

Abstract
Gold(I)-thiolate drugs are compounds that specifically interact with thiol and/or selenol groups and are essentially utilized in the treatment of rheumatoid arthritis.
Considering the importance of thiol groups in regulating mitochondrial membrane permeability, the effects of auranofin (S-triethylphosphinegold(I)-2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside), a second-generation gold drug, were studied on mitochondria isolated from rat liver.
Auranofin, at submicromolar concentrations, was able to induce the mitochondrial membrane permeability transition observed as swelling and loss of membrane potential. Both events are completely inhibited by cyclosporin A, the specific inhibitor of mitochondrial permeability transition. Calcium ions and energization by succinate are required for the occurrence of permeability transition.
By interacting with the active site selenol group, auranofin results as an extremely potent inhibitor of mitochondrial thioredoxin reductase, both isolated and in its mitochondrial environment.
It is concluded that auranofin, in the presence of calcium ions, is a highly efficient inducer of mitochondrial membrane permeability transition, potentially referable to its inhibition of mitochondrial thioredoxin reductase.
Keywords: Auranofin, calcium, gold(I) complexes, mitochondrial permeability transition, selenium, thiol groups, thioredoxin reductase
Introduction
Gold(I)-thiolate drugs are compounds essentially utilized in the treatment of rheumatoid arthritis (Kean et al., 1997). They have also been tested as anticancer agents (Simon et al., 1981). Their mode of action involves the control of oxidative damage (Grootveld et al., 1990) and inhibition of several enzymes or transcription factors (Handel et al., 1995; Daniel et al., 1995).
The molecular mechanism of their inhibitory effects was essentially referred to an interaction with thiol groups with a reactivity depending on the type of ligand associated with the gold (Crooke & Snyder, 1986). A sequential sulfhydryl exchange mechanism was also postulated to explain the cellular distribution of auranofin and related gold complexes (Snyder et al., 1986). However, gold(I) compounds exhibit also a marked and specific reactivity with selenoenzymes such as glutathione peroxidase (GSPx) (Chaudière & Tappel, 1984), iodothyronine deiodinase type I (Berry et al., 1991), and thioredoxin reductase (Hill et al., 1997; Gromer et al., 1998a, b; Smith et al., 1999), an enzyme recently shown to possess selenium at its catalytic site (Tamura & Stadtman, 1996; Zhong et al., 1998). Considering glutathione peroxidase, gold(I) derivatives such as aurothiomalate, aurothioglucose and auranofin have been shown to exert their inhibitory action by forming a glutathionate-gold(I)-selenocysteine glutathione peroxidase ternary complex (GPxSe-Au-SG) (Chaudière & Tappel, 1984; Roberts & Shaw, 1998). Thioredoxin reductase is present in both the cytosol and mitochondria (Rigobello et al., 1998) where exhibits a sequence different from that of the cytosolic isoform but, similarly to the latter enzyme, possess selenium at its active site (Lee et al., 1999). Selenols are able to bind heavy metals more efficiently than thiols (Grootveld et al., 1990) and, therefore, the selenocysteine of thioredoxin reductase appears as the target of organic gold inhibitors (Gromer et al., 1998a; Zhong et al., 1998).
In previous research, it was demonstrated that the redox state of mitochondrial thiols could control the permeability of the mitochondrial membranes (Bindoli et al., 1997; Kowaltowski et al., 2001). In this respect, the thioredoxin system present in mitochondria might play a critical role (Wudarczyk et al., 1996; Rigobello et al., 1998; Kim et al., 1999). We have previously demonstrated (Rigobello et al., 1999) that 13-cis retinoic acid, an inhibitor of thioredoxin reductase (Schallreuter & Wood, 1989) is very effective in inducing mitochondrial permeability transition and release of cytochrome c. Similar effects on mitochondrial membranes permeability increase were observed with arsenite and arsenicals, heavy and transition metal cations, quinones, alloxan and sulfhydryl reagents (Gunter & Pfeiffer, 1990; Zoratti & Szabò, 1995; Sakurai et al., 2001) that are also inhibitors or substrates of thioredoxin reductase. In the latter case they act as electron acceptors (Arnér et al., 1999), therefore, diverting electrons from thioredoxin reductase that are no longer fed to its natural substrate, thioredoxin.
In the present paper we report evidence that auranofin is a potent inducer of mitochondrial permeability transition possibly referable to its interaction with the mitochondrial isoform of thioredoxin reductase.
