

Abstract
The ATP-sensitive potassium channel (KATP) of pancreatic β-cells is composed of the sulphonylurea-binding protein, SUR1, and the inwardly rectifying K+ channel subunit, Kir6.2. We have characterized two novel isoforms of rat SUR1 in the RINm5F insulin-secreting cell line.
SUR1A2 is an allelic variant with a single amino acid change in the first nucleotide-binding domain. Coinjection of SUR1A2 plus Kir6.2 into Xenopus oocytes or expression of a SUR1A2–Kir6.2 tandem in HEK-293 cells yielded large currents with characteristics similar to the wild-type KATP channel.
SUR1BΔ31, detected in several human tissues, is a splice variant of the rat SUR1 gene that lacks exon 31 of the corresponding human SUR1 gene. SUR1BΔ31 lacks the TM16–TM17 transmembrane-spanning helices leading to a protein with a different transmembrane topology. Coinjection of SUR1BΔ31 plus Kir6.2 into Xenopus oocytes or expression of the Kir6.2/SUR1BΔ31 tandem construct in HEK-293 cells did not result in any current, and a surface expression assay indicated that this channel does not reach the plasma membrane.
SUR1A2 and SUR1A1 proteins expressed in HEK-293 cells display similar binding affinities for [3H]-glibenclamide, while SUR1BΔ31 shows a 500-fold lower affinity.
These findings confirm that TM16–TM17 of SUR1 are important for high-affinity glibenclamide binding and that their deletion impairs trafficking of the KATP channel to the surface membrane.
Keywords: KATP channel, sulphonylurea receptor variants, sulphonylurea binding sites, glibenclamide, SUR-Kir trafficking
Introduction
Type 2 diabetes mellitus, a common heterogeneous syndrome, is characterized by both impaired β-cell function and impaired insulin action. Because abnormal insulin secretion plays a major role in this disease, it is important to analyse the genes encoding the key components of the pancreatic β-cell stimulus–secretion coupling pathway. One such protein is the ATP-sensitive potassium (KATP) channel which links the metabolic activity of the pancreatic β-cell to its membrane potential (Ashcroft & Gribble, 1999). This is achieved by metabolically induced changes in intracellular nucleotides (ATP and ADP), which regulate KATP channel activity. Closure of the channel in response to glucose-stimulated changes in ATP and ADP levels stimulates insulin secretion. In addition, KATP channels are the targets for a number of clinically important drugs. The hypoglycaemic sulphonylurea drugs, such as glibenclamide or tolbutamide, bind to KATP channels in the plasma membrane of pancreatic β-cells inducing channel closure and insulin release. They form the mainstay of pharmacological therapy for type 2 diabetes (Ashcroft & Ashcroft, 1992; Ashcroft et al., 1993). A chemically diverse group of drugs known as potassium channel openers (KCOs), like diazoxide, also interact directly with the KATP channel. Those elicit channel opening and thereby act to inhibit insulin release (Sturgess et al., 1988).
At the molecular level, the β-cell KATP channel is an octameric complex of two structurally unrelated types of subunit: the sulphonylurea receptor (SUR1) (Aguilar-Bryan et al., 1995), which is a member of the ATP binding cassette (ABC) family of proteins, and a pore-forming subunit (Kir6.2) (Sakura et al., 1995), which belongs to the inward-rectifier K+ channel family (Reimann & Ashcroft, 1999). There is good evidence that the β-cell KATP channel is a hetero-octamer containing four molecules each of Kir6.2 and SUR1 (Clement et al., 1997; Inagaki et al., 1995). Hydropathy analysis and epitope labelling suggests that SUR1 has 17 transmembrane domains (TMs) (Conti et al., 2001). These may be grouped into three sets (TMD 0,1,2), consisting of 5, 6 and 6 TMs, respectively (Tusnady et al., 1997). Like other ABC transporters, SUR has two large cytosolic loops referred to as nucleotide binding domains (NBD1 and NBD2) that contain consensus sequences for Mg-nucleotide binding and hydrolysis (Gribble et al., 1997b, 1998; Matsuo et al., 2000). Interaction of nucleotides with the NBDs stimulates channel activity. The sulphonylurea receptor SUR1 contains the binding sites for sulphonylureas and KCOs and endows the KATP channel with sensitivity to both groups of drugs (Schwanstecher et al., 1998; D'hahan et al., 1999; Ashfield et al., 1999). Kir6.2 has no obvious consensus site for nucleotide binding; nevertheless, it is now clear that Kir6.2 forms the KATP channel pore and contains the site at which ATP mediates channel inhibition (Tucker et al., 1997, 1998). Moreover, Kir6.2 also has a low-affinity binding site for glibenclamide (Gros et al., 1999).
Given the essential role of SUR in regulated insulin secretion, sequence variations in its gene could play a role in susceptibility to impaired insulin secretion. Consistent with this concept, altered regulation of insulin secretion was found in SUR1 knockout mice (Seghers et al., 2000). Furthermore, several studies have shown that mutations in the SUR1 gene result in loss of functional KATP channels in pancreatic β-cells, and thereby produce congenital hyperinsulinaemia of infancy (CHI), a disease characterized by excessive insulin secretion (Dunne et al., 1997; Otonkoski et al., 1999).
Using the polymerase chain reaction (PCR), we analysed the SUR1 gene transcripts present in the RINm5F insulin secreting cell line and found two novel forms of the rat sulphonylurea receptor. SUR1A2 is an allelic variant characterized by a single amino acid substitution in NBD1 (Thr699Ile). It displays similar functional characteristics to the original SUR1 isoform. SUR1BΔ31 is a splice variant lacking TM16 and TM17, which impairs the sensitivity of the KATP channel to glibenclamide and trafficking of SUR1–Kir6.2 to the surface membrane.
