

Abstract
We have used a range of in vitro electrophysiological techniques to investigate the mechanism of rapid cortisol neuromodulation of parvocellular neurones in the rat paraventricular nucleus.
In our study, we found that cortisol (10 μM) increased spontaneous action–current firing frequency to 193%. This effect was insensitive to the glucocorticoid intracellular-receptor antagonist mifepristone.
Cortisol (0.1–10 μM) had no detectable effects on whole-cell GABA current amplitudes, or GABAA single-channel kinetics.
Cortisol (10 μM) inhibited whole-cell potassium currents in parvocellular neurones by shifting the steady-state activation curve by 14 mV to the right.
Additionally, in a cell line expressing both the glucocorticoid intracellular receptor and recombinant, fast inactivating potassium channels (hKv1.3), cortisol (1 and 10 μM) inhibited potassium currents by shifting their steady-state activation curves to the right by 12 mV (10 μM cortisol). This effect was also insensitive to the cortisol antagonist, mifepristone.
These data suggest that inhibition of voltage-gated potassium channels may contribute to the rapid neuromodulatory effects of cortisol, possibly by direct interaction with the ion channel itself.
Keywords: Cortisol, GABA, hKv1.3, Kv1.3, neuromodulation, paraventricular nucleus, patch clamp, potassium channel, sympathetic
Introduction
Physiological and psychological stress cause activation of the sympathetic nervous system and secretion of glucocorticoids (GCs, mostly cortisol in humans) from the adrenal cortex. The pattern and regulation of glucocorticoid secretion is very complex, although the so-called ‘stress response' is largely co-ordinated by the hypothalamus, and specifically by neurones in the parvocellular subnucleus of the paraventricular nucleus (PVN, Sawchenko et al., 1996).
The parvocellular subnucleus of the hypothalamus is a region of the PVN which contains both neurons with axons that synapse with sympathetic preganglionic neurones, and corticotrophin releasing factor (CRH) neurones which project to the hypothalamic medial eminence (Sawchenko et al., 1996). A variety of stimuli induce the secretion of CRH, which in turn increases the secretion of adrenocorticotrophic hormone (ACTH) and thence GCs (Buckingham, 1979a). Released GCs can exert a classic negative feedback on the hypothalamic–pituitary–adrenal (HPA) axis, suppressing the release of both ACTH and CRH, thus limiting GC's own secretion (Buckingham, 1996).
In addition to these classical effects, GCs exert a number of other effects on neuronal activity. Iontophoretic application of cortisol has been reported to both excite and inhibit action potential firing in rat PVN neurones (Kasai et al., 1988), and to increase neuronal activity in other regions of the hypothalamus and brain (Avanzino et al., 1984;1987; Feldman & Dafny, 1970; Filaretov, 1976; Venero & Borrell, 1999). The precise physiological significance of these effects is unclear, but direct modulation of parvocellular neurones could potentially contribute to HPA-axis feedback, or to the modulation of sympathetic outflow arising from the central administration of cortisol (Takahashi et al., 1983).
Whilst many of the classical actions of GCs involve interaction with an intracellular receptor, transcription of lipocortins and sustained effects (Buckingham, 1996), many neuromodulatory actions occur within a few seconds of exposure to GCs, by some rapid, non-genomic mechanism(s) (reviewed by Falkenstein et al., 2000). The rapid modulatory actions of other neuroactive steroids (e.g., pregnenolone sulphate, tetrahydroxycorticosterone and allopregnanolone) mostly involve interaction with the GABAA receptor (Paul & Purdy, 1992), and there is some evidence to suggest that GCs may act via a similar pathway (GCs reduce GABA binding to rat brain homogenates for example, and reduce the amplitude of GABA responses in amphibian neurones (Ariyoshi & Akasu, 1986; Majewska et al., 1985; Majewska, 1987). Other studies suggest another potential target for the cortisol action is the potassium ion channel. For example, Chen et al. (1991) found neuromodulation of guinea-pig coeliac ganglia neurones to be primarily associated with the modulation of potassium channels. Regulation of neuronal potassium channels by GCs is an attractive hypothesis given that potassium channel modulation is a major pathway for regulating membrane excitability in the CNS (Brown, 1990; Miyake et al., 1989; North, 1989; Stanfield et al., 1985).
In this study, we have used a range of electrophysiological techniques to investigate the mechanism by which cortisol rapidly modulates the activity of parvocellular neurones of the PVN. We find an increase in neuronal activity in response to application of cortisol that was independent of the GC intracellular receptor. We find no modulation of GABAA current by cortisol at either the single channel or whole-cell level, but rather an inhibition of voltage-gated potassium currents, suggesting that cortisol may exert some of its rapid neuronal effects through direct potassium channel modulation.
Methods
In vitro slice experiments
Slices of paraventricular nucleus were prepared using the methods of Barrett-Jolley et al. (2000). Briefly, young adult rats (3–5 weeks) were killed by an overdose of anaesthetic (Sagital) and the brain removed to iced, low-Ca2+ artificial cerebrospinal fluid (ACSF, solution I, Table 1). The general area of the hypothalamus was blocked and glued in the chamber of a Campden Instruments Vibroslice. The chamber was maintained at 0–4°C by frequent addition of iced low-Ca2+-ACSF (solution I, Table 1). Coronal slices, approximately 200 μm, were cut and removed to a multiwell culture dish containing normal ACSF (solution II, Table 1). The culture dish was incubated at 35–37°C and bubbled with 95% O2–5% CO2, for at least 1 h prior to recording.
Table 1.
Composition of solutions (mM)
Neurones were visualized with a Nikon E600FN Eclipse upright microscope equipped with near infrared DIC optics.
Action current frequency experiments (slice)
Action potential propagation through a neurone under cell-attached patch clamp results in a detectable ‘action current'. Calculation of the action–current frequency can then be used as a measure of the underlying action–potential frequency. In this study action currents were measured with the cell-attached patch technique following methods similar to Fenwick et al. (1982) and Wolters et al. (1994).
Neurones were superfused with a normal ACSF solution (solution II, Table 1) at 34–36°C and patch pipettes were filled with solution III (Table 1). Following seal formation (cell-attached patch), the command voltage was permanently set to 0 mV. Measured current is then the sum of the resistive and capacitance currents flowing between the cell interior and the patch pipette. It is theoretically possible to derive absolute membrane potential values for the underlying action potential (Fenwick et al., 1982), however, this requires very accurate discrimination between membrane patch resistance and seal resistance. In the experiments reported in this paper, action currents are used simply to allow precise measurement of underlying action–potential frequency without penetration of the cell, or rupture of the cell membrane. Importantly, this method does not disturb the intracellular environment of the target neurone.
We found a number of parvocellular PVN neurones to be spontaneously active; such spontaneous action potential generation could result from either intrinsic membrane properties (such as Andrew, 1987) or from a net excitatory synaptic input. We found that imposition of synaptic block, by either addition of elevated Mg2+ or a combination of excitatory amino acid antagonists D(−)- 2-amino-5-phosphonovaleric acid (APV) and 6,7-dinitroquinoxaline-2-3-dione (DNQX), greatly reduced, or abolished spontaneous action current firing: Action current firing was reduced to 36±17% (n=3) by 10 mM Mg2+, and to 2±1.4% (n=4) 50 μM APV and 10 μM DNQX (combined). This strongly suggests that spontaneous action potential firing of parvocellular neurones arises from a tonic excitatory synaptic input, consistent with the observations of Bains & Ferguson (1995) who showed that parvocellular neurones receive excitatory glutaminergic input.
Actions of drugs were assessed following at least 2 min of exposure, and expressed relative to the pre-exposure frequency. Neurones with initial action-current frequency of above 0.05 Hz were accepted for study. In some previous studies involving GC action on neuronal activity, each target neurone was designated as being either inhibited, excited or not sensitive to GCs (e.g., Kasai & Yamashita, 1988b; Saphier & Feldman, 1988). The criterion for these classifications required only that the frequency in the presence of steroids was above, below or within 20–30% of the pre-exposure frequency (in each experiment). We found, however, that even in the absence of drug or vehicle exposure, action-current frequency could fluctuate with time. Instead, we chose to calculate the mean activity of neurones in the presence and absence of test substances.
