

Abstract
We recently demonstrated that endothelin-1 (ET-1) activates two types of Ca2+-permeable nonselective cation channels (NSCC-1 and NSCC-2) in Chinese hamster ovary cells expressing endothelinB receptors (CHO-ETBR) that couple with Gq and Gi. The purpose of the present study was to identify the G proteins involved in the activation of these Ca2+ channels by ET-1. For this purpose, we constructed CHO cells expressing an unpalmitoylated (Cys402Cys403 Cys405→Ser402Ser403Ser405) ETBR (CHO-SerETBR) and ETBR truncated at the cytoplasmic tail downstream of Cys403 (CHO-ETBRΔ403).
Based on the data obtained from actin stress fibre formation, CHO-ETBR couple with G13. Therefore, CHO-ETBR couple with Gq, Gi and G13. CHO-SerETBR and CHO-ETBRΔ403 couple with G13 and Gq, respectively.
ET-1 activated NSCC-1 in CHO-ETBR preincubated with phospholipase C (PLC) inhibitor, U73122, and in CHO-SerETBR. On the other hand, ET-1 failed to activate Ca2+ channels in CHO-ETBRΔ403. Microinjection of dominant negative mutants of G13 (G13G225A) abolished activation of NSCC-1 and NSCC-2 in CHO-ETBR and that of NSCC-1 in CHO-SerETBR.
Y-27632, a specific Rho-associated kinase (ROCK) inhibitor, did not affect the ET-1-induced transient and sustained increase in [Ca2+]i in CHO-ETBR.
These results indicate that (1) the cytoplasmic tail downstream of the palmitoylation sites of ETBR, but not the palmitoylation site itself, is essential for coupling with G13, (2) the activation mechanism of each Ca2+ channel by ET-1 is different in CHO-ETBR. NSCC-1 activation depends on G13-dependent cascade, and NSCC-2 activation depends on both Gq/PLC- and G13-dependent cascades. Moreover, ROCK-dependent cascade is not involved in the activation of these channels.
Keywords: Endothelin, endothelinB receptor, G protein, actin stress-fibre formation, nonselective cation channel
Introduction
Endothelin-1 (ET-1) has a wide variety of biological effects on various tissues and cell types (Yanagisawa et al., 1988; Masaki, 1993) that are mediated by specific heterotrimetric guanine nucleotide-binding protein (G-protein)-coupled receptor (GPCR) subtypes, the endothelinA receptor (ETAR) and endothelinB receptor (ETBR) (Arai et al., 1990; Sakurai et al., 1990). When expressed in Chinese hamster ovary (CHO) cells, ETAR couples with members of the Gq and Gs families and stimulates phospholipase C (PLC) and adenylyl cyclase, respectively. ETBR couples with members of the Gq and Gi families and stimulates PLC and inhibits adenylyl cyclase, respectively (Aramori & Nakanishi, 1992; Takagi et al., 1995). Many GPCRs including ETAR and ETBR were shown to be palmitoylated at a cluster of cysteine residues located in the cytoplasmic tail (Horstmeyer et al., 1996; Okamoto et al., 1997). The functional role of palmitoylation and the cytoplasmic tail downstream of the palmitoylation site in coupling with G proteins has been studied for ETAR and ETBR in some detail. In the case of ETBR, palmitoylation is essential for coupling of the receptor with both Gq and Gi, while the cytoplasmic tail downstream of the palmitoylation sites is also required for coupling with Gi (Okamoto et al., 1997). It is reported that both ETAR and ETBR can also couple with the G12 subfamily, consisting of G12 and G13, in NIH3T3 cells (Mao et al., 1998). The G12 subfamily has been shown to mediate important signaling pathways such as Rho/Rho-associated kinase (ROCK)-dependent formation of actin stress fibres (Buhl et al., 1995) and vascular smooth muscle cell contraction (Gohla et al., 2000). These reports suggested that the G12 subfamily may play important roles in several ET-1-induced vascular disorders such as stroke or vasospasm. Thus, the control of G12 subfamily activation may become a new treatment strategy for these conditions. Recently, it was shown that upon treatment with ET-1, ETBR in fibroblast cell lines did not induce stress fiber formation (Gohla et al., 1999), whereas ETBR in HEK 293 cells coupled to G13 (Kitamura et al., 1999). These data imply that in some types of cells, stimulation of ETBR can induce actin stress fibre formation via a member of G12 family, although direct evidence is absent. Therefore, in the present study, we attempt to determine whether stimulation of ETBR actually induces actin stress fibre formation in CHO cells expressing recombinant ETBR (CHO-ETBR), and if so, which subtypes of G proteins are involved in the formation. Furthermore, we analyse the domains of ETBR that are necessary for coupling of the receptor with the G protein. For these purposes, we use dominant negative mutants of G12 and G13 as well as mutated ETBRs that lose the ability to couple with Gq and/or Gi. Previous report demonstrated that CHO cells expressing ETBR truncated at the cytoplasmic tail downstream of Cys403 (CHO-ETBRΔ403) coupled with Gq but not with Gi, whereas CHO cells expressing the unpalmitoylated (Cys402Cys403Cys405→Ser402Ser403Ser405) ETBR (CHO-SerETBR) coupled with neither Gq nor Gi (Okamoto et al., 1997).
