

Abstract
Acetylcholine release at the neuromuscular junction relies on rapid, local and transient calcium increase at presynaptic active zones, triggered by the ion influx through voltage-dependent calcium channels (VDCCs) clustered on the presynaptic membrane. Pharmacological investigation of the role of different VDCC subtypes (L-, N-, P/Q- and R-type) in spontaneous and evoked acetylcholine (ACh) release was carried out in adult mouse neuromuscular junctions (NMJs) under normal and pathological conditions.
ω-Agatoxin IVA (500 nM), a specific P/Q-type VDCC blocker, abolished end plate potentials (EPPs) in normal NMJs. However, when neurotransmitter release was potentiated by the presence of the K+ channel blocker 4-aminopyridine (4-AP), an ω-agatoxin IVA- and ω-conotoxin MVIIC-resistant component was detected. This resistant component was only partially sensitive to 1 μM ω-conotoxin GVIA (N-type VDCC blocker), but insensitive to any other known VDCC blockers. Spontaneous release was dependent only on P/Q-type VDCC in normal NMJs. However, in the presence of 4-AP, it relied on L-type VDCCs too.
ACh release from normal NMJs was compared with that of NMJs of mice passively injected with IgGs obtained from patients with Lambert-Eaton myasthenic syndrome (LEMS), a disorder characterized by a compromised neurotransmitter release. Differently from normal NMJs, in LEMS IgGs-treated NMJs an ω-agatoxin IVA-resistant EPP component was detected, which was only partially blocked by calciseptine (1 μM), a specific L-type VDCC blocker.
Altogether, these data demonstrate that multiple VDCC subtypes are present at the mouse NMJ and that a resistant component can be identified under ‘pharmacological' and/or ‘pathological' conditions.
Keywords: Acetylcholine, autoimmune, Lambert-Eaton myasthenic syndrome, neuromuscular junction, voltage-dependent calcium channels
Introduction
Neurotransmitter release at neuronal synapses is highly dependent on depolarization-induced influx of calcium ions through VDCCs. Different VDCC subtypes have been biophysically and pharmacologically characterized and classified as N-, P/Q-, L-, R- and T-types (Nowycky et al., 1985; Tsien et al., 1988; Zhang et al., 1993). The presence of multiple VDCC subtypes has been highlighted in different synapses throughout the nervous system (Wheeler et al., 1994; Wu & Saggau, 1994; Wu et al., 1998). At the mammalian NMJ, P/Q-type VDCCs are present (Ousley & Froehner, 1994; Westenbroek et al., 1998) and play a major role in evoked ACh release (Uchitel et al., 1992; Protti & Uchitel, 1993; Hong & Chang, 1995; Bowersox et al., 1995). However, other studies have suggested that different VDCC subtypes may be present at mammalian NMJs (Day et al., 1997) and may be involved in both spontaneous (Losavio & Muchnik, 1997) and/or nerve-evoked ACh release (Lin & Lin-Shiau, 1997). L-type VDCCs, for example, can play a role in ACh release at normal NMJs (Atchison, 1989; Urbano & Uchitel, 1999; Correia-de-sá et al., 2000a, b); Urbano et al., 2001) and may also contribute to ACh release under pathological conditions; for example, in amyotrophic lateral sclerosis (Fratantoni et al., 2000) and in botulinum toxic-poisoned mouse motor nerve terminals (Santafé et al., 2000). A role for L-type VDCCs has also been suggested in reinnervating (Katz et al., 1996) and newly formed (Sugiura & Ko, 1997; Santafé et al., 2001) rat NMJs. An involvement of N-type VDCCs in ACh release at newly formed rat NMJs has also been recently described (Rosato Siri & Uchitel, 1999; Santafé et al., 2001). Overall, the question is still open as to how many different VDCC subtypes are present at the mammalian nerve terminal and the conditions under which they contribute to transmitter release.
Lambert-Eaton myasthenic syndrome (LEMS) is an autoimmune disorder of neuromuscular transmission characterized by a decreased release of vesicles in response to a single nerve impulse (quantal content), facilitation following high frequency stimulation and a normal post-synaptic response (Lambert & Elmqvist, 1971). LEMS patients have high titres of anti-VDCC auto-antibodies, mainly anti-P/Q-type (∼ 90% of the patients) and anti-N-type VDCC (∼50% of the patients) antibodies. LEMS immunoglobulins (IgGs) have been shown to down-regulate P/Q-type Ca2+ currents in different cell types (Magnelli et al., 1996; Pinto et al., 1998a, b). Passive transfer of LEMS IgGs into mice is sufficient to reproduce the human disease in animals (Lang et al., 1983; Kim, 1985). However, little is known about the VDCC subtypes involved in synaptic transmission at the LEMS IgGs-treated NMJ. Changes in the pharmacological sensitivity of VDCCs in mice motor nerve terminals after chronic exposure to LEMS IgGs have been demonstrated (Smith et al., 1995; Xu et al., 1998).
In the present study we aimed to characterize, relying on elecrophysiological and pharmacological approaches, which VDCC subtypes play the most relevant role in controlling ACh release at normal and pathological NMJs.
Methods
Patients
Sera from six patients with typical clinical and electromyographic features of LEMS were used in this study. The patients' details are briefly summarized in Table 1. All sera, assayed as previously described (Motomura et al., 1997), had high titres of anti-VDCC auto-antibodies. All patients had high titres of anti-P/Q-type VDCC antibodies and patient 5 also had anti-N-type VDCC antibodies.
Table 1.
Details of the LEMS patients used in this study

IgGs were prepared by the ethacridine lactate-ammonium sulphate method as previously described (Lang et al., 1983). Patients were considered to be positive if their antibodies' titre was three standard deviations above the mean for healthy controls; this value has been determined as >30 pM in the anti-P/Q-type VDCCs assay and >18 pM in the anti-N-type VDCCs assay (Motomura et al., 1997). IgGs were also prepared from three healthy controls; one from a healthy 45 year old woman and the other two from pooled plasma of healthy individuals (n=30).
LEMS patients were also diagnosed by electromyography. Patients were considered to have LEMS when the compound muscle action potential (CMAP) amplitude, measured from a muscle in the hand, was between 1 and 4 mV and when CMAP amplitude increment during tetanic stimulation was >100%.
Animal work
Male albino (MF1) mice (20–40 g) were used and treated in accordance with British Government Home Office guidelines.
For passive transfer experiments, mice were injected daily intraperitoneally (i.p.) with 1 ml of IgGs (concentration=10 mg ml−1) for a total period of 9 days. IgGs from healthy controls (n=3) and LEMS patients (n=6) were coded so that the nature of the IgGs injected into each mouse was unknown until after the data had been analysed. On day 10, mice were sacrificed by raised atmospheric CO2 followed by cervical dislocation. There was no significant difference in weight in animals receiving different treatments (29.8±0.4 g for LEMS IgGs-treated vs 30.7±0.4 g for control mice) and no obvious sign of muscle weakness was detected in LEMS IgGs-treated mice.
Electrophysiology
Mouse phrenic nerve/hemidiaphragm preparations were excised and pinned out in a 2 ml Sylgard-coated Petri dish containing physiological Krebs solution, continuously bubbled with 95% O2/5% CO2 at room temperature (20–23°C). The nerve was stimulated via a suction electrode coupled to a pulse generator (GRASS Instruments S48, solid-state square wave stimulator, Quincy, U.S.A.) with an associated stimulus isolation unit. To block muscle contraction, 2.5 μM μ-conotoxin GIIIB (Peptide Institute Inc., Japan) was added to the bath. Nerve-muscle viability was first tested by nerve stimulation in the absence of μ-conotoxin GIIIB. Recordings were made at room temperature (20–23°C). The recording electrodes were connected to an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA, U.S.A.).
Nerve evoked EPPs and MEPPs were recorded intracellularly with conventional glass microelectrodes filled with 3 M KCl (10–15 MΩ resistance; Clark Electromedical Instruments, U.K.) and filtered at 1 kHz. The recording pipette was brought close to the nerve-terminal region under microscopic visualization. End plates were localized by searching for EPPs with fast rise times (⩽1 ms).