Parvocellular GABA current experiments (slice)
Both whole-cell and outside-out patch experiments followed methods similar to that of Barrett-Jolley (2001). Neurones were held at −60 mV and superfused at 2 mls min−1 with 34–36°C HEPES buffered low chloride solution (solution IV, Table 1). Pipettes contained a HEPES buffered low potassium solution (solution V, Table 1). For whole-cell experiments, the tip of a pressure ejection pipette (∼5 μM) was placed 40–150 μM away from the target neurone. A brief pulse (approximately 1 s) of GABA was then applied at the required final concentration, using a nitrogen gas charged Picospritzer (General Valve Corporation, Fairfield, NJ, U.S.A.). In outside-out patch-clamp experiments, GABA was applied to the flow solution. Previous studies (Barrett-Jolley, 2001) showed these currents to be GABAA, blocked by bicuculline, and reversing around the equilibrium potential for chloride ions (approximately +40 mV under these conditions).
Kinetic analysis followed the procedures of Barrett-Jolley & Davies (1997). Data were initially filtered at 5 kHz (8-pole Bessel) and sampled at 10 kHz. Data were then digitally filtered at 2 kHz and dwell times calculated by an automated 50% threshold crossing protocol. The idealized record was then log binned (Sigworth & Sine, 1987), and fitted with a maximum likelihood probability density function. Bursts were defined by a critical closed time. This was calculated for each patch, by equalizing proportions of misclassified events.
Parvocellular potassium current experiments (slice)
Neurones were held at −80 mV and continually superfused with warm (34–36°C) ACSF (solution II, Table 1). A low chloride, HEPES buffered pipette solution was used (solution III, Table 1) and series resistance was routinely in the range of 5–25 MΩ and always compensated for by at least 80%, with the internal Axopatch 200 (or B) circuitry. The voltage protocols used are given in the figure legends. For current voltage curves, we measured ‘peak outward' currents, specifically a 2 ms average centred on the maximum current point in each trace. Conductance–voltage curves were calculated for each patch using the following transformation:
 |
where G is the conductance (S), I the current (A), Vc the command potential (V) and EK is the equilibrium potential for potassium ions (V). These individual conductance–voltage data were then each fit with the standard Boltzmann equation:
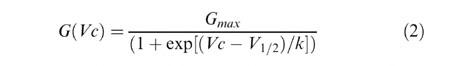 |
where G(Vc) gives the conductance (S) at Vc, Gmax is the maximum conductance (S), V1/2 is the midpoint parameter (V) and k is the slope factor.
Mean Boltzmann parameters were calculated from these individual fits. Statistical tests were performed between populations of control and test patches. Absolute maximum conductance values tend to vary considerably between cells, therefore, for display purposes (only), conductance–voltage data from each individual patch was normalized and meaned before plotting (see Figures 6,7,8). This normalization involved dividing the conductance value at each voltage, by the previously determined mean Gmax value.
Figure 6.
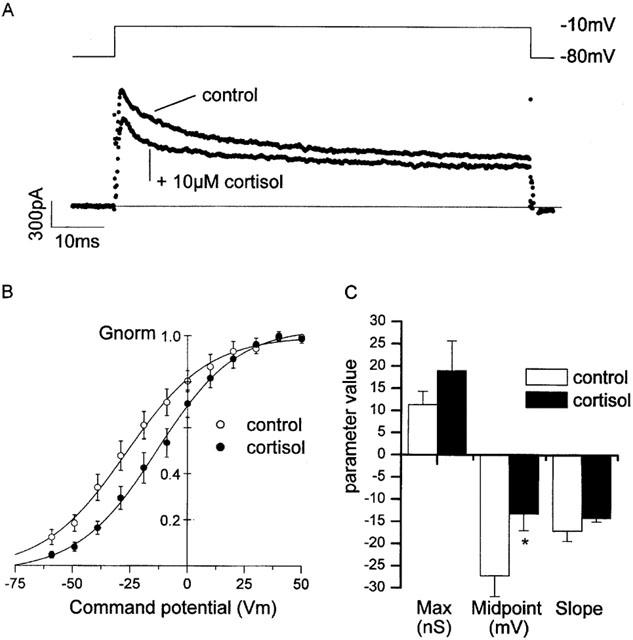
Action of cortisol on native potassium currents in parvocellular PVN neurones. (A) Potassium currents recorded in the absence and presence of 10 μM cortisol. The dotted line shows the zero current level. The voltage protocol is illustrated in the upper panel. Neurones were held at −80 mV and stepped to a command potential of, in this example, −10 mV. (B) Mean normalized Boltzmann curves from a number of experiments similar to that shown in (A). (C) Mean Boltzmann parameters obtained from fits to individual experiments. Only the midpoint cortisol parameter is significantly different from the control value. Control: V1/2 −27.2±4.7 mV, k 7.1±2.3 mV, maximum 11.3±3.0 nS, n=9. 10 μM cortisol: V1/2 −13.2±3.6 mV (P<0.05), k 14.2±0.8 mV, maximum 19.0±6.2 nS, n=8. Pipette solution III, bath solution II (Table 1).
Figure 7.
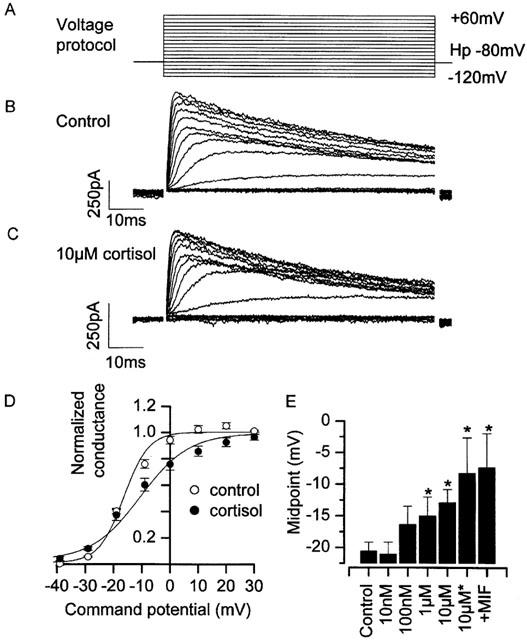
Action of cortisol on recombinant hKv1.3 potassium channels. (A) Illustration of the voltage protocol used; cells were held at −80 mV and stepped to command potentials in the range of −120 mV to +70 mV, in 10 mV increments. Up to 20 s was allowed between each pulse for recovery of inactivation. (B) A family of hKv1.3 potassium currents recorded under control conditions. (C) A family of hKv1.3 potassium currents recorded in the presence of 10 μM cortisol. (D) Mean normalized Boltzmann curves from a number of experiments similar to those shown in (B) & (C). (E) Shift in midpoint parameter was statistically significant at both 1 μM and 10 μM, and greatest when cortisol was included both in the bath, and in the pipette (indicated by ‘10 μM*'). Note that 10 μM mifepristone, applied in conjunction with cortisol (indicated by ‘+MIF') failed to inhibit the midpoint shift by cortisol. Control: V1/2 −20.6±1.5 mV, k 5.7±0.6 mV, maximum 4.5±0.9 nS (n=14). 0.01 μM cortisol: V1/2 −21.1±1.9 mV, k 2.9±1.4 mV, maximum 0.9±0.1 nS (n=4). 0.1 μM cortisol: V1/2 −16.4±3.0 mV, k 5.8±1.0 mV, maximum 2.5±0.3 nS (n=6). 1 μM cortisol: V1/2 −15.0±3.0 mV (P<0.05), k 5.8±1.0 mV, maximum 3.0±0.6 nS (n=7). 10 μM cortisol: V1/2 −12.9±2.3 mV (P<0.05), k 9.4±1.7 mV, maximum 8.0±1.7 nS (n=12). 10 μM cortisol (bath+pipette): V1/2 −8.3±5.6 mV (P<0.05), k 11.2±6.3 mV, maximum 5.5±1.3 nS (n=4). 10 μM cortisol +10 μM mifepristone (bath+pipette): V1/2 −7.4±5.4 mV (P<0.05 compared to control), k 10.7±1.7 mV, maximum 7.1±3.6 nS (n=3). 24°C Pipette solution III, bath solution VI (Table 1).
Figure 8.

