

Abstract
It is known that nonselective cyclo-oxygenase (COX) inhibitors have small but significant effects on blood pressure (BP), most notably in hypertensive patients on antihypertensive medication. Whether selective COX-2 inhibitors also interfere with BP regulation is not well understood. Therefore, we aimed to examine the effect of chronic treatment with a selective COX-2 inhibitor (rofecoxib) on systolic blood pressure (sBP) in normotensive Wistar-Kyoto rats (WKY) and on the developmental changes of sBP in young spontaneously hypertensive rats (SHR). In addition, we investigated a possible influence of salt intake on the effects of COX-2 inhibition on BP in these two rat strains.
Rofecoxib dose dependently increased sBP and decreased plasma levels of 6-keto prostaglandin (PG)F1α in WKY rats fed a normal salt diet (0.6% NaCl, wt wt−1), without affecting serum thromboxane (TX)B2 levels.
Rofecoxib significantly elevated sBP in both rat strains fed normal salt or high salt diet (8% NaCl, wt wt−1), but not in rats on low salt intake (0.02% NaCl, wt wt−1).
Rofecoxib significantly decreased plasma levels of 6-keto PGF1α in both rat strains fed normal or high salt diet, but not in rats during low salt intake.
Rofecoxib exerted no influence on the changes of body weight nor on water intake. Plasma renin activity (PRA) and renocortical renin mRNA abundance were not changed by rofecoxib, but plasma aldosterone concentration (PAC) was significantly reduced.
These results suggest that chronic inhibition of COX-2 causes an increase of blood pressure that depends on prostacyclin synthesis. Furthermore, this increase is independent on genetic predisposition and can be prevented by salt deprivation. Since water intake and body weight gain were not changed by rofecoxib, fluid retention appears not to be a major reason for the development of hypertension. Similarly, an activation of the renin-angiotensin-aldosterone axis appears to be an unlikely candidate mechanism.
Keywords: Hypertension, cyclo-oxygenase-2, aldosterone, renin, SHR, prostaglandin, blood pressure, kidney
Introduction
Prostaglandins (PGs) are of relevance in physiological responses, inflammation and thrombosis (Narumiya et al., 1999; Breyer & Breyer, 2000). They are formed from arachidonic acid by the catalytic activity of cyclo-oxygenases (COX) via the unstable endoperoxide PGH2. Isomerization or reduction by cell-specific isomerases or reductases leads to the formation of the prostaglandins PGE2, PGD2, prostacyclin (PGI2) and thromboxane (Tx) A2 (Smith et al., 1991). Cyclo-oxygenases exist at least in two isoforms (Vane et al., 1998). COX-1 is constitutively expressed in all tissues, whereas COX-2 was thought to be an inducible isoform (Feng et al., 1993), but recently found to be also constitutively expressed in some tissues such as the kidney, albeit to a much smaller extent as COX-1 (Feng et al., 1993; Harris et al., 1994; Yang et al., 1999). Prostacyclin (PGI2) is known to be a potent vasodilator and an inhibitor of platelet aggregation. PGI2 is produced mainly by the vascular endothelium (Bunting et al., 1983) and its potent vasorelaxant properties contribute to the maintenance of normal vascular tone. Moreover, PGI2 stimulates the release of renin from the kidney (Imig, 2000; Harris & Breyer, 2001). Thromboxane (Tx) A2 is the major product of platelet COX-1 activity (Funk et al., 1991), and lowering of serum TxB2, its inactive hydrolysis product, reflects inhibition of this isoform. Furthermore, TxA2 is known to be a potent vasoconstrictor and plays therefore also an important role in the regulation of vascular haemostasis (Bunting et al., 1983). The balance between the formation of PGI2 and of TxA2 is thought to play also a role in the regulation of platelet aggregation and in vascular tone. Inhibition of prostaglandin synthesis is suggested to be responsible for both the therapeutic and adverse effects associated with the administration of conventional nonsteroidal anti-inflammatory drugs (NSAIDs) (Vane & Botting, 1995). Several studies in humans have demonstrated that NSAIDs attenuate the antihypertensive effect by different antihypertensive drugs, but the pathogenic mechanisms underlying being poorly understood (Chrysant et al., 1980; Clive & Stoff, 1984; Patrono & Dunn, 1987; Muscara et al., 1998). Since the cardiovascular, gastrointestinal and other side effects of nonselective NSAIDs have been mainly attributed to COX-1 inhibition, selective COX-2 inhibitors have been developed with the hope to avoid major adverse cardiovascular effects of NSAIDs. There is also emerging evidence for a physiological role of COX-2. Therefore, the possibility arises that selective COX-2 inhibition will result in adverse effects. In humans, it has been demonstrated that the selective COX-2 inhibitors celecoxib and rofecoxib markedly suppressed prostacyclin synthesis in healthy volunteers (McAdam et al., 1999; Catella-Lawson et al., 1999). Furthermore, it has been described that selective COX-2 inhibitors lower PGE2 concentrations in the rat kidney cortex (Höcherl et al., 2001) and decrease renal excretion of PGs as well (Kammerl et al., 2001). Moreover, COX-2 inhibitors have been reported to decrease renal blood flow and to induce sodium retention (Rodriguez et al., 2000; Rossat et al., 1999). Whether and how selective COX-2 inhibitors interfere with BP regulation is yet not clear. In elderly humans, it has been reported that COX-2 inhibitors either do not change BP (Rossat et al., 1999; Catella-Lawson et al., 1999) or increase BP in a yet undefined subpopulation of patients suffering from osteoarthritis (Whelton et al., 2001). In Wistar-Kyoto (WKY) rats, prolonged treatment with a COX-2 inhibitor increased BP (Muscara et al., 2000), whilst in Sprague Dawley rats with renovascular hypertension COX-2 inhibition lowered BP (Wang et al., 1999). Because of this diffuse situation we systematically investigated the effect of selective COX-2 inhibition on sBP in rats. In particular, we were further interested to learn more about the effects of COX-2 inhibition on BP in SHR during development of a genetically fixed hypertension, a period where SHR show a salt sensitivity for blood pressure regulation (Zicha & Kunes, 1999). For this purpose we subjected normotensive WKY rats and spontaneously hypertensive rats (SHRs) to different salt diets and treated them with the selective COX-2 inhibitor rofecoxib.
Methods
Selection of dose
To determine a dose of rofecoxib for further in vivo studies, which selectively inhibits COX-2 isoform, the air pouch model was performed. In brief, an air pouch was induced as described previously (Muscara et al., 1998). Carrageenan (2 ml of a 1% wt vol−1 solution in sterile isotonic saline; Sigma, Germany) was injected into the air pouch. Six hours later, the pouch was opened during sevoflurane anaesthesia (3% vol vol−1) and the exudate was collected for PGE2 measurement as an index for COX-2 activity using a commercially available EIA (Cayman Chemicals, Ann Arbor, U.S.A.). Blood was collected by cardiac puncture and incubated at 37°C for 60 min for measurement of whole blood thromboxane synthesis (Cayman Chemicals, Ann Arbor, U.S.A.) as an index for COX-1 activity. After decapitation, the stomach was removed, cleaned and frozen in liquid nitrogen until determination of PGE2. Rats were pre-treated orally with saline or rofecoxib with increasing doses (1, 3, 10 and 30 mg kg−1 d−1) 30 min before injection of carrageenan. A further control group received an injection of 2 ml saline and saline orally.