Protocols
The nerve was stimulated supramaximally with platinum-wire electrodes using standard protocols. After impalement of a muscle fibre, the nerve was first stimulated at 1 Hz for 30 s before recording 30–50 EPPs at this frequency.
Pulses of 0.1 ms duration and of different intensities, depending on the threshold of each preparation, were used for stimulation. The nerve was then left unstimulated for 1 min followed by a train of 50 pulses at 40 Hz. In order to evaluate MEPPs amplitude and frequency, 30–50 traces were recorded and stored for further analysis. Each drug used in the pharmacological studies was directly added to the bath solution and allowed to achieve the final concentration by diffusion. Two different drug application protocols were used. One protocol (referred to as ‘acute application' in the relevant results paragraphs and figure legends) was used to study the time course of drugs effects on a single end plate. Alternatively, a second protocol (referred to as ‘pre-incubation') consisted in pre-incubating the preparation with the relevant drugs for 1 h and then recording, in the continuous presence of the drug, from many different end plates. Results obtained from this second protocol are expressed as the average of all end plates recorded.
Data analysis
Recordings were rejected if the membrane potential, Vm, was <−60 mV or decreased by more than 5 mV during the recording period or if the 10–90% EPP rise time was >1 ms. The signals were digitized at 12.5 kHz (CED-1401 interface, Science Park Cambridge, U.K.), stored and computer analysed. The software WCP (Whole Cell Program, Strathclyde Electrophysiology Software, John Dempster, 1993–1994) was used for data acquisition and analysis. Each MEPP and EPP was visually inspected before analysis and poor quality traces were discarded.
The mean quantal content (m) was calculated by the direct method (the most accurate method for calculation):
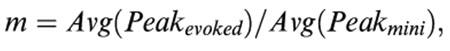 |
where Avg is the average, Peakevoked is the amplitude at the peak of EPP and Peakmini is the amplitude at the peak of MEPP.
Averaged EPPs were corrected for non-linear summation (McLachlan & Martin, 1981) using the formula:
 |
where V1 is the corrected EPP amplitude, V is the uncorrected EPP amplitude, E is the resting membrane potential and 0.8 is the correction factor. Before correction for non-linear summation, all EPPs and MEPPs were corrected to a standardized membrane potential of −80 mV to correct for changes in driving force due to altered postjunctional membrane potential (Katz & Thesleff, 1957).
Data acquisition, analysis, fitting, averaging and presentation were carried out using a combination of WCP, Excel (Microsoft), SigmaPlot (SPSS), GraphPad Software (Prism and Instat), PowerPoint (Microsoft) and Corel Draw (Corel). Values are expressed as means±s.e. Statistical significance (P values in the text and figure legends) was evaluated using the two-tailed Student's t-test. P<0.05 was considered to be statistically significant.
Drugs
The bath buffer (Krebs) for the recordings was the following (in mM): NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 24.9 and glucose 11. 2.5 mM CaCl2 was added to the Krebs solution the day of the experiment.
CdCl2 was obtained from Acros Organics (UK). ω-Agatoxin IVA, ω-conotoxin GVIA, ω-conotoxin MVIIC were purchased from Peptide Institute, Inc. (Japan) and calciseptine from Alomone Labs (Israel). 4-aminopyridine, nifedipine and S-(−) Bay K 8644 were obtained from Sigma. Nifedipine and S-(−) Bay K 8644 were dissolved in 100% ethanol and stored in the dark at 4°C. Experiments in the presence of nifedipine or S-(−) Bay K 8644 were carried out in the absence of direct illumination to the preparation except for the time required to position the recording electrode. SNX482 was generously donated by Dr G. Dayanithi (Montpellier, France). The toxin was left for an hour at room temperature in the recording solution before use in order to be at neutral pH, where it is more effective.
Results
Effects of Cd2+ on ACh release
The effects of Cd2+ (500 μM), a non-specific VDCC blocker, were tested on neuromuscular transmission under control conditions. Figure 1A shows that the block caused by this inorganic ion on evoked ACh release was complete (100% inhibition, n=4) within 20 min. Cd2+ also significantly (P<0.01) decreased MEPP frequency (60.4±7.3% reduction; n=4) but it did not exert any significant effect on MEPP amplitude (5.2±2.6% inhibition; n=4; data not shown), indicating that this inorganic ion has no postsynaptic effect. These results indicate and confirm that both the evoked release and the frequency of spontaneous release are processes dependent on the influx of Ca2+ through VDCCs present at the nerve terminal.
Figure 1.
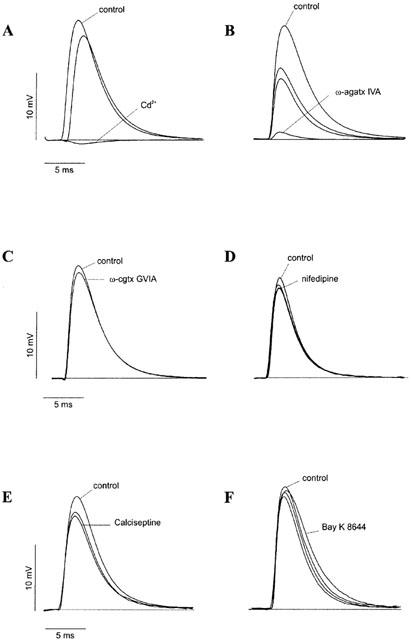
Effect of different VDCCs blockers on EPPs. Representative EPP traces recorded from phrenic nerve/hemidaiphragm preparations in 2.5 mM Ca2+ Krebs solution in the absence or presence of Cd2+ (500 μM; A), ω-agatoxin IVA (500 nM; B), ω-conotoxin GVIA (1 μM; C), nifedipine (5 μM; D), calciseptine (1 μM; E) or S-(−) Bay K 8644 (1 μM; F). Traces show the effect of the VDCCs blockers 10, 20 and 30 min after their application.
Effects of ω-agatoxin IVA on ACh release
As shown in Figure 1B, the application of a relatively high concentration of ω-agatoxin IVA (500 nM), a specific P/Q-type VDCC blocker (Mintz et al., 1992), virtually abolished (94.1%±2.6%; n=8) EPP amplitude, with a maximal block reached within 25–30 min.
Like Cd2+, also ω-agatoxin IVA (500 nM) significantly reduced MEPP frequency (25.9±9.6% reduction; n=8; P<0.05), but had no effect on MEPP amplitude (0.1±5.4% inhibition; n=8; data not shown).
Effects of other VDCC blockers on ACh release
We further investigated whether L- and N-type VDCCs play any role in the control of ACh release at the adult mouse NMJ. As shown in Figure 1C, ω-conotoxin GVIA (1 μM), an N-type VDCC specific blocker, had no significant effect on EPP amplitude (n=3). Nifedipine (5 μM; Figure 1D), a DHP L-type VDCC antagonist, calciseptine (1 μM; Figure 1E), a potent peptide inhibitor of L-type VDCC and S-(−) Bay K 8644 (1 μM; Figure 1F), an L-type agonist, also had not significant effects.
MEPP frequency and MEPP amplitude were also not significantly inhibited by any of these VDCC blockers.
Effects of VDCC blockers on ACh release using the ‘pre-incubation' protocol
Similar results were obtained with the VDCC blockers described above utilizing the ‘pre-incubation' protocol (Figure 2). However, some differences between the two protocols were observed and are highlighted here.
Figure 2.

Effect of pre-incubation with specific VDCC blockers on neurotransmitter release at the mouse neuromuscular junction. Bar graphs showing the effect of pre-incubations (1 h; see Methods for description of the protocol) of phrenic nerve/hemidiaphragm preparations with either 2.5 mM Ca2+ Krebs solution alone (control; n=5) or in the presence of one of the compounds listed below on MEPPs amplitude (A), MEPPs frequency (B), EPPs amplitude (C) and quantal content (D). Results are shown as means+s.e. (*P<0.05; **P<0.01); n indicates the number of phrenic nerve/hemidiaphragm preparations. Nifedipine (5 μM; n=3); S-(−) Bay K8644 (1 μM; n=3); ω-conotoxin GVIA (1 μM; n=3); ω-agatoxin IVA (500 nM or 1 μM; n=3 and n=4, respectively) and cadmium (500 μM; n=3).