Methods
Isolation of total RNA, synthesis of cDNA and plasmid DNAs constructions
Total RNAs from RINm5F and HEK-293 cells were prepared by using Trizol reagent (Gibco BRL) and were treated for 10 min at 37°C with RNase-free DNase I (BRL). DNase I was inactivated by heating the samples for 10 min at 65°C and first strand cDNAs were synthesized using Superscript II RNAase H-reverse transcriptase (BRL) with 1 μg total RNA primed by oligo(dT). PCR was performed with Advantage cDNA polymerase mix (Clontech Laboratories, Inc, Palo Alto, CA, U.S.A.). Three fragments of RINm5F SUR cDNA were obtained using the following oligonucleotides: 5′-CGC GCC ACC ATG CCT TTG GCC TTC TGC G-3′(sense) and 5′-TGA TGC CCC GGA GCA TCT CAT TGG TCT GC-3′ (antisense), 5′-TGG CCA CCA AGC TGT CCC AGG CAC AGC G-3′ (sense) and 5′-TCC TTC TCC AGC TCT TGG TCC T-3′ (antisense), 5′-GAC TCA GAT TGG GGA ACG AGG-3′ (sense) and 5′-TGA GGT GTG GGG TGG CAC TTT GGC GCT GG-3′ (antisense). PCR amplification was carried out using a hot start protocol in a final volume of 50 μl containing 5 ul of each cDNA, 50 pmole of each primer, 1.5 mM MgCl2 and 2.5 U of Taq polymerase (Eurobio, Paris, France) in a Stratagene Thermal Robocycler Gradient 96 with the following protocol: 3 min at 94°C followed by 40 cycles (94°C for 30 s, 60°C for 30 s, 68°C for 5 min) and a final elongation period of 10 min at 72°C. PCR products were purified on 1% agarose gel, sequenced and subcloned with Original TA Cloning Kit (Invitrogen, San Diego, CA, U.S.A.). The three fragments were subsequently cut with the appropriate restriction enzymes, ligated to generate the full SUR cDNAs and cloned as an XbaI/EcoRI fragment into the pcDNA3.1 Zeo(−) vector (Invitrogen, Carlsbad, CA, U.S.A.). Kir6.2 cDNA was obtained and cloned into pcDNA3.1 Zeo(−) vector as previously described (Gros et al., 1999). SUR1A2–Kir6.2 and SUR1BΔ31–Kir6.2 linked in tandem were also cloned into pcDNA3.1 Zeo(−) vector. To link SUR1 and Kir6.2 in a head-to-tail fashion, the nucleotide sequence encoding a stretch of six glycines residues was introduced between the 3′-end of SUR1 cDNA and the 5′-end of mouse Kir6.2 cDNA (Inagaki et al., 1997). As a result, the intervening amino acid sequence (G)6 is present between the SUR1 and Kir6.2 subunits.
PCR analysis
Multiple tissue cDNA panels from different human tissues (Clontech, U.S.A.) are sets of normalized, first-strand cDNA-generated using Clontech's premium RNA. cDNA from the HEK-293 cell line was obtained from total RNAs as previously described. PCR analysis of SUR1A1/2 and SUR1BΔ31 expression in various tissues and cell lines was performed using primers flanking exon 31 of rat: 5′-ACC CCT CGC CGT CGT GTG CTA CTT C-3′ (sense) and 5′-TTT TCA GGA GTG TGT GGA TAC GCT T-3′ (antisense) or human SUR1: 5′-GCC CCT GGC CAT CGT GTG CTA CTT C-3′ (sense) and 5′-TTT TCA GGA GCC CAT GGA TGC GCT T-3′ (antisense). PCR amplification was carried out using a hot start protocol in a final volume of 50 μl containing 5 ul of each cDNA, 50 pmole of each primer, 1.5 mM MgCl2 and 2.5 U of Taq polymerase (Eurobio, Paris, France) in a Stratagene Thermal Robocycler Gradient 96 with the following protocol: 3 min at 94°C followed by 35 cycles for human tissues or 25 cycles for transfected HEK-293 cells (94°C for 30 s, 64°C for 30 s, 72°C for 1 min) and a final elongation period of 10 min at 72°C. The quality of cDNAs of each of the tissues and cells was controlled following the same PCR protocol for 25 cycles with human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers provided with the Clontech's multiple tissue cDNA panels: 5′-TGA AGG TCG GAG TCA ACG GAT TTG GT-3′ (sense) and 5′-CAT GTG GGC CAT GAG GTC CAC CAC-3′ (antisense). PCR products were electrophoresed on 2% agarose gel.
Electrophysiological experiments
HEK-293 cells were transfected with SUR1A2–Kir6.2 or SUR1BΔ31–Kir6.2 tandems together with pEGFP (Clontech, Basingstoke, U.K.), using the Lipofectamine plus system (Life Technologies, Paisley, U.K.). Conventional whole-cell patch clamp recordings were performed on fluorescent cells using an EPC-7 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany). Electrodes were pulled from borosilicate glass (GC 150 TF; Clark Electromedical Instruments, Pangbourne, U.K.). The external solution contained (mM): NaCl 138, KCl 5.6, MgCl2 1.2, CaCl2 2.6, HEPES 10 (pH 7.4). The pipette solution contained (mM): KCl 107, NaCl 10, MgCl2 2, CaCl2 1, EGTA 10, HEPES 10 (pH 7.2). Experiments were carried out at 22–24°C. The resting potential was determined in current clamp mode. Whole-cell KATP currents were recorded in response to ±20 mV voltage pulses from a holding potential of −70 mV. They were filtered at 1 kHz, digitized at 2 kHz using a Digidata 1200 Interface and analysed using pClamp software (Axon Instruments, Burlingame, U.S.A.).