Action of cortisol on recombinant hKv1.1 potassium channels. (A) Illustration of the voltage protocol used; cells were held at −80 mV and stepped to command potentials in the range of −120 mV to +60 mV, in 10 mV increments. Up to 20 s was allowed between each pulse for recovery of inactivation. (C) A family of hKv1.1 potassium currents recorded under control conditions. (C) A family of hKv1.1 potassium currents recorded in the presence of 10 μM cortisol. (D) Mean normalized Boltzmann curves from a number of experiments similar to those shown in (B) & (C). (E) Neither 1 μM or 10 μM cortisol caused significant alteration in midpoint parameters. Control V1/2 −30.0±2.6 mV, k 8.4±1.2 mV, maximum 4.8±1.9 nS (n=8). 1 μM cortisol: V1/2 −30.4±3.3 mV, k 7.1±1.0 mV, maximum 2.0±0.9 nS (n=5). 10 μM cortisol: V1/2 −28.9±1.8 mV, k 8.0±1.2 mV, maximum 5.1±1.1 nS (n=5). 24°C Pipette solution III, bath solution VI (Table 1).
Recombinant potassium channel experiments
hKv 1.1 and 1.3 clones were stably transfected into the murine erythroleukemia (MEL) cell line (Shelton et al., 1993) and expression induced with DMSO 1–3 days before patch-clamp experiments. For patch-clamp experiments, cells were spun at 800 r.p.m. in a bench-top centrifuge for 2 min (Fisons MSE), resuspended in a HEPES buffered bath solution (solution VI, Table 1), and transferred to the recording chamber where they were maintained at room temperature for up to 1 h. Voltage protocols, and potassium–current analysis procedures were identical to those used in slice experiments. In some cases hKv1.3 MEL cells were exposed to 5 mM DTT to restore the phenotypic N-type inactivation (Ruppersberg et al., 1991). Patch pipettes were filled with a low chloride, intracellular solution (solution III, Table 1).
Electrophysiological recording
In all experiments, patch pipettes were fabricated from thick-walled borosilicate glass (Harvard's ‘Clark GC150F') using a Brown-Flaming MP P-80 horizontal puller (Sutter Instrument Co.). When filled, pipettes had resistances of approximately 5 MΩ. Data were recorded with an Axon Axopatch 200 (or 200B) amplifier and low-pass filtered at between 2–10 KHz. In the case of action current measurement, data were additionally high-pass filtered at 0.1 Hz, a level of AC coupling that has no effect on the action–current waveform. Analysis was performed with the Axgox suite of programs, written by Noel Davies, University of Leicester (Davies, 1993), or with custom written software.
Solutions
Six different physiological saline solutions were used in this study, each of these is summarized in Table 1. All experiments were conducted at 34–36°C, except where stated.
Cortisol and mifepristone were dissolved in DMSO (as 100 mM stocks). The final concentration of DMSO never exceeded 1 : 1000, at which level it is inactive in all the systems reported in this paper. All drugs and chemicals were supplied by the Sigma-Aldrich Co. Ltd., Poole, Dorset (U.K.).
Statistics
Means are presented as mean±s.e.mean, n refers to the number of experiments (i.e., cells or patches). ‘P⩽x' values are derived from unpaired Student's t-tests (except where stated) between test and control samples calculated with Microsoft Excel 97, a probability of less than 0.05 is defined as statistically significant. Curve fitting was performed by the non-linear curve fitting algorithms of MicroCal Origin 6.0, confidence limits are those returned by Origin.
Results
Action-current frequency increased by cortisol
We began by investigating the possible action of cortisol on a population of spontaneously active neurones in the parvocellular region of the PVN (see Methods). Following exposure to cortisol (10 μM) for a period of 2–5 min, spontaneous action–current frequency increased to 193±42% (n=5) of the control (pre-application) frequency (Figure 1A,E).
Figure 1.
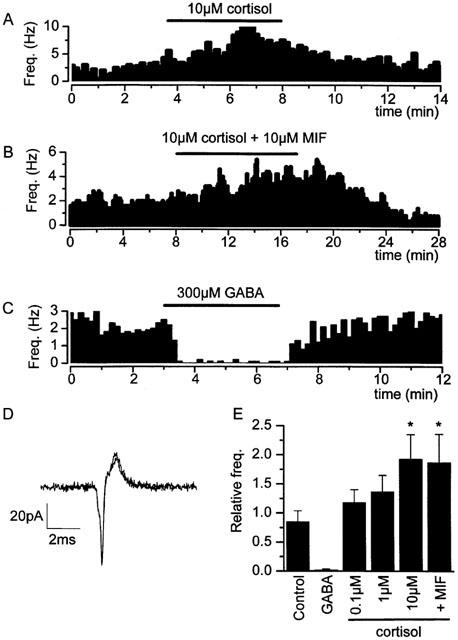
Action of cortisol on spontaneously active parvocellular neurones of the PVN. (A) Application of cortisol (10 μM cortisol) increases action current frequency. (B) A similar increase in action current frequency was observed when the cortisol antagonist mifepristone (RU-486) was applied (10 μM) simultaneously with cortisol. (C) GABA (300 μM) added to the flow solution abolishes the spontaneous action current activity of parvocellular neurones. (D) High resolution, superimposed images action currents in the presence and absence of cortisol (50 μs sample interval). (E) Relative action–current frequencies in the presence of treatment (relative to that prior to the addition of substance) were as follows: Control (vehicle) 0.85±0.16 (n=14); 300 μM GABA: 0.02±0.01% (n=4), P<0.001; 100 nM cortisol: 1.18±0.22% (n=12); 1 μM cortisol: 1.37±0.28 (n=7); 10 μM cortisol: 1.93±0.42% (n=5), P<0.01; 10 μM cortisol +10 μM mifepristone: 1.87±0.48 (n=5), P<0.05. ‘P' values are derived from Mann–Whitney U-tests between relative action current frequencies in the presence of drug, compared with relative action current frequency in the presence of vehicle. Pipette solution III, bath solution II (Table 1).
Effect of cortisol not blocked by mifepristone
Many actions of cortisol are mediated by the glucocorticoid intracellular receptor (GR) and transcription of a family of effector peptides known as lipocortins (Buckingham, 1996). Such actions usually have a latency of several hours. More rapid GR mediated activity has also been reported (Croxtall et al., 2000), but to date, all cortisol responses resulting from interaction with GR have proven sensitive to the GR antagonist, mifepristone (also known as ‘RU38486', or ‘RU486', Gagne et al., 1985; Moguilewsky & Philibert, 1984). Parvocellular PVN neurones express GR in abundance (Agnati et al., 1985; Fuxe et al., 1985a,b), so to investigate whether the activation of parvocellular neurones was mediated by the intracellular GR, we applied 10 μM mifepristone simultaneously to cortisol. This failed to inhibit the action of cortisol (Figure 1B,E. 10 μM mifepristone +10 μM cortisol: 187±48%, n=5).
Investigation of possible GABA receptor interactions with cortisol
In many cases, steroids modulate neuronal activity by interaction with GABAA receptors (Majewska et al., 1986; Paul & Purdy, 1992). Parvocellular PVN neurones express GABAA receptors (Barrett-Jolley, 2001) and spontaneous action currents are virtually abolished by application of 300 μM GABA to the flow solution (residual frequency: 2.4±1.1% n=4, Figure 1C,E).
To investigate the likely interaction of cortisol with GABAA channels, we made both single-channel and whole-cell patch clamp recordings of parvocellular GABA currents in the absence and presence of cortisol. These currents are clearly carried through GABAA ion channel/receptor proteins due to their reversal near ECl− and their sensitivity to the GABAA receptor antagonist bicuculline (Barrett-Jolley, 2001). Bath applied cortisol at concentrations in the range 0.1–10 μM had no effect on the amplitude of transient GABA whole-cell currents evoked by pressure ejection of 300 μM GABA (Figure 2). These data are summarized in Figure 2E.
Figure 2.