Animal treatment and blood pressure measurement
Male WKY and SHRs were obtained from Charles River (Sulzfeld, Germany) at an age of 21±3 days. To determine dose dependent effects of rofecoxib on blood pressure, groups of eight rats each were orally treated each day with rofecoxib (1, 3, 10 or 30 mg kg−1) or vehicle and received a normal salt diet (0.06% NaCl wt wt−1) for 4 weeks. Rats were conscious during gavaging of the drug or vehicle. The volume of vehicle- or drug solution was about 0.2 ml at the beginning of the study and increased to about 1.0 ml at the end of the study.
For subsequent studies groups of eight rats each were treated by gavage with rofecoxib (10 mg kg−1 d−1) or vehicle and placed for 4 weeks on either a low salt diet (0.02% NaCl, wt wt−1), a normal salt diet (0.6% NaCl, wt wt−1) or a high salt diet (8% NaCl, wt wt−1).
Body weight, water and food intake was determined daily before drug administration. Systolic blood pressure and heart rate measurements were performed (tail cuff method) prior to treatment and at 1-week intervals for a total of 4 weeks. Measurements were always performed 18–24 h after administration of rofecoxib or vehicle. At the end of the study period, the final doses were given 2 h prior to decapitation of rats during sevoflourane anaesthesia (3%, vol. vol.−1). Plasma and kidney cortex slices were frozen in liquid nitrogen and stored at −80°C until assay.
Determination of plasma renin activity (PRA), plasma aldosterone concentration (PAC) and plasma 6-keto PGF1α levels
PRA and PAC were determined using commercially available radioimmunoassays (Sorin Biomedical, Düsseldorf, Germany). The reliable detection limit for the PAC RIA was approximately 8.0 pg. The lowest value for the standard curve was 6.25 pg. Concentrations of plasma 6-keto PGF1α were determined by using an EIA kit (Cayman Chemical, Ann Arbor, U.S.A.).
Ribonuclease protection assay for renocortical renin mRNA and β-actin mRNA abundance
Renocortical renin mRNA and β-actin mRNA levels were measured by specific RNase protection assay basically as described previously (Mann et al., 2001). In brief, 40 μg of total renocortical mRNA were used for the determination of renin mRNA and 1 μg for β-actin mRNA.
Determination of plasma potassium levels
Plasma potassium levels were determined in triplicate using Reflotron® (Boehringer Mannheim, Mannheim, Germany).
Statistical analysis
Level of significance was calculated by one-way ANOVA followed by student's t-test. A P value <0.05 was considered significant.
Results
Selection of dose of rofecoxib
Rofecoxib did not significantly affect COX-1 activity, as measured by whole blood thromboxane synthesis, at any dose tested (1–30 mg kg−1) (Figure 1). Furthermore, rofecoxib, at any dose tested, exerted no influence on gastric PGE2 concentration (Figure 1). However, rofecoxib dose-dependently inhibited inflammatory COX-2 activity. Thus, 1 mg kg−1 reduced PGE2 concentration in the pouch exudates to about 60% and 30 mg kg−1 to about 25% compared to vehicle treated rats (Figure 1).
Figure 1.
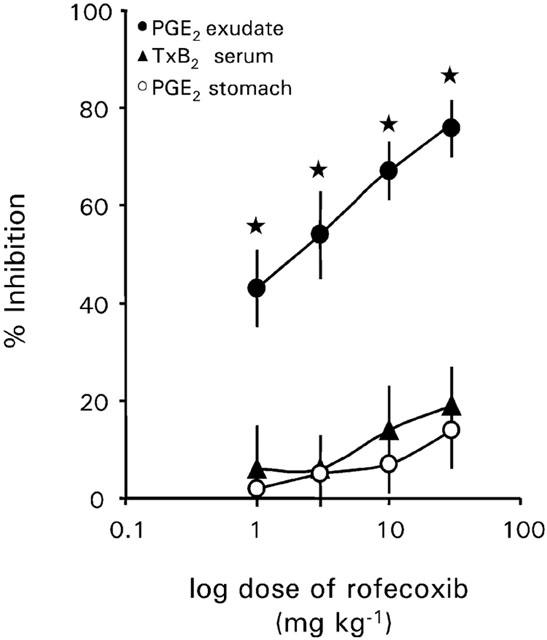
Effect of increasing doses of rofecoxib (0, 1, 3, 10 and 30 mg kg−1 d−1) on the release of PGE2 into inflammatory exudate, on whole blood TxB2 synthesis and gastric PGE2. Rats were pre-treated with rofecoxib 30 min before injection of carrageenan (2 ml of a 1% wt vol−1 solution in sterile saline) into the air-pouch. Data represent the mean±s.e.mean of n=4–5 rats. Asterisk indicates significance of control rats to rofecoxib treated rats.
Dose-dependency of rofecoxib on systolic blood pressure in WKY rats during normal salt intake
In vehicle treated WKY rats sBP developmentally increased from 109±3 mmHg at an age of 3 weeks to about 123±2 mmHg at an age of 7 weeks. Daily oral treatment of rofecoxib at a dose of 10 or 30 mg kg−1 d−1 resulted in a significant increase in systolic blood pressure in normotensive WKY rats after 1 week as compared to vehicle treated rats. Rofecoxib at a dose of 3 mg kg−1 d−1 increased blood pressure after 2 weeks, whereas 1 mg kg−1 d−1 of rofecoxib increased systolic blood pressure after 3 weeks of the beginning with the treatment (Figure 2) as compared to vehicle treated rats. At the end of the treatment the mean sBP of rofecoxib treated rats was 15, 17, 21 and 23 mmHg higher than vehicle treated rats, respectively (Figure 3a).
Figure 2.
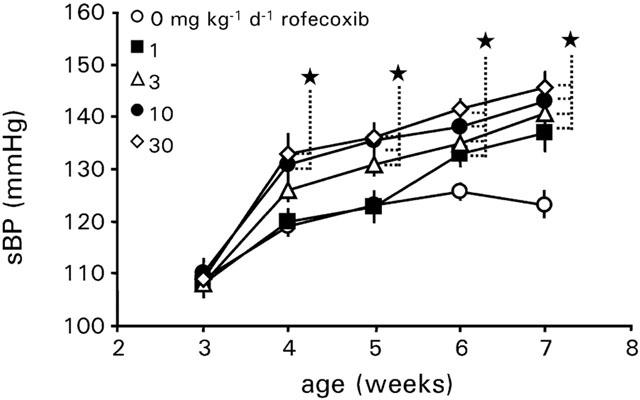
Development of systolic blood pressure of WKY rats treated with vehicle or rofecoxib at increasing doses (1, 3, 10 and 30 mg kg−1 d−1) for 4 weeks during normal salt intake (0.6%, wt wt−1). Data represent the mean±s.e.mean of n=8 rats. Asterisk indicates significance of control rats to rofecoxib treated rats.
Figure 3.
