

Abstract
We have investigated pre- and post-junctional responsiveness in vas deferens from wild-type and α2A/D-adrenoceptor knockout mice. The response to a single stimulus was not significantly different between wild-type and knock-out mice. The isometric contraction to 10 Hz stimulation for 4 s was significantly larger in vas deferens from knockout as compared with wild-type.
The maximum potentiation of 10 Hz stimulation-evoked contractions by yohimbine was to 206.2±38.0% of control in wild-type but to 135.8±13.6% of control in knockout. The α2A/D-adrenoceptor selective antagonist BRL 44408 significantly increased the 10 Hz stimulation-evoked contraction in wild-type but not knockout, and the reverse was true for the α2C-adrenoceptor selective antagonist spiroxatrine. The α2B-adrenoceptor antagonist imiloxan had no effect on the evoked contraction except at high concentrations, and only in wild-type. Following cocaine (3 μM) and BRL 44408 (1 μM), 10 Hz responses were similar in shape and maximum between wild-type and knock-out.
The α2-adrenoceptor agonist xylazine virtually abolished the early component of the contraction to 10 Hz stimulation in the presence of nifedipine (10 μM) in vas deferens from knockout mice in a way consistent with a change of receptor subtype but without clear evidence for a reduced receptor number. However, the late component of the contraction to 10 Hz stimulation was significantly potentiated by xylazine in tissues from knock-out mice.
It is concluded that, although non-α2A/D-adrenoceptors replace α2D-adrenoceptors in this knockout, the α2-adrenoceptor agonist and antagonist data are contradictory. The antagonist data suggest a major loss of prejunctional α2-adrenoceptors, but this is not necessarily supported by the agonist data.
Keywords: α2-adrenoceptors, α2A/D-adrenoceptors, mouse vas deferens
Introduction
α2-Adrenoceptors have been subdivided into three subtypes, α2A-, α2B- and α2C-adrenoceptors, based on ligand binding and molecular cloning studies (Bylund, 1992; Lorenz et al., 1990), and the rat α2D-adrenoceptor is a species orthologue of the human α2A-adrenoceptor (Lanier et al., 1991; Harrison et al., 1991a). Functional prejunctional α2-adrenoceptors in rat submandibular gland (Smith et al., 1992), rat vas deferens (Smith et al., 1992; Smith & Docherty, 1992), rat cerebral cortex (Trendelenburg et al., 1993), pithed rat heart (Smith et al., 1995) and mouse atria (Wahl et al., 1996), resemble the α2D-adrenoceptor ligand binding site, whereas those in rabbit cerebral cortex (Trendelenburg et al., 1993) and human saphenous vein (Molderings & Gothert, 1995) resemble the α2A-adrenoceptor.
However, in addition to the α2D-adrenoceptor, a second subtype of adrenoceptor is found in the rat atrium but not the cerebral cortex and resembles the α2C-adrenoceptor subtype (Ho et al., 1998). Studies in α2-adrenoceptor knockout mice confirm that two subtypes of α2-adrenoceptor are present prejunctionally (Hein & Kobilka, 1998; Scheibner et al., 2001). Therefore, in α2A/D-adrenoceptor knockout mice, the prejunctional α2A/D-adrenoceptor is replaced by a non-α2A/D-adrenoceptor which at least in the heart is an α2C-adrenoceptor (Altman et al., 1999; Trendelenburg et al., 2001).
The advent of knockout technology has led many researchers to turn to the mouse, and in this study we wish to establish the basic parameters of the mouse vas deferens and how these are affected by knockout of the α2A/D-adrenoceptor. It is well established that noradrenaline and ATP are co-transmitters mediating contractions of mouse vas deferens (Allcorn et al., 1986), and acetylcholine may also be involved (Kaschube & Zetler, 1988). In mice lacking P2X1 receptors, contractions of vas deferens to nerve stimulation are reduced to 60% of normal, and male fertility is reduced by 90% (Mulryan et al., 2000).
We have recently shown that the ability of methylenedioxymethamphetamine (MDMA), which is an α2-adrenoceptor agonist (Lavelle et al., 1999), to inhibit stimulation-evoked contractions of mouse vas deferens was similar in tissues from WT and α2A/D-adrenoceptor knockout mice (Rajamani et al., 2001). The object of this study was to investigate whether changes in prejunctional receptors in the α2A/D-adrenoceptor knockout mouse affect responses to agonists and antagonists equally.
Methods
C57BL/6 Black mice (18–28 g) (both WT and homozygous α2A/D-adrenoceptor knockouts) were obtained as breeding pairs from the Jackson Laboratory, Bar Harbor, MA, U.S.A.). The ko was originally constructed on a mixed genetic background (donor strain 129S2; background strain C57BL/6). All experiments were conducted with a congenic ko strain generated by repeated backcrossing of the mutation onto WT C57BL/6 for at least 12 generations.
Mouse vas deferens
Mice were killed by CO2 overdose, and whole vas deferens (approximately 15 mm long) was dissected out. Tissues were attached to myograph transducers, orientated with the epididymal portion uppermost, under 0.5 g tension in organ baths at 37°C in Krebs–Henseleit solution of the following composition: (mM): NaCl 119; NaHCO3 25; D-glucose 11.1; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4 1.0. The solution was bubbled with 5% Co2/95% O2. Cocaine (3 μM) or nifedipine (10 μM) was present in some experiments, as indicated in the text. Data acquisition was carried out using a MacLab system.
Stimulation-evoked isometric contractions
In experiments investigating the ability of competitive agonists or antagonists to inhibit or potentiate the isometric twitch in mouse vas deferens (in the presence of vehicle, nifedipine 10 μM or cocaine 3 μM), tissues were placed between platinum electrodes and stimulated every 5 min with trains of 40 pulses at 10 Hz or a single stimulus pulse (0.5 ms pulses, supramaximal voltage) to produce isometric contractions. Where two tissues from the same animal were used, the average of the two stimulation-evoked responses was taken in calculation of control responses to 10 Hz 4 s or a single stimulus. Agonists, antagonists or vehicle were added cumulatively in one log unit increments at 5 min intervals. An isometric contractile response was obtained following 5 min exposure to each agonist or antagonist concentration, or following exposure to the vehicle. Effects of test drug were not corrected for changes occurring in experiments in which repeated additions of vehicle replaced test drug: after five additions of vehicle the stimulation-evoked contraction was reduced by less than 10% in WT or knockout.