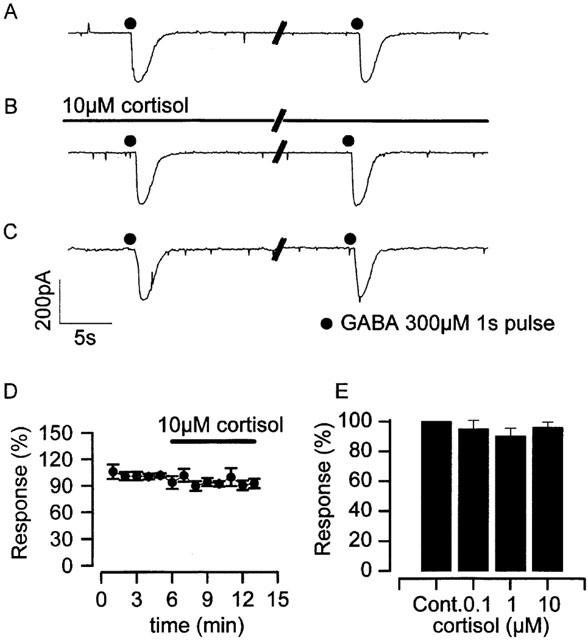
Action of cortisol on parvocellular GABA currents (whole-cell). Recordings (A) to (C) show whole-cell GABA currents, evoked by pressure application of 300 μM GABA to parvocellular neurones held at −60 mV. GABA pulses were applied at 2 min intervals. (A) currents recorded prior to the application of 10 μM cortisol to the flow solution, (B) in the presence of 10 μM cortisol and (C) following exposure to cortisol. (D) Mean 300 μM GABA response amplitude during the application of 10 μM cortisol, there is no significant rapid effect of cortisol (n=3). (E) Mean amplitude of 300 μM GABA evoked currents in the presence of 0.1 μM (95.1±5.8%, n=5), 1 μM (90.4±5.2%, n=3) and 10 μM cortisol (96.2±3.5%, n=3), relative to control current. Parallel diagonal lines in (A), (B) and (C) indicate an approximately 1 min interval. Pipette solution V, bath solution IV (Table 1).
To insure that the lack of effect on GABA currents was not simply due to cortisol's failure to penetrate the surface of the slice, we used the outside-out patch configuration. In this technique membrane patches were pulled from parvocellular neurones and exposed to a continuous stream of flow solution containing either GABA alone, or GABA with cortisol. We found 10 μM cortisol to have no detectable effects on either the steady-state kinetic properties of GABAA channels (Figures 3A,B and 4, and Table 2), or on mean GABAA single channel amplitude (Figure 3).
Figure 3.
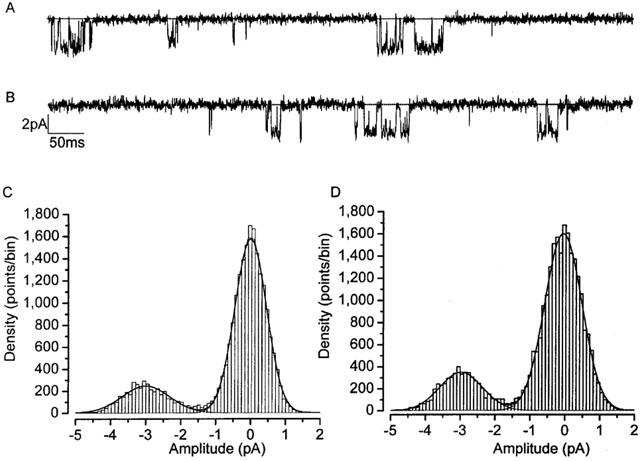
Parvocellular neurone GABAA channel amplitudes unaffected by cortisol. GABA single channel currents (evoked by 300 μM GABA) recorded under control conditions (A), and in the presence of 10 μM cortisol (B). GABA single-channel all point amplitude histograms in the absence (C) and presence (D) of 10 μM cortisol. Single channel amplitudes were not significantly different under these two conditions: Control; 2.5±0.5 pA (n=5), cortisol 10 μM; 2.4±0.4 pA (n=7). Membrane holding potential −60 mV, 36°C, pipette solution V, bath solution IV (Table 1).
Figure 4.
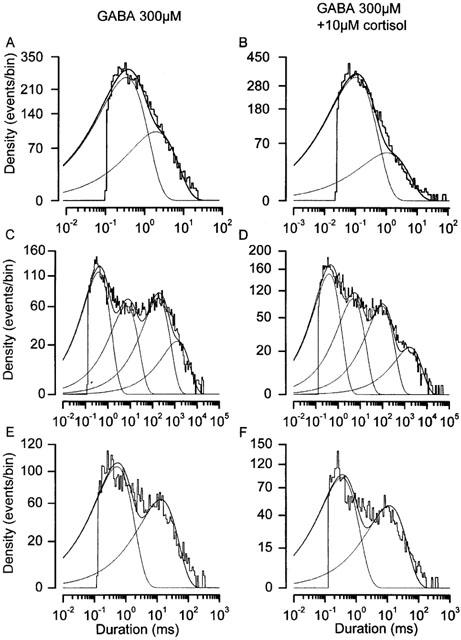
Parvocellular neurone GABAA channel dwell times unaffected by cortisol. Dwell time histograms for GABA channels recorded in the presence of 300 μM GABA (A, C & E) and 300 μM GABA +10 μM cortisol (B, D & F). Dwell times are log binned (see Methods) and event density is displayed on a square root scale. Under this transformation, individual time constants appear as density peaks. The histograms show the aggregate data for four patches analysed in the absence and four patches analysed in the presence of 10 μM cortisol. Mean data for fits to each individual patch are shown in Table 2. Solid lines show the combined fits and dotted lines show the individual components of each fit. Membrane holding potential −60 mV, 36°C, pipette solution V, bath solution IV (Table 1).
Table 2.
GABAA channel dwell times in the presence and absence of cortisol

Cortisol interaction with native potassium conductance in parvocellular neurones
In several cases, where rapid steroid actions appear independent of GABA receptor interaction, steroids have been postulated to modulate potassium channels (Chen et al., 1991; Pennington et al., 1994; Wang et al., 1998; 1999). Any such modulation of potassium channels is likely to alter membrane excitability and action potential firing frequency. Chen et al. (1991) reported a small hyperpolarization of guinea-pig coeliac ganglia by glucocorticoids, and pregnenolone sulphate has been shown to positively modulate recombinant voltage-gated potassium (Kv1.1) channels (Wang et al., 1998) by shifting their half activation potentials to more negative potentials. We therefore investigated the effect of cortisol on native voltage-gated potassium currents of parvocellular neurones. Following inhibition of sodium currents with TTX, depolarizing command potentials evoke outward currents (Figure 5A,B) with both sustained and transient components. Tail current experiments show a reversal potential near to EK+ and demonstrate these voltage-gated currents to be predominantly carried by potassium ions (Figure 5C,D). We constructed steady-state conductance-voltage (Boltzmann) activation curves for these currents and calculated a mean half-activation potential (V1/2) of −27.2±4.7 mV (n=9, Figure 6B,C). In the presence of bath applied cortisol (incubation time <10 min), potassium currents were inhibited, with the Boltzmann activation curves being shifted to more positive values (10 μM cortisol: V1/2 −13.2±3.6 mV, n=8, P⩽0.05) There were no significant changes in either slope or maximum amplitude.
Figure 5.

Potassium currents in parvocellular neurones. (A) A family of voltage-gated currents recorded from parvocellular neurones in brain slice experiments under control conditions. The upper panel illustrates the voltage protocol. Neurones were held at −80 mV and stepped to command potentials in the range of −120 mV to +70 mV, in 10 mV increments. Five seconds were allowed in between each pulse for recovery of inactivation. (B) Mean (peak) current-voltage curves for nine experiments similar to that shown in (A). (C) To measure tail currents a different protocol was used (illustrated in the upper panel). Neurones were held at −80 mV and stepped to a conditioning voltage of +40 mV, before stepping to a test potential of between −30 mV and −130 mV. The dotted line shows the zero current level. The inset shows a blow up of the tail current region of the main trace (x-axis amplified 3×, y-axis amplified 2×). The solid lines are simple exponentials fit to the tail currents. (D) Mean tail current amplitudes for four experiments similar to that in (C), the smooth line represents a Goldman–Hodgkin–Katz current equation fit to the data (Hille, 1984c): Pk/PNa ratio 0.021 (95% confidence limits 0.003–0.03, n=8). Vrev −85 mV. Pipette solution III, bath solution II (Table 1).
Interaction with recombinant voltage-gated (hKv) potassium channels
The mouse erythroleukemia cell line (MEL cells) expresses the glucocorticoid intracellular receptor, GR (Golde et al., 1979; Tsiftsoglou et al., 1986). Therefore, we chose to investigate the specificity of cortisol action on two different subtypes of potassium channels by applying cortisol to MEL cells stably transfected with recombinant human hKv voltage-gated potassium channels (hKv 1.1 and hKv 1.3; Shelton et al., 1993). Potassium currents were recorded using protocols similar to those used for rat parvocellular neurones (above). In some experiments, cortisol or cortisol together with mifepristone was/were applied to the bath prior to recording for period of time not exceeding 30 min (Figures 7 and 8). Conductance–voltage activation curves were constructed for both hKv 1.1 and hKv 1.3 and fitted with Boltzmann curves in both the presence and absence of cortisol.