Methods
Rat liver mitochondria were isolated with differential centrifugation according to Myers & Slater (1957) using a medium containing 220 mM mannitol, 70 mM sucrose, 1 mM EDTA and 5 mM HEPES at pH 7.0. In the homogenization buffer, 1 mM EGTA was also present but was omitted in the subsequent washings and in the final resuspension of mitochondria. Mitochondrial proteins were estimated with the biuret procedure (Gornall et al., 1949). Mitochondrial matrix was prepared from mitochondrial suspension (60 mg ml−1 in 20 mM Tris-HCl, pH 7.5) subjected to freezing and thawing (three times) followed by 30 s of sonication (twice) and 45 min of centrifugation at 100,000×g. Supernatant was dialyzed overnight against the same buffer, in order to remove glutathione, and concentrated in a pressure dialysis system using an Amicon YM10 membrane. Rat liver mitochondria thioredoxin reductase was prepared and assayed as described by Rigobello et al. (1998) and its protein content was measured according to Lowry et al. (1951). Glutathione reductase was assayed according to Carlberg & Mannervik (1985). Mitochondrial swelling was followed spectrophotometrically by the decrease in absorbance at 540 nm (Lehninger, 1962). Membrane potential was assessed by measuring the movements of TPP+ (tetraphenylphosphonium ion) across the mitochondrial membrane with a TPP+ selective electrode (Kamo et al., 1979). The ordinate ΔΨ values of the curves were recalculated from the experimental values in order to obtain a linear scale. Oxygen uptake was measured polarographically utilizing a Clark-type oxygen electrode (Estabrook, 1967) inserted in a water-jacketed chamber (25°C) with constant stirring; the system was connected to a personal computer (Cazzaro et al., 1996).
The data obtained from the various experiments and generated via the spectrophotometer, oxygraph or membrane potential measurement apparatus softwares were stored and converted to ASCII format. The data pairs format was used to transfer the data to numerical analysis and graphics software. Therefore, the data obtained were utilized for averaging the various curves that are the mean of 5–6 experiments.
Statistical analysis
All the values are the means±S.D. of not less than five measurements.
Results
As apparent in Figure 1, auranofin, in the presence of succinate, acts as a potent inducer of mitochondrial membrane permeability transition both in the presence and absence of calcium. By comparing Figure 1A,B, it can be observed that the presence of added calcium to the incubation mixture, although at a relatively low concentration (12 μM), markedly reinforces the action of auranofin. The extent of swelling is concentration-dependent and occurs, in our conditions, at concentrations as low as 16 nM, while, at concentrations around 1 μM, the maximal rate and extent of swelling are reached. Energization by a respiratory substance such as succinate appears critical for the occurrence of swelling since, in its absence, the swelling induced by 1.2 μM auranofin, a concentration more than ten times higher than that observed to give the maximal swelling (Figure 1A, trace e) is inhibited by about 70% (Figure 1C, trace f′). Similarly, de-energization of mitochondria by the uncoupler CCCP (carbonylcyanide m-chlorophenylhydrazone) completely prevents swelling (Figure 1C, trace c′). The lack of effect observed in the absence of succinate or in the presence of the uncoupler is probably related to the prevention of calcium uptake by the mitochondria. This interpretation is reinforced by taking into account the effects of ruthenium red which is an inhibitor of the electrophoretic uniport process of calcium uptake (Moore, 1971), and hence of calcium cycling, and prevents the swelling induced by 1.2 μM auranofin by more than 70% (Figure 1C, trace e′). As expected, EGTA completely inhibits the swelling induced by auranofin, indicating the essentiality of calcium ions for the induction of this change of mitochondrial membrane permeability properties. Swelling is also completely inhibited by cyclosporin A (Figure 1C, trace d′), a drug considered to be a highly specific inhibitor of the ‘pore' involved in the mitochondrial permeability transition (Fournier et al., 1987) indicating that a specific increase of membrane permeability does occur after the addition of auranofin.
Figure 1.
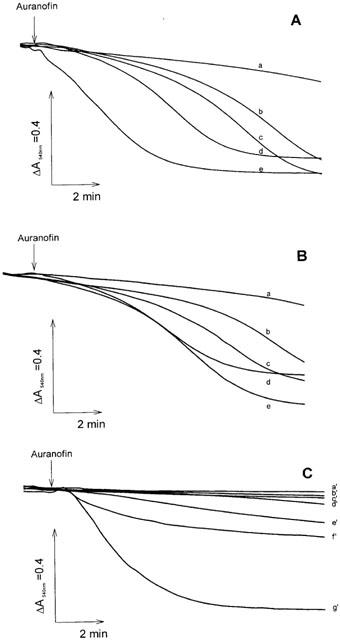
Induction of mitochondrial swelling by auranofin. Rat liver mitochondria (0.25 mg ml−1) were incubated at 25°C in 213 mM mannitol, 71 mM sucrose, 5 mM HEPES-Tris (pH 7.4), 5 mM succinate, 3 μg ml−1 rotenone, and 3 μM oligomycin. In (A) 12 μM CaCl2 was also present. In (A) and (B) swelling was triggered by the addition of auranofin at the following concentrations (nM): a, none; b, 16; c, 33; d, 66; e, 100. In (C) swelling was initiated by 1.2 μM auranofin (g′). Other additions were: a′, 0.5 mM EGTA; b′, no auranofin; c′, 0.3 μM CCCP; d′, 0.5 μM cyclosporin A; e′, 0.6 μM ruthenium red; f′, succinate absent.
The effect of auranofin on the mitochondrial membrane potential is apparent in Figure 2. Rat liver mitochondria, in the presence of succinate as a respiratory substrate, establish a membrane potential of about −180 mV. The addition of a pulse of calcium ions (10 μM) determines a transient decrease, rapidly re-established, of membrane potential due to calcium intake. In standard conditions, this potential is maintained for several minutes. When auranofin is added there is a progressive decrease of membrane potential followed by a rapid and total collapse. The duration of the slow phase of membrane potential decrease is inversely correlated with the concentration of auranofin added. In the presence of 10 μM auranofin (Figure 2, trace f) the slow phase is no longer apparent. The addition of EGTA after swelling partially restores the membrane potential (not shown). The addition of DTT (dithiothreitol), after the onset of swelling, stops the latter without reversing the phenomenon, indicating an involvement of thiol or selenol groups in the action of auranofin (not shown).