Recent reports demonstrated that ET-1-induced extracellular Ca2+ influx through voltage-independent Ca2+ channels (VICCs) plays a critical role for ET-1-induced vasoconstriction and cell proliferation (Zhang et al., 1999; Kawanabe et al., 2002). Thus, it is important to elucidate the activation mechanisms of VICCs by ET-1. We focused on investigating which G-protein subtypes were involved in activation of each Ca2+ channel by ET-1 in CHO-ETBR. Transfection and functional expression of the cDNA clone for wild type or mutant ETBRs into the same cell type provide a model system to study the precise characteristics of signal transduction by a single receptor subtype. We used CHO cells stably expressing wild-type or mutant ETBRs in the present study. A recent report showed that ET-1 activates two types of Ca2+-permeable nonselective cation channels (designated NSCC-1 and NSCC-2) in CHO-ETBR (Kawanabe et al., 2001). In particular, these channels can be distinguished using Ca2+ channel blockers, SK&F 96365 and LOE 908. Thus, NSCC-1 is sensitive to LOE 908 and resistant to SK&F 96365, whereas NSCC-2 is sensitive to both LOE 908 and SK&F 96365 (Kawanabe et al., 2001). In the present study, we used a dominant negative mutant of G13 and two types of mutated ETBRs designated ETBRΔ403 and SerETBR to clarify the involvement of Gq, GI and G13 for Ca2+ channel activation by ET-1.
Methods
Mutagenesis
Wild-type G12 and G13 in pcDNA3(+) were kindly provided by Dr Manabu Negishi (Kyoto University, Japan). G12G228A and G13G225A, which were shown to be the dominant negative types (Gohla et al., 1999), were generated using the QuickChange™ site-directed mutagenesis kit (Stratagene, La Jolla, CA, U.S.A.). Mutations were verified by sequencing.
Cell culture
We used CHO-ETBR, CHO-ETBRΔ403 and CHO-SerETBR, which were constructed as described previously (Okamoto et al., 1997). The KD and Bmax values of selected cell clones that were used in this study were 43±3 pM and 0.98±0.11 pmol (mg protein)−1, respectively, for CHO-ETBR; 122±10 pM and 1.66±0.12 pmol (mg protein)−1, respectively, for CHO-ETBRΔ403; and 38±3 pM and 1.29±0.10 pmol (mg protein)−1, respectively, for CHO-SerETBR. Cells were maintained in Ham's F-12 medium supplemented with 10% foetal calf serum (FCS) under a humidified 5% CO2/95% air atmosphere.
Microinjection
For microinjection, cells were seeded onto glass coverslips coated with fibronectin (Iwaki glass, Chiba, Japan), which were marked with a cross to facilitate the localization of injected cells, and incubated overnight in Ham's F-12 medium containing 1% FCS. Plasmids (100 ng μl−1) encoding for G12G228A and G13G225A were microinjected into cell nuclei. As a control, expression plasmids without inserts were microinjected in an adjacent field on the same coverslip. Microinjection was performed using a manual microinjection system (Eppendorf, Hamburg, Germany) equipped with an Axiovert 100 inverted microscope (Carl-Zeiss, Frankfurt, Germany).
Stress fiber formation
After incubation of the cells with serum-free Ham's F-12 medium for 24 h, ET-1 was added at 37°C for 5 min. Cells were washed three times with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS at room temperature for 15 min. After being washed five times with PBS containing 0.1% Triton-X100 (PBS-Tx), the cells were incubated with fluorescein rhodamin-phalloidin (Molecular Probes, Eugene, OR, U.S.A.) in PBS-Tx (1 : 200) at room temperature for 10 min. After being washed five times with PBS-Tx, the labeled cells were mounted on glass slides and examined with an MRC 1024 laser-scanning confocal microscope (Bio-Rad, Hercules, CA, USA) equipped with an Axiovert 135 M inverted microscope (Carl-Zeiss, Frankfurt, Germany).
Measurement of intracellular free Ca2+ concentration ([Ca2+]i)
[Ca2+]i was measured using a fluorescent probe fluo-3. The measurement of fluorescence by a CAF 110 spectrophotometer (JASCO, Tokyo, Japan) was performed exactly as described in a previous report (Kawanabe et al., 2001).
Microfluorimetry of fluo-3 was done as described previously (Zhang et al., 1999). Briefly, the cells were seeded on 35-mm glass-bottomed plastic dishes (MatTek Corporation, Ashland MA, U.S.A.), which were marked with a cross to facilitate the localization of injected cells, were loaded with fluo-3 by incubating them with Ca2+-free Krebs-HEPES solution containing 10 μM fluo-3/AM for 30 min at 37°C under a reduced light. Ca2+-free Krebs-HEPES solution contained (in mM): NaCl 140, KCl 3, MgCl2 1, glucose 11 and HEPES 10 (pH 7.4, adjusted with NaOH). After washing with Krebs-HEPES solution (2.2 mM CaCl2 was added to Ca2+-free Krebs-HEPES solution), they were kept in fresh Krebs-HEPES solution at 37°C for at least 30 min. Fluo-3 microfluorimetry was done at 25°C by an Attofluor Ratio- Vision real-time digital fluorescence analyser (Atto Instruments, Potomac, MD, U.S.A.) equipped with a Carl-Zeiss Axiovert-100 inverted epifluorescent microscope. A 100-W mercury burner served as the source of excitation. In measurement of [Ca2+]i, fluo-3 was excited at 450–490 nm and fluorescence was detected at 515–565 nm. At the end of the experiment, ionomycin and subsequently EGTA were added at final concentrations of 10 μM and 10 mM, respectively, to obtain the fluorescence intensity maximum (Fmax) and the fluorescence intensity minimum (Fmin). [Ca2+]i was determined from the equilibrium equation, [Ca2+]i=Kd(F - Fmin)/(Fmax-F), where F was the experimental value of fluorescence and Kd was defined as 0.4 μM (Minta et al., 1989).