Cortisol shifted the Boltzmann midpoint parameter of hKv 1.3 currents significantly to the right (Figure 7, control: V1/2 −20.6±1.5 mV, n=14. 10 μM cortisol (bath+pipette): V1/2 −8.3±5.6 mV, n=4, P⩽0.05). This effect was observed with both 1 and 10 μM cortisol, and, importantly, not blocked by the intracellular glucocorticoid-receptor antagonist, mifepristone (Figure 7, 10 μM cortisol +10 μM mifepristone (bath+pipette): V1/2 −7.4±5.4 mV, n=3, P⩽0.05). Interestingly, applied in the concentration range of 1–10 μM, cortisol had no effect on hKv 1.1 currents (Figure 8, V1/2 −30.0±2.6 mV, n=8, 10 μM cortisol: V1/2 −28.9±1.8 mV, n=5).
Discussion
In this study, we sought to investigate the cellular mechanism by which cortisol rapidly modulates action potential firing frequency in the parvocellular subdivision of the paraventricular nucleus (PVN). We found cortisol to increase the frequency of spontaneous action currents in parvocellular PVN neurones. In parallel to this we found native voltage-gated potassium currents of parvocellular neurones to be inhibited. Studies with the glucocorticoid (GC) intracellular-receptor antagonist, mifepristone, suggested that the excitatory effect of cortisol was independent of the GC intracellular receptor. Further experiments with recombinant wild-type human voltage-gated potassium channels (hKv1.3) suggest that this effect may occur through a direct channel/membrane interaction.
Cortisol effects on action current frequency
There have been several prior investigations into the rapid neuronal effects of GCs (Avanzino et al., 1984; Filaretov, 1976; Venero & Borrell, 1999). Most of these studies were conducted in vivo and both excitatory and inhibitory actions of GCs were observed. For example, excitation was observed in experiments on hippocampal CA1 neurones (Venero & Borrell, 1999), neurones in the Raphe nucleus (Avanzino et al., 1984) and hypothalamic neurones (anterior and posterior, Filaretov, 1976). Several early studies, directed specifically into rapid GC action in the PVN, also reported both activation and/or inhibition of neuronal activity by GCs (Kasai et al., 1988; Kasai & Yamashita, 1988a,b; Saphier & Feldman, 1988). In this current work, we report a rapid increase in neuronal activity following application of cortisol. Several factors could explain why GCs produce apparently opposite (rapid) effects when studied under different experimental conditions. Firstly, in complex, multi-neuronal systems, particularly in vivo, it is quite possible that both excitation and inhibition could result from a common action. For example, excitation of an inhibitory input to the impaled (or sealed) neurone would result in apparent inhibition. Secondly, many studies used iontophoretic application of steroid (Avanzino et al., 1984; Saphier & Feldman, 1988; Venero & Borrell, 1999). Whilst this is an excellent system for a rapid and controlled release of ligand at a reasonably precise locus, there is very little knowledge of the concentration which actually reaches the neuronal membrane. Some of the other studies mentioned above used intravenous infusion of steroid (e.g., Filaretov, 1976). Although steroids are lipophilic and most will readily cross the blood brain barrier, it is difficult to calculate the concentration immediately surrounding any particular neuronal membrane. Thirdly, GCs may act by several non-genomic mechanisms in addition to the well characterized genomic mechanisms (Falkenstein et al., 2000).
Lack of cortisol interaction with GABAA channels
There is substantial evidence to suggest that in most cases, rapid modulatory actions of neuroactive steroids involve an interaction with GABAA receptors. For example, GABAA currents are inhibited by pregnenolone sulphate and tetrahydroxycorticosterone (Majewska et al., 1988; Wetzel et al., 1999) and activated by allopregnanolone and tetrahydroxycorticosterone (Paul & Purdy, 1992; Wetzel et al., 1999). Additionally, glucocorticoids modulate GABAA receptor binding in rat brain (Majewska et al., 1985; Majewska, 1987) and GABAA channel current in guinea-pig ileum and amphibian brain (Andres-Trelles et al., 1989; Ariyoshi & Akasu, 1986). Several GABAergic pathways input the PVN (Hermes et al., 1996; Tasker & Dudek, 1993) and PVN parvocellular neurones express GABAA receptors (Barrett-Jolley, 2001) and are inhibited by addition of bath applied GABA (Figure 1C). However, surprisingly, we could detect no acute modulation of GABAA currents at either the whole-cell or the single-channel level. Furthermore, cortisol modulation of GABAA currents in the mammalian brain has never been shown. The study by Andres-Trelles et al. (1989) that found modulation of guinea-pig ileum GABAA currents by cortisol failed to show any similar modulation of GABAA currents in the cuneate nucleus. It is possible that glucocorticoids are only active on certain subtypes of the GABAA receptor. There are no published data specifically on GABA subtypes of parvocellular PVN neurones, however, the PVN as a whole is rich in the GABAA α2-subunit (Cullinan, 2000; Fenelon et al., 1995; Fenelon & Herbison, 1995; 1996; Fritschy & Mohler, 1995), which is necessary for the potentiatory actions of allopregnanolone (Maitra & Reynolds, 1999). Less is known about the possible PVN distribution of the important γ3-subunit (Maitra & Reynolds, 1999). On the other hand, GABAA ‘antagonism' by pregnanolone sulphate appears to involve the TM2 domain of the GABAA α1-subunit (Akk et al., 2001), which is less prevalent in the PVN (Cullinan, 2000; Fenelon et al., 1995; Fenelon & Herbison, 1995; 1996; Fritschy & Mohler, 1995).
Interaction with potassium currents
In a number of studies where the neuromodulatory effects of steroids have been shown not to involve GABA receptors, the primary site of action has been potassium ion channels (Chen et al., 1991). We therefore chose to investigate the possibility that cortisol could alter parvocellular neurone action potential firing frequency via the modulation of potassium channels.
We found that cortisol induced a rightward shift in the potassium conductance voltage curve. This represents potassium current inhibition, through a weakening of voltage sensitivity, rather than by block of the channel pore itself (Hille, 1984a). This response is the opposite to that elicited by the structurally related steroid, pregnenolone sulphate (on Kv 1.1 channels, 10–100 μM; Wang et al., 1998), or the cortisol induced hyperpolarisation observed by Chen et al. (1991), but consistent with the observed increase in action current frequency induced by cortisol (Hille, 1984b). Without some means to block the cortisol action, it is not possible to confirm whether this potassium current modulation causes the action–current frequency increase. To further investigate the potassium current modulation, we tested cortisol on a cell line (mouse erythroleukemia, MEL) expressing both the GC intracellular receptor (GR: Golde et al., 1979; Tsiftsoglou et al., 1986) and recombinant human potassium channels (Shelton et al., 1993). The inhibition of hKv 1.3 by cortisol largely mimicked the effect observed in native tissue. Strikingly, again, the cortisol effect was not blocked by a saturating concentration of mifepristone, despite the MEL cell GC receptor clearly being mifepristone sensitive (Mayeux et al., 1985). hKv 1.3 is widely distributed throughout the CNS (Coetzee et al., 1999), however, and so it seems likely that this effect too, may be quite widespread, rather than limited to just parvocellular neurones. The lack of interaction between cortisol and hKv 1.1 does, however, imply some specificity to the interaction. Although a specific functional role for hKv 1.3 is not known (Coetzee et al., 1999), its somewhat transient nature suggests that it may be involved with control of neuronal firing frequency (Hille, 1984b).
Mechanism and physiological rôles
GCs exert both rapid and delayed feedback actions in the hypothalamus (Buckingham, 1996). The primary mechanism of action of cortisol during the delayed phase being through interaction with the GC intracellular receptor and transcription of lipocortin (Buckingham, 1996). Much less is known about the mechanisms of cortisol rapid action, although Croxtall et al. (2000) recently showed additional rapid actions of GCs mediated by the same intracellular receptor, but independent of lipocortin transcription. Some other rapid actions of GCs, such as those reported here, are however, independent of the GC intracellular receptor, GR (or, at least, mifepristone insensitive, e.g., Venero & Borrell, 1999). Furthermore, a membrane (non-genomic) GC receptor recently isolated from amphibian brain (‘mGR' Evans et al., 2000) is quite distinct from the intracellular receptor, having both a low affinity for dexamethasone and an insensitivity to mifepristone (Orchinik et al., 1991). Whilst we would not expect mGR to be expressed in the erythroleukemia derived MEL cell line, no studies have yet determined this neuronal receptor's distribution across other species or in other tissues. Another, perhaps more likely explanation of cortisol's mechanism of action observed in the several systems studied here is through direct interaction with potassium channels at the lipid-ion channel interface, although this is a difficult hypothesis to examine explicitly.