Figure 2.
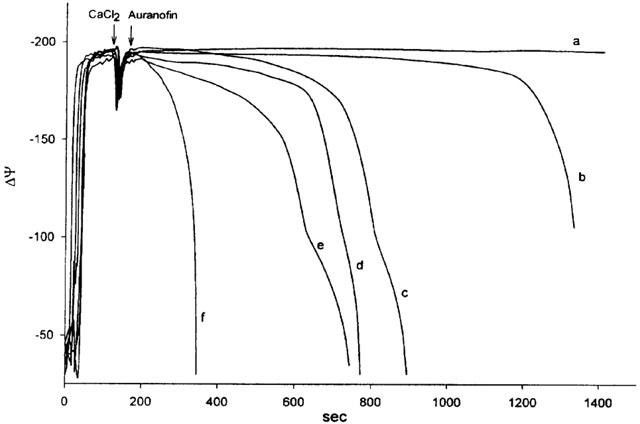
Effect of auranofin on mitochondrial membrane potential. Rat liver mitochondria (1 mg ml−1) were incubated at 25°C in 106 mM mannitol, 35 mM sucrose, 62 mM KCl, 15 mM HEPES-Tris (pH 7.4), 1.4 mM phosphate, 2 μg ml−1 rotenone and 5 μM TPP+. Mitochondria were energized by the addition of 5 mM succinate. After the addition of a pulse of 10 μM CaCl2, auranofin was added at the following concentrations (μM): a, none; b, 0.5; c, 1; d, 2; e, 5; f, 10.
Figure 3 reports the effects of auranofin on mitochondrial respiration in the presence and absence of EGTA. In the latter case (Figure 3B) the addition of 2 μM auranofin determines an increase of succinate-dependent respiration that is not further stimulated by the subsequent addition of ADP. This increase of respiration is dependent on the swelling of mitochondria since it does not occur with the NAD-dependent substrates (not shown) and indicates that no inhibition of respiration is elicited, at least at this concentration, by auranofin. Furthermore, in the presence of EGTA, the ‘uncoupling' effect of auranofin is no longer occurring indicating that the mitochondrial energy conservation mechanism is not affected.
Figure 3.
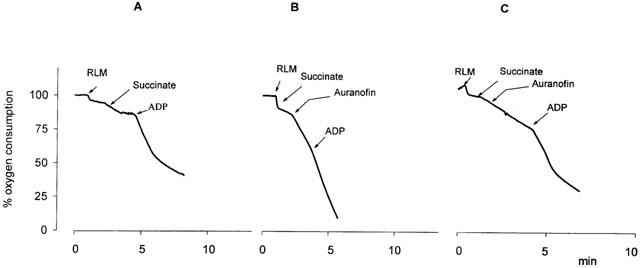
Effect of auranofin on mitochondrial oxygen uptake. Rat liver mitochondria (RLM, 1 mg ml−1) were incubated in 0.1 M sucrose, 50 mM KCl, 1 mM MgCl2, 1 mM NaH2PO4, 20 mM HEPES (pH 7.4), 7 mM succinate, 0.3 mM ADP and, when indicated, 2 μM auranofin. In (C), 0.25 mM EGTA was also present.
As reported before, auranofin is a potent and specific inhibitor of thioredoxin reductase (Gromer et al., 1998a). As apparent in Figure 4A, when a preparation of mitochondrial matrix, able to reduce DTNB (5,5′-dithiobis(2-nitrobenzoic) acid) in the presence of NADPH, was treated with auranofin, the reduction of DTNB was completely inhibited. Further addition of oxidized glutathione restores the reduction of DTNB. This partial sensibility to the inhibition by auranofin indicates the presence of two different reactions, the first dependent on thioredoxin/thioredoxin reductase and sensitive to auranofin, while the second depends on the glutathione/glutathione reductase system and is not inhibited by auranofin. Figure 4B shows that the isolated and purified mitochondrial thioredoxin reductase is completely inhibited by very low concentrations of auranofin (20 nM) in agreement with the effect observed in the crude matrix, therefore confirming the sensitivity of the mitochondrial isoform of the enzyme to this gold complex. A recent paper has shown that the mitochondrial thioredoxin reductase is inhibited by aurothioglucose (Kerimova et al., 2000). In both cases, this is a rather expected result because of the similarity at the active site, between the mitochondrial and cytosolic isoforms of the enzyme. It was further tested if, in condition of complete prevention of swelling, the mitochondrial enzyme can be inhibited by the same concentrations of auranofin previously used to induce the membrane permeability transition. When mitochondria are incubated in the presence of cyclosporin A and EGTA, in order to absolutely avoid swelling, and are treated with increasing concentrations of auranofin, a net inhibition of thioredoxin reductase is apparent (Figure 5A). This result indicates that auranofin is able to easily penetrate the mitochondrial membrane and that the mitochondrial isoform of this enzyme can be inhibited in its matrix environment. In addition, the concentrations of auranofin able to completely inhibit the enzyme, correspond to those inducing, in the absence of EGTA and cyclosporin A, the maximal swelling, thus indicating a direct link between the two events. Interestingly, in the same experimental conditions, glutathione reductase is not modified at all (Figure 5B). Triethylphosphine gold chloride (TEPAu) shows a behaviour superimposable to that of auranofin, while dimethylsulphide gold chloride (DMSAu) is less efficient (Figure 5A).