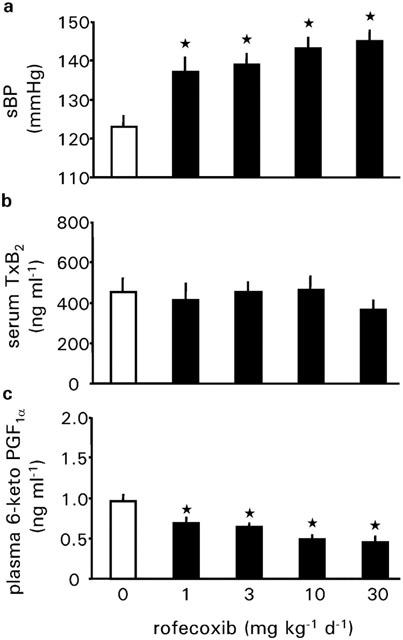
Systolic blood pressure, serum thromboxane B2 concentration and plasma 6-keto prostaglandin F1α concentration of WKY rats treated with vehicle or rofecoxib at increasing doses (1, 3, 10 and 30 mg kg−1 d−1) for 4 weeks during normal salt intake (0.6%, wt wt−1). Data represent the mean±s.e.mean of n=8 rats. Asterisk indicates significance of control rats to rofecoxib treated rats.
Daily treatment with rofecoxib for 4 weeks had no significant effect on serum thromboxane B2 levels. However, rofecoxib dose-dependently decreased plasma 6-keto PGF1α levels. Thus, 30 mg kg−1 d−1 of rofecoxib reduced plasma levels of 6-keto PGF1α to about 45% compared with vehicle treated rats (Figure 3c).
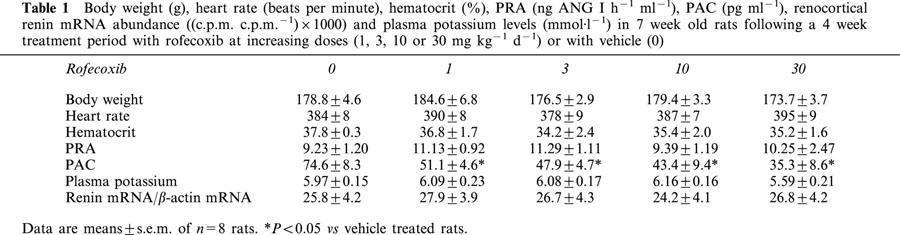
Daily treatment with rofecoxib for 4 weeks had no significant effect on body weight, heart rate, plasma renin activity, renocortical renin mRNA abundance and plasma potassium levels (Table 1). Furthermore, rofecoxib did not affect the hematocrit (Table 1). However, rofecoxib treatment for 4 weeks dose-dependently decreased plasma aldosterone concentration to about 50% of control levels at a dose of 30 mg kg1 d−1 (Table 1).
Table 1.
Body weight (g), heart rate (beats per minute), hematocrit (%), PRA (ng ANG I h−1 ml−1), PAC (pg ml−1), renocortical renin mRNA abundance ((c.p.m. c.p.m.−1)×1000) and plasma potassium levels (mmol·l−1) in 7 week old rats following a 4 week treatment period with rofecoxib at increasing doses (1, 3, 10 or 30 mg kg−1 d−1) or with vehicle (0)
Since treatment with rofecoxib at a dose of 10 mg kg−1 d−1 resulted in similar results as a dose of 30 mg kg −1 d−1, subsequent experiments in spontaneously hypertensive rats compared to normotensive Wistar-Kyoto rats during low, normal and high salt intake for 4 weeks were performed by treating rats with rofecoxib with a dose of 10 mg kg−1 d−1.
Influence of rofecoxib on blood pressure and heart rate during salt intake
In vehicle treated WKY rats sBP developmentally increased from 110±4 mmHg at an age of 3 weeks to 124±4 mmHg at 7 weeks of age during normal salt intake. In WKY rats, different salt intake for 4 weeks did not alter sBP. The mean sBP of WKY rats was 119±5 mmHg during low salt intake and 131±4 mmHg during high salt intake. Administration of rofecoxib resulted in a significant increase in sBP during normal salt and high salt intake, whereas sBP was not influenced by rofecoxib during low salt intake. At an age of 7 weeks, the mean difference in sBP after a 4-week treatment was 23 mmHg during normal salt or 18 mmHg during high salt intake (Figure 4a–c) compared to vehicle treated rats, respectively.
Figure 4.
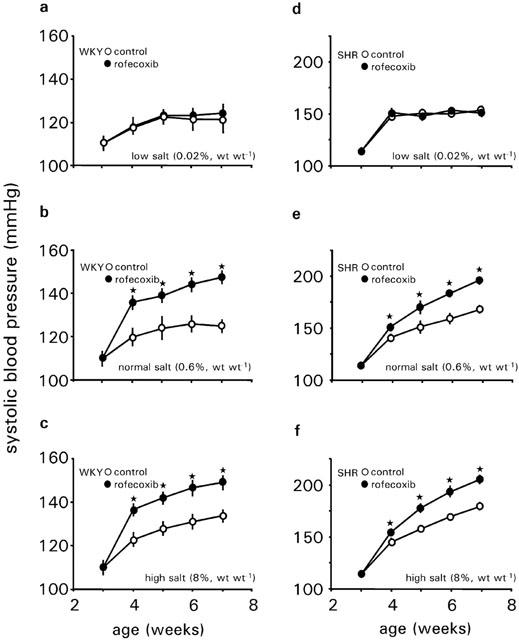
Development of systolic blood pressure of WKY rats (a–c) and SHRs (d–f) treated with vehicle or rofecoxib (10 mg kg−1 d−1) for 4 weeks during low salt intake (0.02%, wt wt−1) (a, d), normal salt intake (0.6%, wt wt−1) (b, e) or high salt intake (8%, wt wt−1) (c, f). Data represent the mean±s.e.mean of n=8 rats. Asterisk indicates significance of control rats to rofecoxib treated rats.
Systolic BP was similar between SHR and WKY at 3 weeks of age. Blood pressure of SHR on normal salt intake increased gradually from age 3 weeks and was significantly higher at ages 4, 5, 6, and 7 weeks compared to those of WKY rats during normal salt intake.
SHRs showed a salt dependent time pattern for the development of hypertension. During normal salt diet sBP increased from 112±3 mmHg at an age of 3 weeks to 166±4 mmHg at an age of 7 weeks. During low salt intake sBP increased to 152±4 mmHg only (P<0.05 vs normal salt intake) and to 180±4 mmHg during high salt intake (P<0.05 vs normal salt intake). Rofecoxib treatment further increased sBP in SHRs fed normal salt diet into the range of 193±4 mmHg and in SHRs fed a high salt diet into the range of 205±5 mmHg, respectively. As for WKY rats, rofecoxib exerted no influence on sBP in SHRs during low salt intake (Figure 4d–f).
Heart rate was not altered either by any salt diet or by rofecoxib treatment in WKY rats.
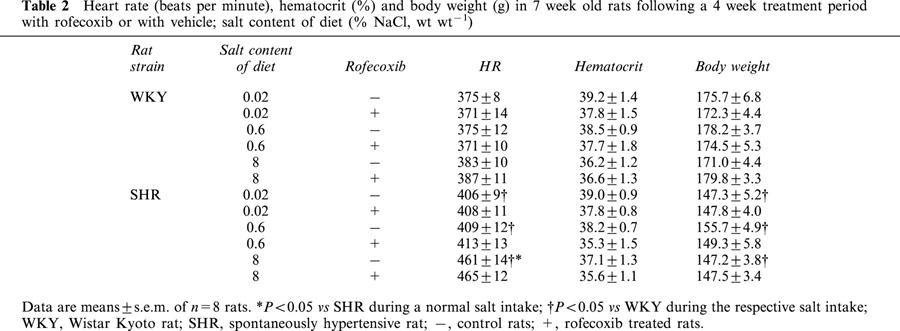
In SHRs, heart rate was significantly higher compared to WKY rats at any given salt diet. High salt intake further enhanced heart rate in SHRs compared to normal salt intake. Rofecoxib treatment did not change heart rate on any salt diet (Table 2).
