

Abstract
The aim of this study was to investigate the binding of a novel GABAB receptor radioligand, [3H]-CGP62349, to human post-mortem control and epileptic hippocampal sections using quantitative receptor autoradiography. Utilizing human control hippocampal sections it was shown that [3H]-CGP62349 bound with high affinity (KD 0.5 nM) to this tissue.
Hippocampal slices from surgical specimens obtained from patients with hippocampal sclerosis (HS) and temporal lobe epilepsy (TLE) were compared with neurologically normal post-mortem control subjects for neuropathology and GABAB receptor density and affinity. Neuronal loss was observed in most of the hippocampal subregions, but in the subiculum no significant difference was detected.
The localization of GABAB receptors with the antagonist [3H]-CGP62349 in human control hippocampal sections supported and extended earlier studies using the agonist ligand [3H]-GABA.
The kinetics of binding to the GABAB receptor in human hippocampus using this novel compound was comparable to previous data obtained in rat hippocampal membranes.
GABAB receptor density (Bmax) was significantly reduced in CA3, hilus, and dentate gyrus (DG); the affinity was increased exclusively in DG. The trend is identical in all the hippocampal subregions with the agonist and the antagonist, although significant differences with the antagonist where recorded in CA3 and hilus, whereas with the agonist a significant reduction was reported in all of the hippocampal subfields.
GABAB receptor expression per remaining neuron appeared significantly increased in CA3 and hilus. These results suggest altered GABAB receptor function may occur in human TLE, possibly as a result of synaptic reorganization, and may contribute to epileptogenesis.
Keywords: GABAB receptors, hippocampus, temporal lobe epilepsy, hippocampal sclerosis, GABAB antagonist
Introduction
γ-Aminobutyric acid GABA is the main inhibitory neurotransmitter in the mammalian brain (Curtis et al., 1974; Krnjevic, 1974), which acts through ionotropic (GABAA/C) and metabotropic (GABAB) receptors to modulate neuronal activity (Hill et al., 1984; Andrade et al., 1986; Kaupmann et al., 1997). Stimulation of postsynaptic GABAB receptors increases neuronal K+ conductance to generate long-lasting inhibitory postsynaptic potentials (Dutar & Nicoll, 1988) and inhibition of adenylate cyclase activity, leading to a reduction of cAMP levels (Wojcik & Neff, 1984). Activation of presynaptic GABAB receptors decreases Ca2+ influx (Takahashi et al., 1998). Each of these events is mediated via G-proteins (Misgeld et al., 1995).
In 1997 cDNA sequences for GABAB1 receptors were published (Kaupmann et al., 1997) and a second GABAB receptor subunit, GABAB2, was identified a year later (Kaupmann et al., 1998; Jones et al., 1998; White et al., 1998) with homology to GABAB1. It then became apparent that the functional receptor comprises a dimeric composition formed by isoforms of these two subunits. Current evidence suggests that whilst both subunits are required for expression of the functional receptor in the neuronal membrane, only GABAB1 provides the binding domain. GABAB2 appears to be responsible for second messenger processing (Kuner et al., 1999; Martin et al., 1999; Ng et al., 1999; Robbins et al., 2001).
Temporal lobe epilepsy (TLE) is a common adult seizure disorder and, when associated with hippocampal sclerosis (HS), is the most refractory to pharmacotherapy (Engel, 1994; Semah et al., 1998). Neuronal loss has been described in HS (Kim et al., 1990; Hopkins et al., 1995), with a specific pattern of cell loss, notably with individual regions being differentially affected (Babb et al., 1984). Evidence from electrophysiological studies of neurones from human hippocampal sclerosis specimens, has indicated a reduction in evoked inhibitory postsynaptic potentials (IPSP) when compared to neurones in specimens obtained from patients with structural lesions (Isokawa et al., 1991; Knowles et al., 1992). This suggests that GABA mediated inhibition in the hippocampus of HS patients may be compromised and alterations in GABAA receptor subtypes have been described in relation to human TLE (Loup et al., 2000). Numerous studies in animal models of TLE have also shown different degrees of impairment in GABAB mediated neurotransmission (Asprodini et al., 1992; Buhl et al., 1996; Mangan & Lothman, 1996; Haas et al., 1996; Wu & Leung, 1997) suggesting that the GABAB receptor may also play an important role in the control of neuronal excitability and seizures.
The aim of the present study was to investigate abnormalities in GABAB receptor binding parameters in HS and TLE, using the antagonist [3H]-CGP62349 for receptor autoradiographic analysis and to relate this to the quantitative neuropathology from the same hippocampal specimens.
Methods
Tissue preparation
Human brain tissue was obtained from patients with medically refractory, unilateral, mesial TLE who were undergoing surgical resection of the hippocampus (Table 1). Following resection, the specimens were immediately frozen and stored at −80°C until further sectioning. The remaining hippocampal tissue was fixed in 4% formalin and subsequently paraffin-embedded for microtome sectioning.
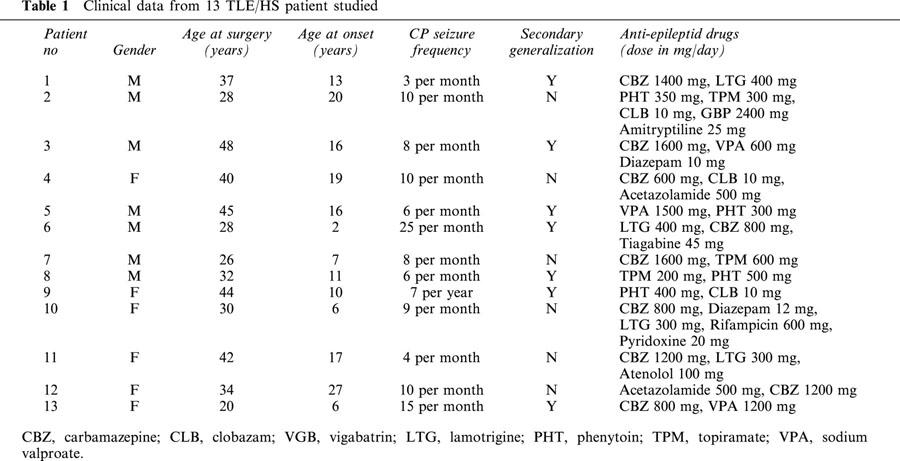
Table 1.