The concentration range we studied (0.1–10 μM) comfortably overlaps the physiological norm. Glucocorticoids, such as corticosterone (mostly in rats) and cortisol (mostly in humans) would normally be maintained in the range of approximately 0.1–0.45 μM (free plasma concentration, Bowman & Rand, 1980; Ekman et al., 1961) and follow a circadian rhythm. Concentrations of most GCs in the brain will then follow concentrations in the plasma, although brain cortisol levels can be up to 30× that of the plasma (cats: Henkin et al., 1968). We found cortisol to be active at 1 μM, a concentration exceeded during high stress (e.g., in rats 3 μM: Bassett et al., 1978), trauma (patients with multiple trauma >1 μM: Vermes et al., 1995), disease (e.g., Cushing's syndrome >1 μM free cortisol (54 μg/100 ml): Ekman et al., 1961) and medication (de vroede et al., 1998; Salem et al., 1994), but possibly above that required for feedback inhibition of the HPA-axis itself (Buckingham, 1979b,c; Buckingham & Hodges, 1977). Parvocellular PVN neurones also synapse with sympathetic preganglionic neurones, and are believed to be important for regulation of cardiovascular control (Badoer, 2001; Coote, 1995; Pyner & Coote, 2000). It is therefore possible that interaction between glucocorticoids and parvocellular neurone Kv channels could partly explain the increase in blood pressure induced by central administration of cortisol (Scheuer & Bechtold, 2001; Takahashi et al., 1983). Furthermore, a more widespread interaction between glucocorticoids and neuronal potassium channels could contribute to many of the psychological effects of hypercortisolism, such as confusion and memory loss (Gustafson et al., 1993; Newcomer et al., 1999).
Acknowledgments
This work was funded by a British Heart Foundation Fellowship awarded to R. Barrett-Jolley. We would also like to express our thanks to Professor John Coote and Dr Caroline Dart for their invaluable help during this work.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- ECl−
equilibrium potential for chloride ions
- GABA
γ-aminobutyric acid
- GC
glucocorticoid
- GR
glucocorticoid intracellular receptor
- hKv
human voltage-gated potassium channel
- Kv
voltage-gated potassium channel
- MEL
mouse erythroleukemia
- mGR
membrane glucocorticoid receptor
- Mif
mifepristone
- PK
potassium ion permeability
- PNa
sodium ion permeability
- PVN
paraventricular nucleus
- Vrev
reversal potential
References
- AGNATI L.F., FUXE K., YU Z.Y., HARFSTRAND A., OKRET S., WIKSTROM A.C., GOLDSTEIN M., ZOLI M., VALE W., GUSTAFSSON J.A. Morphometrical analysis of the distribution of corticotrophin releasing factor, glucocorticoid receptor and phenylethanolamine-N-methyltransferase immunoreactive structures in the paraventricular hypothalamic nucleus of the rat. Neurosci. Lett. 1985;54:147–152. doi: 10.1016/s0304-3940(85)80070-7. [DOI] [PubMed] [Google Scholar]
- AKK G., BRACAMONTES J., STEINBACH J.H. Pregnenolone sulfate block of GABA(A) receptors: mechanism and involvement of a residue in the M2 region of the alpha subunit. J. Physiol. 2001;532:673–684. doi: 10.1111/j.1469-7793.2001.0673e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANDRES-TRELLES F., BIBBY V., LUSTMAN S., SIMMONDS M.A. Effects of cortisol on GABAA receptor-mediated responses compared in guinea-pig ileum and rat cuneate nucleus. Neuropharmacology. 1989;28:705–708. doi: 10.1016/0028-3908(89)90154-8. [DOI] [PubMed] [Google Scholar]
- ANDREW R.D. Isoperiodic bursting by magnocellular neuroendocrine cells in the rat hypothalamic slice. J. Physiol. (Lond). 1987;384:467–477. doi: 10.1113/jphysiol.1987.sp016464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARIYOSHI M., AKASU T. Glucocorticoid modulates the sensitivity of the GABAA receptor on primary afferent neurons of bullfrogs. Brain Research. 1986;367:332–336. doi: 10.1016/0006-8993(86)91613-6. [DOI] [PubMed] [Google Scholar]
- AVANZINO G.L., ERMIRIO R., COGO C.E., RUGGERI P., MOLINARI C. Effects of corticosterone on neurones of the locus coeruleus, in the rat. Neurosci. Lett. 1987;80:85–88. doi: 10.1016/0304-3940(87)90500-3. [DOI] [PubMed] [Google Scholar]
- AVANZINO G.L., ERMIRIO R., RUGGERI P., COGO C.E. Effect of microelectrophoretically applied corticosterone on raphe neurones in the rat. Neurosci. Lett. 1984;50:307–311. doi: 10.1016/0304-3940(84)90504-4. [DOI] [PubMed] [Google Scholar]
- BADOER E. Hypothalamic paraventricular nucleus and cardiovascular regulation. Clin. Exp. Pharmacol. Physiol. 2001;28:95–99. doi: 10.1046/j.1440-1681.2001.03413.x. [DOI] [PubMed] [Google Scholar]
- BAINS J.S., FERGUSON A.V. Paraventricular nucleus neurons projecting to the spinal cord receive excitatory input from the subfornical organ. Am. J Physiol. 1995;268:R625–R633. doi: 10.1152/ajpregu.1995.268.3.R625. [DOI] [PubMed] [Google Scholar]
- BARRETT-JOLLEY R. Nipecotic acid directly activates GABA(A)-like ion channels. Br. J. Pharmacol. 2001;133:673–678. doi: 10.1038/sj.bjp.0704128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRETT-JOLLEY R., DAVIES N.W. Kinetic analysis of the inhibitory effect of glibenclamide on KATP channels of mammalian skeletal muscle. J. Memb. Biol. 1997;155:257–262. doi: 10.1007/s002329900178. [DOI] [PubMed] [Google Scholar]
- BARRETT-JOLLEY R., PYNER S., COOTE J.H. Measurement of voltage-gated potassium currents in identified spinally-projecting sympathetic neurones of the paraventricular nucleus. J. Neurosci. Met. 2000;102:25–33. doi: 10.1016/s0165-0270(00)00271-5. [DOI] [PubMed] [Google Scholar]
- BASSETT J.R., STRAND F.L., CAIRNCROSS K.D. Glucocorticoids, adrenocorticotropichormone and related polypeptides on myocardial sensitivity to noradrenaline. Eur. J. Pharmacol. 1978;49:243–249. doi: 10.1016/0014-2999(78)90099-7. [DOI] [PubMed] [Google Scholar]
- BOWMAN W.C., RAND M.J. Textbook of Pharmacology 1980Oxford, UK: Blackwell Scientific Publishing; 2nd edn [Google Scholar]
- BROWN D.A. G-proteins and potassium currents in neurons. Annu. Rev. Physiol. 1990;52:215–242. doi: 10.1146/annurev.ph.52.030190.001243. [DOI] [PubMed] [Google Scholar]
- BUCKINGHAM J.C. Corticotrophin releasing factor. Pharmacol Rev. 1979a;31:253–275. [PubMed] [Google Scholar]
- BUCKINGHAM J.C. Influence of various steroids on the production in vitro of corticotrophin releasing hormone and corticotrophin. J. Endocrinol. 1979b;83:38P–39P. [PubMed] [Google Scholar]
- BUCKINGHAM J.C. The influence of corticosteroids on the secretion of corticotrophin and its hypothalamic releasing hormone. J. Physiol. 1979c;286:331–342. doi: 10.1113/jphysiol.1979.sp012622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUCKINGHAM J.C. Fifteenth Gaddum Memorial Lecture December 1994. Stress and the neuroendocrine-immune axis: the pivotal role of glucocorticoids and lipocortin 1. Br. J. Pharmacol. 1996;118:1–19. doi: 10.1111/j.1476-5381.1996.tb15360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUCKINGHAM J.C., HODGES J.R. Production of corticotrophin releasing hormone by the isolated hypothalamus of the rat. J. Physiol. 1977;272:469–479. doi: 10.1113/jphysiol.1977.sp012054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Y.Z., HUA S.Y., WANG C.A., WU L.G., GU Q., XING B.R. An electrophysiological study on the membrane receptor-mediated action of glucocorticoids in mammalian neurons. Neuroendocrinol. 1991;53 Suppl:30. doi: 10.1159/000125791. [DOI] [PubMed] [Google Scholar]