Drugs
Boehringer Ingelheim K.G. (Ingelheim, Germany) kindly provided LOE 908. (R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide (Y-27632) was kindly provided by Welfide Corporation (Osaka, Japan). Chemicals were obtained from the following sources: ET-1 from the Peptide Institute (Osaka, Japan); rhodamin-phalloidin from Molecular Probes (Eugene, OR, U.S.A.); pertussis toxin and 1-(6-{[17β-3-methoxyestra-1,3,5(10)-trien-17-yl] amino}hexyl)-1H-pyrrole-2,5-dione (U73122) from Funakoshi Co. Ltd (Tokyo, Japan); SK&F 96365 from Biomol (Plymouth Meeting, PA, USA); and fluo-3/AM from Dojindo Laboratories (Kumamoto, Japan). All other chemicals were of reagent grade and were obtained commercially.
Statistical analysis
All results were expressed as mean±s.e.m.
Results
ET-1-induced actin stress fiber formation in CHO-ETBR
We attempted to determine the structural basis for coupling of ETBR with G12/G13 and subtypes of G proteins involved in ET-1-induced stress fibre formation. For these purposes, we examined the effects of inhibition of either one of the G protein-mediated signalling cascades by blockers and dominant negative mutants of G12 or G13 (G12G228A or G13G225A, respectively) on ET-1-induced actin stress fibre formation in CHO-ETBRΔ403 and CHO-SerETBR as well as CHO-ETBR. As described previously (Gohla et al., 1999), G12G228A inhibited ET-1-induced actin stress fibre formation in CHO cells expressing endothelinA receptors (data not shown).
ET-1 induced actin stress fiber formation in CHO-ETBR (Figure 1B). In contrast, ET-1 failed to induce stress fibre formation in CHO-ETBR that had been preincubated with 10 μM Y-27632, a selective ROCK inhibitor (Figure 1C). Y-27632 was added 15 min before stimulation with ET-1. Stress fiber formation was not affected by pretreatment with 5 μM U73122, a PLC inhibitor, or microinjection of G12G228A or G13G225A in CHO-ETBR (data not shown). When the cells were subjected to microinjection of G13G225A followed by pretreatment with U73122, ET-1 failed to induce stress fiber formation (Figure 1D). In contrast, microinjection of G12G228A in combination with pretreatment by U73122 had no effect on ET-1-induced stress fiber formation (data not shown). These results demonstrate that ETBR couples to G13 but not G12 in CHO cells.
Figure 1.
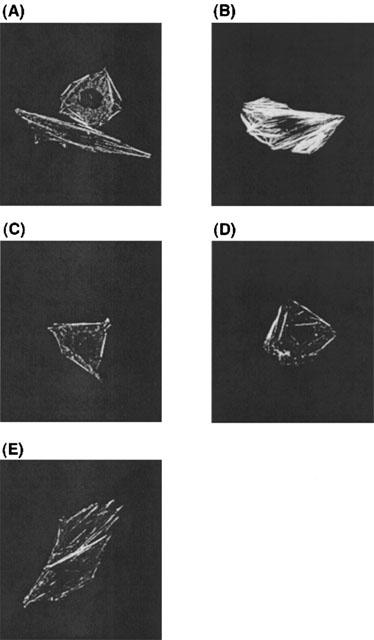
Effects of Y-27632, U73122, G12G228A and G13G225A on the ET-1-induced actin stress fibre formation in CHO-ETBR. Cells were stimulated with (B∼E) or without (A) 10 nM ET-1. The effects of preincubation with 10 μM Y-27632 (C), combination of G13G225A microinjection and 5 μM U73122 preincubation (D) and combination of G13G228A microinjection, PTX (50 ng ml−1) preincubation and 5 μM U73122 preincubation (E) on ET-1-induced stress fibre formation are shown. Y-27632, U73122 and PTX were added 15 min before stimulation with ET-1. Expression plasmids encoding for G12G228A and G13G225A were microinjected into the cell nuclei 24 h before stimulation with ET-1. Actin stress fibers were visualized as described in Methods. Representative examples of stress fibres in individual cells are shown.
A previous report demonstrated that, in preadipocytes,α2-adrenergic receptor activation of Rho was PTX sensitive (Betuing et al., 1998). In contrast, the other previous report showed that Rho-mediated effects on the cytoskeleton are PTX insensitive (Buhl et al., 1995). Therefore, we tried to clarify whether Gi played roles in ET-1-induced actin stress fibre formation in CHO-ETBR using pertussis toxin (PTX; 50 ng ml−1). It is generally accepted that PTX inhibits the effects of Gi (Takagi et al., 1995). The degree of ET-1-induced stress fiber formation in CHO-ETBR that had been treated with combination of G13G225A microinjection, PTX preincubation, and U73122 preincubation (Figure 1E) was similar to that in CHO-ETBR that had been treated with combination of G13G225A microinjection and U73122 preincubation (Figure 1D). These results indicate that Gi may not be involved in ET-1-induced actin stress fiber formation in CHO-ETBR.
ET-1-induced actin stress fiber formation in CHO-SerETBR and CHO-ETBRΔ403
ET-1 induced stress fiber formation in CHO-SerETBR (Figure 2B). Like CHO-ETBR, ET-1 induced stress fiber formation was inhibited by preincubation of CHO-SerETBR with Y-27632 (Figure 2C), but was not affected by microinjection of G12G228A (Figure 2D). Notably, unlike CHO-ETBR, it was inhibited by microinjection of G13G225A (Figure 2E).
Figure 2.