Clinical data from 13 TLE/HS patient studied
Control hippocampi were obtained at autopsy, from individuals with no previous medical history of neurological or psychiatric disease (Table 2). Control brains were dissected into blocks, frozen and stored at −80°C (Kingsbury et al., 1996). The remaining hemisphere was treated as described for the hippocampal sclerosis specimens.
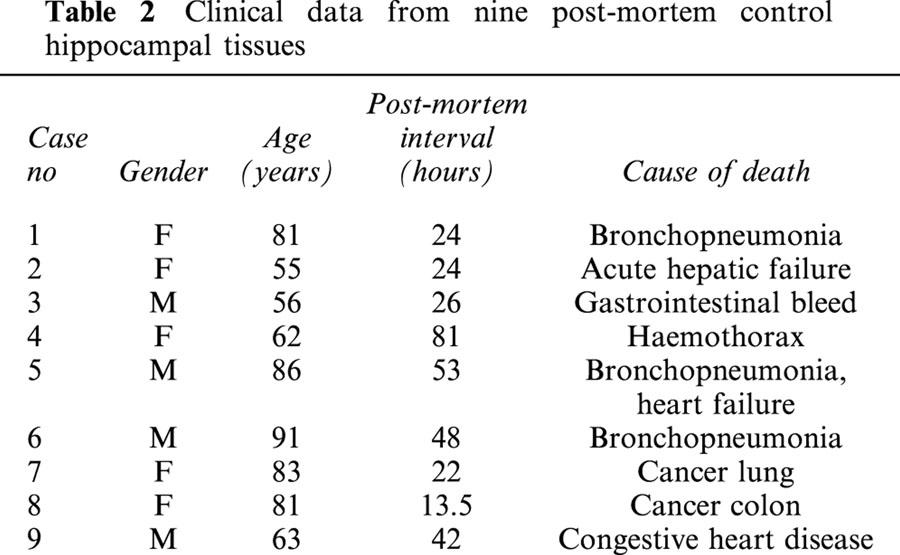
Table 2.
Clinical data from nine post-mortem control hippocampal tissues
Sections (12 μm) of frozen human brain tissue were cut at −20°C and thaw mounted onto charged microscope slides (BDH Superfrost Plus). Tissue sections were allowed to air-dry for up to 30 min in order to ensure adhesion to the microscope slides and stored with desiccant in plastic slide boxes at −80°C.
Tissue sections for quantitative neuropathology were cut from paraffin-embedded blocks of formalin-fixed tissue, using a microtome set at 25 μm. Sections were mounted onto charged microscope slides, counterstained with cresyl-violet/luxol fast blue and stored at room temperature until required.
This study was approved by the Joint Medical Ethics Committee of Institute of Neurology and the National Hospital for Neurology and Neurosurgery.
Quantitative neuropathology
Quantitative neuropathology consists of assessment of neuronal numbers in hippocampal subfields of control and sclerotic hippocampi, was based on three-dimensional counting (3DC) methods published by Williams & Rakic (1988). Briefly, the neuronal densities were estimated in six subregions of human hippocampi (granular layer of the DG, and pyramidal layer of hilus, CA3, CA2, CA1, and subiculum) in neurologically normal post-mortem controls and in tissue from TLE patients, in 20 μm cresyl violet stained sections of paraffin embedded tissues from an adjacent part of the same sample.
Receptor autoradiography
Sections were removed from the freezer on the day of the assay and allowed to equilibrate to room temperature for 1 h. They were then incubated in assay buffer (50 mM Tris/HCl pH 7.4, 2.5 mM CaCl2) at 23°C for 20 min, followed by a further 60 min in fresh assay buffer. Slides were then dried under a stream of cool air for 30 min. Each section was incubated for 60 min in solution containing the receptor ligand, [3H]-CGP62349 (0.125–8 nM). Non-specific binding was determined by the addition of an unlabelled GABAB antagonist, CGP54626A (10 μM). After incubation the solution was aspirated and the slides dipped in assay buffer for 2×1 min followed by 1 min dip in distilled water to remove buffer salt. Slides were allowed to dry under a stream of cool air for 30 min before being apposed to [3H]-sensitive autoradiographic films (Hyperfilm, Amersham, U.K.) in lightproof cassettes for 21 days stored at room temperature. Each cassette contained a slide mounted plastic impregnated microscale along with the experimental slides to allow for subsequent quantitative densitometric analysis.
Quantitative data analysis
Quantification of receptor autoradiography was achieved by film densitometry using an image analysis system (MCID–Microcomputer Imaging Device, Imaging Research Inc., Canada), and optical density was converted to fmol mg−1 of bound ligand. Total binding was assessed in two to four sections per concentration of [3H]-CGP62349. Binding parameters receptor density (Bmax) and affinity (KD) were determined by the use of the Langmuir equation in Prism PC software (GraphPad Software, San Diego, CA, U.S.A.).
Results
Comparison of neuronal density in control and epileptic patients
In hippocampi resected from patients with HS, significant neuronal loss was observed in all hippocampal subregions, except for the subiculum (Figure 1). CA3, hilus, and DG, were the most severely affected, with neuronal density reductions of 72, 77, and 73% respectively. A lower neuronal loss was detected in CA1 and CA2 at 56 and 58%, whilst no significant difference was noted in the subiculum (3%).
Figure 1.