Figure 4.
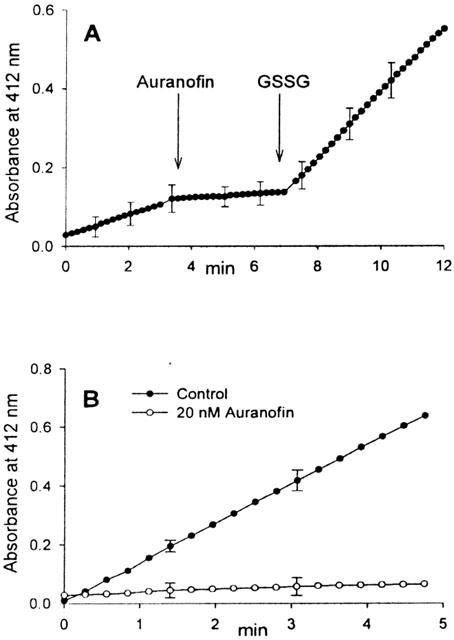
Inhibitory effect of auranofin on mitochondrial thioredoxin reductase measured both in the matrix (A) and with the purified enzyme (B). (A) Aliquots of mitochondrial matrix (0.5 mg ml−1) were incubated with 0.25 mM NADPH in 0.2 M Tris-HCl (pH 8.1) containing 1 mM EDTA. Where indicated, 0.5 μM auranofin and 1 mM GSSG were also added. (B) Aliquots of purified mitochondrial thioredoxin reductase (0.2 μg ml−1) were incubated at 25°C with 0.25 mM NADPH in 0.2 M Tris-HCl (pH 8.1) containing 1 mM EDTA in the presence or absence of auranofin. Reactions were started by the addition of 3 mM DTNB and the absorbance followed at 412 nm.
Figure 5.
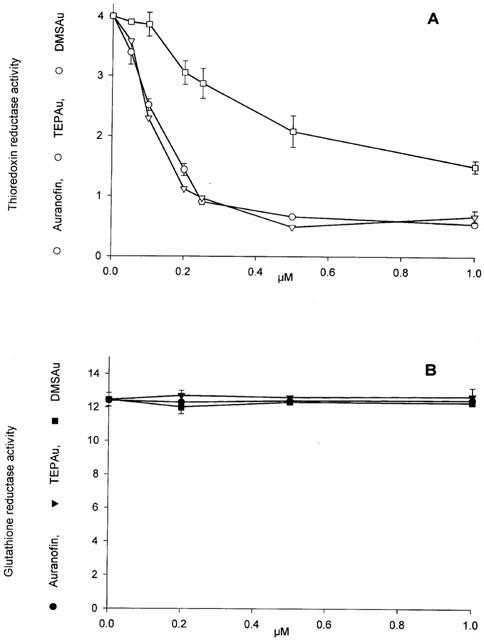
Effects of gold (I) complexes on the activities of thioredoxin reductase (A) and glutathione reductase (B) of intact mitochondria. Rat liver mitochondria (1 mg ml−1) were incubated at 25°C for 5 min in 107 mM mannitol, 35 mM sucrose, 62 mM KCl, 15 mM HEPES-Tris (pH 7.4), 6.5 μM rotenone, 0.25 mM EGTA, 1.5 μM cyclosporin A and the indicated concentrations of auranofin, TEPAu and DMSAu in a final volume of 12 ml. Reactions were quenched by diluting with 24 ml of cold (4°C) incubation medium followed by a rapid centrifugation of mitochondria at 6500×g for 8 min. Mitochondrial pellet was gently resuspended and washed with the same medium. The final pellet was resuspended in 60 μl of 0.3 M CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulphonate), and 300 μl of 0.2 M Tris-HCl (pH 8.1) containing 1 mM EDTA. The activities of thioredoxin reductase (nmoles DTNB reduced min−1 mg−1 protein; (A) and glutathione reductase (nmoles NADPH oxidized min−1 mg−1 protein; (B) of solubilized mitochondria were measured as indicated under Methods.