Radioligand binding studies
Preparation of rat kidney membranes was carried out as described in Connaughton & Docherty (1990), and preparation of rat submandibular gland membranes were as described for rat kidney. Membranes of Sf9 cells expressing human recombinant α2C-adrenoceptors were purchased from Research Biochemicals. The resultant pellets were used immediately or stored at −20°C for later use. Pellets were reconstituted in five volumes (submandibular), 10 volumes (kidney) or 25 volumes (Sf9 cells) of incubation buffer.
In saturation experiments, aliquots of membrane suspension were incubated with various concentrations of [3H]yohimbine (specific activity: 81 Ci mmole−1, Amersham) at 25°C (rat kidney: 0.5–30 nM; Sf9 cells: 0.2–20 nM; rat submandibular gland: 1.0–40 nM; incubation buffer: Tris-HCl 50 mM, EDTA 5 mM, pH 7.4 at 25°C). In competition studies, [3H]yohimbine (5 or 10 nM) was incubated with competing ligands in concentrations from 0.1 nM to 1 mM in 0.5 log unit increments for 30 min. Non-specific binding was determined in the presence of phentolamine (10 μM). Specific binding of [3H]yohimbine was 70–90% of total binding at the concentration used in displacement experiments. Assays were terminated by washing with ice-cold incubation buffer, followed by rapid vacuum filtration through Whatman GF/C filters, using a Brandel Cell Harvester. Radioactivity retained on filters was determined by liquid scintillation spectroscopy.
The inhibition constant (Ki) for inhibition of radiolabelled ligand binding was determined from the formula:
where IC50 is the concentration of competing ligand that inhibits radioligand specific binding by 50%, KD is the dissociation constant for the radioligand yohimbine (rat kidney: 8.80±0.62 nM, n=7; human recombinant α2C-adrenoceptors: 8.7±3.5 nM, n=3; rat submandibular gland: 23.7±2.0 nM, n=4), and [3H] is the concentration of tritiated yohimbine (5 nM: rat kidney; 10 nM: Sf9 cells and rat submandibular gland).
Drugs
BRL 44408 (2-((4,5-dihydro-1H-imidazol-2-yl)methyl)-2,3-dihydro-1-methyl-1H-isoindole; Sigma, Dublin, Ireland); cocaine hydrochloride (Sigma); imiloxan hydrochloride (Tocris); nifedipine (Sigma); prazosin hydrochloride (Sigma); propranolol hydrocholoride (Sigma); spiroxatrine (Tocris); xylazine hydrochloride (Gift: Bayer, Ireland); yohimbine hydrochloride (Sigma).
Drugs were dissolved in distilled water, except for nifedipine (100% ethanol: maximum bath concentration, 0.1%) and spiroxatrine (50 μl HCl prior to addition of distilled water). Drugs were diluted in distilled water.
Statistics
Values are mean±s.e.mean from n experiments. Agonist pD2 (−logEC50) values were compared between groups, and effects of antagonist on nerve stimulation-evoked responses were compared with the effects of vehicle, by Analysis of Variance and Tukey's or Dunnett's post-test. Statistical and graphical analysis was carried out using Instat for MacIntosh and GraphPad Prism for PC.
Results
Stimulation-evoked isometric contractions in mouse vas deferens: effects of antagonists
Stimulation with a single stimulus produced a monophasic isometric contraction of 0.053±0.008 g (n=19) and 0.059±0.020 g (n=11) in vas deferens from WT and α2A/D-adrenoceptor knockout mice, respectively (no significant difference) (see Table 1).
Table 1.
Contractions evoked by electrical stimulation in vas deferens from wild-type or α2A/D-adrenoceptor knockout mice
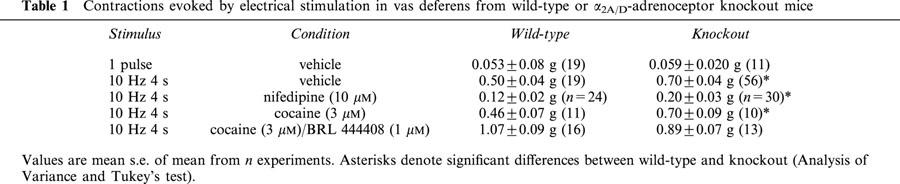
Stimulation with 40 pulses at 10 Hz produced isometric contractions which tended to differ both qualitatively and quantitatively between tissues from WT and α2A/D-adrenoceptor knockout. Responses in WT in the absence of cocaine usually consisted of an early sharp peak, a concave decline in response, and sometimes a late increase in response to a second peak (in 10 from 92 tissues the second peak was greater than the first peak) (see Figure 1a, although the late increase occurs after cocaine in this example). Responses in α2A/D-adrenoceptor knockout in the absence of cocaine usually consisted of a slower rise to peak and a more rounded and often convex decline in response, and a second peak was very rare (see Figure 1a). However, a number of traces from knockout animals did resemble the typical WT response (39 from 102), although the reverse was rarely true (four from 92). Stimulation with 40 pulses at 10 Hz produced a contraction of 0.50±0.04 g (n=60) and 0.70±0.04 g (n=56) in WT and knockout mice, respectively (response was significantly greater in vas from knockout mice: P<0.001) (see Table 1). The time to peak contraction (first peak) was 0.83±0.02 s (n=92) and 1.32±0.04 s (n=102) in tissues from WT and knockout animals, respectively (peak was significantly later in vas from knockout mice: P<0.001).
Figure 1.
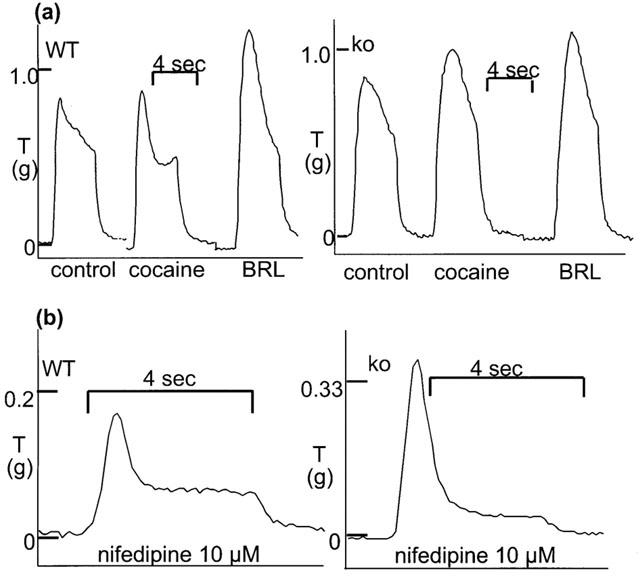
Typical isometric contractions to 10 Hz stimulation for 4 s in vas deferens from wild-type (WT) and α2A/D-adrenoceptor knockout (ko) mice. (a) Control response and response following cocaine (3 μM) and subsequent BRL 44408 (1 μM). (b) Response in the presence of nifedipine (10 μM). Time and tension scales have arbitrary zero points.