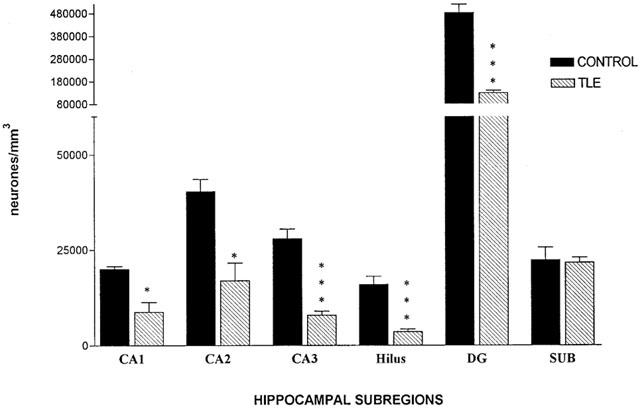
Mean neuronal density in post-mortem control hippocampal samples (n=5) and resected epileptic hippocampal tissues (n=6–11). Three-dimensional cell counts were performed on formaline fixed tissue sections (20 μm thickness). Data are expressed as mean±s.e.mean. Statistical analysis used unpaired Student's t-test (two-tailed), where *P<0.05, **P<0.01, ***P<0.001. SUB, subiculum.
GABAB receptor autoradiography
The association of [3H]-CGP62349 to GABAB receptors at 23°C was rapid, with equilibrium reached within 60 min, and stability maintained for at least a further 60 min. Association at 4°C was much slower with equilibrium reached after 120 min. Analysis performed assuming pseudo-first order association kinetics indicated an apparent association rate at 23°C of=0.082±0.014, and a half-life of t1/2=18.32±3.76 min. An incubation time of 60 min at 23°C was adopted for use in subsequent studies.
Dissociation by infinite dilution of [3H]-CGP62349 for increasing periods of time was demonstrated to be initially rapid for the first 10 min, then very slow with a substantial amount of binding remaining at 120 min. Kinetic analysis yielded a dissociation rate constant k−1=0.45±0.34 min−1 with a half-life of t1/2=1.53±0.31 min at 23°C, and a dissociation rate constant k−1=0.11+0.05 min−1 with a half-life of t1/2=6.24+1.13 min at 4°C. A washing schedule of 2×1 min at 23°C was adopted, as this provided a high percentage of specific binding (approximately 89% at 0.5 nM).
A representative example of a saturation plot from CA1 of a single post-mortem control sample measured in duplicate is shown in Figure 2. Specific binding appeared to be saturable within the concentration range used (0.125–8 nM), and non-specific binding remains linear and constant at the film background levels over the entire concentration range employed. Analysis of specific components of binding using a one site hyperbola fit indicated an equilibrium dissociation constant KD=0.499±0.12 and a receptor density of Bmax=581±42.19 fmol mg−1 tissue.
Figure 2.
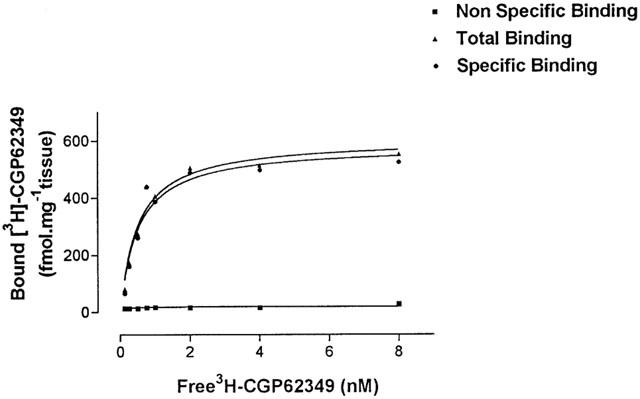
Concentrations of [3H]-CGP62349 were in the range 0.125 to 8 nM. Non-specific binding is indicated by closed squares, and total binding by closed triangles. The difference between total and non-specific gives specific binding denoted by dots. Non-specific binding was determined in the presence of 10 μM CGP54626A. Data shown are from a single tissue sample.
GABAB receptors distribution in the human control hippocampus
Figure 3A is an image of a section of post-mortem control hippocampus showing that GABAB receptors are widely and evenly distributed throughout the human hippocampus, as previously reported (Chu et al., 1987). The greatest density was observed in the granular layer of the dentate gyrus (SGDG), (Bmax value of 868.06±38.52 fmole mg−1). The strata oriens, pyramidale, radiatum and lacunosum of CA1 subregion (654.18±42.10 fmole mg−1), CA2 subregion (708.85±39.59 fmole mg−1), CA3 subregion (653.25± 26.79 fmole mg−1) and hilus (700.5±74.6 fmole mg−1), demonstrated a comparable level of binding between the hippocampal subregions. The subiculum expressed a lower level (429.32±37.62 fmole mg−1) of GABAB binding. Non-specific binding was the same level as the background.
Figure 3.
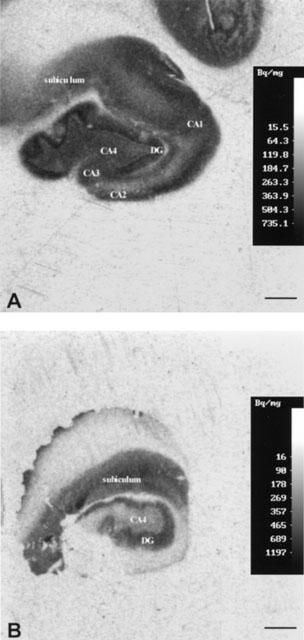
Autoradiographic images illustrating film optical density corresponding to total binding site distribution following autoradiography [3H]-CGP62349 (0.75 nM concentration) in sections of hippocampus from a control post-mortem brain numbered in Table 2 case no 5 (A) and an epileptic patient affected by HS (B). An apparent loss of GABAB receptor can be seen mainly in CA4, DG. The control and TLE hippocampi were processed at the same time in the same conditions. In TLE CA1, CA2 and CA3 were missed because of the surgical procedure. Scale bars represent 4 mm.
Comparison of receptor affinity (KD) between control and HS patients
Data obtained from saturation receptor autoradiography studies of hippocampi slices from control and HS patients describing the affinity of [3H]-CGP62349 for its binding site on GABAB receptors are summarized in Figure 4. In the control group the hilus had the lowest affinity for [3H]-CGP62349 and the subiculum demonstrated the highest affinity (KD values of 0.626±0.065 and 0.428±0.049 respectively). Binding affinity for [3H]-CGP62349 was similar for DG, CA1, CA2, CA3 subregions with KD values of 0.585±0.066, 0.527±0.044, 0.538±0.073, and 0.449±0.035 respectively. The affinity of [3H]-CGP62349 receptor sites increased significantly in the DG of HS patients as compared to post-mortem controls with a decrease in KD value of 53.51±4%. There were no significant differences in the other hippocampal subregions.