Table 2.
Heart rate (beats per minute), hematocrit (%) and body weight (g) in 7 week old rats following a 4 week treatment period with rofecoxib or with vehicle; salt content of diet (% NaCl, wt wt−1)
Influence of rofecoxib on body weight, water intake and hematocrit during salt intake
To detect a possible effect on fluid retention induced by rofecoxib, we measured body weight gain as an indirect marker. Treatment with rofecoxib did not change age dependent body weight gain compared to vehicle-treated rats in any rat strain or for any salt diet (Table 2).
For WKY rats, daily water intake normalised to body weight was dependent on the salt content of the diet. During a normal salt intake, the ratio of water intake per day to body weight decreased slightly with increasing age. Low salt diet significantly reduced the values and a high salt diet enhanced it (Figure 5). Also at low or high salt intake the decline of water intake during development was observed. Treatment with rofecoxib did not alter water intake at any salt diet.
Figure 5.
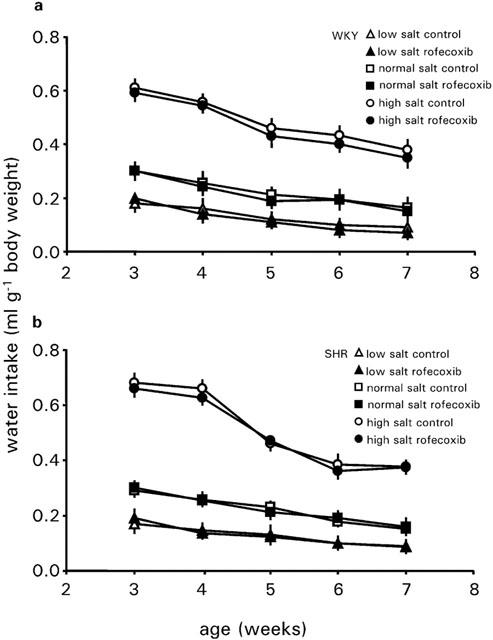
Development of water intake of WKY rats and SHRs treated with vehicle or rofecoxib (10 mg kg−1 d−1) for 4 weeks during low salt intake (0.02%, wt wt−1), normal salt intake (0.6%, wt wt−1) or high salt intake (8%, wt wt−1). Data represent the mean±s.e.mean of n=8 rats.
SHRs behaved very similar to WKY rats with regard to the water intake at any salt diet. Also in these animals, rofecoxib exerted no influence on the water intake at any salt diet (Figure 5). The hematocrit of WKY rats and SHRs was not dependent on salt intake or on rofecoxib treatment (Table 3).
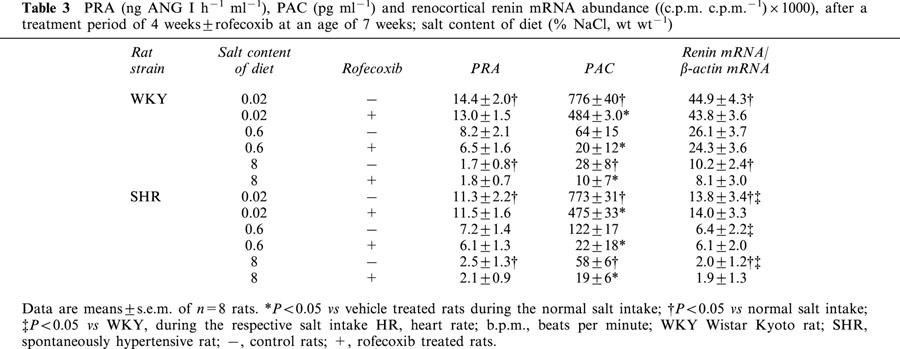
Table 3.
PRA (ng ANG I h−1 ml−1), PAC (pg ml−1) and renocortical renin mRNA abundance ((c.p.m. c.p.m.−1)×1000), after a treatment period of 4 weeks±rofecoxib at an age of 7 weeks; salt content of diet (% NaCl, wt wt−1)
Influence of rofecoxib treatment on plasma renin activity, plasma aldosterone concentration and renocortical renin mRNA during salt intake
The renin-angiotensin-aldosterone system (RAAS) plays an important role for the regulation of blood pressure and also for salt- and water-homeostasis. Its activity depends primarily on the activity of plasma renin. Therefore, we determined PRA which was found to be inversely related to the salt intake. Low salt diet increased PRA about 2.0 fold and high salt diet decreased PRA 2.5 fold compared with a normal salt intake in WKY-rats (Table 3). Similar data were obtained for SHRs. Treatment with the COX-2 inhibitor rofecoxib (10 mg kg−1 d−1) did not influence PRA at any salt diet and in any strain (Table 3).
In the same way as PRA, renin mRNA abundance changed inversely with the salt content of the diet. Thus in WKY rats, a low salt diet increased renin mRNA abundance about 2 fold and a high salt diet decreased renin mRNA abundance about 2.5 fold compared with a normal salt diet (Table 3). SHRs behaved very similar to WKY rats, with the exception, that SHRs had significantly lower renin mRNA levels under all diets (P<0.05). Rofecoxib treatment did not influence renocortical renin mRNA (Table 3).
Furthermore, we measured PAC, another important component of the RAAS. PAC was also inversely correlated to the salt intake. In WKY rats, low salt diet increased PAC about 12 fold and a high salt diet decreased PAC about 2.0 fold compared with animals during normal salt intake (Table 3). SHRs showed a similar behaviour of PAC in response to the salt diet as WKY rats.
Treatment with rofecoxib reduced PAC to about the half of the values at any given diet and in both rat strains (Table 3).
Influence of rofecoxib treatment on plasma levels of 6-keto PGF1α during salt intake
Plasma levels of 6-keto PGF1α changed in both rat strains with the salt intake. SHRs had increased levels during normal and high salt intake as compared to WKY rats. Additional treatment with rofecoxib did not affect plasma levels of 6-keto PGF1α in both rat strains during low salt intake. However, during normal and high salt intake rofecoxib treatment decreased plasma levels of 6-keto PGF1α in both rat strains to about the levels found for low salt intake (Figure 6).
Figure 6.
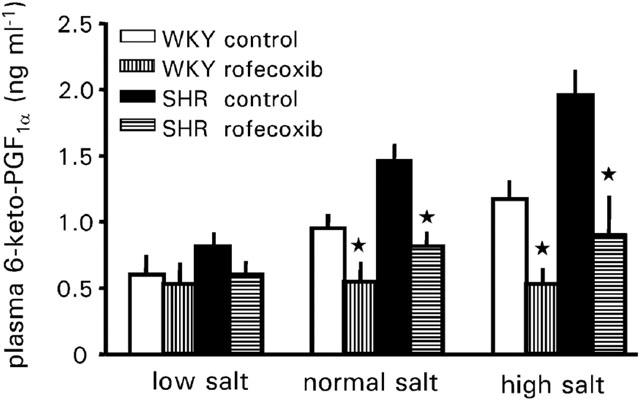
Plasma 6-keto PGF1α levels of WKY-rats and SHRs treated with vehicle or rofecoxib (10 mg kg−1 d−1) for four weeks during low salt intake (0.02%, wt wt−1), normal salt intake (0.6%, wt wt−1) or high salt intake (8%, wt wt−1). Data represent the mean±s.e.mean of n=8 rats. Asterisk indicates significance of control rats to rofecoxib treated rats.