Cd2+ (500 μM; n=2) reduced EPP amplitude from 17.2±1.4 mV to 0.29±0.1 mV (Figure 2C), without affecting MEPP amplitude (from 0.96±0.1 mV to 0.99±0.06 mV; n=5; Figure 2A). However, in contrast with the experiments where Cd2+ was acutely applied and reduced MEPP frequency, after 1 h of pre-incubation Cd2+ effects reversed almost to normal values (0.65±0.1 s−1 in Cd2+ vs 0.8±0.1 s−1 in normal 2.5 mM Ca2+ Krebs solution; Figure 2B).
In the ‘pre-incubation' experiments, ω-agatoxin IVA inhibited EPP amplitude from a control value of 17.2±1.4 mV to a value of 1.64±0.6 mV (P<0.01) and 0.58±0.2 mV (P<0.01) at 500 nM (n=3) and 1 μM (n=4) concentrations, respectively (Figure 2C). The toxin also caused a reduction in MEPP frequency in these ‘pre-incubation' experiments from a control value of 0.8±0.1 s−1 to values of 0.53±0.1 s−1 and 0.44±0.07 s−1 at 500 nM and 1 μM toxin (P<0.05), respectively (Figure 2B).
The other compounds tested (1 μM ω-conotoxin GVIA, 5 μM nifedipine and 1 μM S-(−) Bay K 8644) were unable to inhibit either evoked or spontaneous ACh release. Only nifedipine significantly increased evoked release from a control value of 17.2±1.4 mV to a value of 24.8±2.2 mV (Figure 2C; n=3; P<0.05), an effect not seen during the acute applications (Figure 1D).
Finally, we also tested the effect of ω-conotoxin MVIIC (2 μM) which, at this concentration, is known to inhibit both the N- and the P/Q-type VDCCs. ω-Conotoxin MVIIC reduced EPP amplitude from a control value of 22.9±0.7 mV (n=4) to a value of 6.9±1.3 mV (n=4; P<0.0001; data not shown).
In summary, most of the drugs effects were the same if applied acutely or by using the pre-incubation protocol. We therefore chose to use the pre-incubation protocol for the rest of this study in order to optimize the number of experiments performed while using less of these precious toxins. Only Cd2+ and nifedipine showed some differential effects that must be taken in consideration when interpreting the data, as when comparing data from different laboratories (see Discussion).
Effects of 4-AP on ACh release
4-Aminopyridine (4-AP; 300 μM) prevents repolarization of the presynaptic membrane and, as a consequence, drastically increases the amplitude and prolongs the duration of EPPs by blocking K+ channels located at the nerve terminal (Lundh, 1978; Thomsen & Wilson, 1983). 4-AP increased EPP amplitude and area by 42±7.4% and of 80±6% (n=7), respectively.
MEPP amplitude was not affected by 4-AP (0.5±3% inhibition; n=3), thus excluding a direct post-synaptic effect of this compound.
Interestingly, in the continuous presence of 4-AP (300 μM), ω-agatoxin IVA (500 nM; n=6) only partially inhibited EPPs area (45.9±9.3% inhibition; Figure 3A), confirming that under these conditions of ‘pharmacological' enhanced ACh release, an ω-agatoxin IVA-resistant component of release becomes highly relevant (Lin & Lin-Shiau, 1997).
Figure 3.

Effect of ω-agatoxin IVA and ω-conotoxin MVIIC in the presence of 4-aminopyridine on EPPs. (A) Representative EPP traces recorded from mouse phrenic nerve/hemidiaphragm preparation in 2.5 mM Ca2+ Krebs solution (control), 20 min after the application of 4-aminopyridine (4-AP; 300 μM) and 30 min after the application of ω-agatoxin IVA (500 nM) in the continuous presence of 4-AP (4-AP+ω-agatx IVA). (B) Representative EPP traces recorded from mouse phrenic nerve/hemidiaphragm preparation in 2.5 mM Ca2+ Krebs solution and 4-aminopyridine (4-AP; 300 μM) and 10, 20 and 30 min after the application of ω-agatoxin IVA (ω-agatx IVA; 500 nM) and ω-conotoxin MVIIC (ω-cgtx MVIIC; 2 μM) in the continuous presence of 4-AP. Each trace is the average of 20–30 recordings.
A partial inhibition was also found when ω-agatoxin IVA (500 nM) and ω-conotoxin MVIIC (2 μM) were co-applied in the continuous presence of 4-AP (50.2±8% inhibition; n=2; Figure 3B), suggesting that, even when all P/Q-type VDCCs are effectively blocked, a component of Ca2+ influx sustaining ACh release is still available and belongs to VDCCs other than the P/Q-type.
We studied further this ω-agatoxin IVA- and ω-conotoxin MVIIC-resistant component utilizing the ‘pre-incubation' protocol. Phrenic nerve/hemidiaphragm preparations were incubated for 1 h in either 4-AP alone or 4-AP plus one of the above described L-, N- and P/Q-type VDCC specific blockers or a combination of them. SNX 482 (200 nM), a toxin that has been shown to block at least some subtypes of the R-type VDCCs family (Newcomb et al., 1998; Wang et al., 1999; Tottene et al., 2000; Wilson et al., 2000), was also tested in these experiments.
In agreement with the above finding in ‘acute' experiments, ω-agatoxin IVA inhibited EPP area only by ∼50% with respect to the EPPs measured in 4-AP alone (Figure 4A; 147.1±18.3 mV×ms vs 300.1±22.6 mV×ms; P<0.01, n=3). In the presence of 4-AP, the non-specific VDCC blocker Cd2+ completely abolished evoked release (Figure 4A; 5.7±1.1 mV×ms (n=2) vs 300.1±22.6 mV×ms (n=3); P<0.01). This finding crucially demonstrates that also under the extreme release conditions caused by 4-AP, ACh release still relies completely on Ca2+ influx via VDCCs. Interestingly, in the presence of 4-AP, none of the other specific VDCC blockers was able to significantly reduce nerve-evoked release, when applied singularly.
Figure 4.

Effect of pre-incubation with specific VDCCs blockers in the presence of 4-aminopyridine on EPPs. Bar graphs showing the effect of pre-incubation of mouse phrenic nerve/hemidiaphragm preparation with 2.5 mM Ca2+ Krebs solution alone (control, n=5), with 4-aminopyridine (4-AP, 300 μM; n=3) or with 4-aminopyridine and one (A) or more (B) of the compounds listed below on EPPs area. Results are shown as means+s.e. (*P<0.05; **P<0.01); n indicates the number of hemidiaphragm preparations. Nifedipine (5 μM; n=4); calciseptine (calc., 1 μM; n=3); S-(−) Bay K 8644 (1 μM; n=3); ω-conotoxin GVIA (ω-cgtx GVIA; 1 μ M; n=3); cadmium (500 μM; n=2); ω-agatoxin IVA (ω-agatx IVA, 500 nM; n=3); ω-agatoxin IVA and SNX 482 (200 nM; n=2); ω-agatoxin IVA and ω-conotoxin GVIA (n=3); ω-agatoxin IVA and nifedipine (n=3); ω-agatoxin IVA and calciseptine (n=3); ω-agatoxin IVA, calciseptine and ω-conotoxin GVIA (n=5); ω-agatoxin IVA, calciseptine, ω-conotoxin GVIA and SNX 482 (n=2).