The non-selective α2-adrenoceptor antagonist yohimbine (0.01–1 μM) significantly increased the evoked contraction in both WT and knockout, but the maximum potentiation was much greater in WT (Figure 2). The α2A/D-adrenoceptor antagonist BRL 44408 (0.1–1 μM) significantly increased contractions evoked by 10 Hz stimulation in WT but not in knockout animals; the reverse was true for the α2C-adrenoceptor antagonist spiroxatrine (spiroxatrine 0.01–0.1 μM significantly increased contractions in tissues from knockout only) (Figure 2). The α2B-adrenoceptor selective antagonist imiloxan significantly increased contractions only in tissues from WT and only at 10 μM, a concentration at which it acts on α2D-adrenoceptors. Following yohimbine (1 μM), the time to peak contraction (first peak) was 1.65±0.09 s (n=6) and 1.80±0.14 s (n=6) in tissues from WT and knockout animals, respectively (no significant difference between tissues from WT and knockout, but in both cases significantly later in the presence than absence of yohimbine).
Figure 2.
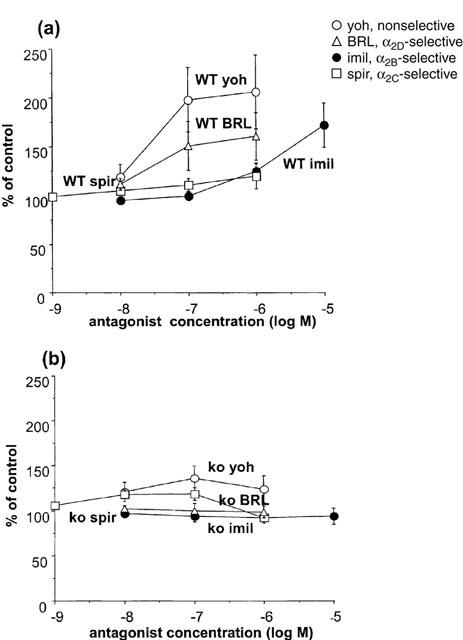
Effects of the α2-adrenoceptor antagonists yohimbine (yoh, non-selective), BRL 44408 (BRL, α2D-selective), imiloxan (imil, α2B-selective) and spiroxatrine (spir, α2C-selective) on isometric contractions evoked by 10 Hz electrical stimulation for 4 s in vas deferens from wild-type (a) or α2A/D-adrenoceptor knockout (b) mice. Vertical bars indicate s.e. of mean from four (BRL 44408, imiloxan) or six (other antagonists) experiments.
The effects of cocaine on responses to a single stimulus and 10 Hz 4 s were also examined. Cocaine (3 μM) significantly increased the response to a single stimulus to 298.9±44.6% of control (n=12) in WT, and to 400.1±51.0% of control (n=11) in knockout (no significant difference between WT and knockout). Cocaine (3 μM) slightly, but non-significantly increased the peak response to 10 Hz 4 s to 109.4±3.1% of control (n=8) (vehicle: 101.6=3.1%, n=6) in WT, and significantly increased the response to 118.9±4.6% of control (n=6) (vehicle: 101.8±2.1%, n=8) in knockout (Figure 3). The addition of cocaine (3 μM) produced a clear accentuation of the convex shape and late peak in WT, but had little effect on shape of the response in knockout (see right inset to Figure 1a). The effect could be quantified by expressing the response 1.5 s after beginning stimulation as a percentage of the response at 1 s: this was 86.4±3.8% (n=11) in vas from WT prior to cocaine, but significantly reduced to 68.6±6.2% of control (n=11) following cocaine (cocaine had no significant effect in knockout; vehicle had no effect in WT or knockout). In both WT and knockout, cocaine prolonged the time taken for the response to decline to baseline at the end of stimulation (Figure 1a). The addition of BRL 44408 (1 μM) produced a large significant increase in contraction in WT to 158.1±7.2% of control (n=8) (vehicle: 93.4±2.3%, n=6), but no further increase in contraction in knockout to 121.0±7.0% of control (n=6) (vehicle: 98.7±4.2%, n=8) (Figure 3). However, the shapes and maximum of the stimulation-evoked contraction were now similar in tissues from WT and knockout (Figure 1a), with similar times to peak (1.64±0.08 s, n=16 and 1.69±0.11 s, n=10, in WT and knockout, respectively). In WT following BRL 44408 (1 μM), cocaine (3 μM) had no further effect on the shape or maximum response (n=3).
Figure 3.
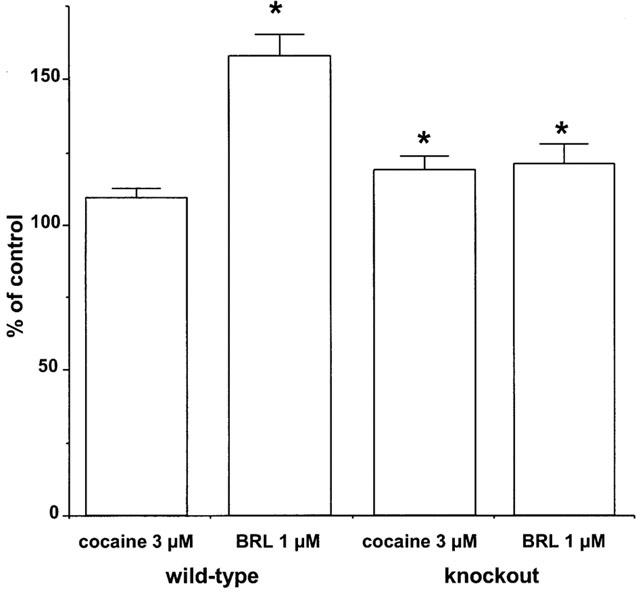
Effects of cocaine (3 μM) and the α2-adrenoceptor antagonist BRL 44408 (1 μM) on isometric contractions evoked by 10 Hz electrical stimulation for 4 s in vas deferens from wild-type or α2A/D-adrenoceptor knockout mice. Vertical bars indicate s.e. of mean from at least five experiments. Asterisks denote significance of difference from control responses (* P<0.05).