- COETZEE W.A., AMARILLO Y., CHIU J., CHOW A., LAU D., MCCORMACK T., MORENO H., NADAL M.S., OZAITA A., POUNTNEY D., SAGANICH M., VEGASAENZ D.M., RUDY B. Molecular diversity of K+ channels. Ann. N.Y. Acad. Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- COOTE J.H. Cardiovascular function of the paraventricular nucleus of the hypothalamus. Biol. Signals. 1995;4:142–149. doi: 10.1159/000109434. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., FLOWER R.J. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 2000;130:289–298. doi: 10.1038/sj.bjp.0703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CULLINAN W.E. GABA(A) receptor subunit expression within hypophysiotropic CRH neurons: a dual hybridization histochemical study. J. Comp. Neurol. 2000;419:344–351. doi: 10.1002/(sici)1096-9861(20000410)419:3<344::aid-cne6>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- DAVIES N.W. A suite of programs for acquisition and analysis of voltage-clamp and patch-clamp data developed using the axobasic library. J. Physiol. 1993;111:459. [Google Scholar]
- DE VROEDE, M., BEUKERING R., SPIT M., JANSEN M. Rectal hydrocortisone during stress in patients with adrenal insufficiency. Arch. Dis. Child. 1998;78:544–547. doi: 10.1136/adc.78.6.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EKMAN H., HAKANSSON B., MCCARTHY J.D., LEHMANN J., SJOGREN B. Plasma 17-hydroxycorticosteroids in Cushing's syndrome. J. Clin. Endocrinol. Metab. 1961;21:684–694. doi: 10.1210/jcem-21-6-684. [DOI] [PubMed] [Google Scholar]
- EVANS S.J., MURRAY T.F., MOORE F.L. Partial purification and biochemical characterization of a membrane glucocorticoid receptor from an amphibian brain. J. Steroid Biochem. Mol. Biol. 2000;72:209–221. doi: 10.1016/s0960-0760(00)00031-5. [DOI] [PubMed] [Google Scholar]
- FALKENSTEIN E., TILLMANN H.C., CHRIST M., FEURING M., WEHLING M. Multiple actions of steroid hormones - A focus on rapid, nongenomic effects. Pharmacol. Rev. 2000;52:513–555. [PubMed] [Google Scholar]
- FELDMAN S., DAFNY N. Changes in single cell responsiveness in the hypothalamus in cats following cortisol administration. Brain Res. 1970;20:369–377. doi: 10.1016/0006-8993(70)90167-8. [DOI] [PubMed] [Google Scholar]
- FENELON V.S., HERBISON A.E. Characterisation of GABAA receptor gamma subunit expression by magnocellular neurones in rat hypothalamus. Brain Res. Mol. Brain Res. 1995;34:45–56. doi: 10.1016/0169-328x(95)00130-k. [DOI] [PubMed] [Google Scholar]
- FENELON V.S., HERBISON A.E. Plasticity in GABAA receptor subunit mRNA expression by hypothalamic magnocellular neurons in the adult rat. J. Neurosci. 1996;16:4872–4880. doi: 10.1523/JNEUROSCI.16-16-04872.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FENELON V.S., SIEGHART W., HERBISON A.E. Cellular localization and differential distribution of GABAA receptor subunit proteins and messenger RNAs within hypothalamic magnocellular neurons. Neurosci. 1995;64:1129–1143. doi: 10.1016/0306-4522(94)00402-q. [DOI] [PubMed] [Google Scholar]
- FENWICK E.M., MARTY A., NEHER E. A patch-clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. J. Physiol. 1982;331:577–597. doi: 10.1113/jphysiol.1982.sp014393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILARETOV A.A. The afferent input and functional organization of the hypothalamus in reactions regulating pituitary-adreno-cortical activity. Brain Res. 1976;107:39–54. doi: 10.1016/0006-8993(76)90094-9. [DOI] [PubMed] [Google Scholar]
- FRITSCHY J.M., MOHLER H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- FUXE K., HARFSTRAND A., AGNATI L.F., YU Z.Y., CINTRA A., WIKSTROM A.C., OKRET S., CANTONI E., GUSTAFSSON J.A. Immunocytochemical studies on the localization of glucocorticoid receptor immunoreactive nerve cells in the lower brain stem and spinal cord of the male rat using a monoclonal antibody against rat liver glucocorticoid receptor. Neurosci. Lett. 1985a;60:1–6. doi: 10.1016/0304-3940(85)90372-6. [DOI] [PubMed] [Google Scholar]
- FUXE K., WIKSTROM A.C., OKRET S., AGNATI L.F., HARFSTRAND A., YU Z.Y., GRANHOLM L., ZOLI M., VALE W., GUSTAFSSON J.A. Mapping of glucocorticoid receptor immunoreactive neurons in the rat tel- and diencephalon using a monoclonal antibody against rat liver glucocorticoid receptor. Endocrinol. 1985b;117:1803–1812. doi: 10.1210/endo-117-5-1803. [DOI] [PubMed] [Google Scholar]
- GAGNE D., PONS M., PHILIBERT D. RU 38486: a potent antiglucocorticoid in vitro and in vivo. J. Steroid Biochem. 1985;23:247–251. doi: 10.1016/0022-4731(85)90401-7. [DOI] [PubMed] [Google Scholar]
- GOLDE D.W., BERSCH N., LIPPMAN M.E., FRIEND C. Detection of glucocorticoid receptors on Friend erythroleukemia cells. Proc. Natl. Acad. Sci. U.S.A. 1979;76:3515–3517. doi: 10.1073/pnas.76.7.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUSTAFSON Y., OLSSON T., ASPLUND K., HAGG E. Acute confusional state (delirium) soon after stroke is associated with hypercortisolism. Cerebrovasc. Dis. 1993;3:33–38. [Google Scholar]
- HENKIN R.I., CASPER A.G., BROWN R., HARLAN A.B., BARTTER F.C. Presence of corticosterone and cortisol in the central and peripheral nervous system of the cat. Endocrinol. 1968;82:1058–1061. doi: 10.1210/endo-82-5-1058. [DOI] [PubMed] [Google Scholar]
- HERMES M.L., CODERRE E.M., BUIJS R.M., RENAUD L.P. GABA and glutamate mediate rapid neurotransmission from suprachiasmatic nucleus to hypothalamic paraventricular nucleus in rat. J. Physiol. 1996;496:749–757. doi: 10.1113/jphysiol.1996.sp021724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILLE B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates Inc; 1984a. Classic Biophysics of the Squid Giant Axon; pp. 23–57. [Google Scholar]
- HILLE B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates Inc; 1984b. Potassium Channels and Chloride Channels; pp. 99–116. [Google Scholar]
- HILLE B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates Inc; 1984c. Selective Permeability:Independence; pp. 226–248. [Google Scholar]
- KASAI M., KANNAN H., UETA Y., OSAKA T., INENAGA K., YAMASHITA H. Effects of iontophoretically applied cortisol on tuberoinfundibular neurons in hypothalamic paraventricular nucleus of anesthetized rats. Neurosci. Lett. 1988;87:35–40. doi: 10.1016/0304-3940(88)90141-3. [DOI] [PubMed] [Google Scholar]
- KASAI M., YAMASHITA H. Cortisol suppresses noradrenaline-induced excitatory responses of neurons in the paraventricular nucleus; an in vitro study. Neurosci. Lett. 1988a;91:65–70. doi: 10.1016/0304-3940(88)90250-9. [DOI] [PubMed] [Google Scholar]
- KASAI M., YAMASHITA H. Inhibition by cortisol of neurons in the paraventricular nucleus of the hypothalamus in adrenalectomized rats; an in vitro study. Neurosci. Lett. 1988b;91:59–64. doi: 10.1016/0304-3940(88)90249-2. [DOI] [PubMed] [Google Scholar]