Figure 4.
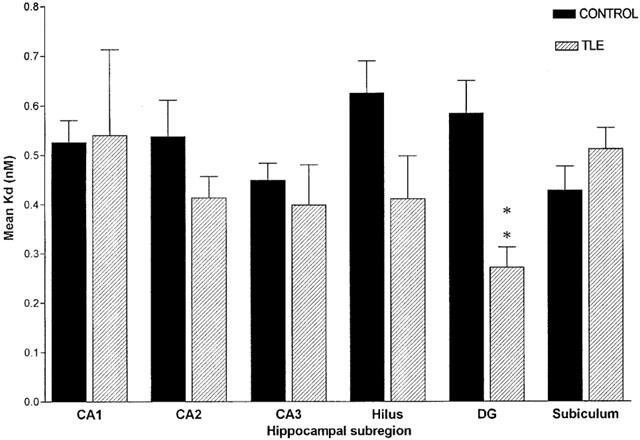
Mean receptor affinity (KD) in post mortem control hippocampal tissue (n=5–8) and resected epileptic hippocampal sample (n=8–13). Data are represented as mean±s.e.mean. Statistical analysis used unpaired Student's t-test (two-tailed), where **P<0.01. SUB, subiculum.
Comparison of Bmax between control and epileptic patients
Deficits in [3H]-CGP62349 binding to GABAB receptors are evident in the images of Figure 3. The comparison between the two images of [3H]-CGP62349 binding in control (Figure 3A) and HS (Figure 3B) hippocampi revealed an apparent loss of GABAB binding sites in the subregions in HS. The greatest loss was present in the hilus and DG with respective decreases of 52 and 47%. A minor deficit was observed in CA1 and CA3 with a reduction of 39 and 37%, whereas CA2 did not show a significant deficit with a reduction of 16% and the subiculum was unaltered (Figure 3).
The value of Bmax showed a reduction of [3H]-CGP62349 binding to GABAB, which was statistically significant in the DG 48±4% (P<0.001), in hilus 52±3% (P<0.001) and in CA3 36±8% (P<0.05), CA1 40±5% (P<0.05) (Figure 5).
Figure 5.
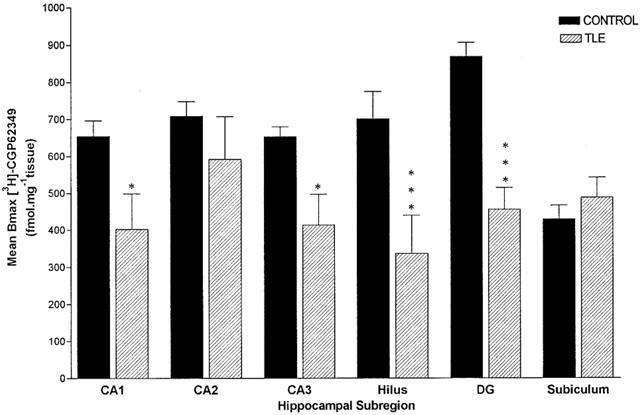
Mean receptor population (Bmax) in post mortem control hippocampal tissue (n=5–8) and resected epileptic hippocampal sample (n=8–13). Data are represented as mean±s.e.mean. Statistical analysis used unpaired Student's t-test (two-tailed), where *P<0.05, **P<0.01, ***P<0.001. SUB, subiculum.
It should be noted that binding data for CA3, CA2, and sometimes CA1, were not available in some of the HS patients due to damage to these areas occurring during resections.
[3H]-CGP62349 binding corrected for neuronal density
To correct for the alteration in the neuronal numbers in HS, we calculated the ratio of GABAB binding sites per neurone by dividing the Bmax values relating to each subregion by the corresponding neuronal density for that subregion (Figure 6). There was no significant change in the ratio of Bmax to neuronal density in four of the six subregions examined. However, in CA3 and hilus the ratio of Bmax to neuronal density in HS demonstrated an increment of 85 and 123% over mean control values and these increases in binding sites on surviving neurones were both statistically significant (P<0.05, P<0.001). Trends toward changes in binding within the different regions of the TLE hippocampus compared favourably with those observed in the previous study by Billinton et al. (2001) using the agonist ligand, [3H]-GABA.
Figure 6.
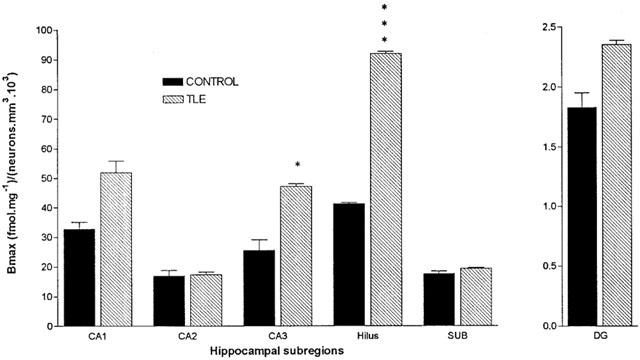
Histogram representing ratio of Bmax to neuronal density in tissue sections from post-mortem control and patients with HS. Histogram bars represent the mean ratio from hippocampal subregion and error bars represent s.e.mean. Groups were compared using an unpaired Student's t-test (two-tailed) where *P<0.05, ***P<0.001. SUB, subiculum.
Discussion
It is generally accepted that GABAA receptor mechanisms play a crucial role in the manifestation of partial and generalized tonic-clonic seizures (Möhler et al., 1997; Loup et al., 2000) whereas the contribution of GABAB receptors is unclear. Whilst GABAB sites have been implicated in absence seizures (Marescaux et al., 1992; Vergnes et al., 1997) there has been little to suggest that they are involved in the generation of partial and generalized tonic-clonic seizures (Sato et al., 1990; Kofler et al., 1994).