Effects of xylazine on stimulation-evoked contractions: cocaine experiments
Stimulation with 40 pulses at 10 Hz in the presence of cocaine (3 μM) produced a contraction of 0.46±0.07 g (n=11) and 0.70±0.09 g (n=10) in WT and knockout mice, respectively (response was significantly greater in vas from knockout mice: P<0.001). In the presence of cocaine, xylazine produced a concentration-dependent inhibition of the stimulation-evoked contraction in tissues from WT animals, but potentiated the response in tissues from knockout animals (Figure 4). However, the effects of xylazine were more complex than the graph suggests: the early part of the nerve stimulation-evoked contraction was virtually abolished by xylazine (the response at 0.5 s after beginning stimulation was reduced by xylazine, 0.1 μM, to 23.9±4.6% and 51.8±4.9% of control, and by xylazine, 1 μM, to 14.9±5.8% and 18.8±5.7% of control, in tissues from WT and knockout animals, respectively), and the later part was more resistant (WT) or even potentiated (knockout) (see Figure 5). The insets to Figure 5 demonstrate typical responses in tissues from a WT and a knockout mouse: note how the early component of the contraction is markedly reduced by xylazine (1 or 10 μM) in both WT and knockout, but the late component is reduced by less than 50% in WT and is actually potentiated in knockout. The α2D-adrenoceptor antagonist BRL 44408 (1 μM) significantly increased the stimulation-evoked contraction in WT, but had no significant effect in knockout (see Figure 3). However, BRL 44408 (1 μM) significantly reduced the potency of xylazine in WT but not knockout. Indeed, BRL 44408 prevented the potentiation by xylazine of contractions to 10 Hz stimulation in knockout mice, presumably by an α1-adrenoceptor antagonist action to prevent postjunctional actions of xylazine. Given the postjunctional actions of xylazine in knockout animals in the presence of cocaine, we carried out experiments in the presence of nifedipine which virtually eliminates postjunctional actions of agonists in rat vas deferens (see Docherty, 1984).
Figure 4.
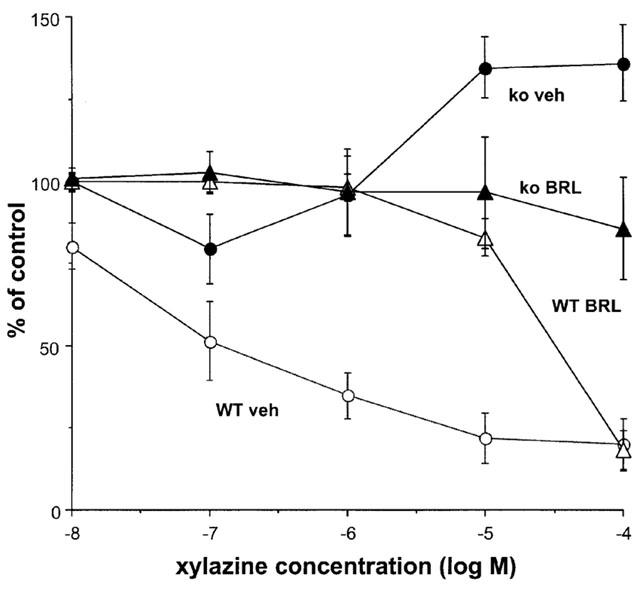
Effects of the α2-adrenoceptor agonist xylazine, in the presence of vehicle (veh) or the α2-adrenoceptor antagonist BRL 44408 1 μM (BRL, α2D-selective), on isometric contractions evoked by 10 Hz electrical stimulation for 4 s in vas deferens from wild-type (WT) or α2A/D-adrenoceptor knockout (ko) mice in the presence of cocaine (3 μM). Vertical bars indicate s.e. of mean from at least four experiments.
Figure 5.
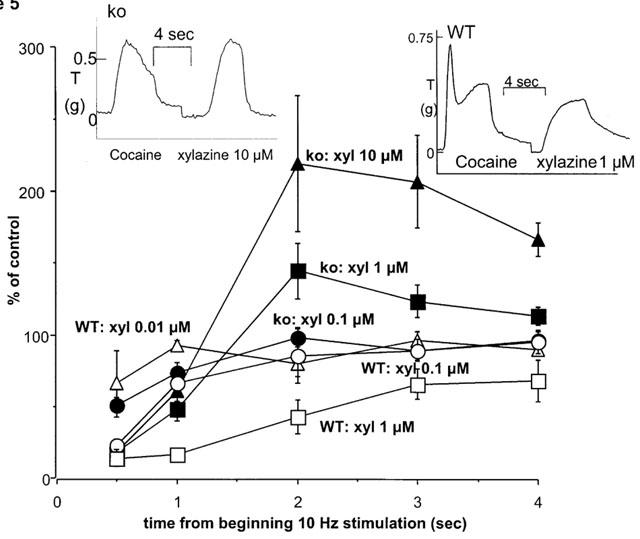
Effects of the α2-adrenoceptor agonist xylazine, in the presence of vehicle on isometric contractions evoked by 10 Hz electrical stimulation for 4 s in vas deferens from wild-type (WT) or α2A/D-adrenoceptor knockout (ko) mice in the presence of cocaine (3 μM). Effects of xylazine 0.01–1 μM (WT) or 0.1–10 μM (ko) on contractions are shown at various times (0.5, 1, 2, 3, 4 s) from beginning of 10 Hz stimulation. Note that xylazine inhibits at all time points in WT, but inhibits at 0.5–1 s and potentiates at 2–4 s in ko (at least with 1 and 10 μM). Vertical bars indicate s.e. of mean from at least four experiments. The insets show typical isometric responses to 10 Hz stimulation before and after xylazine (1 or 10 μM) in tissues from a knockout (ko) and a wild-type (WT) mouse. Time and tension scales have arbitrary zero points.