Discussion
Alterations of mitochondria by gold-based compounds, essentially used as anti-arthritic drugs, have been known for a long time (Stuve & Galle, 1970; Abou-Khalil et al., 1981). It was previously reported that gold complexes of bidentate phosphine determine an alteration of mitochondrial functions possibly acting as uncouplers of oxidative phosphorylation (Hoke et al., 1988). Their action appears to facilitate the exchange of H+ and K+ and result of a gross increase of membrane permeability (Hoke et al., 1988). Although with a different mechanism, in isolated mitochondria, triethylphosphine gold chloride causes a rapid dissipation of membrane potential, inhibits state three respiration and decreases respiratory control ratio (Rush et al., 1987b; Hoke et al., 1989). It causes a concentration-dependent inhibition of uncoupler-stimulated respiration that, however, can be reversed by 2 mM dithiothreitol (Rush et al., 1987b; Hoke et al., 1989) indicating that its inhibitory action is exerted through a reversible binding to sulphhydryl groups. Electron microscopy of hepatocytes exposed to triethylphosphine gold chloride reveals that mitochondria are the first organelles to show an altered ultrastructure. After 60 min of incubation, they undergo swelling with disruption of the cristae (Rush et al., 1987a). At variance with the previously reported gold derivatives, less attention was paid to the interaction of auranofin with mitochondria. According to Dong et al. (1997), in some human tumour cell lines, cisplatin resistance is associated with sensitivity to the killing by a large number of inhibitors of mitochondrial functions including auranofin. The latter is effective at very low concentrations and its action is potentially attributable to the inhibition of complex III or to an alteration of the membranes.
In the present paper, we report that auranofin acts as a potent inducer of mitochondrial permeability transition since it is effective at very low concentrations. Permeability transition indicates an increase of the mitochondrial inner membrane permeability possibly dependent on the opening of an unselective pore, that can be elicited by calcium accumulation and several inducing agents apparently unrelated from the chemical or functional point of view (Gunter & Pfeiffer, 1990; Zoratti & Szabò, 1995). The occurrence of an effective permeability transition is demonstrated by the marked inhibition exerted by cyclosporin A, the specific inhibitor of this process. EGTA is a strong inhibitor of the auranofin-induced permeability transition while, on the contrary, the presence of calcium ions in the incubation medium reinforces the action of auranofin, indicating that calcium acts as the critical factor in the induction of permeability transition as pointed out by Gunter & Pfeiffer (1990). The presence of a respiratory substrate appears relevant for eliciting the effect of auranofin that is no longer apparent in its absence. In addition, the calcium transport inhibitor ruthenium red and uncouplers inhibit the auranofin-induced swelling effect. Altogether, these observations indicate that calcium ions play a fundamental role in eliciting the auranofin-dependent swelling. Auranofin, at the concentrations used in this study, is completely ineffective in inhibiting the flux of electrons along the respiratory chain (Figure 3) at variance with triethylphosphine gold chloride (Rush et al., 1987b; Hoke et al., 1989). However, at concentrations higher than 10 μM, auranofin is also able to inhibit the respiratory chain. Although with different reactivity, the action of both compounds should be essentially referred to their interaction with thiol groups. Interestingly, auranofin completely inhibits thioredoxin reductase at concentrations scarcely affecting thiols (not shown) indicating a specificity of this compound towards the selenol moiety.
The redox state of soluble and membrane-bound mitochondrial thiol groups appears critical for controlling the permeability conditions of the membrane since, in general, swelling is stimulated by thiol blocking or oxidizing agents and is prevented by thiol reducing agents (Siliprandi et al., 1978; Kowaltowski et al., 2001). However, in the presence of concentrations of auranofin able to induce permeability transition, the redox state of mitochondrial glutathione is not modified while total thiol groups are only slightly decreased (not shown). This is at variance with the effect exhibited by triethylphosphine gold chloride that determines a marked oxidation of glutathione both in hepatocytes (Rush et al., 1987a) and in isolated mitochondria (Rush et al., 1987b; Hoke et al., 1989). Up to now, the thiols involved in permeability transition are not clearly defined. We show that mitochondrial thioredoxin reductase is invariably inhibited also at very low concentrations of auranofin indicating a potential correlation between its inhibition and permeability transition. It appears that the inhibition of mitochondrial thioredoxin reductase, associated with a proper amount of endogenous calcium, can create the conditions for the increase of membrane permeability since calcium ions are essential triggers of this condition (Gunter & Pfeiffer, 1990). We have previously reported that an inhibitor of thioredoxin reductase such as 13-cis retinoic acid is also an inducer of permeability transition and apoptosis (Rigobello et al., 1999). Similar considerations can be drawn for arsenicals, menadione and alloxan (Gunter & Pfeiffer, 1990; Zoratti & Szabò, 1995; Sakurai et al., 2001; Arnèr et al., 1999). More recently it was reported that ebselen, an antioxidant selenazol drug and glutathione peroxidase mimic (Sies, 1993), is able to induce swelling and calcium release (Gogvadze et al., 2000) acting also as a direct substrate for thioredoxin reductase (Holmgren et al., 2000).
As previously reported (Chaudière & Tappel, 1984; Berry et al., 1991; Hill et al., 1997; Gromer et al., 1998a, b; Smith et al., 1999), gold(I) compounds show a specific reactivity with selenoenzymes such as glutathione peroxidase, iodothyronine deiodinase type I and thioredoxin reductase. The latter enzyme appears extremely sensitive to this inhibition (Gromer et al., 1998a). Therefore, auranofin, at the mitochondrial level, allows to establish a link between a specific enzyme inhibition and a function such as permeability transition. As pointed out by Nordberg & Arnèr (2001) the inhibition of thioredoxin reductase activity leads to an impaired regeneration of the large number of the substrates of this enzyme and a decrease of the total antioxidant capacity. In particular, the regeneration of reduced thioredoxin is drastically hampered. All this brings to a shift towards a more oxidized state of thiols and to an increased production of reactive oxygen species (Nordberg & Arnèr, 2001). This is particularly important at the mitochondrial level since this organelle plays also a crucial role in controlling cellular apoptosis. A recent paper on the effects of auranofin on neutrophil apoptosis (Liu et al., 2000) reports contrasting effects since low concentrations of auranofin delay spontaneous apoptosis, while higher concentrations shorten cell survival. In preliminary experiments (not reported) we have observed that auranofin is able to induce, in isolated mitochondria, at all the concentrations tested, the release of cytochrome c, indicating a potential enhancement of cell apoptosis.