The present study was designed to test whether the hippocampus of patients with HS and TLE contains an altered density of GABAB receptors, which might contribute to the increased excitatory activity, observed in this tissue. HS encompasses a wide range of neuronal loss and this necessitated using quantitative 3D cell counting to obtain precise neurone densities together with an accurate diagnosis of the severity of HS in each case.
A relatively new high affinity antagonist ligand was used for this study. Several studies have previously been performed in rat brain utilizing the agonist ligands, [3H]-GABA and [3H]-baclofen (Hill & Bowery, 1981; Gehlert et al., 1985) which showed a high level of non-specific binding and a lower affinity compared to the new antagonist [3H]-CGP62349 which has a high affinity for GABAB sites with low non-specific binding (Bittiger et al., 1996). This ligand also associates rapidly, and dissociates slowly from the receptor. The distribution of GABAB receptors in rat brain, as displayed by [3H]-CGP62349, was similar to that obtained with [3H]-GABA and [3H]-baclofen (Wilkin et al., 1981; Gehlert et al., 1985; Bowery et al., 1987; Chu et al., 1990; Billinton et al., 2001).
Factors such as post-mortem interval (PMI) or sample storage times of 4–10 years have been reported to influence GABAB stimulated GTPase activity, and may affect agonist binding parameters (Odagaki et al., 1998), but we have bypassed these limitations using the antagonist ligand [3H]-CGP62349. The binding of the ligand would not be influenced by these factors because the antagonist binds to both the uncoupled receptor whilst the agonist is capable of only binding to the G-protein coupled receptor. After very long storage times e.g. 6 years it is suggested (Lloyd & Dreksler, 1979) that agonist binding may well be reduced. All of the tissue samples used in this study have been stored for less than 4 years. Interestingly, Lloyd & Dreksler (1979) have demonstrated that even [3H]-GABA binding in human tissue is independent of age, PMI, sex or storage time up to 6 years. If the agonist binding is not affected by any of these factors it is even less likely that antagonist binding would be affected.
GABAB receptor autoradiography
A previous study from our laboratory, utilizing the agonist [3H]-GABA, demonstrated a wide distribution of GABAB binding sites in all of the human hippocampal subregions. A significant decrease in receptor density in CA1, CA2, CA3, hilus, and DG regions of hippocampi from TLE patients was reported, whereas the subiculum showed an apparent increase in receptor density (Billinton et al., 2001). In the current study [3H]-CGP62349 also demonstrated a significant reduction in receptor density in CA1, CA3, hilus, and DG, thus confirming the previous results obtained with the agonist ligand in these four subregions. However, in CA2 no significant reduction was noted and there also appeared to be no change in the subiculum. As pointed out above this may be due to the agonist [3H]-GABA binding exclusively to the G-protein coupled receptor, whilst the antagonist, [3H]-CGP62349, binds to both the coupled and uncoupled receptor, giving a better indication of the total number of GABAB receptors. The former study (Billinton et al., 2001) also showed an increase in the receptor affinity, for the [3H]-GABA agonist, in CA3 and hilus, whereas the data obtained with the antagonist [3H]-CGP62349 demonstrated a significant augmentation in affinity only in the DG. The underlying reason(s) for the discrepancy is unclear but the 100-fold greater affinity of the antagonist over the agonist, [3H]-GABA, may contribute.
The data from the present study indicate that any decrease in receptor density, between controls and patients with HS and TLE, was minor in most of the hippocampal subregions compared to the study with [3H]-GABA. In the CA3 hippocampal subregion the reduction in receptor density reported for the agonist [3H]-GABA was greater than that obtained with the antagonist [3H]-CGP62349. For the DG hippocampal subfield the decrease in receptor density appeared to be less marked with the agonist, [3H]-GABA, than with the antagonist [3H]-CGP62349.
The patients whose tissue was used in this study were all undergoing chronic anti-epileptic drug therapy, and none of drugs administered have been reported to interact directly or indirectly with GABAB receptors. Comparative analysis of the data from individual subjects in the present study indicated that the variations in drug therapy had no influence on the outcome of the binding studies.
Quantitative neuropathology
The advanced technique of three dimensional (3D) cell counting was utilized in this study, as it is believed to be more accurate method than any previously used, which usually involve a correction factor, such as that introduced by Abercombie (1946). According to Williams & Rakic (1988), the other methods could rarely claim accuracy greater than ±10% and the correction factors introduced bias in the form of size, shape and orientation of cells, which have to be taken into account. The 3D method defines a counting area within the section, and the main limit to its use is section thickness. The method is direct, does not make use of any correction factors and any splitting of cells by the microtome does not influence it.
Three dimensional cell counts were performed in CA1, CA2, CA3, hilus, the subiculum pyramidal layer, and dentate gyrus granule cell layer (gl). Counts were obtained from four to five post-mortem controls and from six to eleven epileptic patients, though some hippocampal subregions occasionally were not available for quantification due to the loss of some subfields through the surgical process. The data obtained from 3D cell counting confirmed a significant neuronal loss in CA1, CA3, hilus and DG (c.f. Amaral & Insausti, 1990), but not in the subiculum and only partially in CA2, which has been reported to be usually preserved under these conditions (Margerison & Corsellis, 1966).
The necessity of counting the neurones in the hippocampi from control and subjects with epilepsy represents an important task, firstly to confirm the histopathology of the patients; and secondly because the receptor density measured by the Bmax parameter, needs to be interpreted in the light of the characteristic neuronal cell loss in HS.
Comparison of Bmax corrected for neuronal counts
The Bmax value obtained from the autoradiography was corrected for neuronal density to obtain the Bmax values in the remaining neurones.