Effects of xylazine on stimulation-evoked contractions: nifedipine experiments
Nifedipine (10 μM) significantly reduced the contraction evoked by 10 Hz stimulation in both wild-type and knockout (see Figure 1b). Stimulation with 40 pulses at 10 Hz in the presence of nifedipine (10 μM) produced a contraction of 0.12±0.02 g (n=24) and 0.20±0.03 g (n=30) in WT and knockout mice, respectively (response was significantly greater in vas from knockout mice: P<0.05). in the presence of nifedipine, xylazine significantly inhibited stimulation-evoked contractions in tissues from both types of animal, but the potency and/or maximum response of xylazine at inhibiting contractions evoked by 10 Hz stimulation was significantly reduced in knockout (4.54±0.17, n=4) as compared with WT mice (7.01±0.48, n=4) (Figure 6). However, even in the presence of nifedipine, the actions of xylazine in vas from knockout mice were complex, with again a reduction in the early part and an increase in the later part of the contraction (data not shown). BRL 44408 (1 μM) significantly increased the stimulation-evoked contraction to 270.8±65.3% of control (n=4) in WT, but had no significant effect in knockout (97.2±12.0% of control, n=4). BRL 44408 actually increased the ability of xylazine to inhibit contractions in vas from knockout mice presumably by an α1-adrenoceptor antagonist action to prevent post-junctional actions of xylazine, while reducing the potency of xylazine in vas from WT (Figure 6). However, prazosin (0.01 μM), a concentration which reduces the stimulation-evoked contraction to 10 Hz 4 s in WT in the absence of nifedipine below 50% of control (authors, unpublished observations) did not affect the actions of xylazine post-nifedipine in knockout mice (Figure 6). This suggests that even in the presence of nifedipine, xylazine has some postjunctional actions which mask its true pre-junctional action, and these post-junctional actions may be at α2-adrenoceptors, at least in knockout animals.
Figure 6.
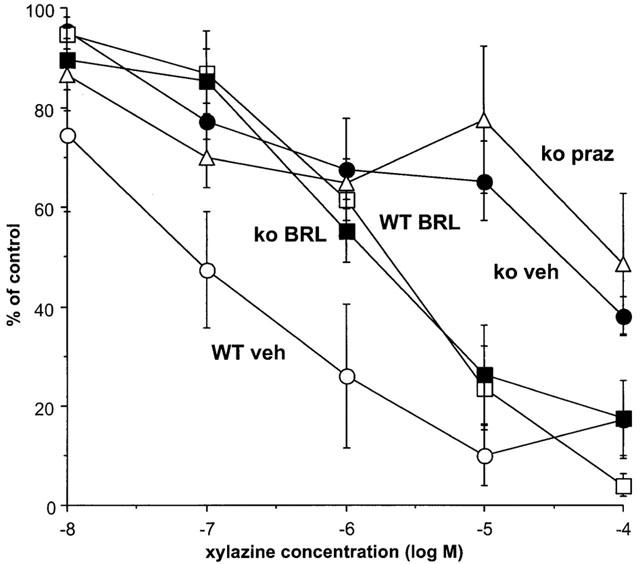
Effects of the α2-adrenoceptor agonist xylazine, in the presence of vehicle (veh), the α2-adrenoceptor antagonist BRL 44408 1 μM (BRL, α2D-selective) or the α1-adrenoceptor antagonist prazosin (praz), on isometric contractions evoked by 10 Hz electrical stimulation for 4 s in vas deferens from wild-type (WT) or α2A/D-adrenoceptor knockout (ko) mice in the presence of nifedipine (10 μM). Vertical bars indicate s.e. of mean from at least four experiments. Effects of xylazine in the presence of vehicle are taken from Rajamani et al. (2001).
Ligand binding studies
Table 2 shows the affinities of antagonists and the agonist xylazine used in this study for α2B-, α2C- and α2D-adrenoceptor ligand binding sites. Yohimbine shows 3 fold selectivity for α2B- or α2C-adrenoceptors over α2D-adrenoceptors. Spiroxatrine shows 500 fold and 30 fold selectivity for α2C- or α2B-adrenoceptors, respectively, whereas BRL 44408 showed 30 fold selectivity for α2D sites (Table 2).
Table 2.
Affinity of antagonists and agonists for α2B-, α2C- and α2D-adrenoceptor ligand binding sites
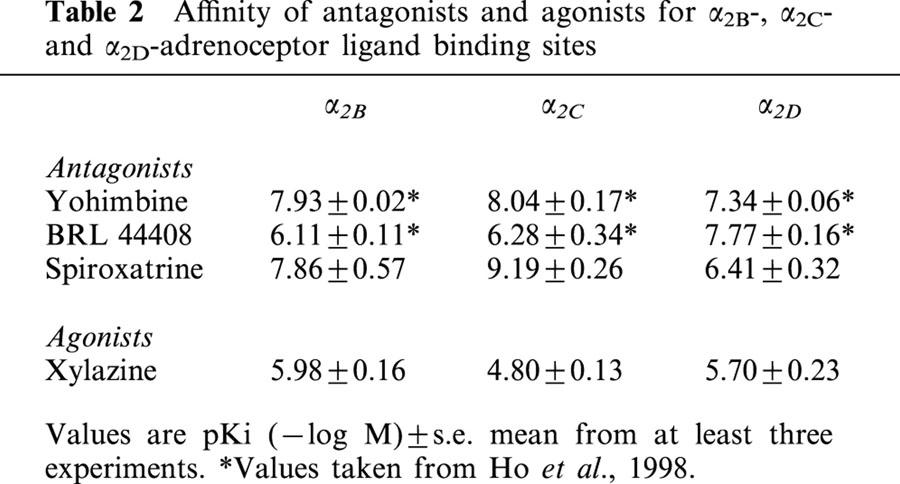
Xylazine showed approximately 10 fold selectivity for α2D over α2C, but no selectivity over α2B (Table 2).
Discussion
In this study, we have looked at α2-adrenoceptor agonist and antagonist actions against nerve stimulation-evoked contractions in mouse vas deferens and how these are altered in α2A/D-adrenoceptor knockout mice. Let us first consider baseline responses to nerve stimulation: noradrenaline and ATP are co-transmitters mediating contractions of mouse vas deferens (Allcorn et al., 1986), and acetylcholine may also be involved (Kaschube & Zetler, 1988). Contractions of mouse vas deferens to nerve stimulation involve both α1-adrenoceptors and purinergic P2X1 receptors, so that deletion of the purinergic receptors greatly reduces the magnitude of the contraction and the residual response is blocked by prazosin (Mulryan et al., 2000). In our studies of mouse vas deferens, prazosin in low concentrations markedly reduced contractions. In some studies, a residual atropine sensitive contraction remaining after α1-adrenoceptor and purinergic blockade suggested a cholinergic component to responses (Kaschube & Zetler, 1988), but in our studies, the residual response following prazosin and the purinergic antagonist suramin was not affected by atropine (authors unpublished results; see also Mulryan et al., 2000).