Female Xenopus laevis were anaesthetised with MS222 (2 g l−1 added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Once the wound had completely healed, the second ovary was removed in a similar operation and the animal was then killed by decapitation whilst under anaesthesia. Immature stage V–VI Xenopus oocytes were incubated for 60 min with 1.0 mg ml−1 collagenase (Sigma, type V) and manually defolliculated. Oocytes were injected with ∼2 ng of mRNA encoding Kir6.2/SUR1BΔ31 or were coinjected with ∼0.1 ng Kir6.2 and ∼2 ng of SUR1A2 or SUR1BΔ31 (giving a 1 : 20 ratio). The final injection volume was ∼50 nl per oocyte. Isolated oocytes were maintained in tissue culture and studied 1–4 days after injection (Gribble et al., 1997a). Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of 0 mV and at 20–24°C (Gribble et al., 1997a). The pipette solution contained (mM): KCl 140, MgCl2 1.2, CaCl2 2.6, HEPES 10 (pH 7.4 with KOH) and the internal (bath) solution contained (mM): KCl 110, MgCl2 2, CaCl2 1, KOH 30, EGTA 10, HEPES 10 (pH 7.2 with KOH). Currents were evoked by repetitive 3 s voltage ramps from −110 mV to +100 mV and recorded using an EPC7 patch-clamp amplifier (HEKA Elektronik). They were filtered at 0.2 kHz, digitized at 0.5 kHz using a Digidata 1200 Interface and analysed using pClamp software (Axon Instruments).
Surface expression assay
Xenopus oocytes were co-injected with Kir6.2-HA (haemagglutinin epitope) and either SUR1BΔ31 or SUR1 (Zerangue et al., 1999). Experiments were performed two days after mRNA injection. Oocytes were incubated for 30 min in ND96 (in mM: NaCl 96, KCl 2, CaCl2 1.8, MgCl2 1, HEPES 5, adjusted to pH 7.4) with 1% Bovine Serum Albumin (BSA) at 4°C to block non-specific antibody binding. Subsequently, oocytes were incubated for 60 min at 4°C with 1 μg ml−1 rat monoclonal anti-HA antibody (3F10, Boehringer, Lewes, U.K.) in 1% BSA/ND96, washed six times at 4°C with 1% BSA/ND96, and then incubated with 2 μg ml−1 horseradish peroxidase-coupled secondary antibody (goat anti-rat fab fragments, Jackson Immunoresearch, West Grove, PA, U.S.A.), for 40 min. Oocytes were thoroughly washed, initially in 1% BSA/ND96 (4°C, 60 min) and then in ND96 alone (4°C, 60 min). Individual oocytes were then added to 50 μ l Power Signal Elisa solution (Pierce, Chester, U.K.) and, after an equilibration period of around 10 s, chemiluminescence was quantified in a Turner TD-20/20 luminometer (Sunnyvale, CA, U.S.A.) by integrating the signal over a period of 15 s. Results are given in relative light units (RLU).
Cell culture and DNA transfection experiments
The RINm5F cell line is derived from a radiation-induced rat insulin tumour (Gazdar et al., 1980). MIN6 cells, a mouse β cell line bearing the transforming SV-40 large T antigen, kindly provided by Dr H. Ishihara (Tokyo, Japan) were grown in DMEM containing 25 mM glucose, supplemented with 15% foetal calf serum, 100 u ml−1 penicillin sulphate and 100 μg ml−1 streptomycin. The HEK-293 cell line is a permanent line of primary human embryonic kidney transformed by sheared human adenovirus type 5 DNA (ATCC, Manassas, VA, U.S.A.). RINm5F and HEK-293 cell lines were cultured in DMEM media (BRL Life Technologies, Inc, Gaithersburg, MD, U.S.A.) containing 25 mmol l−1 glucose supplemented with 10% foetal calf serum, 100 u ml−1 penicillin sulphate and 100 μg ml−1 streptomycin (BRL Life Technologies, Inc, Gaithersburg, MD, U.S.A.). HEK-293 cells were plated in 10 cm culture dishes. After reaching 30% confluence, SUR1A2 and SUR1BΔ31 plasmid constructs (15 μg of plasmid DNA) were transiently transfected into HEK-293 cells using the Transfast™ transfection reagent (Promega, Madison, WI, U.S.A.) according to the manufacturer's instructions. HEK-293 cells transfected with the pcDNA3.1 vector alone are considered as control.
Immunoblotting
Twenty-four hours after transfection, cells were resuspended in 1 ml ice-cold membrane preparation buffer (10 mmol l−1 Tris-HCl, 30 mmol l−1 NaCl, 1 mmol l−1 dithiotreitol, and 5 μmol l−1 phenylmethylsulphonylfluoride, pH 7.5), disrupted using a Dounce homogenizer and centrifuged for 30 min at 20,000 g. The pellet was recovered and resuspended in 200 μl of membrane preparation buffer and 10 μg of total proteins were fractionated by 7.5% SDS–PAGE. Resolved proteins were electrophoretically transferred to 100% pure nitro-cellulose membranes (Protran®, Schleicher & Schuell, Germany). Non-specific binding sites were blocked by immersing the membranes during 1 h at room temperature in PBS containing 5% dry milk and 0.1% Tween-20. Membranes were then immunoblotted overnight at 4°C with a polyclonal anti SUR1 antiserum (1/1000) (Hernandez-Sanchez et al., 1999). After washes membranes were then incubated 1 h at room temperature with Horseradish peroxydase-conjugated donkey anti-rabbit secondary antibodies (1/2000, Amersham Pharmacia Biotech, France). Immunoreactive proteins were detected by chemiluminescence (ECL, Amersham Pharmacia Biotech, France) using X-ray Kodak films BioMax MR1 (Sigma, U.S.A.).