Discussion
Our study aimed to investigate the influence of a prolonged inhibition of COX-2 on the salt dependent developmental regulation of sBP in young SHRs compared with normotensive WKY rats, as genetic control.
In line with previous findings we found an increase in sBP in both rat strains during development (for review see Zicha & Kunes, 1999). This increase was more pronounced in SHR resulting in significant higher levels than at an age of 4 weeks. Furthermore, the increase of sBP in SHR is attenuated by sodium deficient diet and enhanced by high sodium diet, whereas different salt intake has no significant effect on sBP in WKY rats (for review see Zicha & Kunes, 1999). Confirming previous observations, heart rate of SHRs was increased during development at any time when compared to WKY (Dickhout & Lee, 1998). A chronic high salt diet further enhanced heart rate in young SHRs but not in WKY, suggesting enhanced activity of the sympatho-adrenal system in SHRs (Gradin et al., 1986).
Our data now show that COX-2 inhibition for 4 weeks dose dependently increased sBP in WKY rats during normal salt intake, whereas plasma 6-keto PGF1α concentration, a stable metabolite of prostacyclin, decreased in parallel. A low dose of rofecoxib, comparable to the recommended dose of rofecoxib for treatment of human inflammatory disease did increase sBP only after 3 weeks of treatment. This finding would be compatible with recent data obtained with the COX-2 inhibitor NS-398 using a dose of 1 mg kg−1 d−1, where no increase in sBP was found after acute or during a 2-weeks treatment (Harding et al., 1997, 2000) and with nimesulide (Llinas et al., 2000; Rodriguez et al., 2000; Roig et al., 2001), a less selective COX-2 inhibitor than rofecoxib (Warner et al., 1999). At doses of 10 or 30 mg kg−1 d−1 blood pressure increase was achieved already after 1 week of treatment with rofecoxib and remained constantly elevated further on. In line with previous observations, we found that during low salt intake COX-2 inhibition did not increase sBP (Harding et al., 1997, 2000; Rossat et al., 1999). Further, a marked increase of salt intake did not enhance the hypertensive response. With regard to human studies, the VIGOR study has revealed that rofecoxib treatment is associated with an increase of systolic and diastolic blood pressure which is more pronounced than that observed with the non-selective COX-inhibitor naproxen (Mukherjee et al., 2001). Furthermore, a trend towards a rise in sBP was observed by the administration of MK-966 after 14 days (Catella-Lawson et al., 1999). However, it should be noted that in contrast to animal studies, humans respond less sensitive to increases in blood pressure by COX-2 inhibition.
The two prostanoids TxA2 and PGI2 play an essential role in the maintenance of vascular homeostasis. Prostacyclin is the main product of macrovascular endothelial cells and it is a potent vasodilator. Recent studies in healthy volunteers showed that treatment with selective COX-2 inhibitors decreases systemic prostacyclin production without affecting platelet derived TxA2 synthesis (Catella-Lawson et al., 1999; McAdam et al., 1999). This data suggest that during normal physiologic conditions, COX-2 is expressed in the vasculature and is the main source of prostacyclin production. However, the expression of COX-2 seemed to be barely in endothelial cells under static conditions in vitro, but it has been found that laminar shear stress is an inductor of COX-2 (Topper et al., 1996). This finding suggests that COX-2 may be constitutively activated in arterial endothelium in vivo. In the meanwhile, other investigators have shown that COX-2 protein is constitutively expressed in the vasculature and aorta (Matrougui et al., 2001; Arbabi et al., 2000; Garcia-Cohen et al., 2000).
Therefore, we determined plasma levels of 6-keto PGF1α in SHRs and WKY rats during different salt intakes and found that these levels were increased during normal and high salt intake in comparison to low salt intake. This increase was clearly attenuated by COX-2 inhibition, suggesting that vascular COX-2 increases with enhanced salt intake. This conclusion is in good accordance with the recent observations that COX-2 protein in rat mesenteric arteries or in human umbilical vein endothelial cells increases in parallel with the NaCl content of the diet or of the media (Matrougui et al., 2001; Arbabi et al., 2000). Furthermore, the increased amount of 6-keto PGF1α in SHR fits with the recent finding, that COX-2 protein is increased in aorta from SHRs (Garcia-Cohen et al., 2000). Our data also confirm those studies, which report that the vascular endothelium produces increased amounts of the vasodilator prostanoids PGE2 and PGI2 as a compensatory anti-hypertensive mechanism in response to high salt intake and one may conclude that particular PGI2 is of importance in vascular homeostasis during salt load (Falardeau & Martineau, 1983; Uehara et al., 1987; Ishimitsu et al., 1991) and those studies in humans, which suggest that COX-2 is a major source of vascular endothelial prostacyclin biosynthesis (McAdam et al., 1999; Catella-Lawson et al., 1999).
In summary, our data strongly suggest that blood pressure regulation depends on the balance of PGI2 and TxA2, since we did not observe an effect of rofecoxib on serum TxB2 levels. Thus, direct inhibition of PGI2 by COX-2 inhibitors may be a causal factor for the increase in blood pressure.
Our data further show that rofecoxib increased sBP on normal and high salt diet, but not on low salt diet This pattern of response is therefore not typical for a primary salt induced hypertension. For SHRs we obtained very similar findings as with WKY rats, and it appeared as if the increase of sBP in response to COX-2 inhibition in WKY rats was almost additive to the known developmental changes of blood pressure in SHRs. Therefore, one may conclude that COX-2 inhibition neither mimics nor potentiates the mechanisms that cause high blood pressure in SHR.
Nevertheless, with regard to BP, similar data were previously obtained by Muscara et al. (2000), who reported that the COX-2 inhibitor celecoxib increased sBP in WKY rats on normal salt diet nearly to the same extent as we found in this study and who also reported the same additive increase of sBP in WKY rats made hypertensive by pre-treatment with the NO-synthase inhibitor L-NAME. These authors speculated from their findings that the increase of sBP might be causally related to volume retention. Although we cannot definitively rule out such a linkage, our data do not support such a mechanism for two reasons. Firstly, we found no effect of the COX-2 inhibitor on fluid intake, or body weight gain, or on hematocrit suggesting that a possible volume expansion has to be rather minute. Secondly, we found no inhibitory effect of rofecoxib treatment on PRA or on renocortical renin expression, which are known to be sensitively suppressed by volume expansion.