Responses to a single stimulus were unaltered, but 40 pulses at 10 Hz were significantly larger in tissues from knockout mice as compared to WT following vehicle, cocaine or nifedipine. Although there were exceptions, the shape of contractile responses to 10 Hz tended to have characteristic differences between WT and knockout. In general, a sharp early peak was obtained in tissues from WT, followed by a concave decline and sometimes a late increase in response. In tissues from knockout mice, the response was generally more ‘rounded' with a significantly later first peak, and a more convex decline from peak: there was rarely a second peak, although some responses in vas from knockout did resemble the typical WT response. In the presence of yohimbine (1 μM) responses in WT and knockout had similar times to peak and resembled the typical response in knockout tissues in the absence of yohimbine. The noradrenaline re-uptake blocker cocaine (3 μM) changed the shape of the response in WT tissues, making the response more concave and biphasic by reducing the magnitude of contraction at 1.5–2 s, presumably by increasing the amount of noradrenaline available to activate the prejunctional inhibitory α2-adrenoceptors. In contrast, in tissues from knockout animals, cocaine did not significantly affect shape of response but caused a small potentiation of the contraction, presumably by increased availability of noradrenaline at the post-junctional receptors. Following cocaine and BRL 44408, responses were generally similar in shape and magnitude in tissues from WT and knockout, and difficult to distinguish. These results with α2-adrenoceptor antagonists with or without cocaine might suggest that diminished prejunctional control of neurotransmission in tissues from knockout animals explains the prolonged time to peak and increased maximum contraction. Negative feedback by noradrenaline influences contraction beginning at about 0.7–0.8 s in WT and this inhibition is amplified by cocaine and diminished by α2-adrenoceptor antagonists. In knockout, there is still some negative feedback so that α2-adrenoceptor antagonists could still significantly delay the peak response and increase the maximum. It is also possible that α2A/D-adrenoceptors differ from α2C-adrenoceptors in their temporal characteristics and this may contribute to differences between WT and knockout. Returning to cocaine, in vas deferens from both WT and knockout mice, cocaine prolonged the response to nerve stimulation by slowing the rate of decline of the response at the end of stimulation (see Figures 1 and 5).
The ability of α2-adrenoceptor antagonists to increase stimulation-evoked contractions to 40 pulses at 10 Hz by block of endogenous feedback was investigated. The relatively non-selective antagonist yohimbine significantly increased stimulation-evoked contractions in vas deferens from both WT and knockout, but the degree of potentiation was much greater in WT. The α2D-adrenoceptor selective antagonist BRL 44408 significantly increased stimulation-evoked contractions in WT but not knockout. In ligand binding studies, BRL 44408 is approximately 30 times more potent at α2D (7.77) than at α2B/α2C (6.11/6.28), so that BRL 44408 is likely to act in low concentrations by blocking only α2D-adrenoceptors. The α2C-adrenoceptor antagonist spiroxatrine 0.01 μM significantly increased stimulation-evoked contractions in knockout but not WT. In ligand binding studies, spiroxatrine is approximately 30–300 times more potent at α2B (7.86) and α2C (9.19) than at α2D (6.41), so that spiroxatrine is likely to act in low concentrations by blocking only α2B/α2c-adrenoceptors. A significant effect of spiroxatrine at 0.01 μM might suggest an α2C-adrenoceptor action, but we cannot entirely exclude an α2B-action. Imiloxan shows selectivity for α2B- over α2A/D-adrenoceptor subtypes (5–30 fold: Michel et al., 1990; Devedjian et al., 1994; Jasper et al., 1998) and to a lesser extent over α2C-adrenoceptors (3–10 fold: Devedjian et al., 1994; Jasper et al., 1998). However, imiloxan produced effects only in WT and at a concentration consistent with α2D-adrenoceptor antagonism. The lack of effect of imiloxan in knockout animals might suggest that α2c-adrenoceptors, and not α2B-adrenoceptors, replace the α2D-adrenoceptors in vas deferens of knockout. From these results we can conclude two things. Firstly, the prejunctional α2D-adrenoceptor in vas deferens from WT is replaced by an α2B/2C-adrenoceptor (probably an α2C-adrenoceptor) in vas deferens from knockout. Indeed, other authors have shown that an α2C-adrenoceptor replaces the α2D in the heart of the α2A/D knockout (Altman et al., 1999), and found no evidence for prejunctional α2B-adrenoceptors in knockout studies of mouse hippocampus (Scheibner et al., 2001). Secondly, the ability of antagonists to potentiate contractions is greatly reduced in vas from knockout mice (approximately 25% increase versus 60–100% increase in wild-type; present results). This may suggest that endogenous feedback by noradrenaline is reduced in tissues from knockout mice and explains why the contractile response to 10 Hz stimulation is significantly greater in vas from knockout, except following α2-adrenoceptor blockade.
Studies with the α2-adrenoceptor agonist xylazine produced contradictory results. First of all, let us consider the different experimental conditions employed in agonist experiments: cocaine or nifedipine. In the presence of cocaine, there was no large change in the nerve-stimulation evoked response, but postjunctional actions of agonists are possible. In studies of responses to a single pulse stimulation in the presence of cocaine, potency of xylazine was reduced in vas deferens of knockout mice, whereas potency of MDMA (methylenedioxymethamphetamine) was unchanged (Rajamani et al., 2001). In the present study, the effects of xylazine on 10 Hz stimulation-evoked responses in the presence of cocaine in vas from knockout mice was complex: inhibition of an early component of the contraction, but potentiation of the later component. The potentiation was probably by a postjunctional action. In contrast, in tissues from WT animals, xylazine inhibited all components of the response although the later component was inhibited less than the early component.