However, some synergistic effects were observed. The simultaneous application of ω-agatoxin IVA (500 nM) with ω-conotoxin GVIA (1 μM) caused a further reduction compared with the reduction caused by ω-agatoxin IVA alone (Figure 4B; 59.2±12.8 mV×ms (n=3) in ω-agatoxin IVA and ω-conotoxin GVIA vs 147.1±18.3 mV×ms (n=3) in ω-agatoxin IVA alone; P<0.05). Neither calciseptine alone nor the combination of calciseptine and SNX 482 were able to inhibit the ω-agatoxin IVA- and ω-conotoxin GVIA-resistant components in the presence of 4-AP (Figure 4B; 84.3±10 mV×ms (n=5) and 54.7±18 mV×ms (n=2) 59.2±12.8 mV×ms (n=3)). We also tested the effect of pre-incubation of ω-conotoxin MVIIC (2 μM) in the absence and presence of 4-AP and found that, similarly to ω-agatoxin IVA, this toxin was much less effective in inhibiting the evoked release in the presence of 4-AP than in its absence, as shown in Figure 5. EPPs area was indeed reduced by ω-conotoxin MVIIC, in the absence of 4-AP, by ∼76% (133.3±4.1 mV×ms in normal 2.5 mM Ca2+ Krebs solution vs 31.4±7 mV×ms in ω-conotoxin MVIIC; n=4; P<0.0001) and only by ∼36% in its presence (372.7±27.8 mV×ms in 4-AP vs 237.7±44.8 mV×ms in 4-AP+ω-conotoxin MVIIC; n=4; P<0.05).
Figure 5.
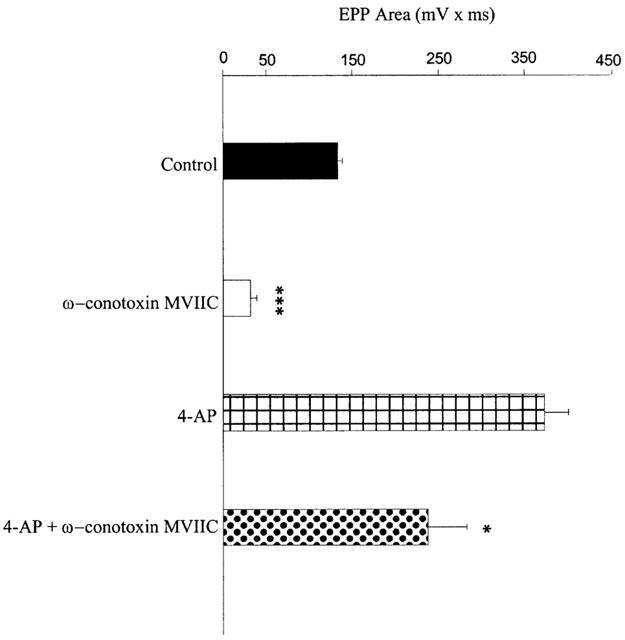
Effect of pre-incubation with ω-conotoxin MVIIC in the absence and in the presence of 4-aminopyridine on EPPs. Bar graph showing the effect of 1 h pre-incubations of mouse phrenic nerve/hemidiaphragm preparations with ω-conotoxin MVIIC (2 μM) in the absence (2.5 mM Ca2+ Krebs solution alone; control) or presence of 4-aminopyridine (300 μM; 4-AP) on EPPs area. Results are shown as means+s.e. (*P<0.05; ***P<0.0001; n=4).
ACh release at LEMS IgGs-treated NMJs
Table 2 summarizes the pooled data of the electrophysiological findings on NMJs of control IgGs- (n=20) and LEMS IgGs-injected (n=26) mice.
Table 2.
Electrophysiological properties of NMJs from mice immunized with control or LEMS IgGs

As shown in previous studies, LEMS IgGs reduced EPP amplitude. Figure 6 shows representative traces of EPPs from mice injected with a control IgG (A1) or with a LEMS IgG (A2; LEMS 4; see Table 1 for details). While EPPs recorded from single end plates in control mice appeared to be homogeneous in size, EPPs in LEMS IgGs-treated mice were found to be highly variable.
Figure 6.
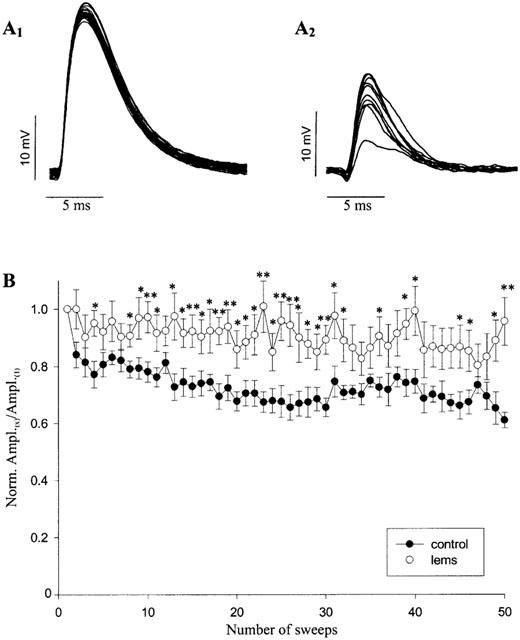
Effect of LEMS IgGs on neurotransmitter release at the mouse neuromuscular junction. Representative EPP traces recorded from control IgGs-treated (A1) and LEMS IgGs-treated (A2, LEMS 4; see Table 1) mouse phrenic nerve/hemidiaphragm preparations in 2.5 mM Ca2+ Krebs solution. Note that, in each experiment, traces are recordings from a single end plate. (B) Graph plotting the normalized mean EPP amplitude against a train of 50 sweeps delivered at 40 Hz. Before averaging across experiments, EPPs amplitude of each recording (Ampl.(X)) was normalized by that obtained in the first recording (Ampl.(1)). Recordings were obtained from phrenic nerve/hemidiaphragm preparations of mice immunized with either control IgGs (n=20) or LEMS IgGs (n=26). Error bars represent s.e.; n indicates the number of experiments. *P<0.05; **P<0.01.
Quantal content, calculated with the ‘direct method' as the ratio between the averaged EPP amplitude and the averaged MEPP amplitude, was also reduced by injections of LEMS IgGs (Table 2). Moreover, EPP amplitude depression during a train of 50 consecutive stimuli given at high frequency (40 Hz) was significantly less marked in LEMS IgGs-treated preparations than in control preparations (Table 2). The time course of EPP amplitude depression during the train also differed between control and LEMS IgGs-treated preparations (Figure 6B). EPP amplitude decreased gradually during the whole protocol in control NMJs (t1/2=0.25 s), whereas it reached its maximum level during the first part of the train in LEMS IgGs-treated NMJs (t1/2=0.087 s).
MEPP amplitude was not affected by LEMS IgGs (Table 2). Although electrophysiological recordings showed that neuromuscular transmission was compromised in LEMS IgGs-treated mice, no visible signs of muscle weakness were observed. This may be due to the high safety factor for this species.
Effects of ω-agatoxin IVA on ACh release at the LEMS IgGs-treated NMJ
Figure 7A shows representative EPP traces recorded from LEMS IgGs-treated NMJs (LEMS 4; see Table 1 for details); ω-agatoxin IVA (500 nM) only partially inhibited EPPs amplitude (48.5±5%; n=7; P<0.001).
Figure 7.

Effect of ω-agatoxin IVA on EPPs of LEMS IgGs-treated mouse neuromuscular junctions. (A) Representative EPP traces recorded from a LEMS-treated mouse phrenic nerve/hemidiaphragm preparation (LEMS 4; see Table 1) in 2.5 mM Ca2+ Krebs solution (control), 15 and 30 min after the application of ω-agatoxin IVA (500 nM). Each trace is the average of 20–30 traces. (B1 and B2) Bar graphs showing the effect of pre-incubations (1 h; see Methods for description of the protocol) of mouse phrenic nerve/hemidiaphragm preparations treated with LEMS IgGs (LEMS 5 and LEMS 6; see Table 1) or with healthy control IgGs (control). Recordings of EPPs amplitude (B1) and quantal content (B2) were carried out in the presence of 2.5 mM Ca2+ Krebs solution alone (empty bars; n=7) or with ω-agatoxin IVA (500 nM, black bars, n=3). Results are shown as means+s.e. (**P<0.01); n indicates the number of hemidiaphragm preparations.
The inhibition was time-dependent with maximal block reached after 30 min from the start of toxin application. The partial block of EPPs caused by ω-agatoxin IVA indicates that in LEMS IgGs-treated NMJs, as in 4-AP-treated NMJs, ω-agatoxin IVA-insensitive Ca2+ channels contribute to the evoked ACh release.