Membranes preparation and binding studies
Twenty-four hours after transfection, cells were detached by adding PBS containing 0.7 mM EDTA, pelleted by centrifugation (500 g, 5 min) and resuspended in ice-cold homogenization buffer (mM): Tris-HCl 10, NaCl 30, dithiothreitol 1 and 5 μmol l−1 phenylmethylsulphonylfluoride, pH 7.5). Cells were disrupted using a Dounce homogenizer and the homogenate was centrifuged at 12,000×g for 5 min at 4°C. The pellet containing total membrane extracts was frozen in liquid nitrogen and stored at −80°C until use. On the day of the experiment, membranes were resuspended in 50 mM Tris-HCl, pH 7.5. Membranes (200 μg protein ml−1) were incubated for 1 h at room temperature in a final volume of 500 μl with 3.5 nmol liter−1 [3H]-glibenclamide (50 Ci mmol, Du Pont-NEN, Boston, MA, U.S.A.), in the presence of various concentrations of glibenclamide. Non-specific binding was determined in the presence of 10 mM glibenclamide. Bound and free radiolabeled ligands were separated by filtration under vacuum on GF/B glass fibre filters (Whatman, Maidstone, U.K.) before 3H-counting in a scintillation medium (ACS II, Amersham, Buckinghamshire, U.K.).
Data analysis and statistics
Data are expressed as the mean±s.e.mean. The differences between experimental values were analysed by the Student's t-test. Differences were considered statistically significant when P<0.05.
Results
cDNA cloning of SUR1 isoforms
The polymerase chain reaction was used to clone cDNAs encoding SUR1 gene transcripts from rat RINm5F cells. Two novel cDNAs were obtained which differed in length. The longer cDNA contained an open reading frame of 4749 nucleotides encoding a 1582 amino acid protein with a calculated molecular weight of 177.2 kDa. The coding nucleotide sequence had 99.6% identity with the previously cloned rat SUR1 gene transcript and the protein sequence had 99.9% identity. In this article, we call the original SUR1 gene transcript (Genbank AB052294) SUR1A1 and the novel isoform SUR1A2 (Genbank X97279). The SUR1A2 cDNA displays a single nucleotide polymorphism and the corresponding protein sequence differs from SUR1A1 by the substitution of an isoleucine for the threonine at position 699 (Thr699Ile) (Figure 1). This mutation is located in the first nucleotide-binding domain (NBD1), just before the Walker A motif. We call the shorter cDNA SUR1BΔ31 because it lacks exon 31. The reading frame remains unchanged by deletion of exon 31. Thus SUR1BΔ31 contains a 4635 nucleotide open reading frame encoding a 1544 amino acid protein with a calculated molecular weight of 173.3 kDa (Genbank AF039595). Hydropathy analysis of the region surrounding the deletion of exon 31 suggests that the last two TMs are deleted, but NBD2 is left intact (Figure 1).
Figure 1.
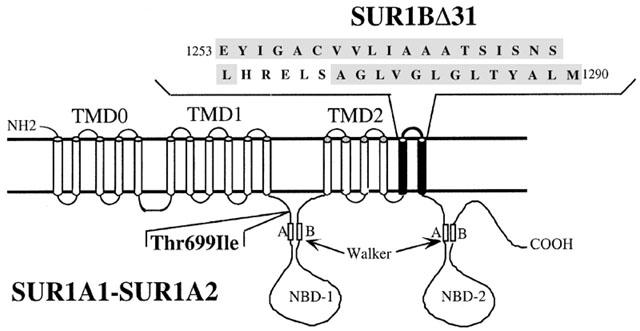
Predicted membrane topology of SUR1A1/2 and SUR1BΔ31. The figure depicts the site of the single amino acid substitution at position 699 (threonine to isoleucine) which distinguishes the SUR1A1 and SUR1A2 allelic variants. The highlighted TMs are absent in the SUR1BΔ31 splice variant and the insert indicates the deleted amino acid sequence.
Tissue distribution of SUR1A1/2 and SUR1BΔ31 isoforms
Human and rat SUR1 specific PCR primers spanning exon 31 were used to determine, by RT–PCR analysis, the tissue distributions of both SUR1A1/SUR1A2 (SUR1A1/2) and SUR1BΔ31 mRNAs. We amplified two PCR products of 456 bp and 341 bp, corresponding to SUR1A1/2 and SUR1BΔ31 respectively. As shown in Figure 2, SUR1A1/2 and SUR1BΔ31 are co-expressed in human brain, heart, pancreas, thymus, testis and in the rat insulin-secreting cell line RINm5F. In addition SUR1A1/2 is expressed in human skeletal muscle, and SUR1BΔ31 is expressed independently of SUR1A1/2 in human kidney, spleen and intestine. Lung, liver, ovary and HEK-293 cells lacked both SUR isoforms. The two PCR fragments observed in the RINm5F cell line were subcloned and sequenced: their sequences corresponded to those of SUR1A1/2 (456 bp fragment) and SUR1BΔ31 (341 bp fragment). The PCR fragments observed in human skeletal muscle and kidney were also sequenced and corresponded to those of SUR1A1/2 and SUR1BΔ31, respectively. As expected, the control GAPDH cDNA was ubiquitously expressed in all tissues and cell lines analysed (Figure 2).
Figure 2.