Abbreviations
- Auranofin
(S-triethylphosphinegold(I)-2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside)
- CCCP
carbonylcyanide m-chlorophenylhydrazone
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulphonate
- DMSAu
dimethylsulphide gold chloride
- DTNB
5,5′-dithiobis(2-nitrobenzoic) acid
- DTT
dithiothreitol
- GSPx
glutathione peroxidase
- TEPAu
triethylphosphine gold chloride
- TPP+
tetraphenylphosphonium ion
- ΔΨ
transmembrane electrical potential
References
- ABOU-KHALIL W.H., YUNIS A.A., ABOU-KHALIL S. Discriminatory effects of gold compounds and carriers on mitochondria isolated from different tissues. Biochem. Pharmacol. 1981;30:3181–3186. doi: 10.1016/0006-2952(81)90516-5. [DOI] [PubMed] [Google Scholar]
- ARNÉR E.S.J., ZHONG L., HOLMGREN A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 1999;300:226–239. doi: 10.1016/s0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- BERRY M.J., BANU L., LARSEN P.R. Type I iodothyronine deiodinase is a selenocysteine-containing enzyme. Nature. 1991;349:438–440. doi: 10.1038/349438a0. [DOI] [PubMed] [Google Scholar]
- BINDOLI A., CALLEGARO M.T., BARZON E., BENETTI M., RIGOBELLO M.P. Influence of the redox state of pyridine nucleotides on mitochondrial sulfhydryl groups and permeability transition. Arch. Biochem. Biophys. 1997;342:22–28. doi: 10.1006/abbi.1997.9986. [DOI] [PubMed] [Google Scholar]
- CARLBERG I., MANNERVIK B. Glutathione reductase. Methods Enzymol. 1985;113:484–490. doi: 10.1016/s0076-6879(85)13062-4. [DOI] [PubMed] [Google Scholar]
- CAZZARO F., RIGOBELLO M.P., BINDOLI A. Personal computer control of electrochemical detectors utilized for mitochondrial studies. Comput. Methods Progr. Biomed. 1996;51:141–151. doi: 10.1016/s0169-2607(96)01736-1. [DOI] [PubMed] [Google Scholar]
- CHAUDIÈRE J., TAPPEL A.L. Interaction of gold(I) with the active site of selenium-glutathione peroxidase. J. Inorg. Biochem. 1984;20:313–325. doi: 10.1016/0162-0134(84)85030-8. [DOI] [PubMed] [Google Scholar]
- CROOKE S.T., SNYDER R.M. The cellular and molecular pharmacology of auranofin and related gold complexes. Scand. J. Rheumatol. 1986;63 Suppl.:1–18. [PubMed] [Google Scholar]
- DANIEL L.W., CIVOLI F., ROGERS M.A., SMITHERMAN P.K., RAJU P.A., ROEDERER M. ET-18-OCH3 inhibits nuclear factor kappa B activation by 12-o-tetradecanoylphorbol-13-acetate but not by tumor necrosis factor-alpha or interleukin 1 alpha. Cancer Res. 1995;55:4844–4849. [PubMed] [Google Scholar]
- DONG Y., BERNERS-PRICE S.J., THORBURN D.R., ANTALIS T., DICKINSON J., HURST T., QIU L., KHOO S.K., PARSONS P.G. Serine protease inhibition and mitochondrial dysfunction associated with cisplatin resistance in human tumor cell lines: targets for therapy. Biochem. Pharmacol. 1997;53:1673–1682. doi: 10.1016/s0006-2952(97)00015-4. [DOI] [PubMed] [Google Scholar]
- ESTABROOK R.W. Mitochondrial respiratory control and the polarographic measurement of ADP:O ratios. Methods Enzymol. 1967;10:41–47. [Google Scholar]
- FOURNIER N., DUCET G., CREVAT A. Action of cyclosporine on mitochondrial calcium fluxes. J. Bioenerg. Biomemb. 1987;19:297–303. doi: 10.1007/BF00762419. [DOI] [PubMed] [Google Scholar]
- GOGVADZE V., KLEIN S.D., SHIGENAGA M., AMES B.N., RICHTER C. Effect of ebselen on Ca2+ transport in mitochondria. Redox Report. 2000;5:359–363. doi: 10.1179/135100000101535924. [DOI] [PubMed] [Google Scholar]
- GORNALL A.G., BARDAWILL C.J., DAVID M.M. Determination of serum proteins by means of the biuret reaction. J. Biol. Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- GROMER S., ARSCOTT L.D., WILLIAMS C.H., JR, SCHIRMER R.H., BECKER K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady-state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 1998a;273:20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- GROMER S., WISSING J., BEHNE D., ASHMAN K., SCHIRMER R.H., FLOHÉ L., BECKER K. A hypothesis on the catalytic mechanism of the selenoenzyme thioredoxin reductase. Biochem. J. 1998b;332:591–592. doi: 10.1042/bj3320591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROOTVELD M., BLAKE D.R., SAHINOGLU T., CLAXSON A.W.D., MAPP P., STEVENS C., ALLEN R.E., FURST A. Control of oxidative damage in rheumatoid arthritis by Gold(I)-thiolate drugs. Free Rad. Res. Comms. 1990;10:199–220. doi: 10.3109/10715769009149889. [DOI] [PubMed] [Google Scholar]