ω-Agatoxin IVA had no significant effects on MEPP amplitude (0.9±0.1 mV in control 2.5 mM Ca2+ Krebs solution vs 1±0.2 mV in ω-agatoxin IVA; n=6) and frequency (0.9±0.03 s−1 in control 2.5 mM Ca2+ Krebs solution vs 0.8±0.16 s−1 in ω-agatoxin IVA; n=6).
The same reduction in the blocking activity of ω-agatoxin IVA was observed in NMJs of mice treated with IgGs from two other LEMS patients (LEMS 5 and 6; see Table 1 for details). In these two cases, the blocking effects of ω-agatoxin IVA were not tested ‘acutely' as for the first case described above, but with the ‘pre-incubation' protocol. Interestingly, NMJs treated with LEMS 6 IgG showed a more marked reduction in EPP amplitude (35%; P<0.01; n=6) and these EPPs (Figure 7B1) were only slightly blocked (∼28% inhibition) by ω-agatoxin IVA (9.9±1.2 mV in ω-agatoxin IVA (n=3) vs 13.8±1.1 mV in control (n=6)). On the other hand, NMJs treated with LEMS 5 IgG showed only a 23% reduction of EPP amplitude (P<0.05; n=7) and these EPPs (Figure 7B1) were significantly blocked (∼71.3% inhibition) by agatoxin IVA (4.7±1 mV in ω-agatoxin IVA (n=3) vs 16.4±1.2 mV in control (n=7); P<0.01).
It is worth noting that LEMS 6 IgG was also more effective in inducing the physiological changes of the passive transfer paradigm. In NMJs from mice treated with control IgGs, EPPs amplitude (Figure 7B1) and quantal content (Figure 7B2) were almost completely abolished (∼93% inhibition) by ω-agatoxin IVA (1.5±0.6 mV in ω-agatoxin IVA (n=7) vs 21.2±1.9 mV in control (n=3); P<0.01), as in non-injected mice.
Effects of other VDCC blockers on ACh release at the LEMS IgGs-treated NMJ
The sensitivity of LEMS IgGs-treated NMJs to other VDCC blockers was then examined (Figure 8). Nifedipine (5 μM; Figure 8A) and calciseptine (1 μM; Figure 8B), two L-type VDCC specific blockers, caused no statistically significant inhibition of EPP amplitude (17.6±5.3% (n=3) and 27.5±13% (n=4), respectively). The amplitude and the frequency of the MEPPs were not significantly affected by these drugs (data not shown). However, the simultaneous application of calciseptine (1 μM) and ω-agatoxin IVA (500 nM) to a LEMS IgGs-treated NMJ caused a larger reduction in EPP amplitude (64±0.8% (n=5; P<0.001; Figure 8C) compared to ω-agatoxin IVA alone and this effect reached statistical significance (Figures 7A and 8B). The amplitude and the frequency of MEPPs were not significantly affected even by the simultaneous application of these drugs (amplitude: 0.97±0.1 mV to 0.95±0.2 mV (n=2); frequency: 0.79±0.1 s−1 to 0.7±0.2 s−1 (n=4)).
Figure 8.

Effect of nifedipine, calciseptine and ω-agatoxin IVA on EPPs of LEMS IgGs-treated mouse neuromuscular junctions. Representative EPP traces recorded from mouse phrenic nerve/hemidiaphragm preparation (LEMS 5; see Table 1 for details) in 2.5 mM Ca2+ Krebs solution (control) and 10, 20 and 30 min after application on (A) nifedipine (5 μM) or (B) calciseptine (1 μM) or (C) calciseptine (1 μM) and ω-agatoxin IVA (500 nM). Each trace is the average of 20–30 traces.
Discussion
In this study, we have investigated the presence of different VDCC subtypes at the adult mouse NMJ and their role in the control of spontaneous and nerve-evoked ACh release. This work is a comprehensive pharmacological study of a wide range of VDCC blockers effect on either normal or pathological phrenic nerve/hemidiaphragm preparations.
In agreement with previous findings in mammalian NMJs (Protti & Uchitel, 1993; Hong & Chang, 1995; Protti et al., 1996), we have found P/Q-type VDCC to be the predominant Ca2+ channel subtype coupled to the evoked release process under control conditions. In contrast, neither N- nor L-type specific VDCC blockers had any significant effect in reducing nerve-evoked ACh release. Under these conditions, however, the ‘pre-incubation' of the preparation with nifedipine caused a small but significant increase in EPP amplitude compared to control recordings. This surprising result of potentiation rather than inhibition of transmitter release after blocking L-type VDCCs has been previously reported by Sugiura & Ko (1997) in immature mammalian and amphibian NMJs. These authors hypothesized that Ca2+ entry through L-type VDCCs may trigger the release of a neuromodulator which, by binding to a receptor coupled to a PTX-sensitive G-protein, could mediate the inhibition of ACh release. The blockade of L-type VDCC with nifedipine would prevent this G-protein-mediated inhibition and result in an increased transmitter release. Nifedipine could also cause the blockade of K+ channels present at the nerve terminal (like 4-AP) and this in turn would cause depolarization and ACh release. Indeed, nifedipine has previously been reported to block cloned K+ channels (Grissmer et al., 1994) as well as K+ channels expressed in the heart (Zhang et al., 1997).
The role of VDCC subtypes in spontaneous release was also investigated. It is known that both nerve-evoked release and MEPP frequency are, in different ways, dependent on intracellular Ca2+ concentration (Matthews & Wickelgren, 1977).
Our results, obtained under physiological conditions, of ω-agatoxin IVA causing a significant reduction of MEPP frequency, strongly suggest that the P/Q-type VDCC is the major Ca2+ channel subtype involved also in spontaneous release. This result is in contrast with the findings of Losavio & Muchnik (1997), who showed a role for L- and N-type, but not P/Q-type VDCCs in the regulation of spontaneous release. The reasons for this discrepancy are unclear but might include the use of different animals (mice vs rats) and of slightly different concentrations of VDCC blockers (Protti et al., 1991).
The inorganic ion Cd2+, a non-specific VDCC blocker which completely abolished nerve-evoked release, significantly reduced MEPP frequency 20 min after its application, but the effect apparently reversed over time and no significant changes in MEPP frequency could be recorded 1 h after its application. This finding is in line with previous reports on the effects of Cd2+ on spontaneous release at mammalian NMJs. MEPP frequency has been shown either to be not significantly affected (Forshaw, 1977; Porter & Wray, 1996), or to be increased (Nishimura et al., 1984; Braga & Rowan, 1994) by Cd2+ ions. Part of the mechanism by which Cd2+ influences spontaneous ACh release is thought to be intracellular, possibly interfering with intracellular Ca2+ stores (Nishimura et al., 1984). The differential effects of Cd2+ on spontaneous release in these published papers may be explained by the different protocols used. During acute exposure, Cd2+ mainly binds to the pore of the channel hence preventing the passage of Ca2+ ions and ACh release (Nachshen, 1984). In contrast, after pre-incubation, Cd2+ might also permeate the cells and cause ACh release either directly or by releasing intracellular Ca2+ from stores.
The above results confirm previous findings suggesting that only Cd2+- and ω-agatoxin IVA-sensitive (P/Q-type) Ca2+ channels contribute to both spontaneous and nerve-evoked ACh release at the mouse NMJ (Protti & Uchitel, 1993; Hong & Chang, 1995; Protti et al., 1996). However, electrophysiological studies have demonstrated that multiple types of VDCCs exist in neuron somata (Dunlap et al., 1995) as well as in central synapses (Takahashi & Momiyama, 1993; Wheeler et al., 1994). In particular, the presence of multiple VDCC subtypes has been demonstrated in mature mouse (Carlin et al., 2000) and rat (Magnelli et al., 1998) spinal motoneurons. Furthermore, Penner & Dreyer (1986) have previously suggested the presence of at least two distinct subtypes of Ca2+ channels in mouse motor nerve terminals.