It has been found that renal renin mRNA of SHR is, either elevated (Samani et al., 1989), unchanged (Pratt et al., 1989) or, in confirmation to our data, decreased (Yu & Di Nicolantonio, 1996) when compared to age matched WKY. These contrary findings may be caused by the genetic heterogenity of SHR from different sources affecting gene expression (Nabika et al., 1991; Zicha & Kunes, 1999). Similar findings as for renal renin mRNA have been observed for plasma renin activity (Yu & Di Nicolantonio, 1996; Samani et al., 1989; Sen et al., 1972). However, we could not observe a decrease in PRA in 7 weeks old SHR compared to age matched WKY. It should be noted, that our study takes place during a period of dramatic shifts in the endogenous renin-angiotensin system (Solomon et al., 1977; Gomez et al., 1988), which may mask strain related differences in PRA.
In contrast to our study, Wang et al. (1999) found that the COX-2 inhibitor SC-58236 lowers blood pressure in a model of renovascular hypertension. They found further that SC-58236 also decreases renin content and plasma renin activity in this study, which led to the suggestion that COX-2 derived prostanoids mediate the increase in the renin-angiotensin system and are also responsible for the increase in blood pressure. This finding and other investigations have led to the concept, that mainly COX-2 derived prostanoids are of relevance for the stimulation of the renin system during low salt intake (Harding et al., 2000), by angiotensin-II antagonists (Cheng et al., 1999) and by renal hypoperfusion (Wang et al., 1999). With regard to the data obtained with the COX-2 selective antagonist NS-398 (Harding et al., 2000) it seems that COX-2 is not an essential link in the dietary NaCl restriction response of PRA, because acute administration had no effect on PRA, and long term administration only partially inhibited renin secretion. Furthermore, the evidence that COX-2 derived prostanoids may be essential stimulators of the renin system was obtained under conditions of strong activation of the renin system (Cheng et al., 1999; Wang et al., 1999). However, our study was not performed during such a situation and it has also been previously shown that the degree of renin stimulation by low salt intake is maximal after 1 week, followed by a decline (Holmer et al., 1993). This decline and the fact that our study takes place during a period of dramatic shifts in the endogenous renin-angiotensin system (Solomon et al., 1977; Gomez et al., 1988) may be explanations, why we could not observe an effect of rofecoxib treatment on renin mRNA or on PRA in both rat strains. However, it should be noted that there are also studies questioning a causal involvement of COX-2 derived prostanoids in the regulation of the renin system by salt intake (Kammerl et al., 2001; Rossat et al., 1999). In summary, our data do not support a major role for COX-2 derived prostanoids in the stimulation of renin by low salt intake. However, this question needs to be further evaluated.
Conversely, it appears also unlikely that a stimulation of the renal renin-angiotensin-aldosterone system itself is the primary event by which COX-2 inhibition increases blood pressure, since renin was normal and aldosterone was even decreased under COX-2 inhibition. That NSAIDs can lower PAC has already previously reported (Miller et al., 1980; Stokes et al., 1991; Campbell & Gomez-Sanchez, 1985) and probably reflects a direct stimulatory effect of prostanoids on aldosterone production in the adrenal gland as previously suggested by in vitro (Csukas et al., 1998) and in vivo (Golub et al., 1976) observations. Since the typical inverse relationship between salt intake and renin was not changed, and the relationship between salt intake and plasma aldosterone was only attenuated but not blunted by the COX-2 inhibitor, we may infer that COX-2 mediated prostanoids are not essentially involved in the control of the RAAS by salt. This conclusion would be in harmony with the idea that the regulation of aldosterone production by salt intake is primarily mediated by angiotensin-II (Mulrow, 1999), which in turn is generated by the renin activity. In conclusion, our data do not support an impact of the RAAS on the increase in sBP. However, from the present data we cannot exclude an influence of rofecoxib on paracrine renin systems, e.g. in the adrenal gland, which are considered to be important in the development of cardiovascular diseases.
Taken together, our data suggest that prolonged treatment with selective COX-2 inhibitors results in an increase on blood pressure, which could be due to decreased prostacyclin levels. Furthermore, the rise in blood pressure induced by COX-2 inhibition can be prevented by sodium deprivation. In SHRs, which are considered as a human model for essential hypertension (Doggrell & Brown, 1998), COX-2 inhibition did not interfere with the typical temporal development of hypertension but produced a more constant additional increment of blood pressure increase.
Acknowledgments
The expert technical assistance provided by Astrid Seefeld and Gertraud Wilberg is gratefully acknowledged. This study was in part supported by a grant from the Deutsche Forschungsgemeinschaft (Ku 859/13-1).
Abbreviations
- ANG
angiotensin
- BP
blood pressure
- COX
cyclo-oxygenase
- NSAID
non steroidal anti-inflammatory drug
- PAC
plasma aldosterone concentration
- PG
prostaglandin
- PRA
plasma renin activity
- RAAS
renin-angiotensin aldosterone system
- sBP
systolic blood pressure
- SHR
spontaneously hypertensive rat
- WKY
Wistar Kyoto rat
References
- ARBABI S., ROSENGART M.R., GARCIA I., MAIER R.V. Hypertonic saline solution induces prostacyclin production by increasing cyclooxygenase-2 expression. Surgery. 2000;128:198–205. doi: 10.1067/msy.2000.107606. [DOI] [PubMed] [Google Scholar]
- BREYER M.D., BREYER R.M. Prostaglandin E receptors and the kidney. Am. J. Physiol. Renal Physiol. 2000;279:F12–F23. doi: 10.1152/ajprenal.2000.279.1.F12. [DOI] [PubMed] [Google Scholar]
- BUNTING S., MONCADA S., VANE J.R. The prostacyclin-thromboxane A2 balance: pathophysiological and therapeutic implications. Br. Med. Bull. 1983;39:271–276. doi: 10.1093/oxfordjournals.bmb.a071832. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GOMEZ-SANCHEZ C.E. Absence of prostacyclin involvement in angiotensin-induced aldosterone secretion in rat adrenal cells. Endocrinology. 1985;117:279–286. doi: 10.1210/endo-117-1-279. [DOI] [PubMed] [Google Scholar]
