Biochim. Biophys. Acta. 1995;1269:307–315. doi: 10.1016/0167-4889(95)00120-0. [DOI] [PubMed] [Google Scholar]
- BEIL W., MANNSCHEDEL W., SEWING K.F. Protein kinase C and parietal cell function. Biochem. Biophys. Res. Commun. 1987;149:720–728. doi: 10.1016/0006-291x(87)90427-x. [DOI] [PubMed] [Google Scholar]
- BROWN M.R., CHEW C.S. Multiple effects of phorbolester on secretory activity in rabbit gastric glands and parietal cells. Can. J. Physiol. Pharmacol. 1987;65:1840–1847. doi: 10.1139/y87-286. [DOI] [PubMed] [Google Scholar]
- BROWN M.R., CHEW C.S. Carbachol-induced protein phosphorylation in parietal cells: regulation by (Ca2+)i. Am. J. Physiol. 1989;257:G99–G110. doi: 10.1152/ajpgi.1989.257.1.G99. [DOI] [PubMed] [Google Scholar]
- CHEW C.S., ZHOU C.-J., PARENTE J.A., JR. Ca2+-independent protein kinase C isoforms may modulate parietal cell HCl secretion. Am. J. Physiol. 1997;272:G246–G256. doi: 10.1152/ajpgi.1997.272.2.G246. [DOI] [PubMed] [Google Scholar]
- CHIBA T., FISHER S.K., AGRANOFF B.W., YAMADA T. Autoregulation of muscarinic and gastrin receptors on gastric parietal cells. Am. J. Physiol. 1989;256:G356–G363. doi: 10.1152/ajpgi.1989.256.2.G356. [DOI] [PubMed] [Google Scholar]
- DAVID P.D., HILL C.H., KEECH E., LAWTON G., NIXON J.S., SEDGWICK A.D., WADSWORTH J., WESTMACOTT D., WILKINSON S.E. Potent selective inhibitors of protein kinase C. FEBS Lett. 1989;259:61–63. doi: 10.1016/0014-5793(89)81494-2. [DOI] [PubMed] [Google Scholar]
- FÄHRMANN M. The role of protein kinase C in the acid secretion of mammalian gastric parietal cells. Trends Comp. Biochem. Physiol. 2000;6:139–150. [Google Scholar]
- FÄHRMANN M., PFEIFFER A. Protein kinase C modulates peripheral calcium/calmodulin-dependent protein kinase II activity by direct phosphorylation. Eur. J. Cell Biol. 1999;78:57–. [Google Scholar]
- FÄHRMANN M., PFEIFFER A. Co-purification of two holoenzyme-forming calcium/calmodulin-dependent protein kinase II isoforms as holoenzyme from porcine stomach. Arch. Biochem. Biophys. 2000;380:151–158. doi: 10.1006/abbi.2000.1910. [DOI] [PubMed] [Google Scholar]
- FÄHRMANN M., HEINZMANN A., SEIDLER U. CaMKII is activated and translocated to the secretory apical membrane during cholinergically conveyed gastric acid secretion. Cell. Signal. 2002;14:161–168. doi: 10.1016/s0898-6568(01)00231-5. [DOI] [PubMed] [Google Scholar]
- FÄHRMANN M., JACOB P., SEIDLER U., OSTERHOFF M., MÖHLIG M., PFEIFFER A. Ca2+/calmodulin-dependent protein kinase II isoenzymes γ and δ are both present in H+/K+-ATPase containing rabbit gastric tubulovesicles. Eur. J. Biochem. 1999;266:1036–1042. doi: 10.1046/j.1432-1327.1999.00959.x. [DOI] [PubMed] [Google Scholar]
- FÄHRMANN M., MÖHLIG M., SCHATZ H., PFEIFFER A. Purification and characterization of a Ca2+/calmodulin-dependent protein kinase II from hog gastric mucosa using a protein-protein affinity chromatographic technique. Eur. J. Biochem. 1998;255:516–525. doi: 10.1046/j.1432-1327.1998.2550516.x. [DOI] [PubMed] [Google Scholar]
- FORTE J.G., LY B., RONG Q., OGIHARA S., RAMILO M., AGNEW B., YAO X. State of actin in gastric parietal cells. Am. J. Physiol. 1998;274:C97–C104. doi: 10.1152/ajpcell.1998.274.1.C97. [DOI] [PubMed] [Google Scholar]