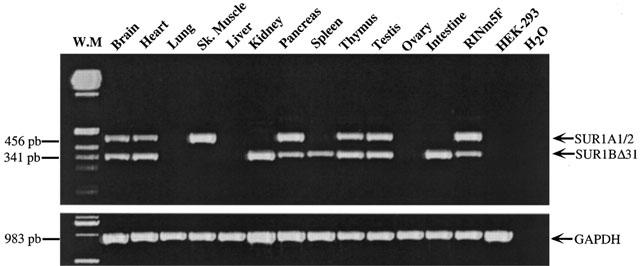
RT–PCR analysis of SUR1A1/2 and SUR1BΔ31 transcripts from human tissues and cell lines (upper panel). The expected size of the PCR products is indicated. GAPDH transcripts were analysed in the same tissues and cell lines as an internal control (lower panel). The results are representative of three similar experiments.
Electrophysiological experiments
HEK-293 cells were transiently transfected with SUR1A2–Kir6.2, or SUR1BΔ31–Kir6.2, and green fluorescent protein (pEGFP). Expressing cells were selected by their fluorescence. After establishment of the whole-cell configuration SUR1A2–Kir6.2 currents increased in amplitude, as ATP dialysed out of the cell into the pipette (Figure 3A,Bi). In contrast, SUR1BΔ31–Kir6.2 currents did not increase with time. Switching to current clamp after 5 min of recording revealed a resting membrane potential of −72±1 mV (n=5) for SUR1A2–Kir6.2 and of −22±4 mV (n=5) for SUR1BΔ31–Kir6.2 (Figure 3Bii).
Figure 3.
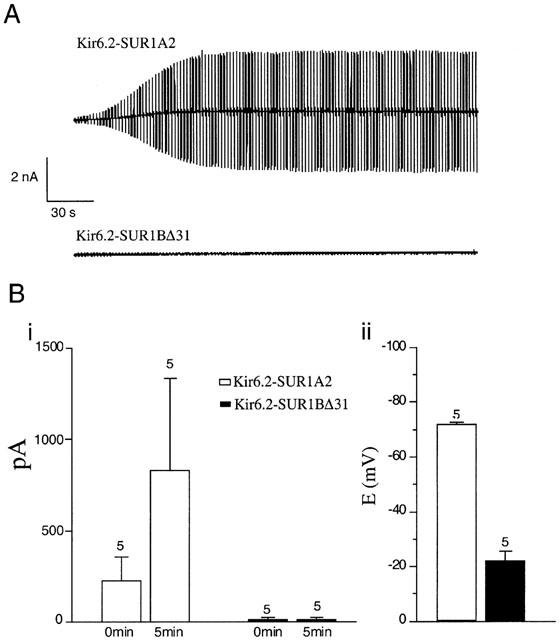
Functional analysis of Kir6.2/SUR1A2 and Kir6.2/SUR1BΔ31 channels expressed in HEK-293 cells. (A) Washout currents recorded with ATP-free pipette solution. VH was −70 mV and ±20 mV voltage pulses were applied to monitor membrane resistance. (B) Left panel: mean data from experiments as performed in A. The mean current response to +20 mV pulses at t=0 and t=5 min is plotted for SUR1A2–Kir6.2 and SUR1BΔ31–Kir6.2 currents. Right panel: Mean resting potential measured in current clamp after 5 min perfusion with ATP-free pipette solution.
There are two possible explanations for the lack of measurable KATP currents in SUR1BΔ31–Kir6.2 transfected HEK-293 cells. First, the protein might not be present in the plasma membrane. Second, the protein might be present at the cell surface but exhibit impaired regulation, so that it remains closed even when ATP levels fall. To explore which was the case, SUR1A2 or SUR1BΔ31 were coexpressed with Kir6.2 in Xenopus oocytes. Oocytes co-injected with mRNA encoding Kir6.2 and SUR1A2 exhibited small currents in cell-attached patches, but large inwardly rectifying currents in excised inside-out patches (Figure 4A,B). This reflects the fact that KATP channels are blocked in intact oocytes, but activate upon excision into nucleotide-free solution. Oocytes co-injected with mRNA encoding Kir6.2 and SUR1BΔ31 showed no increase in current upon patch excision (Figure 4A,B). Likewise, no currents were recorded from patches excised from oocytes injected with mRNA encoding the SUR1BΔ31–Kir6.2 tandem (data not shown). This would be consistent with a lack of KATP channels in the plasma membrane.
Figure 4.
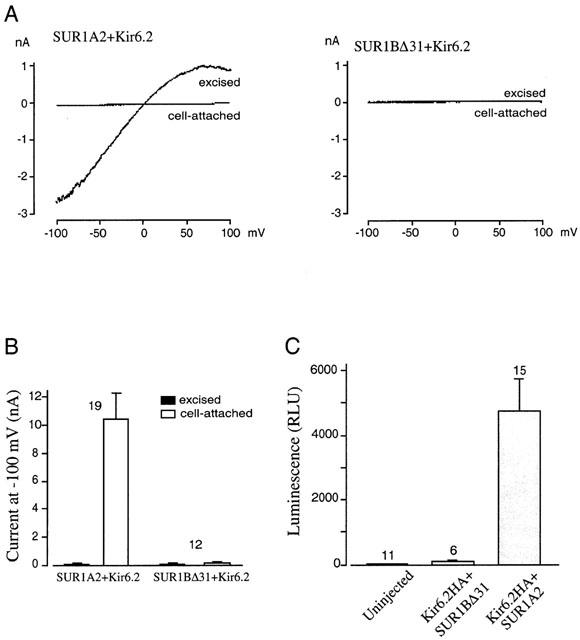
Functional expression essays for Kir6.2/SUR1A2 and Kir6.2/SUR1BΔ31 channels in Xenopus oocytes. (A) Macroscopic currents recorded in response to voltage ramps from −110 to +100 mV before and after excision of inside-out patches. (B) Mean current at −100 mV measured in experiments as shown in A. (C) Surface expression measured from Kir6.2HA/SUR1BΔ31 channels, Kir6.2/SUR1 channels and uninjected oocytes. R.L.U.: relative light units.