- CATELLA-LAWSON F., MCADAM B., MORRISON B.W., KAPOOR S., KUJUBU D., ANTES L., LASSETER K.C., QUAN H., GERTZ B.J., FITZGERALD G.A. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J. Pharmacol. Exp. Ther. 1999;289:735–741. [PubMed] [Google Scholar]
- CHENG H.F., WANG J.L., ZHANG M.Z., MIYAZAKI Y., ICHIKAWA I., MCKANNA J.A., HARRIS R.C. Angiotensin II attenuates renal cortical cyclooxygenase-2 expression. J. Clin. Invest. 1999;103:953–961. doi: 10.1172/JCI5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHRYSANT S.G., MANDAL A.K., NORDQUIST J.A. Renal functional and organic changes induced by salt and prostaglandin inhibition in spontaneously hypertensive rats. Nephron. 1980;25:151–155. doi: 10.1159/000181772. [DOI] [PubMed] [Google Scholar]
- CLIVE D.M., STOFF J.S. Renal syndromes associated with nonsteroidal antiinflammatory drugs. N. Engl. J. Med. 1984;310:563–572. doi: 10.1056/NEJM198403013100905. [DOI] [PubMed] [Google Scholar]
- CSUKAS S., HANKE C.J., REWOLINSKI D., CAMPBELL W.B. Prostaglandin E2-induced aldosterone release is mediated by an EP2 receptor. Hypertension. 1998;31:575–581. doi: 10.1161/01.hyp.31.2.575. [DOI] [PubMed] [Google Scholar]
- DICKHOUT J.G., LEE R.M.K.W. Blood pressure and heart rate development in young spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 1998;274:H794–H800. doi: 10.1152/ajpheart.1998.274.3.H794. [DOI] [PubMed] [Google Scholar]
- DOGGRELL S.A., BROWN L. Rat models of hypertension, cardiac hypertrophy and failure. Cardiovasc Res. 1998;39:89–105. doi: 10.1016/s0008-6363(98)00076-5. [DOI] [PubMed] [Google Scholar]
- FALARDEAU P., MARTINEAU A. In vivo production of prostaglandin I2 in Dahl salt-sensitive and salt-resistant rats. Hypertension. 1983;5:701–705. doi: 10.1161/01.hyp.5.5.701. [DOI] [PubMed] [Google Scholar]
- FENG L., SUN W., XIA Y., TANG W.W., CHANMUGAM P., SOYOOLA E., WILSON C.B., HWANG D. Cloning two isoforms of rat cyclooxygenase: differential regulation of their expression. Arch. Biochem. Biophys. 1993;307:361–368. doi: 10.1006/abbi.1993.1601. [DOI] [PubMed] [Google Scholar]
- FUNK C.D., FUNK L.B., KENNEDY M.E., PONG A.S., FITZGERALD G.A. Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomal assignment. FASEB J. 1991;9:2304–2312. [PubMed] [Google Scholar]
- GARCIA-COHEN E.-C., MARIN J., DIEZ-PICAZO L.D., BAENA A.B., SALAICES M., RODRIGUEZ-MARTINEZ M. Oxidative stress induced by tert-butyl hydroperoxide causes vasoconstriction in the aorta from hypertensive and aged rats: role of cyclooxygenase-2 isoform. J. Pharmacol. Exp. Ther. 2000;293:75–81. [PubMed] [Google Scholar]
- GOLUB M.S., SPECKART P.F., ZIA P.K., HORTON R. The effect of prostaglandin A1 on renin and aldosterone in man. Circ. Res. 1976;39:574–579. doi: 10.1161/01.res.39.4.574. [DOI] [PubMed] [Google Scholar]
- GOMEZ R.A., LYNCH K.R., CHEVALIER R.L., WILFONG N., EVERETT A., CAREY R.M., PEACH M.J. Renin and angiotensinogen gene expression in maturing rat kidney. Am. J. Physiol. 1988;254:F582–F587. doi: 10.1152/ajprenal.1988.254.4.F582. [DOI] [PubMed] [Google Scholar]
- GRADIN K., DAHLÖF C., PERSSON B. A low dietary sodium intake reduces neuronal noradrenaline release and the blood pressure in spontaneously hypertensive rats. Naunyn Schmiedebergs Arch. Pharmacol. 1986;332:364–369. doi: 10.1007/BF00500088. [DOI] [PubMed] [Google Scholar]
- HARDING P., SIGMON D.H., ALFIE M.E., HUANG P.L., FISHMAN M.C., BEIERWALTES W.H., CARRETERO O.A. Cyclooxygenase-2 mediates increased renal renin content induced by low-sodium diet. Hypertension. 1997;29:297–302. doi: 10.1161/01.hyp.29.1.297. [DOI] [PubMed] [Google Scholar]
- HARDING P., CARRETERO O.A., BEIERWALTES W.H. Chronic cyclooxygenase-2 inhibition blunts low sodium-stimulated renin without changing renal haemodynamics. J. Hypertens. 2000;18:1107–1113. doi: 10.1097/00004872-200018080-00016. [DOI] [PubMed] [Google Scholar]
- HARRIS R.C., BREYER M.D. Physiological regulation of cyclooxygenase-2 in the kidney. Am. J. Physiol. Renal. Physiol. 2001;281:F1–F11. doi: 10.1152/ajprenal.2001.281.1.F1. [DOI] [PubMed] [Google Scholar]
- HARRIS R.C., MCKANNA J.A., AKAI Y., JACOBSON H.R., DUBOIS R.N., BREYER M.D. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J. Clin. Invest. 1994;94:2504–2510. doi: 10.1172/JCI117620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HÖCHERL K., WOLF K., CASTROP H., ITTNER K.P., BUCHER M., KEES F., GROBECKER H.F., KURTZ A. Renocortical expression of renin and of cyclooxygenase-2 in response to angiotensin II AT1 receptor blockade is closely coordinated but not causally linked. Pflügers Arch. 2001;442:821–827. doi: 10.1007/s004240100615. [DOI] [PubMed] [Google Scholar]
- HOLMER S., ECKARDT K.U., LEHIR M., SCHRICKER K., RIEGGER G., KURTZ A. Influence of dietary NaCl intake on renin gene expression in the kidneys and adrenal glands of rats. Pflügers Arch. 1993;425:62–67. doi: 10.1007/BF00374504. [DOI] [PubMed] [Google Scholar]
- IMIG J.D. Eicosanoid regulation of the renal vasculature. Am. J. Physiol. Renal Physiol. 2000;279:F965–F981. doi: 10.1152/ajprenal.2000.279.6.F965. [DOI] [PubMed] [Google Scholar]
- ISHIMITSU T., UEHARA Y., IWAI J., SUGIMOTO T., HIRATA Y., MATSUOKA H., SUGIMOTO T. Vascular eicosanoid production in experimental hypertensive rats with different mechanisms. Prostaglandins Leukot. Essent. Fatty Acids. 1991;43:179–184. doi: 10.1016/0952-3278(91)90166-3. [DOI] [PubMed] [Google Scholar]
- KAMMERL M.C., NÜSING R.M., SEYBERTH H.W., RIEGGER G.A.J., KURTZ A., KRÄMER B.K. Inhibition of cyclooxygenase-2 attenuates urinary prostanoid excretion without affecting renal renin expression. Pflügers Arch. 2001;442:842–847. doi: 10.1007/s004240100616. [DOI] [PubMed] [Google Scholar]
- LLINAS M.T., RODRIGUEZ F., MORENO C., SALAZAR F.J. Role of cyclooxygenase-2-derived metabolites and nitric oxide in regulating renal function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000;279:R1641–R1646. doi: 10.1152/ajpregu.2000.279.5.R1641. [DOI] [PubMed] [Google Scholar]
- MANN B., HARTNER A., JENSEN B.L., HILGERS K.F., HOCHERL K., KRAMER B.K., KURTZ A. Acute upregulation of COX-2 by renal artery stenosis. Am. J. Physiol. Renal Physiol. 2001;280:F119–F125. doi: 10.1152/ajprenal.2001.280.1.F119. [DOI] [PubMed] [Google Scholar]