- GUNTER T.E., PFEIFFER D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- HANDEL M.L., WATTS C.K.W., DEFAZIO A., DAY R.O., SUTHERLAND R.L. Inhibition of AP-1 binding and transcription by gold and selenium involving conserved cysteine residues in Jun and Fos. Proc. Natl. Acad. Sci. U.S.A. 1995;92:4497–4501. doi: 10.1073/pnas.92.10.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILL K.E., MCCOLLUM G.W., BOEGLIN M.E., BURK R.F. Thioredoxin reductase activity is decreased by selenium deficiency. Biochem. Biophys. Res. Comm. 1997;234:293–295. doi: 10.1006/bbrc.1997.6618. [DOI] [PubMed] [Google Scholar]
- HOKE G.D., RUSH G.F., BOSSARD G.E., MCARDLE J.V., JENSEN B.D., MIRABELLI C.K. Mechanism of alterations in isolated rat liver mitochondrial function induced by gold complexes of bidentate phosphines. J. Biol. Chem. 1988;263:11203–11210. [PubMed] [Google Scholar]
- HOKE G.D., RUSH G.F., MIRABELLI C.K. The mechanism of acute cytotoxicity of triethylphosphine gold(I) complexes. III. Chlortriethylphosphine gold(I)-induced alterations in isolated rat liver mitochondrial functions. Toxicol. Appl. Pharmacol. 1989;99:50–60. doi: 10.1016/0041-008x(89)90110-5. [DOI] [PubMed] [Google Scholar]
- HOLMGREN A., ZHONG L., ARNÉR E., AMIRI M.H., MASAYASU H.Structure and mechanism of mammalian selenium-dependent thioredoxin reductases Selenium in Biology and Medicine 2000217th International Symposium pVenice (Italy)
- KAMO N., MURATSUGU M., HONGOH R., KOBATAKE Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membrane Biol. 1979;49:105–121. doi: 10.1007/BF01868720. [DOI] [PubMed] [Google Scholar]
- KEAN W.F., HART L., BUCHANAN W.W. Auranofin. Brit. J. Rheum. 1997;36:560–572. doi: 10.1093/rheumatology/36.5.560. [DOI] [PubMed] [Google Scholar]
- KERIMOVA A.A., ATALAY M., YUSIFOV E.Y., KUPRIN S.P., KERIMOV T.M. Antioxidant enzymes; possible mechanism of gold compound treatment in rheumatoid arthritis. Pathophysiology. 2000;7:209–213. doi: 10.1016/s0928-4680(00)00050-x. [DOI] [PubMed] [Google Scholar]
- KIM K.J., JANG Y.Y., HAN E.S., LEE C.S. Modulation of brain mitochondrial membrane permeability and synaptosomal Ca2+ transport by dopamine oxidation. Mol. Cell. Biochem. 1999;201:89–98. doi: 10.1023/a:1007008417342. [DOI] [PubMed] [Google Scholar]
- KOWALTOWSKI A.J., CASTILHO R.F., VERCESI A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- LEE S.-R., KIM J.-R., KWON K.-S., YOON H.W., LEVINE R.L., GINSBURG A., RHEE S.G. Molecular cloning and characterization of a mitochondrial selenocysteine-containing thioredoxin reductase from rat liver. J. Biol. Chem. 1999;274:4722–4734. doi: 10.1074/jbc.274.8.4722. [DOI] [PubMed] [Google Scholar]
- LEHNINGER A.L. Water uptake and extrusion by mitochondria in relation to oxidative phosphorylation. Physiol. Rev. 1962;42:467–517. doi: 10.1152/physrev.1962.42.3.467. [DOI] [PubMed] [Google Scholar]
- LIU J., AKAHOSHI T., NAMAI R., MATSUI T., KONDO H. Effect of auranofin, an antirheumatic drug, on neutrophil apoptosis. Inflamm. Res. 2000;49:445–451. doi: 10.1007/s000110050615. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MOORE C.L. Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochem. Biophys. Res. Comm. 1971;42:298–305. doi: 10.1016/0006-291x(71)90102-1. [DOI] [PubMed] [Google Scholar]
- MYERS D.K., SLATER E.C. The enzymatic hydrolysis of adenosine triphosphate by rat liver mitochondria. I. Activities at different pH values. Biochem. J. 1957;67:558–572. doi: 10.1042/bj0670558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NORDBERG J., ARNÉR E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- RIGOBELLO M.P., CALLEGARO M.T., BARZON E., BENETTI M., BINDOLI A. Purification of mitochondrial thioredoxin reductase and its involvement in the redox regulation of membrane permeability. Free Rad. Biol. Med. 1998;24:370–376. doi: 10.1016/s0891-5849(97)00216-5. [DOI] [PubMed] [Google Scholar]