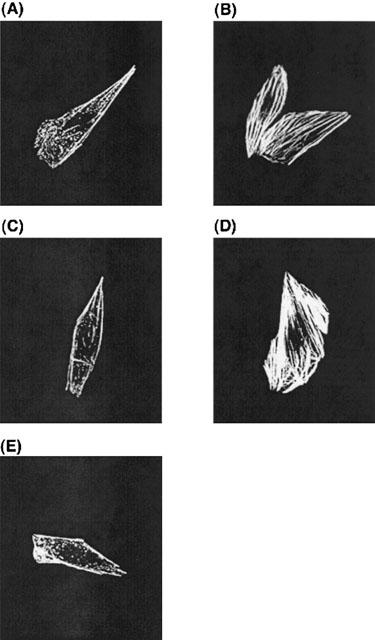
Effects of Y27632, G12G228A and G13G225A on ET-1-induced actin stress fiber formation in CHO-SerETBR. Cells were stimulated with (B∼E) or without (A) 10 nM ET-1. (C) Y-27632 at 10 μM was added 15 min before stimulation with ET-1. Expression plasmids encoding for G12G228A (D) and G13G225A (E) were microinjected into cell nuclei 24 h before stimulation with ET-1. Actin stress fibres were visualized as described in Methods. Representative examples of stress fibres in individual cells are shown.
ET-1 induced stress fiber formation in CHO-ETBRΔ403, in which coupling of the receptor with Gq but not Gi was retained (Figure 3B). Like CHO-ETBR, ET-1-induced stress fiber formation was inhibited by preincubation of CHO-ETBRΔ403 with Y-27632 (Figure 3C). Notably, unlike CHO-ETBR, it was inhibited by preincubation with U73122 (Figure 3D). In contrast, it was not affected by microinjection of G12G228A or G13G225A (Figure 3E,F).
Figure 3.
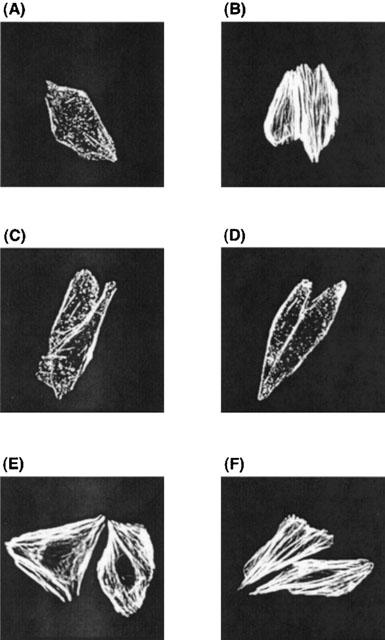
Effects of Y27632, U73122, G12G228A and G13G225A on ET-1-induced actin stress fiber formation in CHO-ETBRΔ403. Cells were stimulated with (B∼F) or without (A) 10 nM ET-1. Y-27632 at 10 μM (C) and U73122 at 5 μM (D) were added 15 min before stimulation with ET-1. Expression plasmids encoding G12G228A (E) and G13G225A (F) were microinjected into the cell nuclei 24 h before stimulation with ET-1. Actin stress fibres were visualized as described in Methods. Representative examples of stress fibres in individual cells are shown.
These results indicate that SerETBR, but not ETBRΔ403, couples with G13.
Basic properties of the ET-1-induced increase in [Ca2+]i in CHO-ETBR, CHO-ETBRΔ403 and CHO-SerETBR
ET-1 induced a biphasic increase in [Ca2+]i in CHO-ETBR, consisting of an initial transient phase and a subsequent sustained phase (Figure 4A). Both the transient and sustained increase in [Ca2+]i were dependent on the concentrations of ET-1, and reached the maximal value at concentrations ⩾1 nM (Figure 4D,E).
Figure 4.
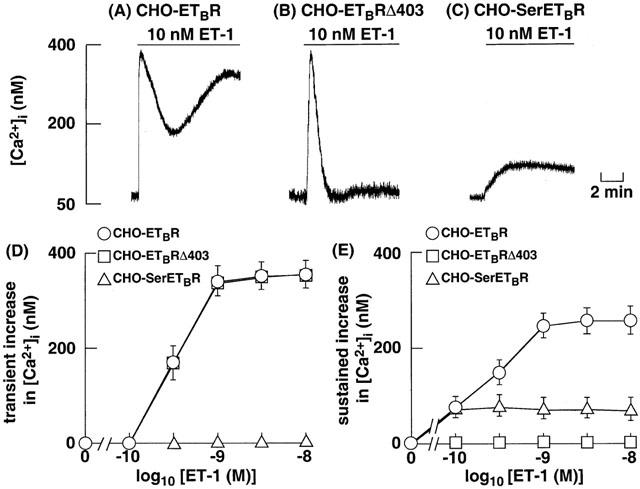
Original tracings illustrating the effects of ET-1 on the increase in [Ca2+]i in CHO-ETBR (A), CHO-ETBRΔ403 (B) and CHO-SerETBR (C). The cells were loaded with fluo-3 and stimulated with 10 nM ET-1 at the time indicated by arrows. Effects of various concentrations of ET-1 on the transient increase in [Ca2+]i (D) and the sustained increase in [Ca2+]i (E) in CHO-ETBR, CHO-ETBRΔ403 and CHO-SerETBR. The values for CHO-ETBR, CHO-ETBRΔ403 and CHO-SerETBR were represented by circles, squares and triangles, respectively. Each point represents the mean±s.e.m. of five experiments.
In CHO-ETBRΔ403, which are coupled with Gq alone, stimulation with 1 nM ET-1 caused a transient peak but not sustained increase in [Ca2+]i (Figure 4B). The magnitude of the transient increase in [Ca2+]i in CHO-ETBRΔ403 was essentially similar to that in CHO-ETBR (Figure 4D).