- MATROUGUI K., LOUFRANI L., LEVY B.I., HENRION D. High NaCl intake decreases both flow-induced dilation and pressure-induced myogenic tone in resistance arteries from normotensive rats: involvement of cyclooxygenase-2. Pharmacol. Toxicol. 2001;89:183–187. doi: 10.1111/j.0901-9928.2001.890407.x. [DOI] [PubMed] [Google Scholar]
- MCADAM B.F., CATELLA-LAWSON F., MARDINI I.A., KAPOOR S., LAWSON J.A., FITZGERALD G.A. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc. Natl. Acad. Sci. U.S.A. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER R.T., DOUGLAS J.G., DUNN M.J. Dissociation of aldosterone and prostaglandin biosynthesis in the rat adrenal glomerulosa. Prostaglandins. 1980;20:449–456. doi: 10.1016/0090-6980(80)90032-5. [DOI] [PubMed] [Google Scholar]
- MUKHERJEE D., NISSEN S.E., TOPOL E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- MULROW P.J. Angiotensin II and aldosterone regulation. Regul. Pept. 1999;80:27–32. doi: 10.1016/s0167-0115(99)00004-x. [DOI] [PubMed] [Google Scholar]
- MUSCARA M.N., MCKNIGHT W., DEL SOLDATO P., WALLACE J.L. Effect of a nitric oxide-releasing naproxen derivative on hypertension and gastric damage induced by chronic nitric oxide inhibition in the rat. Life Sci. 1998;62:235–240. doi: 10.1016/s0024-3205(98)00072-1. [DOI] [PubMed] [Google Scholar]
- MUSCARA M.N., VERGNOLLE N., LOVREN F., TRIGGLE C.R., ELLIOTT S.N., ASFAHA S., WALLACE J.L. Selective cyclo-oxygenase-2 inhibition with celecoxib elevates blood pressure and promotes leukocyte adherence. Br. J. Pharmacol. 2000;129:1423–1430. doi: 10.1038/sj.bjp.0703232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NABIKA T., NARA Y., IKEDA K., ENDO J., YAMORI Y. Genetic heterogeneity of the spontaneously hypertensive rat. Hypertension. 1991;18:12–16. doi: 10.1161/01.hyp.18.1.12. [DOI] [PubMed] [Google Scholar]
- NARUMIYA S., SUGIMOTO Y., USHIKUBI F. Prostanoid Receptors: Structures, Properties, and Functions. Physiol. Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- PATRONO C., DUNN M.J. The clinical significance of inhibition of renal prostaglandin synthesis. Kidney Int. 1987;32:1–12. doi: 10.1038/ki.1987.164. [DOI] [PubMed] [Google Scholar]
- PRATT R.E., ZOU W.M., NAFTILAN A.J., INGELFINGER J.R., DZAU V.J. Altered sodium regulation of renal angiotensinogen mRNA in the spontaneously hypertensive rat. Am. J. Physiol. 1989;256:F469–F474. doi: 10.1152/ajprenal.1989.256.3.F469. [DOI] [PubMed] [Google Scholar]
- RODRIGUEZ F., LLINAS M.T., GONZALEZ J.D., RIVERA J., SALAZAR F.J. Renal changes induced by a cyclooxygenase-2 inhibitor during normal and low sodium intake. Hypertension. 2000;36:276–281. doi: 10.1161/01.hyp.36.2.276. [DOI] [PubMed] [Google Scholar]
- ROIG F., LLINAS M.T., LOPEZ R., RODRIGUEZ F., SALAZAR F.J.Renal effects of chronic cyclooxygenase-2 inhibition changes with sodium intake Hypertension 200138982(abstract) [Google Scholar]
- ROSSAT J., MAILLARD M., NUSSBERGER J., BRUNNER H.R., BURNIER M. Renal effects of selective cyclooxygenase-2 inhibition in normotensive salt-depleted subjects. Clin. Pharmacol. Ther. 1999;66:76–84. doi: 10.1016/S0009-9236(99)70056-1. [DOI] [PubMed] [Google Scholar]
- SAMANI N.J., SWALES J.D., BRAMMAR W.J. A widespread abnormality of renin gene expression in the spontaneously hypertensive rat: modulation in some tissues with the development of hypertension. Clin. Sci. (Lond.) 1989;77:629–636. doi: 10.1042/cs0770629. [DOI] [PubMed] [Google Scholar]
- SEN S., SMEBY R.R., BUMPUS M. Renin in rats with spontaneous hypertension. Circ. Res. 1972;31:876–880. doi: 10.1161/01.res.31.6.876. [DOI] [PubMed] [Google Scholar]
- SMITH W.L., MARNETT L.J., DE WITT D.L. Prostaglandin and thromboxane biosynthesis. Pharmacol. Ther. 1991;49:153–179. doi: 10.1016/0163-7258(91)90054-p. [DOI] [PubMed] [Google Scholar]
- SOLOMON S., IAINA A., ELIAHOU H., SERBAN I. Postnatal changes in plasma and renal renin of the rat. Biol. Neonate. 1977;32:237–242. doi: 10.1159/000241024. [DOI] [PubMed] [Google Scholar]
- STOKES G.S., BROOKS P.M., JOHNSTON H.J., MONAGHAN J.C., OKORO E.O., KELLY D. The effects of sulindac and diclofenac in essential hypertension controlled by treatment with a beta blocker and/or diuretic. Clin. Exp. Hypertens A. 1991;13:1169–1178. doi: 10.3109/10641969109042120. [DOI] [PubMed] [Google Scholar]
- TOPPER J.N., CAI J., FALB D., GIMBRONE M.A., JR Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10417–10422. doi: 10.1073/pnas.93.19.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UEHARA Y., TOBIAN L., IWAI J., ISHII M., SUGIMOTO T. Alterations of vascular prostacyclin and thromboxane A2 in Dahl genetical strain susceptible to salt-induced hypertension. Prostaglandins. 1987;33:727–738. doi: 10.1016/0090-6980(87)90038-4. [DOI] [PubMed] [Google Scholar]
- VANE J.R., BOTTING R.M. A better understanding of anti-inflammatory drugs based on isoforms of cyclooxygenase (COX-1 and COX-2) Adv. Prostaglandin. Thromboxane Leukot. Res. 1995;23:41–48. [PubMed] [Google Scholar]
- VANE J.R., BAKHLE Y.S., BOTTING R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- WANG J.L., CHENG H.F., HARRIS R.C. Cyclooxygenase-2 inhibition decreases renin content and lowers blood pressure in a model of renovascular hypertension. Hypertension. 1999;34:96–101. doi: 10.1161/01.hyp.34.1.96. [DOI] [PubMed] [Google Scholar]
- WARNER T.D., GIULIANO F., VOJNOVIC I., BUKASA A., MITCHELL J.A., VANE J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHELTON A., FORT J.G., PUMA J.A., NORMANDIN D., BELLO A.E., VERBURG K.M. Cyclooxygenase-2–specific inhibitors and cardiorenal function: a randomized, controlled trial of celecoxib and rofecoxib in older hypertensive osteoarthritis patients. Am. J. Ther. 2001;8:85–95. doi: 10.1097/00045391-200103000-00003. [DOI] [PubMed] [Google Scholar]
- YANG T., SCHNERMANN J.B., BRIGGS J.P. Regulation of cyclooxygenase-2 expression in renal medulla by tonicity in vivo and in vitro. Am. J. Physiol. 1999;277:F1–F9. doi: 10.1152/ajprenal.1999.277.1.F1. [DOI] [PubMed] [Google Scholar]
- YU H., DI NICOLANTONIO R. Altered age-dependent modulation of tissue renin messenger RNA levels in the spontaneously hypertensive rat. J. Hypertens. 1996;14:871–880. doi: 10.1097/00004872-199607000-00010. [DOI] [PubMed] [Google Scholar]
- ZICHA J., KUNES J. Ontogenetic aspects of hypertension development: analysis in the rat. Physiol. Rev. 1999;79:1227–1282. doi: 10.1152/physrev.1999.79.4.1227. [DOI] [PubMed] [Google Scholar]