In the presence of nifedipine (10 μM), postjunctional actions of α1-adrenoceptor agonists are virtually abolished in rat vas deferens (see Docherty, 1984), but the present results suggest that some post-junctional α-adrenoceptor actions remain in mouse vas deferens. This may suggest differences between rat and mouse vas deferens in the adrenoceptors targeted by exogenous agonists in terms of the involvement of entry of calcium. In studies of contractions to 10 Hz stimulation in the presence of nifedipine, xylazine and oxymetazoline (present results and Rajamani et al., 2001) were significantly less potent at inhibiting stimulation-evoked contractions in vas deferens from knockout mice, but, perhaps surprisingly, MDMA had similar potency in knockout and WT (Rajamani et al., 2001). The change in receptor subtype was confirmed by the effects of BRL 44408: the antagonist reduced potency of xylazine in wild-type but not in knockout (present results). In fact, xylazine potency was increased in knockout, but this was probably due to blockade by BRL 44408 of some of the postjunctional actions of high concentrations of xylazine. However, the α1-adrenoceptor antagonist prazosin at a concentration which significantly affects nerve-stimulation-evoked contractions, did not increase xylazine potency, suggesting that the above actions of BRL 44408 in knockout animals is by action at α2-adrenoceptors, α2B- or α2C-adrenoceptors. Hence, even in the presence of nifedipine, there is a postjunctional component to the response to xylazine in knockout animals, which may even be α2-adrenoceptor mediated. There is previous evidence that α2-adrenoceptors are involved in contractions of mouse vas deferens (Bultmann et al., 1991). Indeed, in vas from knockout, xylazine inhibited the early component (0.5 s) but tended to increase the later component (up to 4 s) of the contraction to 10 Hz stimulation. The lower potency of xylazine in knockout and the similar potency of MDMA can be explained in terms of differing ligand binding affinity for α2B/2C and α2D. Hence, agonist results could be taken to indicate that number of pre-junctional α2-adrenoceptors is relatively unchanged in knockout in relation to wild-type. This contrasts with the antagonist data.
How do we reconcile antagonist and agonist data. Antagonist results clearly show a change from α2D- to α2B/2C-adrenoceptors and an apparently diminished negative feedback, suggesting fewer receptors as targets for endogenous noradrenaline. The agonist results are more consistent with a change in receptor subtype but similar numbers of receptors. The reduced endogenous feedback by noradrenaline in knockout can be explained by a decreased number of prejunctional receptors or by a diminished ability of noradrenaline to interact with the new receptors, either due to reduced affinity/efficacy for the new receptors, or to limited access to the receptors due to change of location. In ligand binding studies, noradrenaline has a 2–3 fold higher affinity for α2B/2C than α2D (Harrison et al., 1991b), tending to rule out the former possibility. However, in cultured sympathetic neurons from 3 day old mice nerve terminal receptors were found to be α2D, whereas soma-dendritic receptors were a mixture of α2C and α2D (Trendelenburg et al., 2001). Although the soma-dendritic α2C-adrenoceptors reduced depolarization-evoked calcium currents, they did not seem to be involved in presynaptic inhibition of transmitter release (Trendelenburg et al., 2001). However, the situation may be different in adult sympathetic neurons, so that soma-dendritic α2C-adrenoceptors may increase in number in α2A/D-knockout mice and additionally contribute to agonist but not antagonist actions in the mouse vas deferens due to their location distal to the synapse.
How do the results of this study differ from those of previous studies. Firstly, Altman et al. (1999) found that the ability of the α2-adrenoceptor agonist dexmedetomide to reduce stimulation-evoked contractions was greatly reduced in vas deferens from α2A/D-adrenoceptor knock-out mice, with a much smaller reduction in its ability to inhibit transmitter release. The present study demonstrates that, in the absence of nifedipine, prejunctional inhibitory actions of α2-adrenoceptor agonists are masked by post-junctional actions in α2A/D-adrenoceptor knockout mice, resulting in an overestimation of the loss of prejunctional function. However, like Altman et al. (1999), we found that the ability of antagonists to potentiate evoked contractions was reduced in tissues from knockout animals. This clearly demonstrates a decreased number of prejunctional α2-adrenoceptors, or that the receptors are less accessible to endogenous neurotransmitter while remaining accessible to exogenous agonists.
It is concluded that the prejunctional α2A/D-adrenoceptor is replaced by the α2B/2C-adrenoceptor (probably the α2C-adrenoceptor) in vas deferens from this knockout mouse, resulting in diminished prejunctional modulation of neurotransmission in terms of α2-adrenoceptor antagonist but not agonist actions. The reasons for this require further investigation.
Acknowledgments
Supported by The Health Research Board (Ireland), and the Irish Heart Foundation.
Abbreviations
- BRL 44408
2-((4,5-dihydro-1H-imidazol-2-yl)methyl)-2,3-dihydro-1-methyl-1H-isoindole
- KD
dissociation constant
- Ki
inhibition constant
- ko
knockout (α2A/D-adrenoceptor knockout)
- MDMA
methylenedioxymethamphetamine
- pD2
−logEC50
- WT
wild-type
References
- ALLCORN R.J., CUNNANE T.C., KIRKPATRICK K. Actions of a,b-methylene ATP and 6-hydroxydopamine on sympathetic neurotransmission in the vas deferens of the guinea-pig, rat and mouse: support for co-transmission. Br. J. Pharmacol. 1986;89:647–659. doi: 10.1111/j.1476-5381.1986.tb11169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALTMAN J.D., TRENDELENBURG A.U., MACMILLAN L., BERNSTEIN D., LIMBIRD L., STARKE K., KOBILKA B.K., HEIN L. Abnormal regulation of the sympathetic nervous system in α2A-adrenoceptor knockout mice. Mol. Pharmacol. 1999;56:154–161. doi: 10.1124/mol.56.1.154. [DOI] [PubMed] [Google Scholar]