- HATT J.F., HANSON P.J. Sites of action of protein kinase C on secretory activity in rat parietal cells. Am. J. Physiol. 1989;256:G129–G128. doi: 10.1152/ajpgi.1989.256.1.G129. [DOI] [PubMed] [Google Scholar]
- HIDAKA H., YOKOKURA H. Molecular and cellular pharmacology of a calcium/calmodulin-dependent protein kinase II (CaM kinase II) inhibitor, KN-62, & proposal of CaM kinase phosphorylation cascades. Adv. Pharmacol. 1996;36:193–219. doi: 10.1016/s1054-3589(08)60583-9. [DOI] [PubMed] [Google Scholar]
- HONG D.H., FORSTNER J.F., FORSTNER G.G. Protein kinase C-epsilon is the likely mediator of mucin exocytosis in human colonic cell lines. Am. J. Physiol. 1997;272:G31–G37. doi: 10.1152/ajpgi.1997.272.1.G31. [DOI] [PubMed] [Google Scholar]
- JOHANNES F.J., PRESTLE J., EIS S., OBERHAGEMANN P., PFIZENMAIER K. PKCμ is a novel, atypical member of the protein kinase C family. J. Biol. Chem. 1994;269:6140–6148. [PubMed] [Google Scholar]
- KAJIMURA M., REUBEN M.A., SACHS G. The muscarinic receptor gene expressed in rabbit, parital cells is the m3 subtype. Gastroenterology. 1992;103:870–875. doi: 10.1016/0016-5085(92)90019-u. [DOI] [PubMed] [Google Scholar]
- KOPP R., PFEIFFER A. Effects of phorbolester treatment on dibutyryl cyclic adenosine-5′monophosphate- and carbachol-stimulated aminopyrine accumulation in isolated rat parietal cells. Scand. J. Gastroenterol. 2000;35:686–693. doi: 10.1080/003655200750023336. [DOI] [PubMed] [Google Scholar]
- KRAUSS S., BRAND M.D. Quantitation of signal transduction. FASEB J. 2000;14:2581–2588. doi: 10.1096/fj.00-0064com. [DOI] [PubMed] [Google Scholar]
- MARTINY-BARON G., KAZANIETZ M.G., MISCHAK H., BLUMBERG P.M., KOCHS G., HUG H., MARMÉ D., SCHÄCHTELE C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J. Biol. Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- MAYER P., MÖHLIG M., SEIDLER U., ROCHLITZ H., FÄHRMANN M., SCHATZ H., HIDAKA H., PFEIFFER A. Characterization of γ- and δ-subunits of Ca2+/calmodulin-dependent protein kinase II in rat mucosal cell populations. Biochem. J. 1994;297:157–162. doi: 10.1042/bj2970157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKENNA J.P., HANSON P.J. Inhibition by Ro 31-8220 of acid secretory activity induced by carbachol indicates a stimulatory role for protein kinase C in the action of muscarinic agonists on isolated rat parietal cells. Biochem. Pharmacol. 1993;46:583–588. doi: 10.1016/0006-2952(93)90541-4. [DOI] [PubMed] [Google Scholar]
- MUALLEM S., FIMMEL C.J., PANDOL S.J., SACHS G. Regulation of free cytosolic Ca2+ in the peptic and parietal cells of rabbit gastric glands. J. Biol. Chem. 1986;261:2660–2667. [PubMed] [Google Scholar]
- MUALLEM S., KWIATKOWSKA K., XU X., YIN H.L. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J. Cell Biol. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NANDI J., BOSCHE M.C., LEVINE R.A. Effects of a phorbol ester and isoquinoline sulfonamides on rabbit parietal cell function. J. Pharm. Exp. Therap. 1996;279:97–105. [PubMed] [Google Scholar]
- NEWTON A.C. Regulation of protein kinase C. Curr. Opin. Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- NISHIZUKA Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- OANCEA E., MEYER T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- PFEIFFER A., KOPP R., ROCHLITZ H. Stimulation of inositol phosphate and diacylglycerol production by RHC 80267, a diacylglycerol-lipase inhibitor, in gastric parietal cells: effect on hydrogen ion secretion. Biochem. Biophys. Acta. 1989;1001:191–195. doi: 10.1016/0005-2760(89)90147-1. [DOI] [PubMed] [Google Scholar]