Surface expression analysis
The level of surface expression was assessed directly using an enzyme linked immunosorbance assay (ELISA) developed by Zerangue et al. (1999). An HA epitope (YPYDVPDYA) was introduced at residue 102 in the extracellular loop between TM1 and TM2 of Kir6.2. An anti HA antibody was used to detect the presence of this epitope at the cell surface. Because Kir6.2 does not reach the plasma membrane in the absence of its regulatory partner protein SUR, this technique also measures surface expression of SUR. Oocytes co-injected with mRNA encoding Kir6.2–HA and SUR1BΔ31 exhibited immuno-luminescence levels that were not substantially different from those of uninjected oocytes suggesting that Kir6.2–HA/SUR1BΔ31 is retained within internal membranes and does not reach the plasma membrane. In contrast, luminescence from oocytes expressing wild-type KATP channels was 50 times greater, indicative of cell surface expression (Figure 4C).
Analysis of [3H]-glibenclamide binding on total membrane preparations from HEK-293 cells expressing SUR1A2 and SUR1BΔ31 isoforms
HEK-293 cells were transiently transfected with SUR1A2 or SUR1BΔ31 cDNAs. Twenty-four hours after transfection, SUR1A2 and SUR1BΔ31 proteins were detected in HEK-293 transfected cells but not in HEK-293 cells transfected with the vector alone (Figure 5A). Thus both novel SUR proteins are translated. A band of 175-kDa protein corresponding to the SUR1 subunit, was also detected in MIN6 and RINm5F β-cells (Figure 5A).
Figure 5.
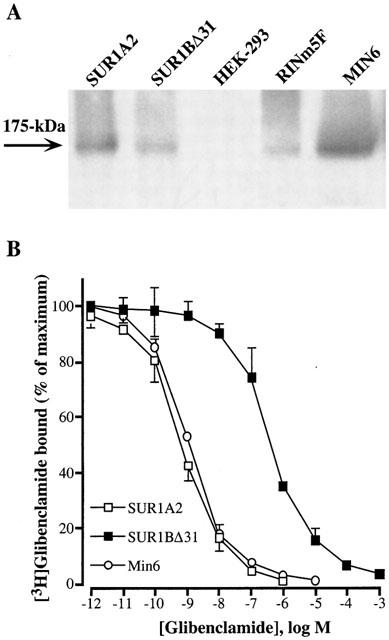
(A) Western blots of SUR1A2 (lane 1) and SUR1BΔ31 (lane 2) proteins expressed in HEK-293 cells. Lane (3) untransfected HEK-293 cells. Lanes (4) and (5) endogenous SUR in RINm5F and MIN6 cells, respectively. The results are representative of three similar experiments. (B) Inhibition of specific [3H]-glibenclamide binding by increasing concentrations of unlabelled glibenclamide. Total membrane extracts from MIN6 cells and HEK-293 cells transiently transfected with SUR1A2 or SUR1BΔ31 were incubated with [3H]-glibenclamide in the presence of various concentrations of unlabelled glibenclamide. Specific binding was expressed as a percentage of the maximal specific binding measured with labelled glibenclamide alone. Each point is the mean±s.e.mean of three different experiments, each performed in triplicates.
Comparison of [3H]-glibenclamide binding to total membranes fractions isolated from MIN6 β-cells or HEK-293 cells expressing SUR1A2 or SUR1BΔ31 isoforms showed 21.8, 19.7 and 3.5% of specific binding, respectively, whereas non-specific binding represented 0.9, 0.8 and 0.6%, respectively, of the total radioactivity added. [3H]-glibenclamide binding observed on HEK-293 cells transfected with the vector alone represented 0.8% of the total radioactivity added. Glibenclamide inhibited [3H]-glibenclamide binding to HEK-293 membranes expressing SUR1A2 with a Kd of 1.1±0.1 nM, similar to that found for MIN6 cells (Figure 5B). In contrast, HEK-293 cells expressing SUR1BΔ31 isoform showed a 500 times loss of glibenclamide affinity, with a Kd of 498±26 nM (Figure 5B).
Discussion
We have identified three variants of the sulphonylurea receptor SUR1 in the RINm5F cell line: SUR1A1, SUR1A2 and SUR1BΔ31. SUR1A1 corresponds to the original rat SUR1 gene transcript (Genbank AB052294), and SUR1A2 (Genbank X97279) differs from it by a single nucleotide, which results in a single amino acid change threonine 699 to isoleucine. Interestingly, the human SUR1 receptor (Swissprot Q09428) has an isoleucine at position 699. This supports the view that SUR1A2 represents a genuine polymorphism rather than a RT–PCR or sequencing artefact. In future publications, both SUR1A1 and SUR1A2 should be referred to as SUR1 since they differ only by a single nucleotide. The tissue distribution of SUR1A seems to be wider than that previously reported for Northern blot analysis (Aguilar-Bryan et al., 1995; Sakura et al., 1995), although SUR1 mRNA has been detected in heart by RT–PCR (Sakura et al., 1999). The differences in tissue expression reported in different studies probably reflects the relative sensitivity of the methods used.
When SUR1A2 was coexpressed with Kir6.2 in Xenopus oocytes, large inwardly rectifying K+ currents were observed on patch excision into nucleotide-free solution, consistent with the presence of KATP channels that are blocked in the cell-attached configuration by intracellular ATP. Likewise, when the tandem construct Kir6.2–SUR1A2 was expressed in HEK-293 cells KATP washout currents similar to those found in native mouse β-cells were observed. These results suggest that SUR1A2 is capable of forming functional KATP channels when coexpressed with Kir6.2.