In CHO-SerETBR, which are coupled with G13 (Figure 2), ET-1 failed to induce an initial transient increase in [Ca2+]i, and it induced only a sustained increase in [Ca2+]i (Figure 4C). The magnitude of the sustained increase in [Ca2+]i in CHO-SerETBR was lower than that in CHO-ETBR (Figure 4E).
Pharmacological identification of Ca2+ channels activated by ET-1 in CHO-ETBR and CHO-SerETBR
As described previously (Kawanabe et al., 2001), in CHO-ETBR, the ET-1-induced sustained increase in [Ca2+]i was completely suppressed by the maximally effective concentration (10 μM) of LOE 908, whereas it was partially inhibited by the maximally effective concentration (10 μM) of SK&F 96365 (Figure 5A,B,E). PTX at 50 ng ml−1 failed to affect the resting [Ca2+]i and the ET-1-induced increase in [Ca2+]i in CHO-ETBR (data not shown). In CHO-SerETBR, the ET-1-induced sustained increase in [Ca2+]i was completely inhibited by 10 μM LOE 908, whereas SK&F 96365 at concentrations up to 10 μM had no effects (Figure 5C,D,F). These results demonstrate that ET-1 activates only NSCC-1 (LOE 908-sensitive and SK&F 96365 resistant) in CHO-SerETBR.
Figure 5.
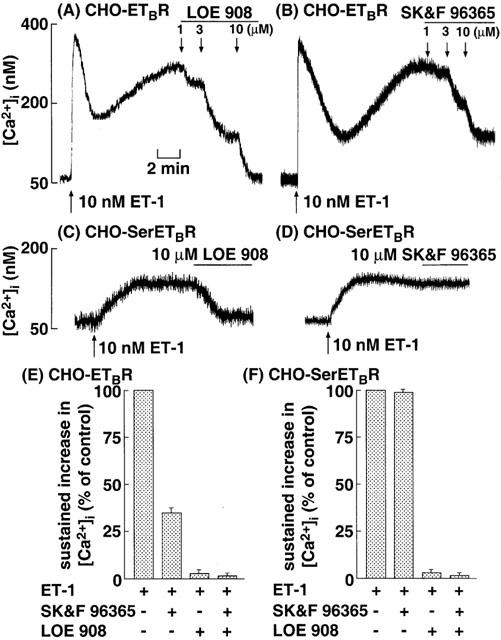
Original tracings illustrating the effects of various concentrations of LOE 908 and SK&F 96365 on the ET-1-induced sustained increase in [Ca2+]i in CHO-ETBR (A,B) and CHO-SerETBR (C,D). The cells were loaded with fluo-3 and stimulated with 10 nM ET-1 at the time indicated by arrows. After [Ca2+]i reached a steady-state, various concentrations of LOE 908 or SK&F 96365 was added as indicated by arrows. Effects of maximally effective concentration of LOE 908, SK&F 96365 and their combination on the ET-1-induced sustained increase in [Ca2+]i in CHO-ETBR (E) and CHO-SerETBR (F). The experimental protocols were described in Methods, and the values of [Ca2+]i following addition of 10 μM LOE 908 and/or 10 μM SK&F 96365 were determined. Each point represents the mean±s.e.m. of five experiments.
Effects of inhibition of PLC on the species of ET-1-activated Ca2+ channels in CHO-ETBR
In CHO-SerETBR, coupling between the receptor and Gq is missing and hence PLC as an effector of Gq cannot be activated upon stimulation of the receptor. To mimic the stimulation in CHO-SerETBR and confirm that PLC actually acts as an effector for activation of Ca2+ channels, we used U73122 in CHO-ETBR stimulated by ET-1. ET-1 at 1 nM induced only the sustained increase in [Ca2+]i in CHO-ETBR treated with 5 μM U73122 (Figure 6A,B). The magnitude of the sustained increase in [Ca2+]i was about 35% of that in the absence of U73122 (Figure 6C). This sustained increase in [Ca2+]i was completely inhibited by 10 μM LOE 908, whereas SK&F 96365 at concentrations up to 10 μM had no effects (Figure 6). These results indicate that ET-1 activates only NSCC-1 in CHO-ETBR treated with U73122.
Figure 6.
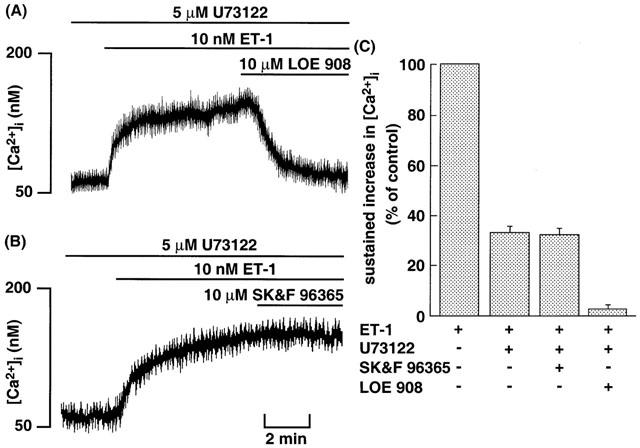
Original tracings illustrating the effects of maximally effective concentration of LOE 908 (A) and SK&F 96365 (B) on the ET-1-induced sustained increase in [Ca2+]i in CHO-ETBR pretreated with U73122. The cells loaded with fluo-3 were incubated with 5 μM U73122 for 10 min before 10 nM ET-1 stimulation. After [Ca2+]i reached a steady-state, 10 μM LOE 908 or 10 μM SK&F 96365 was added at the time indicated by horizontal bars. (C) Effects of maximally effective concentration of LOE 908 and SK&F 96365 on the ET-1-induced sustained increase in [Ca2+]i in CHO-ETBR pretreated with U73122. The experimental protocols were described in Methods, and the values of [Ca2+]i following addition of 10 μM LOE 908 or 10 μM SK&F 96365 were determined. Each point represents the mean±s.e.m. of five experiments.