- PFEIFFER A., ROCHLITZ H., NOELKE B., TACKE R., MOSER U., MUTSCHLER E., LAMBRECHT G. Muscarinic receptors mediating acid secretion in isolated rat gastric parietal cells are of M3 type. Gastroenterology. 1990;98:218–222. doi: 10.1016/0016-5085(90)91314-v. [DOI] [PubMed] [Google Scholar]
- PREKERIS R., HERNANDEZ R.M., MAYHEW M.W., WHITE M.K., TERRIAN D.M. Molecular analysis of the interactions between protein kinase C-ε and filamentous actin. J. Biol. Chem. 1998;273:26790–26798. doi: 10.1074/jbc.273.41.26790. [DOI] [PubMed] [Google Scholar]
- REDMAN R.S., SEARL T.J., HIRSH J.K., SILINSKY E.M. Opposing effects of phorbolesters on transmitter release and calcium currents at frog motor nerve endings. J. Physiol. 1997;501:41–48. doi: 10.1111/j.1469-7793.1997.041bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAITOH N., HORI T., TAKAHASHI T. Activation of the epsilon isoform of protein kinase C in the mammalian nerve terminal. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14017–14021. doi: 10.1073/pnas.241333598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAKAI N., SASAKI K., IKEGAKI N., SHIRAI Y., ONO Y., SAITOH N. J. Cell Biol. 1997. pp. 1465–1476. [DOI] [PMC free article] [PubMed]
- SEIDLER U., PFEIFFER A. Formation of polyphospho-inositides and intracellular calcium-changes in rabbit gastric mucous cells during secretagogue-induced stimulation. Am. J. Physiol. 1991;260:G133–G141. doi: 10.1152/ajpgi.1991.260.1.G133. [DOI] [PubMed] [Google Scholar]
- SHIRAI Y., KASHIWAGI K., YAGI K., SAKAI N., SAITO N. Distnct effects of fatty acids on translocation of γ- and ε-subspecies of protein kinase C. J. Cell Biol. 1998;143:511–521. doi: 10.1083/jcb.143.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SODERLING T.R., CHANG B., BRICKEY D. Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 2001;276:3719–3722. doi: 10.1074/jbc.R000013200. [DOI] [PubMed] [Google Scholar]
- STOSCHEK C.M. Increased uniformity in the response of the coomassie blue G protein assay to different proteins. Anal. Biochem. 1990;184:111–116. doi: 10.1016/0003-2697(90)90021-z. [DOI] [PubMed] [Google Scholar]
- TSUNODA Y., FUNASAKA M., MODLIN I.M., HIDAKA H., FOX L.M., GOLDENRING J.R. An inhibitor of Ca2+/calmodulin-dependent protein kinase II, KN- 62, inhibits cholinergic-stimulated parietal cell secretion. Am. J. Physiol. 1992;262:G118–G122. doi: 10.1152/ajpgi.1992.262.1.G118. [DOI] [PubMed] [Google Scholar]
- VITALE M.L., SEWARD E.P., TRIFARO J.M. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron. 1995;14:353–363. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- YAO X., CHAPONNIER C., GABBIANI G., FORTE J.G. Polarized distribution of actin isoforms in gastric parietal cells. Mol. Biol. Cell. 1995;6:541–557. doi: 10.1091/mbc.6.5.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YEDOVITZKY M., MOCHLY-ROSEN D., JOHNSON J.A., GRAY M.O., RON D., ABRAMOVITCH E., CERASI E., NESHER R. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic β-cells. J. Biol. Chem. 1997;272:1417–1420. doi: 10.1074/jbc.272.3.1417. [DOI] [PubMed] [Google Scholar]