As a consequence of this correction a significant up-regulation was found in CA3 and hilus, which might represent an increase in the number of GABAB receptors at pre- and/or post-synaptic sites. This might be part of the mechanism involved in the reduction of inhibition produced in TLE patients, contributing to epileptogenesis (Misgeld et al., 1995). An increase in both GAD65 and GAD67 at the gene and protein levels occurs in the remaining neurons of HS/TLE patients (Esclapez & Houser, 1999), with a reduction in GAT-1 and an increase in excitatory amino acid transporter, EAAT3, immunoreactivity in the granular and pyramidal hippocampal layers (Mathern et al., 1999). The increase in the enzymes synthesizing GABA coupled with a reduction of the transporter might suggest that there is an inadequacy of GABA and thus an increase in receptor numbers might be a compensatory mechanism, which is still insufficient to compensate for the hyperexcitation induced by an increase in glutamate, as reflected in the change in EAAT3. An increase in glutamate receptors such as NMDA and AMPA has previously been reported in TLE tissue (Brines et al., 1997). Changes in GABAA receptor mechanisms have also been demonstrated to be involved in the reduction of inhibition, which is considered to underlie most forms of epilepsy. Loup et al. (2000) have analysed the expression of six different GABAA receptor subunits in human TLE using immunocytochemistry, and each of these subunits appeared to be altered in the different hippocampal subregions of HS/TLE. There was no correlation between the changes that were observed in this study and our present data.
The data might suggest that changes in GABAB receptor function occur in human TLE, possibly as a result of synaptic reorganization. It is impossible at present to distinguish between pre- and post-synaptic receptor forms by binding, as antagonists to achieve this purpose are not yet available. Immunocytochemical studies using the antibodies raised against the two different GABAB1 isoforms and GABAB2 subunit should help clarify the involvement of GABAB receptor in human TLE.
Even though a deficit of GABAergic inhibition is a basic hypothesis to verify in epileptic tissue it has revealed very difficult. One of the reasons is represented by the multiplicity of GABAergic inhibitory pathways and the multiplicity of the variables characterizing inhibition within any different inhibitory pathway. What is emerging from recent studies (Esclapez & Houser, 1999; Mathern et al., 1999; Brines et al., 1997; Loup et al., 2000) GABAergic inhibition may appear increased, decrease, or unaffected, and these alterations are brain area and inhibitory pathway-specific.
Acknowledgments
This work was supported by ‘Community Fund' grant No. 000218562. We are grateful to Mr William F.J. Harkness, National Hospital for Neurology and Neurosurgery, for his expert hippocampal resections, and to the Parkinson's Disease Society Bank (Institute of Neurology, London) for providing frozen and fixed postmortem control brain tissue. We are also grateful to Novartis Pharma (Basel, Switzerland) for the generous gift of [3H]-CGP62349.
Abbreviations
- GABA
γ-Aminobutyric acid
- cAMP
cyclic AMP
- 3D
three dimensional
- 3DC
three dimensional cell counting
- HS
hippocampal sclerosis
- PMI
post-mortem interval
- TLE
temporal lobe epilepsy
- SGDG
stratum granulare dentate gyrus
- SUB
subiculum
References
- ABERCOMBIE M. Estimation of nuclear populations from microtome sections. Anat. Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- AMARAL D.G., INSAUSTI R.Hippocampal formation The human nervous system 1990New York: Accademic Press; 711–752.ed. Paxinios, G pp [Google Scholar]
- ANDRADE R., MALENKA R.C., NICOLL R.A. A G protein couples serotonin and GABAB receptors to the same channel in hippocampus. Science. 1986;234:1261–1265. doi: 10.1126/science.2430334. [DOI] [PubMed] [Google Scholar]
- ASPRODINI E.K., RAINNIE D.G., SHINNICK-GALLAGHER P. Epileptogenesis reduces the sensitivity of presynaptic GABAB receptors on glutamatergic afferents in the amigdala. J. Pharmacol. Exp. Ther. 1992;262:1011–1021. [PubMed] [Google Scholar]
- BABB T.L., BROWN W.J., PRETORIUS J.K., DAVENPORT C., LIEB J.P., CRANDALL P.H. Temporal lobe volumetric cell densities in temporal lobe epilepsy. Epilepsia. 1984;25:729–793. doi: 10.1111/j.1528-1157.1984.tb03484.x. [DOI] [PubMed] [Google Scholar]
- BILLINTON A., BAIRD V.H., DUNCAN J.S., UPTON N., BOWERY N.G. GABAB receptor autoradiography in hippocampal sclerosis associated with human temporal lobe epilepsy. Br. J. Pharmacol. 2001;132:475–480. doi: 10.1038/sj.bjp.0703854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BITTIGER H., BELLOUIN C., FROESTL W., HEID J., SCHUMUTZ M., STAMPF P. [3H]- CGP62349: a new high affinity antagonist radioligand for GABAB receptors. Pharmacol. Rev. Comm. 1996;8:97–98. [Google Scholar]
- BOWERY N.G., HUDSON A.L., PRICE G.W. GABAA and GABAB receptor site distribution in the rat central nervous system. Neurosci. 1987;20:365–383. doi: 10.1016/0306-4522(87)90098-4. [DOI] [PubMed] [Google Scholar]