We therefore conducted further experiments in order to evaluate if different VDCC subtypes contribute to ACh release from mouse motor nerve terminals under ‘modified' and autoimmune (LEMS auto-antibodies) conditions. Indeed, in LEMS, patients producing VDCC auto-antibodies release less ACh from their nerve terminals; furthermore, this condition is treated with 4-AP, which potentiates ACh release. We indeed found, in both these physiological conditions, an ω-agatoxin IVA- and ω-conotoxin MVIIC-resistant Ca2+ channel component contributing to ACh release from mouse motor nerve terminals.
Although we could not confirm the results by Lin & Lin-Shiau (1997), showing a synergistic effect of ω-agatoxin IVA and ω-conotoxin MVIIC, we did find a synergistic effect of ω-agatoxin IVA and ω-conotoxin GVIA in their ability to depress EPPs in the presence of 4-AP.
These results support the hypothesis that VDCCs other than the P/Q-type become involved in ACh release in the presence of 4-AP; in particular, our results suggest that both N- and ‘R'-type VDCCs become important under this pharmacological condition.
N-type VDCCs presence in normal mammalian nerve terminals has already been previously reported and these channels are identified by their sensitivity in ω-conotoxin GVIA (Protti et al., 1991; Rossoni et al., 1994; Santafé et al., 2000). The presence of Ca2+ channels resistant to all the known VDCC specific blockers (‘R'-type) contributing to evoked ACh release, as shown here and by Lin and Lin-Shiau, is less established and characterized. Interestingly, these ‘R'-type VDCCs appear to be insensitive to SNX 482 under our experimental conditions. SNX 482 has been previously reported to selectively block some but not all R-type VDCCs (Newcomb et al., 1998; Wang et al., 1999; Tottene et al., 2000) present in several different systems. We are not aware of other reports where this toxin has been tested on NMJs.
In the LEMS IgGs-treated mice, EPP amplitude was significantly reduced compared to that in control NMJs and ω-agatoxin IVA was much less effective in inhibiting EPP amplitude in these LEMS IgGs-treated NMJs. Interestingly, the extent of EPP inhibition caused by LEMS IgGs seems to be inversely correlated to the extent of EPP inhibition caused by ω-agatoxin IVA in the same junctions. The simultaneous application of ω-agatoxin IVA and calciseptine to the LEMS IgGs-treated NMJs caused greater EPP inhibition than that caused by the individual toxins, indicating that L-type VDCCs do play a role in the control of nerve-evoked release under this pathological condition.
The ‘recruitment' of a DHP-sensitive L-type VDCC component in LEMS IgGs-treated mice NMJs as well as in LEMS IgGs-treated mice motoneurons has been previously reported (Smith et al., 1995; Garcia & Beam, 1996; Xu et al., 1998). However, DHP drugs are known to be non-selective Ca2+ channel blockers when used at micromolar concentrations (Diochot et al., 1995). For this reason, we tested the possible involvement of L-type VDCCs in ACh release using calciseptine, a more selective L-type VDCC blocker (de weille et al., 1991).
Our results with calciseptine confirm the hypothesis that L-type VDCCs become involved in the control of synaptic transmission at LEMS IgGs-treated mouse NMJ. L-type VDCCs in mammalian motor nerve terminals (Atchison & O'leary, 1987; Atchison, 1989; Katz et al., 1996; Sugiura & Ko, 1997; Urbano & Uchitel, 1999; Correia-de-sá et al., 2000a, b; Santafé et al., 2000) have been previously identified; moreover, a role of these channels in the control of ACh release during development (Gray et al., 1992; Fu & Huang, 1994) and pathological conditions (Fratantoni et al., 2000) has been demonstrated. Our results show for the first time that, in LEMS IgGs-treated NMJs, both L- and ‘R'-type VDCCs are ‘recruited' for a functional role when a deficit in P/Q-type VDCCs is occurring, like in LEMS.
Compensatory increases in specific components of Ca2+ currents have already been described. An increase in the proportion of a residual ‘R'-type Ca2+ current and a correspondent decrease in the proportion of P-type current after 15–22 h of incubation of cerebellar Purkinje neurons in the presence of LEMS IgGs has been reported by Pinto et al. (1998a). Gillard et al. (1997) found similar compensatory mechanisms using anti-P/Q-type antisense oligonucleotides in the same type of cells.
The physiological significance of the expression of multiple VDCC subtypes is still not completely clear. Ca2+ channel subtypes differ from each other for their distinct biophysical properties, differential modulation and variable coupling to intracellular processes (Neveu et al., 1994; Arnot et al., 2000).
The existence of different Ca2+ channel subtypes at the nerve terminal would allow a more flexible and versatile regulation of ACh release, especially under non-physiological conditions. Different VDCC subtypes may play important and selective roles during development and regeneration of NMJs and in pathological conditions when transmitter release is compromised.
Finally, the presence of multiple VDCCs at the NMJ is likely to represent the basis for the clinical efficiency of compounds such as 4-AP, that rely on the ‘recruitment' of ‘spared' VDCCs in order to potentiate release from compromised NMJs. A direct potentiation of selective VDCC subtypes to treat secretory deficits is another approach worth considering.
Acknowledgments
We would like to thank Dr Paola Pedarzani (Department of Physiology, UCL, U.K.) for reading the manuscript and offering valuable suggestions. We thank Dr G. Dayanithi (Montpellier, France) for the kind donation of the toxin SNX 482. This work has been financially supported by Eli Lilly and Research Centre Lim (Surrey, U.K.).
Abbreviations
- 4-AP
4-aminopyridine
- ACh
acetylcholine
- EPP
end plate potential
- IgG
immunoglobulin
- LEMS
Lambert-Eaton myasthenic syndrome
- MEPP
miniature end plate potential
- NMJ
neuromuscular junction
- VDCC
voltage-dependent calcium channel
References
- ARNOT M.I., STOTZ S.C., JARVIS S.E., ZAMPONI G.W. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms. J. Physiol. 2000;527:203–212. doi: 10.1111/j.1469-7793.2000.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATCHISON W.D., O'LEARY S.M. BAY K 8644 increases release of acetylcholine at the murine neuromuscular junction. Brain Res. 1987;419:315–319. doi: 10.1016/0006-8993(87)90599-3. [DOI] [PubMed] [Google Scholar]
- ATCHISON W.D. Dihydropyridine-sensitive and -insensitive components of acetylcholine release from rat motor nerve terminals. J. Pharmacol. Exp. Ther. 1989;251:672–678. [PubMed] [Google Scholar]
- BOWERSOX S.S., MILJANICH G.P., SUGIURA Y., LI C., NADASDI L., HOFFMAN B.B., RAMACHANDRAN J., KO C.P. Differential blockade of voltage-sensitive calcium channels at the mouse neuromuscular junction by novel omega-conopeptides and omega-agatoxin-IVA. J. Pharmacol. Exp. Ther. 1995;273:248–256. [PubMed] [Google Scholar]
- BRAGA M.F., ROWAN E.G. The pharmacological effects of cadmium on skeletal neuromuscular transmission. Gen. Pharmacol. 1994;25:1729–1739. doi: 10.1016/0306-3623(94)90379-4. [DOI] [PubMed] [Google Scholar]
- CARLIN K.P., JIANG Z., BROWNSTONE R.M. Characterization of calcium currents in functionally mature mouse spinal motoneurons. Eur. J. Neurosci. 2000;12:1624–1634. doi: 10.1046/j.1460-9568.2000.00050.x. [DOI] [PubMed] [Google Scholar]