- RIGOBELLO M.P., SCUTARI G., FRISO A., BARZON E., ARTUSI S., BINDOLI A. Mitochondrial permeability transition and release of cytochrome c induced by retinoic acids. Biochem. Pharmacol. 1999;58:665–670. doi: 10.1016/s0006-2952(99)00149-5. [DOI] [PubMed] [Google Scholar]
- ROBERTS J.R., SHAW C.F., III Inhibition of erythrocyte selenium-glutathione peroxidase by auranofin analogues and metabolites. Biochem. Pharmacol. 1998;55:1291–1299. doi: 10.1016/s0006-2952(97)00634-5. [DOI] [PubMed] [Google Scholar]
- RUSH G.F., SMITH P.F., ALBERTS D.W., MIRABELLI C.K., SNYDER R.M., CROOKE S.T., SOWINSKI J., JONES H.B., BUGELSKI P.J. The mechanism of acute cytotoxicity of triethylphosphine gold (I) complexes. I. Characterization of triethylphosphine gold chloride-induced biochemical and morphological changes in isolated hepatocytes. Toxicol. Appl. Pharmacol. 1987a;90:377–390. doi: 10.1016/0041-008x(87)90130-x. [DOI] [PubMed] [Google Scholar]
- RUSH G.F., SMITH P.F., HOKE G.D., ALBERTS D.W., SNYDER R.M., MIRABELLI C.K. The mechanism of acute cytotoxicity of triethylphosphine gold(I) complexes. II. Triethylphosphine gold chloride-induced alterations in mitochondrial function. Toxicol. Appl. Pharmacol. 1987b;90:391–400. doi: 10.1016/0041-008x(87)90131-1. [DOI] [PubMed] [Google Scholar]
- SAKURAI K., KATOH M., FUJIMOTO Y. Alloxan-induced mitochondrial permeability transition triggered by calcium, thiol oxidation, and matrix ATP. J. Biol. Chem. 2001;276:26942–26946. doi: 10.1074/jbc.M102029200. [DOI] [PubMed] [Google Scholar]
- SCHALLREUTER K.U., WOOD J.M. The stereospecific suicide inhibition of human melanoma thioredoxin reductase by 13-cis-retinoic acid. Biochem. Biophys. Res. Comm. 1989;160:573–579. doi: 10.1016/0006-291x(89)92471-6. [DOI] [PubMed] [Google Scholar]
- SIES H. Ebselen, a selenoorganic compound as glutathione peroxidase mimic. Free Radic. Biol. Med. 1993;14:313–323. doi: 10.1016/0891-5849(93)90028-s. [DOI] [PubMed] [Google Scholar]
- SILIPRANDI N., SILIPRANDI D., BINDOLI A., TONINELLO A.Effect of oxidation of glutathione and membrane thiol groups on mitochondrial functions Functions of Glutathione in Liver and Kidney 1978Berlin-Heidelberg: Springer-Verlag; 139–147.ed. Sies, H. & Wendel, A. pp [Google Scholar]
- SIMON T.M., KUNISHIMA D.H., VIBERT G.J., LORBER A. Screening trial with the coordinated gold compound auranofin using mouse lymphocyte leukemia p388. Cancer Res. 1981;41:94–97. [PubMed] [Google Scholar]
- SMITH A.D., GUIDRY C.A., MORRIS V.C., LEVANDER O.A. Aurothioglucose inhibits murine thioredoxin reductase in vivo. J. Nutrition. 1999;129:194–198. doi: 10.1093/jn/129.1.194. [DOI] [PubMed] [Google Scholar]
- SNYDER R.M., MIRABELLI C.K., CROOKE S.T. Cellular association, intracellular distribution, and efflux of auranofin via sequential ligand exchange reactions. Biochem. Pharmacol. 1986;35:923–932. doi: 10.1016/0006-2952(86)90078-x. [DOI] [PubMed] [Google Scholar]
- STUVE J., GALLE P. Role of mitochondria in the handling of gold by the kidney. J. Cell. Biol. 1970;44:667–676. doi: 10.1083/jcb.44.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAMURA T., STADTMAN T.C. A new selenoprotein from human lung adenocarcinoma cells: purificatoin, properties, and thioredoxin reductase activity. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1006–1011. doi: 10.1073/pnas.93.3.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WUDARCZYK J., DEBSKA G., LENARTOWICZ E. Relation between the activities of reducing disulfides and the protection against membrane permeability transition in rat liver mitochondria. Arch. Biochem. Biophys. 1996;327:215–221. doi: 10.1006/abbi.1996.0112. [DOI] [PubMed] [Google Scholar]
- ZHONG L., ARNÉR E.S.J., LUNG J., ÅSLUND F., HOLMGREN A. Rat and calf thioredoxin reductase are homologous to glutathione reductase with a carboxyl-terminal elongation containing a conserved catalytically active penultimate selenocysteine residue. J. Biol. Chem. 1998;273:8581–8591. doi: 10.1074/jbc.273.15.8581. [DOI] [PubMed] [Google Scholar]
- ZORATTI M., SZABÒ I. The mitochondrial permeability transition. Biochim. Biophys. Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]