Effects of G13 on the ET-1-induced sustained increase in [Ca2+]i in CHO-ETBR or CHO-SerETBR
To assess the involvement of G13 in the activation of Ca2+ channels, we investigated the effects of G13G225A on the ET-1-induced increase in [Ca2+]i in CHO-ETBR and CHO-SerETBR. In this experiment, G13G225A was microinjected into CHO-ETBR and CHO-SerETBR, and the ET-1-induced increase in [Ca2+]i in these cells was analysed using microfluorimetry.
ET-1 failed to induce sustained increase in [Ca2+]i in CHO-ETBR microinjected with G13G225A (Figure 7A) and CHO-SerETBR microinjected with G13G225A (Figure 7B). These results indicate that G13 plays critical roles in the activation of NSCC-1 and NSCC-2 caused by ET-1.
Figure 7.
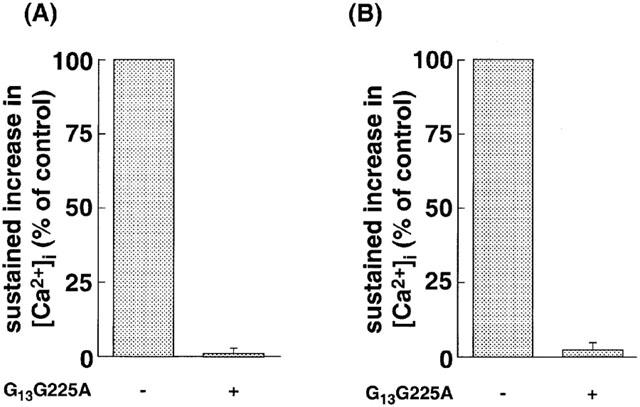
Effects of G13G225A on ET-1-induced sustained increase in [Ca2+]i in CHO-SerETBR (A) and CHO-ETBR (B). The cells loaded with fluo-3 were stimulated by 10 nM ET-1. The sustained increase in [Ca2+]i in the presence of G13G225A is represented as a percentage of values in its absence. Data are presented as mean±s.e.m. of three experiments.
Effects of Y-27632 on the ET-1-induced sustained increase in [Ca2+]i in CHO-ETBR
It is generally accepted that Rho/Rho-kinase (ROCK) pathway is a downstream target of G13 (Seasholtz et al., 1999). We examined the effects of ROCK on ET-1-induced increase in [Ca2+]i in CHO-ETBR. In this experiment, Y-27632 was used as a specific inhibitor of ROCK (Uehata et al., 1997). Y-27632 was added 15 min before stimulation with ET-1. Y-27632 at 10 μM did not affect the ET-1-induced transient and sustained increase in [Ca2+]i (Figure 8).
Figure 8.
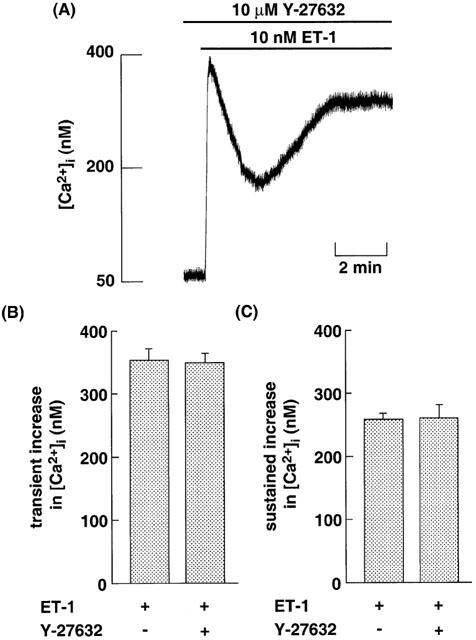
(A) Original tracing illustrating the effects of Y-27632 on the ET-1-induced increase in [Ca2+]i in CHO-ETBR. The cells loaded with fluo-3 were incubated with 10 μM Y-27632 for 15 min before 10 nM ET-1 stimulation. (B, C) Effects of 10 μM Y-27632 on 10 nM ET-1-induced transient (B) and sustained (C) increase in [Ca2+]i in CHO-ETBR. The experimental protocols were described in Methods. Data are presented as mean±s.e.m. of three experiments.
Discussion
As reported previously (Kawanabe et al., 2001), the ET-1-induced sustained increase in [Ca2+]i in CHO-ETBR results from extracellular Ca2+ influx through two types of Ca2+-permeable NSCC: NSCC-1 and NSCC-2 (Figures 4 & 5). The most important novelty of this study is that ET-1-induced NSCC-1 activation depends on G13-dependent cascade, and NSCC-2 activation depends on both Gq/PLC- and G13-dependent cascades in CHO-ETBR.