- BRINES M.L., SUNDARESAN S., SPENCER D.D., DE LANEROLLE N.C. Quantitative autoradiographic analysis of ionotropic glutamate receptor subtypes in human temporal lobe epilepsy: up-regulation in reorganized epileptogenic hippocampus. Eur. J. Neurosci. 1997;9:2035–2044. doi: 10.1111/j.1460-9568.1997.tb01371.x. [DOI] [PubMed] [Google Scholar]
- BUHL E.H., OTIS T.S., MODY I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- CHU D.C., ALBIN R.L., YOUNG A.B. Distribution and kinetics of GABAB binding sites in the rat central nervous system: a quantitative autoradiographic study. Neurosci. 1990;34:341–357. doi: 10.1016/0306-4522(90)90144-s. [DOI] [PubMed] [Google Scholar]
- CHU D.C., PENNEY J.B., JR, YOUNG A.B. Quantitative autoradiography of hippocampal GABAB and GABAA receptor changes in Alzheimer's disease. Neurosci. Lett. 1987;82:246–252. doi: 10.1016/0304-3940(87)90264-3. [DOI] [PubMed] [Google Scholar]
- CURTIS D.R., GAME C.G., JOHNSTON G.A., MCCULLOCH R.M. Central effects of beta-(para-chlorophenyl)-gamma-aminobutyric acid. Brain Res. 1974;70:493–499. doi: 10.1016/0006-8993(74)90257-1. [DOI] [PubMed] [Google Scholar]
- DUTAR P., NICOLL R.A. A physiological role for GABAB receptors in the central nervous system. Nature. 1988;322:156–158. doi: 10.1038/332156a0. [DOI] [PubMed] [Google Scholar]
- ENGEL J., JR Epilepsy surgery. Curr. Opin. Neurol. 1994;7:140–147. doi: 10.1097/00019052-199404000-00010. [DOI] [PubMed] [Google Scholar]
- ESCLAPEZ M., HOUSER C.R. Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. J. Comp. Neurol. 1999;412:488–505. [PubMed] [Google Scholar]
- GEHLERT D.R., YAMAMURA H.I., WAMSLEY J.K. gamma-Aminobutyric acidB receptors in the rat brain: quantitative autoradiographic localization using [3H](−)-baclofen. Neurosci. Lett. 1985;56:183–188. doi: 10.1016/0304-3940(85)90126-0. [DOI] [PubMed] [Google Scholar]
- HAAS K.Z., SPERBER E.F., MOSHE S.L., STANTON P.K. Kainik acid induced-seizures enhance dentate gyrus inhibition by down regulation of GABAB receptors. J. Neurosci. 1996;16:4250–4260. doi: 10.1523/JNEUROSCI.16-13-04250.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILL D.R., BOWERY N.G. [3H]-Baclofen and [3H]-GABA bind to bicuculline-insensitive GABAB site in rat brain. Nature. 1981;290:149–152. doi: 10.1038/290149a0. [DOI] [PubMed] [Google Scholar]
- HILL D.R., BOWERY N.G., HUDSON A.L. Inhibition of GABAB receptor binding by guanyl nucleotides. J. Neurochem. 1984;42:652–657. doi: 10.1111/j.1471-4159.1984.tb02732.x. [DOI] [PubMed] [Google Scholar]
- HOPKINS A., SHORVON S.D., CASCINO G.D. Epilepsy. London: Chapman & Hall Medical; 1995. [Google Scholar]
- ISOKAWA M., AVANZINI G., FINCH D.M., BABB T.L., LEVESQUE M.F. Physiologic properties of human dentate granule cells in slices prepared from epileptic patients. Epilepsy Res. 1991;9:242–250. doi: 10.1016/0920-1211(91)90058-n. [DOI] [PubMed] [Google Scholar]
- JONES K.A., BOROWSKY B., TAMM J.A., CRAIG D.A., DURKIN M.M., DAU M., YAO W.Y., JOHNSON M., GUNWALDSEN C., HUANG L.Y., TANG C., SHEN Q., SALON J.A., MORSE K., LAZ T., SMITH K.E., NAGARATHNAM D., NOBLE S.A., BRANCHEK T.A., GERALD C. GABAB receptor functional a heterodimeric assembly of the subunits GABABR1 and GABABR2. Nature. 1998;396:674–678. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- KAUPMANN K., HUGGEL K., HEID J., FLOR P.J., BISHOFF S., MICKEL S.J., MCMASTER G., ANGST C., BITTIGHER H., FRESTL W., BETTLER B. Expression cloning of GABAB receptors uncovers similarity to metabotropic glutamate receptors. Nature. 1997;368:239–246. doi: 10.1038/386239a0. [DOI] [PubMed] [Google Scholar]
- KAUPMANN K., MALITSCHEK B., SHULER V., HEID J., FROESTL W., BECK P., MOSBACHER J., BISHOFF S., KULIK A., SHIGEMOTO R., KARSCHIN A., BETTLER B. GABAB receptor subtypes assemble into functional heterodimeric complex. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- KIM J.H., GUIMARAES P.O., SHEN M.Y., MUSUKAWA L.M., SPENCER D.D. Hippocampal neuronal density in temporal lobe epilepsy with and without gliomas. Acta Neuropathol. 1990;80:41–45. doi: 10.1007/BF00294220. [DOI] [PubMed] [Google Scholar]
- KINGSBURY A.E., BRAY E.L., FOSTER O.J. A simplified and rapid procedure for in situ hybridization on human, flash-frozen, post-mortem brain and its combination with immunohistochemistry. J. Neurosci. Methods. 1996;69:213–227. doi: 10.1016/S0165-0270(96)00086-6. [DOI] [PubMed] [Google Scholar]
- KNOWLES W.D., AWAD I.A., NAYEL M.H. Differences of in vitro electrophysiology of hippocampal neurons from epileptic patients with mesiotemporal sclerosis versus structural lesions. Epilepsia. 1992;33:601–609. doi: 10.1111/j.1528-1157.1992.tb02335.x. [DOI] [PubMed] [Google Scholar]
- KOFLER M., KRONENBERG M.F., RIFICI C., SALTUARI L., BAUER G. Epileptic seizures associated with intrathecal baclofen application. Neurology. 1994;44:25–27. doi: 10.1212/wnl.44.1.25. [DOI] [PubMed] [Google Scholar]
- KRNJEVIC K. Chemical nature of synaptic transmission in vertebrates. Physiol. Rev. 1974;54:418–450. [Google Scholar]
- KUNER R., KÖHR G., GRÜNWALD S., EISENHARDT G., BACH A., KORNAU H.C. Role of heteromer in receptor Function. Science. 1999;283:74–77. doi: 10.1126/science.283.5398.74. [DOI] [PubMed] [Google Scholar]