- MAITRA R., REYNOLDS J.N. Subunit dependent modulation of GABAA receptor function by neuroactive steroids. Brain Res. 1999;819:75–82. doi: 10.1016/s0006-8993(98)01316-x. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA M.D. Antagonist-type interaction of glucocorticoids with the GABA receptor- coupled chloride channel. Brain Res. 1987;418:377–382. doi: 10.1016/0006-8993(87)90107-7. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA M.D., BISSERBE J.C., ESKAY R.L. Glucocorticoids are modulators of GABAA receptors in brain. Brain Res. 1985;339:178–182. doi: 10.1016/0006-8993(85)90641-9. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA M.D., HARRISON N.L., SCHWARTZ R.D., BARKER J.L., PAUL S.M. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science. 1986;232:1004–1007. doi: 10.1126/science.2422758. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA M.D., MIENVILLE J.M., VICINI S. Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci. Lett. 1988;90:279–284. doi: 10.1016/0304-3940(88)90202-9. [DOI] [PubMed] [Google Scholar]
- MAYEUX P., FELIX J.M., BILLAT C., JACQUOT R. Effect of the antiglucocorticoid agent RU 38486 on the dexamethasone inhibition of Friend cell differentiation. Biochim. Biophys. Acta. 1985;846:413–417. doi: 10.1016/0167-4889(85)90014-x. [DOI] [PubMed] [Google Scholar]
- MIYAKE M., CHRISTIE M.J., NORTH R.A. Single potassium channels opened by opioids in rat locus ceruleus neurons. Proc. Natl. Acad. Sci. U.S.A. 1989;86:3419–3422. doi: 10.1073/pnas.86.9.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOGUILEWSKY M., PHILIBERT D. RU 38486: potent antiglucocorticoid activity correlated with strong binding to the cytosolic glucocorticoid receptor followed by an impaired activation. J. Steroid Biochem. 1984;20:271–276. doi: 10.1016/0022-4731(84)90216-4. [DOI] [PubMed] [Google Scholar]
- NEWCOMER J.W., SELKE G., MELSON A.K., HERSHEY T., CRAFT S., RICHARDS K., ALDERSON A.L. Decreased memory performance in healthy humans induced by stress-level cortisol treatment. Arch. Gen. Psychiatry. 1999;56:527–533. doi: 10.1001/archpsyc.56.6.527. [DOI] [PubMed] [Google Scholar]
- NORTH R.A. Twelfth Gaddum memorial lecture. Drug receptors and the inhibition of nerve cells. Br. J. Pharmacol. 1989;98:13–28. doi: 10.1111/j.1476-5381.1989.tb16855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ORCHINIK M., MURRAY T.F., MOORE F.L. A corticosteroid receptor in neuronal membranes. Science. 1991;252:1848–1851. doi: 10.1126/science.2063198. [DOI] [PubMed] [Google Scholar]
- PAUL S.M., PURDY R.H. Neuroactive steroids. FASEB J. 1992;6:2311–2322. [PubMed] [Google Scholar]
- PENNINGTON A.J., KELLY J.S., ANTONI F.A. Selective enhancement of an A type potassium current by dexamethasone in a corticotroph cell line. J. Neuroendocrinol. 1994;6:305–315. doi: 10.1111/j.1365-2826.1994.tb00587.x. [DOI] [PubMed] [Google Scholar]
- PYNER S., COOTE J.H. Identification of branching paraventricular neurons of the hypothalamus that project to the rostroventrolateral medulla and spinal cord. Neurosci. 2000;100:549–556. doi: 10.1016/s0306-4522(00)00283-9. [DOI] [PubMed] [Google Scholar]
- RUPPERSBERG J.P., STOCKER M., PONGS O., HEINEMANN S.H., FRANK R., KOENEN M. Regulation of fast inactivation of cloned mammalian Ik(A) channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- SALEM M., TAINSH JR., BROMBERG J., LORIAUX D.L., CHERNOW B. Perioperative glucocorticoid coverage. A reassessment 42 years after emergence of a problem. Ann. Surg. 1994;219:416–425. doi: 10.1097/00000658-199404000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAPHIER D., FELDMAN S. Iontophoretic Application of Glucocorticoids Inhibits Identified Neurons in the Rat Paraventricular Nucleus. Brain Res. 1988;453:183–190. doi: 10.1016/0006-8993(88)90157-6. [DOI] [PubMed] [Google Scholar]
- SAWCHENKO P.E., BROWN E.R., CHAN R.K., ERICSSON A., LI H.Y., ROLAND B.L., KOVACS K.J. The paraventricular nucleus of the hypothalamus and the functional neuroanatomy of visceromotor responses to stress. Prog. Brain Res. 1996;107:201–222. doi: 10.1016/s0079-6123(08)61866-x. [DOI] [PubMed] [Google Scholar]
- SCHEUER D.A., BECHTOLD A.G. Glucocorticoids potentiate central actions of angiotensin to increase arterial pressure. Am. J. Physiol Regul. Integr. Comp Physiol. 2001;280:R1719–R1726. doi: 10.1152/ajpregu.2001.280.6.R1719. [DOI] [PubMed] [Google Scholar]
- SHELTON P.A., DAVIES N.W., ANTONIOU M., GROSVELD F., NEEDHAM M., HOLLIS M., BRAMMAR W.J., CONLEY E.C. Regulated expression of K+ channel genes in electrically silent mammalian cells by linkage to beta-globin gene-activation elements. Receptors. Channels. 1993;1:25–37. [PubMed] [Google Scholar]
- SIGWORTH F.J., SINE S.M. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys. J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STANFIELD P.R., NAKAJIMA Y., YAMAGUCHI K. Substance P raises neuronal membrane excitability by reducing inward rectification. Nature. 1985;315:498–501. doi: 10.1038/315498a0. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI H., TAKEDA K., ASHIZAWA H., INOUE A., YONEDA S., YOSHIMURA M., IJICHI H. Centrally induced cardiovascular and sympathetic responses to hydrocortisone in rats. Am. J. Physiol. 1983;245:H1013–H1018. doi: 10.1152/ajpheart.1983.245.6.H1013. [DOI] [PubMed] [Google Scholar]
- TASKER J.G., DUDEK F.E. Local inhibitory synaptic inputs to neurones of the paraventricular nucleus in slices of rat hypothalamus. J. Physiol. 1993;469:179–192. doi: 10.1113/jphysiol.1993.sp019810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSIFTSOGLOU A., HOUSMAN D., WONG W. The inhibition of commitment of mouse erythroleukemia cells by steroids involves a glucocorticoid-receptor mediated process(es) acting at the nuclear level. Biochim. Biophys. Acta. 1986;889:251–261. doi: 10.1016/0167-4889(86)90111-4. [DOI] [PubMed] [Google Scholar]
- VENERO C., BORRELL J. Rapid glucocorticoid effects on excitatory amino acid levels in the hippocampus: a microdialysis study in freely moving rats. Eur. J. Neurosci. 1999;11:2465–2473. doi: 10.1046/j.1460-9568.1999.00668.x. [DOI] [PubMed] [Google Scholar]
- VERMES I., BEISHUIZEN A., HAMPSINK R.M., HAANEN C. Dissociation of plasma adrenocorticotropin and cortisol levels in critically ill patients: possible role of endothelin and atrial natriuretic hormone. J. Clin. Endocrinol. Metab. 1995;80:1238–1242. doi: 10.1210/jcem.80.4.7714094. [DOI] [PubMed] [Google Scholar]
- WANG L., FENG Z.P., DUFF H.J. Glucocorticoid regulation of cardiac K+ currents and L-type Ca2+ current in neonatal mice. Circ. Res. 1999;85:168–173. doi: 10.1161/01.res.85.2.168. [DOI] [PubMed] [Google Scholar]
- WANG Q., WANG L., WARDWELL-SWANSON J. Modulation of cloned human neuronal voltage-gated potassium channels (hKv1.1 and hKv2.1) by neurosteroids. Pflugers Arch. 1998;437:49–55. doi: 10.1007/s004240050745. [DOI] [PubMed] [Google Scholar]
- WETZEL C.H., VEDDER H., HOLSBOER F., ZIEGLGANSBERGER W., DEISZ R.A. Bidirectional effects of the neuroactive steroid tetrahydrodeoxycorticosterone on GABA-activated Cl- currents in cultured rat hypothalamic neurons. Br. J. Pharmacol. 1999;127:863–868. doi: 10.1038/sj.bjp.0702597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLTERS H., WALLINGA W., YPEY D.L., BOOM H.B. Ionic currents during action potentials in mammalian skeletal muscle fibers analyzed with loose patch clamp. Am. J. Physiol. 1994;267:C1699–C1706. doi: 10.1152/ajpcell.1994.267.6.C1699. [DOI] [PubMed] [Google Scholar]