To identify the G proteins involved in the activation of these Ca2+ channels by ET-1, we assessed whether G12 and G13 are coupled with CHO-ETBR using actin stress fibre formation. A previous report demonstrated that Gq and G12/G13 play important roles in the majority of GPCR-induced, Rho-mediated effects on the cytoskeleton (Seasholtz et al., 1999). Based on sensitivity to U73122, G12G228A and G13G225A (Figure 1), we conclude that ET-1-induced stress fiber formation is mediated via two signalling pathways in CHO-ETBR (i.e., the Gq/PLC- and G13-dependent pathways), and also that only one of the two may be sufficient for actin stress fibre formation. Moreover, Gi may not be involved in ET-1-induced stress fiber formation, because treatment of the cells with sufficient concentrations of PTX did not affect ET-1-induced actin stress fiber formation in CHO-ETBR (Figure 1E). This result is in agreement with the previous indication that Rho-mediated effects on the cytoskeleton are PTX insensitive (Buhl et al., 1995). On the other hand, based on sensitivity to Y-27632, the Rho/ROCK pathway plays important roles in ET-1-induced stress fiber formation in CHO-ETBR (Figure 1C) as reported for a variety of cells (Mao et al., 1998; Gohla et al., 1999; Seasholtz et al., 1999). Therefore, Rho/ROCK pathway may be downstream of both the Gq/PLC- and G13-dependent pathways. We deduced the structural determinant for coupling of ETBR with G13 based on data from experiments using mutated ETBRs. That is, loss of coupling of ETBRΔ403 with G13 (Figure 3F) and retention of coupling of SerETBR with G13 (Figure 2E) clearly show that the cytoplasmic tail downstream of Cys403 but not the palmitoylation site of ETBR is essential for coupling with G13. Judging from these data, we conclude that the cytoplasmic tail downstream of the palmitoylation sites of ETBR, but not the palmitoylation site itself, is essential for coupling with G13.
Given that the subtypes of G proteins that are coupled with CHO-ETBR has become clear, we analysed G proteins involved in the activation of NSCCs caused by ET-1. Judging from the data using U73122-treated CHO-ETBR and CHO-ETBRΔ403 (Figures 4 & 6), Gq/PLC-independent pathways as well as Gq/PLC-dependent pathways are involved in NSCC-2 activation, while only Gq/PLC-independent pathways are involved in NSCC-1 activation. To elucidate the mechanism of NSCC-1 activation, we examined the effects of G13 using CHO-SerETBR. ET-1-induced sustained increase in [Ca2+]i is abrogated in CHO-SerETBR microinjected with G13G225A (Figure 7A). These results indicate that the G13-dependent pathway seems to play an essential role for activation of NSCC-1 caused by ET-1. Moreover, these results support the indication that the cytoplasmic tail downstream of the palmitoylation sites of ETBR, but not the palmitoylation site itself, is essential for coupling with G13. Next, we examined the mechanisms of NSCC-2 activation using CHO-ETBR microinjected with G13G225A. ET-1 failed to induce sustained increase in [Ca2+]i in these cells (Figure 7B). These results indicate that a G13-dependent pathway plays an important role for activation of NSCC-2 as well as NSCC-1. In contrast, judging from the results using PTX, activation of NSCC-1 and NSCC-2 by ET-1 seems not to involve Gi-dependent pathway. In conclusion, NSCC-2 activation by ET-1 involves G13-dependent pathway as well as Gq/PLC-dependent pathway. Recent reports demonstrated that G13 regulates cell growth (Seasholtz et al., 1999). Moreover, extracellular Ca2+ influx through NSCCs is involved in ET-1-induced cell proliferation in a variety of cells (Kawanabe et al., 2001; 2002). Therefore, G13 may be an effector for ET-1-induced cell proliferation by stimulating NSCCs. Collectively, among the subtypes of Gα proteins, Gq and G13 play important roles for activation of NSCCs as follows; (1) NSCC-1 activation involves a G13-dependent pathway. (2) NSCC-2 involves both Gq/PLC- and G13-dependent pathways.
It is important to understand the mechanisms of ET-1 activation for each Ca2+ channel downstream of Gq and/or G13. Because Rho/ROCK pathway is a downstream target of G13 (Seasholtz et al., 1999), we investigated the effects of ROCK on the activation of Ca2+ channels by ET-1 using a selective ROCK inhibitor, Y-27632. However, Y-27632 did not affect ET-1-induced increase in [Ca2+]i (Figure 8). This result indicates that G13 activates NSCC-1 and NSCC-2 via ROCK-independent signaling pathways. Therefore, G13 may have another intracellular signalling pathway for activating NSCCs. Further study is needed to identify the effectors downstream of G13 for activation of NSCC-1 and NSCC-2.
Acknowledgments
We thank Boehringer Ingelheim K.G. for the kind donation of LOE 908. We also thank Welfide Corporation for the kind donation of Y-27632.