- CORREIA-DE-SÁ P., TIMOTEO M.A., RIBEIRO J.A. A(2A) adenosine receptor facilitation of neuromuscular transmission: influence of stimulus paradigm on calcium mobilization. J. Neurochem. 2000a;74:2462–2469. doi: 10.1046/j.1471-4159.2000.0742462.x. [DOI] [PubMed] [Google Scholar]
- CORREIA-DE-SÁ P., TIMOTEO M.A., RIBEIRO J.A. Influence of stimulation on Ca(2+) recruitment triggering [3H]acetylcholine release from the rat motor-nerve endings. Eur. J. Pharmacol. 2000b;406:355–362. doi: 10.1016/s0014-2999(00)00686-5. [DOI] [PubMed] [Google Scholar]
- DAY N.C., WOOD S.J., INCE P.G., VOLSEN S.G., SMITH W., SLATER C.R., SHAW P.J. Differential localization of voltage-dependent calcium channel alpha1 subunits at the human and rat neuromuscular junction. J. Neurosci. 1997;17:6226–6235. doi: 10.1523/JNEUROSCI.17-16-06226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE WEILLE J.R., SCHWEITZ H., MAES P., TARTAR A., LAZDUNSKI M. Calciseptine, a peptide isolated from black mamba venom, is a specific blocker of the L-type calcium channel. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2437–2440. doi: 10.1073/pnas.88.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIOCHOT S., RICHARD S., BALDY-MOULINIER M., NARGEOT J., VALMIER J. Dihydropyridines, phenylalkylamines and benzothiazepines block N-, P/Q- and R-type calcium currents. Pflugers Arch. 1995;431:10–19. doi: 10.1007/BF00374372. [DOI] [PubMed] [Google Scholar]
- DUNLAP K., LUEBKE J.I., TURNER T.J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- FORSHAW P.J. The inhibitory effect of cadmium on neuromuscular transmission in the rat. Eur. J. Pharmacol. 1977;42:371–377. doi: 10.1016/0014-2999(77)90171-6. [DOI] [PubMed] [Google Scholar]
- FRATANTONI S.A., WEISZ G., PARDAL A.M., REISIN R.C., UCHITEL O.D. Amyotrophic lateral sclerosis IgG-treated neuromuscular junctions develop sensitivity to L-type calcium channel blocker. Muscle Nerve. 2000;23:543–550. doi: 10.1002/(sici)1097-4598(200004)23:4<543::aid-mus13>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- FU W.M., HUANG F.L. L-type Ca2+ channel is involved in the regulation of spontaneous transmitter release at developing neuromuscular synapses. Neuroscience. 1994;58:131–140. doi: 10.1016/0306-4522(94)90160-0. [DOI] [PubMed] [Google Scholar]
- GARCIA K.D., BEAM K.G. Reduction of calcium currents by Lambert-Eaton syndrome sera; motoneurons are preferentially affected, and L-type currents are spared. J. Neurosci. 1996;16:4903–4913. doi: 10.1523/JNEUROSCI.16-16-04903.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILLARD S.E., VOLSEN S.G., SMITH W., BEATTIE R.E., BLEAKMAN D., LODGE D. Identification of pore-forming subunit of P-type calcium channels: an antisense study on rat cerebellar Purkinje cells in culture. Neuropharmacology. 1997;36:405–409. doi: 10.1016/s0028-3908(97)00046-4. [DOI] [PubMed] [Google Scholar]
- GRAY D.B., BRUSES J.L., PILA G.R. Developmental switch in the pharmacology of Ca2+ channels coupled to acetylcholine release. Neuron. 1992;18:715–724. doi: 10.1016/0896-6273(92)90092-r. [DOI] [PubMed] [Google Scholar]
- GRISSMER S., NGUYEN A.N., AIYAR J., HANSON D.C., MATHER R.J., GUTMAN G.A., KARMILOWICZ M.J., AUPERIN D.D., CHANDY K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- HONG S.J., CHANG C.C. Inhibition of acetylcholine release from mouse motor nerve by a P-type calcium channel blocker, omega-agatoxin IVA. J. Physiol. 1995;482:283–290. doi: 10.1113/jphysiol.1995.sp020517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATZ B., THESLEFF S. On the factors which determine the amplitude of the ‘miniature end-plate potential'. J. Physiol. 1957;137:267–278. doi: 10.1113/jphysiol.1957.sp005811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATZ E., FERRO P.A., WEISZ G., UCHITEL O.D. Calcium channels involved in synaptic transmission at the mature and regenerating mouse neuromuscular junction. J. Physiol. 1996;497:687–697. doi: 10.1113/jphysiol.1996.sp021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM Y.I. Passive transfer of the Lambert-Eaton myasthenic syndrome: neuromuscular transmission in mice injected with plasma. Muscle Nerve. 1985;8:162–172. doi: 10.1002/mus.880080213. [DOI] [PubMed] [Google Scholar]
- LAMBERT E.H., ELMQVIST D. Quantal components of end-plate potentials in the myasthenic syndrome. Ann. N.Y. Acad. Sci. 1971;183:183–199. doi: 10.1111/j.1749-6632.1971.tb30750.x. [DOI] [PubMed] [Google Scholar]
- LANG B., NEWSOM-DAVIS J., PRIOR C., WRAY D. Antibodies to motor nerve terminals: an electrophysiological study of a human myasthenic syndrome transferred to mouse. J. Physiol. 1983;344:335–345. doi: 10.1113/jphysiol.1983.sp014943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN M.J., LIN-SHIAU S.Y. Multiple types of Ca2+ channels in mouse motor nerve terminals. Eur. J. Neurosci. 1997;9:817–823. doi: 10.1111/j.1460-9568.1997.tb01431.x. [DOI] [PubMed] [Google Scholar]
- LOSAVIO A., MUCHNIK S. Spontaneous acetylcholine release in mammalian neuromuscular junctions. Am. J. Physiol. 1997;273:C1835–C1841. doi: 10.1152/ajpcell.1997.273.6.C1835. [DOI] [PubMed] [Google Scholar]
- LUNDH H. Effects of 4-aminopyridine on neuromuscular transmission. Brain Res. 1978;153:307–318. doi: 10.1016/0006-8993(78)90409-2. [DOI] [PubMed] [Google Scholar]
- MAGNELLI V., BALDELLI P., CARBONE E. Antagonists-resistant calcium currents in rat embryo motoneurons. Eur. J. Neurosci. 1998;10:1810–1825. doi: 10.1046/j.1460-9568.1998.00178.x. [DOI] [PubMed] [Google Scholar]
- MAGNELLI V., GRASSI C., PARLATORE E., SHER E., CARBONE E. Down-regulation of non-L-, non-N-type (Q-like) Ca2+ channels by Lambert-Eaton myasthenic syndrome (LEMS) antibodies in rat insulinoma RINm5F cells. FEBS Lett. 1996;387:47–52. doi: 10.1016/0014-5793(96)00465-6. [DOI] [PubMed] [Google Scholar]
- MATTHEWS G., WICKELGREN W.O. On the effect of calcium on the frequency of miniature end-plate potentials at the frog neuromuscular junction. J. Physiol. 1977;266:91–101. doi: 10.1113/jphysiol.1977.sp011757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCLACHLAN E.M., MARTIN A.R. Non-linear summation of end-plate potentials in the frog and mouse. J. Physiol. 1981;311:307–324. doi: 10.1113/jphysiol.1981.sp013586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINTZ I.M., ADAMS M.E., BEAN B.P. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- MOTOMURA M., LANG B., JOHNSTON I., PALACE J., VINCENT A., NEWSOM-DAVIS J. Incidence of serum anti-P/Q-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J. Neurol. Sci. 1997;147:35–42. doi: 10.1016/s0022-510x(96)05303-8. [DOI] [PubMed] [Google Scholar]
- NACHSHEN D.A. Selectivity of the Ca binding site in synaptosome Ca channels. Inhibition of Ca influx by multivalent metal cations. J. Gen. Physiol. 1984;83:941–967. doi: 10.1085/jgp.83.6.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEVEU D., QUIGNARD J.F., FERNANDEZ A., RICHARD S., NARGEOT J. Differential beta-adrenergic regulation and phenotypic modulation of voltage-gated calcium currents in rat aortic myocytes. J. Physiol. 1994;479:171–182. doi: 10.1113/jphysiol.1994.sp020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEWCOMB R., SZOKE B., PALMA A., WANG G., CHEN XH., HOPKINS W., CONG R., MILLER J., URGE L., TARCZY-HORNOCH K., LOO J.A., DOOLEY D.J., NADASDI L., TSIEN R.W., LEMOS J., MILJANICH G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. doi: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- NISHIMURA M., TSUTSUI I., YAGASAKI O., YANAGIYA I. Transmitter release at the mouse neuromuscular junction stimulated by cadmium ions. Arch. Int. Pharmacodyn. Ther. 1984;271:106–121. [PubMed] [Google Scholar]