The second novel SUR1 variant found in RINm5F cells lacked exon 31: we propose to call this SUR1BΔ31. SUR1BΔ31 has been reported in cultured neonatal rat atrial cardiomyocytes after PCR analysis (Baron et al., 1999) but neither tissue distribution pattern nor pharmacological and functional characterization of this isoform have been performed up to now. We showed that SUR1BΔ31 is expressed in human brain, heart, pancreas and other visceral organs, suggesting that it is a common splice variant expressed in normal human tissues. Its expression in several tissues of different species suggests it may have a specific biological role. We were unable to detect KATP currents in HEK-293 cells or Xenopus oocytes expressing either SUR1BΔ31-Kir6.2 or Kir6.2 plus SUR1BΔ31. Furthermore, no currents were detected on patch excision into nucleotide-free solution, or in a surface expression assay, using Xenopus oocytes. Western blot analysis performed on HEK-293 cells transiently transfected with SUR1BΔ31 cDNA showed that the protein is expressed at a level similar to that observed in MIN6 β cells. This suggests that deletion of exon 31 may induce a conformational change in SUR1 that impairs membrane trafficking. Recently a trafficking defect caused by a SUR1 mutation has been described (Cartier et al., 2001). It is not yet clear whether SUR1BΔ31-Kir6.2 has any physiological role in the intracellular membranes of those cells in which it is expressed. However, we point out that the sulphonylurea receptor has been postulated to play a role in exocytosis (Thévenod et al., 1992) and in mitochondria (Paucek et al., 1992).
The Kd for glibenclamide displacement of [3H]-glibenclamide binding to SUR1A2 (1.1 nM) is similar to that found for SUR1 (Aguilar-Bryan et al., 1995). This indicates that the Thr699Ile mutation does not affect glibenclamide binding. Similar affinities have also been reported for native KATP channels in insulinoma cells (Aguilar-Bryan et al., 1992; Gros et al., 1999), cardiac myocytes (French et al., 1990) and central neurons (Schwanstecher et al., 1994). We also showed the presence of a specific binding on total membrane preparations of HEK cells expressing the SUR1BΔ31 isoform. The fact that SUR1BΔ31 binds [3H]-glibenclamide but shows no surface expression confirms that the SUR1BΔ31 isoform is translated but retained within the intracellular membrane compartment. [3H]-glibenclamide binds on SUR1BΔ31 subunit with a 500 times lower affinity, which suggests that TM16 and TM17 are important for high-affinity glibenclamide binding. A number of previous studies have also indicated that the region containing TMs 15–17 is important for sulphonylurea binding and channel inhibition. In particular, tolbutamide block is abolished by a mutation within the TM15–16 linker of SUR1, and tolbutamide sensitivity can be conferred on SUR2A by transfer of TMs 14–16 from SUR1 (Ashfield et al., 1999). Glibenclamide binding appears to involve both the cytosolic loop between TMs 15 and 16 and that between TMs 5 and 6 (Mikhailov et al., 2001). In SUR1BΔ31, the cytosolic loop between TMs 15 and 16 connects directly to the NBD2. It seems plausible that a conformational change influences the ability of [3H]-glibenclamide to bind to the loop between TMs 15 and 16. Other regions of the protein must also contribute to the glibenclamide binding site since SUR1BΔ31 still retains the ability to bind the sulfonylurea with lower affinity. This low affinity binding that we measure on SUR1BΔ31 could reflect the affinity of glibenclamide for the TM 5–6 linker. However, it is also possible that the lack of the two last transmembrane domains may induce a change in the subunit conformation which may reduce binding to a site elsewhere in the protein.
In conclusion, these results confirm that TM16–17 of SUR1 are important for high affinity glibenclamide binding. Moreover, we show that this two last TMs are required for interaction with Kir6.2 and normal trafficking of the KATP channel to the surface membrane. The tissue distribution of SUR1BΔ31 also raises the question of whether it has a novel physiological role.
Acknowledgments
We thank Dr Jorge Ferrer (Endocrinology and Hormonal Biochemistry units, Hospital Clinic, Institut d'Investigacions Biomediques, Barcelona, Spain) for his generous gift of polyclonal antibody against the SUR1 subunit of the sulphonylurea receptor.