- BULTMANN R., VON KUGELGEN I., STARKE K. Contraction-mediating α2-adrenoceptors in the mouse vas deferens. Naunyn-Schmiedeberg's Arch. Pharmacol. 1991;343:623–632. doi: 10.1007/BF00184294. [DOI] [PubMed] [Google Scholar]
- BYLUND D.B. Subtypes of α1- and α2-adrenergic receptors. FASEB J. 1992;6:832–839. doi: 10.1096/fasebj.6.3.1346768. [DOI] [PubMed] [Google Scholar]
- CONNAUGHTON S., DOCHERTY J.R. No evidence for differences between pre- and postjunctional alpha-2 adrenoceptors in the periphery. Br. J. Pharmacol. 1990;99:97–102. doi: 10.1111/j.1476-5381.1990.tb14660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVEDJIAN J.C., ESCLAPEZ F., DENIS-POUXVIEL C., PARIS H. Further characterization of human α2-adrenoceptor subtypes: [3H]RX821002 binding and definition of additional selective drugs. Eur. J. Pharmacol. 1994;252:43–49. doi: 10.1016/0014-2999(94)90573-8. [DOI] [PubMed] [Google Scholar]
- DOCHERTY J.R. An investigation of presynaptic alpha-adrenoceptor subtypes in the pithed rat heart and in the rat isolated vas deferens. Br. J. Pharmacol. 1984;82:15–23. doi: 10.1111/j.1476-5381.1984.tb16437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRISON J.K., D'ANGELO D.D., ZENG D., LYNCH K.R. Pharmacological characterization of rat α2-adrenergic receptors. Mol. Pharmacol. 1991b;40:407–412. [PubMed] [Google Scholar]
- HARRISON J.K., PEARSON W.R., LYNCH K.R. Molecular characterization of α1- and α2-adrenoceptors. Trends Pharmacol. Sci. 1991a;12:62–67. doi: 10.1016/0165-6147(91)90499-i. [DOI] [PubMed] [Google Scholar]
- HEIN I., KOBILKA B.K. Clinical and molecular pharmacology of adrenergic receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358:R575. [Google Scholar]
- HO S.L., HONNER V., DOCHERTY J.R. Investigation of the subtypes of α2-adrenoceptor mediating prejunctional inhibition in rat atrium and cerebral cortex. Naunyn-Schmeideberg's Arch. Pharmacol. 1998;357:634–639. doi: 10.1007/pl00005218. [DOI] [PubMed] [Google Scholar]
- JASPER J.R., LESNICK J.D., CHANG L.K., YAMANISHI S.S., CHANG T.K., HSU S.A., DAUNT D.A., BONHAUS D.W., EGLEN R.M. Ligand efficacy and potency at recombinant α2-aadrenergic receptors: agonist-mediated [35S]GTPγS binding. Biochem. Pharmacol. 1998;55:1035–1043. doi: 10.1016/s0006-2952(97)00631-x. [DOI] [PubMed] [Google Scholar]
- KASCHUBE M., ZETLER G. Noradrenergic, purinergic and cholinergic transmission in the mouse vas deferens: influence of field-stimulation parameters, reserpinization, 6-hydroxydopamine and 4-aminopyridine. J. Neural Transmission. 1988;76:39–53. doi: 10.1007/BF01244990. [DOI] [PubMed] [Google Scholar]
- LANIER S.M., DOWNING S., DUZIE E., HOMEY C.J. Isolation of rat genomic clones encoding subtypes of the α2-adrenergic receptor. J. Biol. Chem. 1991;266:10470–10478. [PubMed] [Google Scholar]
- LAVELLE A., HONNER V., DOCHERTY J.R. Investigation of the prejunctional alpha2-adrenoceptor mediated actions of MDMA in rat atrium and vas deferens. Br. J. Pharmacol. 1999;128:975–980. doi: 10.1038/sj.bjp.0702875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LORENZ W., LOMASNEY J.W., COLLINS S., REGAN J.W., CARON M.G., LEFKOWITZ R.J. Expression of three α2-adrenergic receptor subtypes in rat tissues: Implications for α2-receptor classification. Mol. Pharmacol. 1990;38:599–603. [PubMed] [Google Scholar]
- MICHEL A.D., LOURY D.N., WHITING R.L. Assessment of imiloxan as a selective α2B-adrenoceptor antagonist. Br. J. Pharmacol. 1990;99:560–564. doi: 10.1111/j.1476-5381.1990.tb12968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLDERINGS G.J., GOTHERT M. Subtype determination of presynaptic α2-adrenoceptors in the rabbit pulmonary artery and human saphenous vein. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;352:483–490. doi: 10.1007/BF00169381. [DOI] [PubMed] [Google Scholar]
- MULRYAN K., GITTERMAN D.P., LEWIS C.J., VIAL C., LECKIE B.J., COBB A.L., BROWN J.E., CONLEY E.C., BUELL G., PRITCHARD C.A., EVANS R.J. Reduced vas deferens contraction and male infertility in mice lacking P2X1 receptors. Nature. 2000;403:86–89. doi: 10.1038/47495. [DOI] [PubMed] [Google Scholar]
- RAJAMANI K., LEONG S., LAVELLE A., DOCHERTY J.R. Prejunctional actions of methylenedioxymethamphetamine in vas deferens from wild-type and alpha2A/D knockout mice. Eur. J. Pharmacol. 2001;423:223–228. doi: 10.1016/s0014-2999(01)01118-9. [DOI] [PubMed] [Google Scholar]
- SCHEIBNER J., TRENDELENBURG A.-U., HEIN L., STARKE K. α2-Adrenoceptors modulating neuronal serotonin release: a study in α2-adrenoceptor subtype-deficient mice. Br. J. Pharmacol. 2001;132:925–933. doi: 10.1038/sj.bjp.0703882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH K., CONNAUGHTON S., DOCHERTY J.R. Investigations of prejunctional α2-adrenoceptors in rat atrium, vas deferens and submandibular gland. Eur. J. Pharmacol. 1992;211:251–256. doi: 10.1016/0014-2999(92)90536-d. [DOI] [PubMed] [Google Scholar]
- SMITH K., DOCHERTY J.R. Are the prejunctional α2-adrenoceptors of the rat vas deferens and submandibular gland of the α2A- or α2D-subtype. Eur. J. Pharmacol. 1992;219:203–210. doi: 10.1016/0014-2999(92)90297-h. [DOI] [PubMed] [Google Scholar]
- SMITH K., GAVIN K., DOCHERTY J.R. Investigation of the subtype of α2-adrenoceptor mediating prejunctional inhibition of cardio-acceleration in the pithed rat heart. Br. J. Pharmacol. 1995;115:316–320. doi: 10.1111/j.1476-5381.1995.tb15879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRENDELENBURG A.-U., LIMBERGER N., STARKE K. Presynaptic α2-autoreceptors in brain cortex: α2D in the rat and α2A in the rabbit. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;348:35–45. doi: 10.1007/BF00168534. [DOI] [PubMed] [Google Scholar]
- TRENDELENBURG A.-U., NORENBERG W., HEIN L., MEYER A., STARKE K. α2-Adrenoceptor-mediated inhibition of cultured sympathetic neurons: changes in α2A/D-adrenoceptor-deficient mice. Naunyn-Schmiedeberg's Arch. Pharmacol. 2001;363:110–119. doi: 10.1007/s002100000331. [DOI] [PubMed] [Google Scholar]
- WAHL C.A., TRENDELENBURG A.U., STARKE K. Presynaptic alpha-2 autoreceptors in mouse heart atria: evidence for the alpha 2D subtype. Naunyn-Schmiedeberg's Arch. Pharmacol. 1996;354:253–261. doi: 10.1007/BF00171055. [DOI] [PubMed] [Google Scholar]