- LLOYD K.G., DREKSLER S. An analysis of [3H]gamma-aminobutyric acid (GABA) binding in the human brain. Brain Res. 1979;163:77–87. doi: 10.1016/0006-8993(79)90152-5. [DOI] [PubMed] [Google Scholar]
- LOUP F., WIESER H.-G., YONEKAWA Y., AGUZZI A., FRITSCHY J.-M. Selective alterations in GABAA subtypes in human temporal lobe epilepsy. J. Neurosci. 2000;20:5401–5419. doi: 10.1523/JNEUROSCI.20-14-05401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANGAN P.S., LOTHMAN E.W. Profound disturbence in pre- and postsynaptic GABAB receptor-mediated processes in region CA1 in a chronic model of temporal lobe epilepsy. J. Neurophysiol. 1996;76:1282–1296. doi: 10.1152/jn.1996.76.2.1282. [DOI] [PubMed] [Google Scholar]
- MARESCAUX C., VERGNES M., LIU Z., DEPAULIS A., BERNASCONI R. GABAB receptor involvement in the control of genetic absence seizure in rats. Epilepsy Res. 1992;9 Suppl:131–139. [PubMed] [Google Scholar]
- MARGERISON J.H., CORSELLIS J.A.N. Epilepsy and temporal lobe. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- MARTIN S.C., RUSSEK S.J., FARB D.H. Molecular identification of the human GABABR2: cell surface expression and coupling to adenylyl cyclase in the absence of GABABR1. Mol. Cell. Neurosci. 1999;13:180–191. doi: 10.1006/mcne.1999.0741. [DOI] [PubMed] [Google Scholar]
- MATHERN G.W., MENDOZA D., LOZADA A., PRETORIUS J.K., DEHNES Y., DANBOLT N.C., NELSON N., LEITE J.P., CHIMELLI L., BORN D.E., SAKAMOTO A.C., ASSIRATI J.A., FRIED I., PEACOCK W.J., OJEMANN G.A., ADELSON P.D. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:453–472. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- MISGELD U., BIJAK M., JAROLIMEK W. A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog. Neurobiol. 1995;46:423–462. doi: 10.1016/0301-0082(95)00012-k. [DOI] [PubMed] [Google Scholar]
- MÖHLER H., BENKE D., BENSON J., LÜSCHER B., RUDOLPH U., FRITSCHY J.M.Diversity in structure, pharmacology, and regulation of GABAA receptors The GABA Receptors. 1997Totowa, NJ: Humana Press Inc; 11–36.ed. ENNA, S.J., BOWERY, N.G. pp [Google Scholar]
- NG G.Y.K., CLARK J., COULOMBE N., ETHIER N., HERBERT T.E., SULLIVAN R., KARGMAN S., CHATEAUNEUF A., TSUKAMOTO N., MEZEY E., JOHNSON M.P., LIU Q., KOLAKOWSKI Jr. L.F., EVANS J.F., BONNER T.I., O'NEILL G.P. Identification of a GABAB receptor subunit, gb2, required for functional GABAB receptor activity. J. Biol. Chem. 1999;274:7607–7610. doi: 10.1074/jbc.274.12.7607. [DOI] [PubMed] [Google Scholar]
- ODAGAKI Y., NISHI N., OZAWA H., SAITO T., TAKAHATA N., RIEDERER P., KOYAMA T. Measurement of receptor mediated functional; activation of G-protein in postmortem human brains membranes. Brain Res. 1998;789:84–91. doi: 10.1016/s0006-8993(98)00019-5. [DOI] [PubMed] [Google Scholar]
- ROBBINS M.J., CALVER A.R., FILIPPOV A.K., HIRST W.D., RUSSEL R.B., WOOD M.D., NASIR S., COUVE A., BROWN D.A., MOSS S.J., PNAGALOS M.N. GABAB2 is essential for G-protein coupling of the GABAB receptor heterodimer. J. Neurosci. 2001;21:8043–8052. doi: 10.1523/JNEUROSCI.21-20-08043.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATO K., MORIMOTO K., OKAMOTO M., NAKAMURA Y., OTSUKI S., SATO M. An analysis of anticonvulsant actions of GABA agonists (progabide and baclofen) in the kindling model of epilepsy. Epilepsy Res. 1990;5:117–124. doi: 10.1016/0920-1211(90)90027-s. [DOI] [PubMed] [Google Scholar]
- SEMAH F., PICOT M.C., ADAM C., BROGLIN D., ARZIMANOGLOU A., BAZIN B., CAVALCANTI D., BAULAC M. Is the underlying cause of epilepsy a major prognostic factor for recurrence. Neurology. 1998;51:1256–1262. doi: 10.1212/wnl.51.5.1256. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI T., KAJIKAWA Y., TSUJIMOTO T. G-protein-coupled modulation of presynaptic calcium currents and transmitter release by GABAB receptor. J. Neurosci. 1998;18:3138–3146. doi: 10.1523/JNEUROSCI.18-09-03138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERGNES M., BOEHRER A., SIMLER S., BERNASCONI R., MARESCAUX C. Opposite effects of GABAB receptor antagonists on absences and convulsive seizures. Eur. J. Pharmacol. 1997;332:245–255. doi: 10.1016/s0014-2999(97)01085-6. [DOI] [PubMed] [Google Scholar]
- WHITE J.H., WISE A., MAIN M.J., GREEN A., FRASER N.J., DISNEY G.H., BARNES A.A., EMSON P., FOORD S.M., MARSHALL F.H. Heterodimerization is required for the formation of a functional GABAB receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- WILKIN G.P., HUDSON A.L., HILL D.R., BOWERY N.G. Autoradiographic localisation of GABAB receptors in rat cerebellum. Nature. 1981;294:584–587. doi: 10.1038/294584a0. [DOI] [PubMed] [Google Scholar]
- WILLIAMS R.W., RAKIC P. Three-dimensional counting: an accurate and direct method to estimated numbers of cells in sectioned material. J. Comp. Neurol. 1988;278:344–352. doi: 10.1002/cne.902780305. [DOI] [PubMed] [Google Scholar]
- WOJCIK W.J., NEFF N.H. Gamma-aminobutyric acid B receptors are negatively coupled to adenylate cyclase in brain, and in the cerebellum these receptors may be associated with granule cells. Mol. Pharmacol. 1984;25:24–28. [PubMed] [Google Scholar]
- WU C., LEUNG L.S. Partial hippocampal kindling decease efficacy of presynaptic GABAB autoreceptors in CA1. J. Neurosci. 1997;17:9261–9269. doi: 10.1523/JNEUROSCI.17-23-09261.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]