Abbreviations
- [Ca2+]i
intracellular free Ca2+ concentration
- CHO-ETBR
Chinese hamster ovary cells that stably express human endothelinB receptor
- CHO-ETBRΔ403
Chinese hamster ovary cells that express human endothelinB receptor truncated at the carboxyl-terminal downstream of Cys403
- CHO-SerETBR
Chinese hamster ovary cells that express the unpalmitoylated (Cys402Cys403 Cys405 → Ser402Ser403 Ser405) human endothelinB receptor
- ET-1
endothelin-1
- FCS
foetal calf serum
- Fmax
fluorescence intensity maximum
- Fmin
fluorescence intensity minimum
- G12G228A
dominant negative type of G12
- G13G225A
dominant negative type of G13
- GPCR
heterotrimetric guanine nucleotide-binding protein (G-protein)-coupled receptor
- NSCC
nonselective cation channel
- IPs
inositol phosphates
- PBS
phosphate-buffered saline
- PBS-Tx
phosphate-buffered saline containing 0.1% Triton-X100
- PLC
phospholipase C
- PTX
pertussis toxin
- ROCK
Rho-associated kinase
- U73122
1-(6-{[17β-3-methoxyestra-1,3,5(10)-trien-17-yl]amino}hexyl)-1H-pyrrole-2,5-dione
- VICC
voltage-independent Ca2+ channel
- Y-27632
(R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide
References
- ARAI H., HORI S., ARAMORI I., OHKUBO H., NAKANISHI S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348:730–732. doi: 10.1038/348730a0. [DOI] [PubMed] [Google Scholar]
- ARAMORI I., NAKANISHI S. Coupling of two endothelin receptor subtypes to differing signal transduction in transfected Chinese hamster ovary cells. J. Biol. Chem. 1992;267:12468–12474. [PubMed] [Google Scholar]
- BETUING S., DAVIAUD D., PAGES C., BONNARD E., VALET P., LAFONTAN M., SAULNIER-BLACHE J.S. Gβγ-independent coupling of α2-adrenergic receptor to p21rhoA in preadipocytes. J. Biol. Chem. 1998;273:15804–15810. doi: 10.1074/jbc.273.25.15804. [DOI] [PubMed] [Google Scholar]
- BUHL A.M., JOHNSON N.L., DHANASEKARAN N., JOHNSON G.L. G alpha 12 and G alpha 13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 1995;270:24631–24634. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- GOHLA A., OFFERMANNS S., WILKIE T.M., SCHULTZ G. Differential involvement of Galpha12 and Galpha13 in receptor-mediated stress fiber formation. J. Biol. Chem. 1999;274:17901–17907. doi: 10.1074/jbc.274.25.17901. [DOI] [PubMed] [Google Scholar]
- GOHLA A., SCHULTZ G., OFFERMANNS S. Role of G12/G13 in agonist-induced vascular smooth muscle cell contraction. Circ. Res. 2000;87:221–227. doi: 10.1161/01.res.87.3.221. [DOI] [PubMed] [Google Scholar]
- HORSTMEYER A., CRAMER H., SAUER T., MULLER-ESTERL W., SCHROEDER C. Palmitoylation of endothelin receptor A. Differential modulation of signal transduction activity by post-translational modification. J. Biol. Chem. 1996;271:20811–20819. doi: 10.1074/jbc.271.34.20811. [DOI] [PubMed] [Google Scholar]
- KAWANABE Y., HASHIMOTO N., MASAKI T. Ca2+ channels involved in endothelin-induced mitogenic response in carotid artery vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2002;282:C330–C337. doi: 10.1152/ajpcell.00227.2001. [DOI] [PubMed] [Google Scholar]
- KAWANABE Y., OKAMOTO Y., ENOKI T., HASHIMOTO N., MASAKI T. Ca2+ channels activated by endothelin-1 in CHO cells expressing endothelin-A or endothelin-B receptors. Am. J. Physiol. Cell Physiol. 2001;281:C1676–C1685. doi: 10.1152/ajpcell.2001.281.5.C1676. [DOI] [PubMed] [Google Scholar]
- KITAMURA K., SHIRAISHI N., SINGER W.D., HANDLOGTEN M.E., TOMITA K., MILLER R.T. Endothelin-B receptors activate Gα13. Am. J. Physiol. Cell Physiol. 1999;276:C930–C937. doi: 10.1152/ajpcell.1999.276.4.C930. [DOI] [PubMed] [Google Scholar]
- MAO J., YUAN H., XIE W., SIMON M.I., WU D. Specific involvement of G proteins in regulation of serum response factor-mediated gene transcription by different receptors. J. Biol. Chem. 1998;273:27118–27123. doi: 10.1074/jbc.273.42.27118. [DOI] [PubMed] [Google Scholar]
- MASAKI T. Endothelins: homeostatic and compensatory actions in the circulatory and endocrine systems. Endocr. Rev. 1993;14:256–268. doi: 10.1210/edrv-14-3-256. [DOI] [PubMed] [Google Scholar]
- MINTA A., KAO J.P.Y., TSIEN R.Y. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 1989;264:8171–8178. [PubMed] [Google Scholar]
- OKAMOTO Y., NINOMIYA H., TANIOKA M., SAKAMOTO A., MIWA S., MASAKI T. Palmitoylation of human endothelinB. J. Biol. Chem. 1997;272:21589–21596. doi: 10.1074/jbc.272.34.21589. [DOI] [PubMed] [Google Scholar]
- SAKURAI T., YANAGISAWA M., TAKUWA Y., MIYAZAKI H., KIMURA S., GOTO K., MASAKI T. Cloning of cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature. 1990;348:732–735. doi: 10.1038/348732a0. [DOI] [PubMed] [Google Scholar]
- SEASHOLTZ T.M., MAJUMDAR M., BROWN J.H. Rho as a mediator of G protein-coupled receptor signaling. Mol. Pharmacol. 1999;55:949–956. doi: 10.1124/mol.55.6.949. [DOI] [PubMed] [Google Scholar]
- TAKAGI Y., NINOMIYA H., SAKAMOTO A., MIWA S., MASAKI T. Structural basis of G protein specificity of human endothelin receptors. A study with endothelinA/B chimeras. J. Biol. Chem. 1995;270:10072–10078. doi: 10.1074/jbc.270.17.10072. [DOI] [PubMed] [Google Scholar]
- UEHATA M., ISHIZAKI T., SATOH H., ONO T., KAWAHARA T., MORISHIMA T., TAMAKAWA H., YAMAGAMI K., INUI J., MAEKAWA M., NARUMIYA S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- YANAGISAWA M., KURIHARA S., KIMURA S., TOMOBE Y., KOBAYASHI M., MITSUI Y., YAZAKI Y., GOTO K., MASAKI T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- ZHANG X.F., IWAMURO Y., ENOKI T., OKAZAWA M., LEE K., KOMURO T., MINOWA T., OKAMOTO Y., HASEGAWA H., FURUTANI H., MIWA S., MASAKI T. Pharmacological characterization of Ca2+ entry channels in endothelin-1-induced contraction of rat aorta using LOE 908 and SK&F 96365. Br. J. Pharmacol. 1999;127:1388–1398. doi: 10.1038/sj.bjp.0702661. [DOI] [PMC free article] [PubMed] [Google Scholar]