- NOWYCKY M.C., FOX A.P., TSIEN R.W. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- OUSLEY A.H., FROEHNER S.C. An anti-peptide antibody specific for the class A calcium channel alpha 1 subunit labels mammalian neuromuscular junction. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12263–12267. doi: 10.1073/pnas.91.25.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENNER R., DREYER F. Two different presynaptic calcium currents in mouse motor nerve terminals. Pflugers Arch. 1986;406:190–197. doi: 10.1007/BF00586682. [DOI] [PubMed] [Google Scholar]
- PINTO A., GILLARD S., MOSS F., WHYTE K., BRUST P., WILLIAMS M., STAUDERMAN K., HARPOLD M., LANG B., NEWSOM-DAVIS J., BLEAKMAN D., LODGE D., BOOT J. Human autoantibodies specific for the alpha1A calcium channel subunit reduce both P-type and Q-type calcium currents in cerebellar neurons. Proc. Natl. Acad. Sci. U.S.A. 1998a;95:8328–8333. doi: 10.1073/pnas.95.14.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINTO A., MOSS F., LANG B., BOOT J., BRUST P., WILLIAMS M., STAUDERMAN K., HARPOLD M., NEWSOM-DAVIS J. Differential effect of Lambert-Eaton myasthenic syndrome immunoglobulin on cloned neuronal voltage-gated calcium channels. Ann. N.Y. Acad. Sci. 1998b;841:687–690. doi: 10.1111/j.1749-6632.1998.tb11003.x. [DOI] [PubMed] [Google Scholar]
- PORTER V.A., WRAY D. Relative potencies of metal ions on transmitter release at mouse motor nerve terminals. Br. J. Pharmacol. 1996;118:27–32. doi: 10.1111/j.1476-5381.1996.tb15362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PROTTI D.A., REISIN R., MACKINLEY T.A., UCHITEL O.D. Calcium channel blockers and transmitter release at the normal human neuromuscular junction. Neurology. 1996;46:1391–1396. doi: 10.1212/wnl.46.5.1391. [DOI] [PubMed] [Google Scholar]
- PROTTI D.A., SZCZUPAK K., SCORNIK F.S., UCHITEL O.D. Effect of omega-conotoxin GVIA on neurotransmitter release at the mouse neuromuscular junction. Brain Res. 1991;557:336–339. doi: 10.1016/0006-8993(91)90156-p. [DOI] [PubMed] [Google Scholar]
- PROTTI D.A., UCHITEL O.D. Transmitter release and presynaptic Ca2+ currents blocked by the spider toxin omega-Aga-IVA. Neuroreport. 1993;5:333–336. doi: 10.1097/00001756-199312000-00039. [DOI] [PubMed] [Google Scholar]
- ROSATO SIRI M.D., UCHITEL O.D. Calcium channels coupled to neurotransmitter release at neonatal rat neuromuscular junctions. J. Physiol. 1999;514:533–540. doi: 10.1111/j.1469-7793.1999.533ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSSONI G., BERTI F., LA MAESTRA L., CLEMENTI F. Omega-Conotoxin GVIA binds to and blocks rat neuromuscular junction. Neurosci. Lett. 1994;176:185–188. doi: 10.1016/0304-3940(94)90078-7. [DOI] [PubMed] [Google Scholar]
- SANTAFÉ M.M., GARCIA N., LANUZA M.A., UCHITEL O.D., TOMÁS J. Calcium channels coupled to neurotransmitter release at dually innervated neuromuscular junctions in the newborn rat. Neuroscience. 2001;102:697–708. doi: 10.1016/s0306-4522(00)00507-8. [DOI] [PubMed] [Google Scholar]
- SANTAFÉ M.M., URBANO F.J., LANUZA M.A., UCHITEL O.D. Multiple types of calcium channels mediate transmitter release during functional recovery of botulinum toxin type A-poisoned mouse motor nerve terminals. Neuroscience. 2000;95:227–234. doi: 10.1016/s0306-4522(99)00382-6. [DOI] [PubMed] [Google Scholar]
- SMITH D.O., CONKLIN M.W., JENSEN P.J., ATCHISON W.D. Decreased calcium currents in motor nerve terminals of mice with Lambert-Eaton myasthenic syndrome. J. Physiol. 1995;487:115–123. doi: 10.1113/jphysiol.1995.sp020865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUGIURA Y., KO C.P. Novel modulatory effect of L-type calcium channels at newly formed neuromuscular junctions. J. Neurosci. 1997;17:1101–1111. doi: 10.1523/JNEUROSCI.17-03-01101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKAHASHI T., MOMIYAMA A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- THOMSEN R.H., WILSON D.F. Effects of 4-aminopyridine and 3,4-diaminopyridine on transmitter release at the neuromuscular junction. J. Pharmacol. Exp. Ther. 1983;227:260–265. [PubMed] [Google Scholar]
- TOTTENE A., VOLSEN S., PIETROBON D. alpha(1E) subunits from the pore of three cerebellar R-type calcium channels with different pharmacological and permeation properties. J. Neurosci. 2000;20:171–178. doi: 10.1523/JNEUROSCI.20-01-00171.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSIEN R.W., LIPSCOMBE D., MADISON D.V., BLEY K.R., FOX A.P. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- UCHITEL O.D., PROTTI D.A., SANCHEZ V., CHERKSEY B.D., SUGIMORI M., LLINAS R. P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3330–3333. doi: 10.1073/pnas.89.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- URBANO F.J., DEPRETIS R.S., UCHITEL O.D. Coupling of L-type calcium channels to neurotransmitter release at mouse nerve terminals. Pflugers Arch. 2001;441:824–831. doi: 10.1007/s004240000489. [DOI] [PubMed] [Google Scholar]
- URBANO F.J., UCHITEL O.D. L-type calcium channels unmasked by cell-permeant Ca2+ buffer at mouse motor nerve terminals. Pflugers Arch. 1999;437:523–528. doi: 10.1007/s004240050813. [DOI] [PubMed] [Google Scholar]
- WANG G., DAYANITHI G., NEWCOMB R., LEMOS J.R. An R-type Ca(2+) current in neurohypophysial terminals preferentially regulates oxytocin secretion. J. Neurosci. 1999;19:9235–9241. doi: 10.1523/JNEUROSCI.19-21-09235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WESTENBROEK R.E., HOSKINS L., CATTERALL W.A. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J. Neurosci. 1998;18:6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHEELER D.B., RANDALL A., TSIEN R.W. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- WILSON S.M., TOTH P.T., OH S.B., GILLARD S.E., VOLSEN S., REN D., PHILIPSON L.H., LEE E.C., FLETCHER C.F., TESSAROLLO L., COPELAND N.G., JENKINS N.A., MILLER R.J. The Status of Voltage-Dependent Calcium Channels in alpha 1E Knock-Out Mice. J. Neurosci. 2000;20:8566–8571. doi: 10.1523/JNEUROSCI.20-23-08566.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU L.G., BORST J.G., SAKMANN B. R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4720–4725. doi: 10.1073/pnas.95.8.4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU L.G., SAGGAU P. Pharmacological identification of two types of presynaptic voltage-dependent calcium channels at CA3-CA1 synapses of the hippocampus. J. Neurosci. 1994;14:5613–5622. doi: 10.1523/JNEUROSCI.14-09-05613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU Y.F., HEWETT S.J., ATCHISON W.D. Passive transfer of Lambert-Eaton myasthenic syndrome induces dihydropyridine sensitivity of ICa in mouse motor nerve terminals. J. Neurophysiol. 1998;80:1056–1069. doi: 10.1152/jn.1998.80.3.1056. [DOI] [PubMed] [Google Scholar]
- ZHANG X., ANDERSON J.W., FEDIDA D. Characterization of nifedipine block of the human heart delayed rectifier, hKv1.5. J. Pharmacol. Exp. Ther. 1997;281:1247–1256. [PubMed] [Google Scholar]
- ZHANG J.F., RANDALL A.D., ELLINOR P.T., HORNE W.A., SATHER W.A., TANABE T., SCHWARZ T.L., TSIEN R.W. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology. 1993;32:1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]