Abbreviations
- CHI
congenital hyperinsulinemia of infancy
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HA
haemagglutinin
- HEK
human embryonic kidney
- KATP
ATP-sensitive potassium channel
- KCO
potassium channel opener
- Kir
inward-rectifier potassium channel
- NBD
nucleotide binding domain
- SUR
sulphonylurea receptor
- TM
transmembrane domain
References
- AGUILAR-BRYAN L., NICHOLS C.G., RAJAN A.S., PARKER C., BRYAN J. Co-expression of sulfonylurea receptors and KATP channels in hamster insulinoma tumor (HIT) cells. Evidence for direct association of the receptor with the channel. J. Biol. Chem. 1992;267:14934–14940. [PubMed] [Google Scholar]
- AGUILAR-BRYAN L., NICHOLS C.G., WECHSLER S.W., CLEMENT IV, J.P., BOYD III A.E., GONZALEZ G., HERRERA-SOSA H., NGUY K., BRYAN J., NELSON D.A. Cloning of the β-cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., GRIBBLE F.M. ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia. 1999;42:9903–9919. doi: 10.1007/s001250051247. [DOI] [PubMed] [Google Scholar]
- ASHCROFT S.J.H., ASHCROFT F.M. The sulfonylurea receptor. Biochim. Biophys. Acta. 1992;1175:45–49. doi: 10.1016/0167-4889(92)90008-y. [DOI] [PubMed] [Google Scholar]
- ASHCROFT S.J., NIKI I., KENNA S., WENG L., SKEER J., COLES B., ASHCROFT F.M. The beta-cell sulfonylurea receptor. Adv. Exp. Med. Biol. 1993;334:47–61. doi: 10.1007/978-1-4615-2910-1_4. [DOI] [PubMed] [Google Scholar]
- ASHFIELD R., GRIBBLE F.M., ASHCROFT S.J.H., ASHCROFT F.M. Identification of the high affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- BARON A., VAN BEVER L., MONNIER D., ROATTI A., BAERTSCHI A.J. A novel KATP current in cultured neonatal rat atrial appendage cardiomyocytes. Circ. Res. 1999;85:707–715. doi: 10.1161/01.res.85.8.707. [DOI] [PubMed] [Google Scholar]
- CARTIER E.A., CONTI L.R., VANDENBERG C.A., SHYNG S.L. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycaemia of infancy. Proc. Natl. Acad. Sci. 2001;98:2882–2887. doi: 10.1073/pnas.051499698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLEMENT IV, J.P., KUNJILWAR K., GONZALEZ G., SCHWANSTECHER M., PANTEN U., AGUILAR-BRYAN L., BRYAN J. Association and stoichiometry of K(ATP) channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- CONTI L.R., RADEKE C.M., SHYNG S.-L., VANDENBERG C.A. Transmembrane topology of the sulfonylurea receptor SUR1. J. Biol. Chem. 2001;276:41270–41278. doi: 10.1074/jbc.M106555200. [DOI] [PubMed] [Google Scholar]
- D'HAHAN N., JACQUET H., MOREAU C., CATTY P., VIVAUDOU M. A transmembrane domain of the sulfonylurea receptor mediates activation of ATP-sensitive K(+) channels by K(+) channel openers. Mol. Pharmacol. 1999;56:308–315. doi: 10.1124/mol.56.2.308. [DOI] [PubMed] [Google Scholar]
- DUNNE M.J., KANE C., SHEPHERD R.M., SANCHEZ J.A., JAMES R.F., JOHNSON P.R., AYNSLEY-GREEN A., LU S., CLEMENT IV, J.P., LINDLEY K.J., SEINO S., AGUILAR-BRYAN L. Familial persistent hyperinsulinemic hypoglycaemia of infancy and mutations in the sulfonylurea receptor. New Engl. J. Med. 1997;336:703–706. doi: 10.1056/NEJM199703063361005. [DOI] [PubMed] [Google Scholar]
- FRENCH J.F., RIERA L.C., SARMIENTO J.G. Identification of high and low (GTP-sensitive) affinity [3H]-glibenclamide binding sites in cardiac ventricular membranes. Biochem. Biophys. Res. Commun. 1990;167:1400–1405. doi: 10.1016/0006-291x(90)90678-g. [DOI] [PubMed] [Google Scholar]
- GAZDAR A.F., CHICK W.L., OIE H.K., SIMS H.L., KING D.L., WEIR G.C., LAURIS V. Continuous, clonal, insulin- and somatostatin-secreting cell lines established from a transplantable rat islet cell tumor. Proc. Natl. Acad. Sci. 1980;77:3519–3523. doi: 10.1073/pnas.77.6.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIBBLE F.M., ASHFIELD R., AMMALA C., ASHCROFT F.M. Properties of cloned ATP-sensitive K= currents expressed in Xenopus oocytes. J. Physiol. 1997a;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIBBLE F.M., TUCKER S.J., ASHCROFT F.M. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J. 1997b;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIBBLE F.M., TUCKER S.J., HAUG T., ASHCROFT F.M. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc. Natl. Acad. Sci. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROS L., VIRSOLVY A., SALAZAR G., BATAILLE D., BLACHE P. Characterisation of low-affinity binding sites for glibenclamide on the Kir6.2 subunit of the beta-cell KATP channel. Biochem. Biophys. Res. Commun. 1999;257:766–770. doi: 10.1006/bbrc.1999.0529. [DOI] [PubMed] [Google Scholar]
- HERNANDEZ-SANCHEZ C., ITO Y., FERRER J., REITMAN M., LEROITH D. Characterization of the mouse sulfonylurea receptor 1 promoter and its regulation. J. Biol. Chem. 1999;274:18261–18270. doi: 10.1074/jbc.274.26.18261. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT IV J.P., NAMBA N., INAZAWA J., GONZALEZ G., AGUILAR-BRYAN L., SEINO S., BRYAN J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., SEINO S. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- MATSUO M., TANABE K., KIOKA N., AMACHI T., UEDA K. Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J. Biol. Chem. 2000;275:28757–28763. doi: 10.1074/jbc.M004818200. [DOI] [PubMed] [Google Scholar]
- MIKHAILOV M.V., MIKHAILOVA E.A., ASHCROFT S.J.H. Molecular structure of the glibenclamide binding site of the β-cell KATP channel. FEBS Lett. 2001;499:154–160. doi: 10.1016/s0014-5793(01)02538-8. [DOI] [PubMed] [Google Scholar]
- OTONKOSKI T., AMMALA C., HUOPIO H., COTE G.J., CHAPMAN J., COSGROVE K., ASHFIELD R., HUANG E., KOMULAINEN J., ASHCROFT F.M., DUNNE M.J., KERE J., THOMAS P.M. A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycaemia of infancy in Finland. Diabetes. 1999;48:408–415. doi: 10.2337/diabetes.48.2.408. [DOI] [PubMed] [Google Scholar]
- PAUCEK P., MIRONOVA G., MAHDI F., BEBÁIS A.D., WOLDEGIORGIS G., GARLID K.D. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent potassium channel from rat liver and beef heart mitochondria. J. Biol. Chem. 1992;267:26062–26069. [PubMed] [Google Scholar]
- REIMANN F., ASHCROFT F.M. Inwardly rectifying potassium channels. Curr. Opin. Cell Biol. 1999;11:503–508. doi: 10.1016/S0955-0674(99)80073-8. [DOI] [PubMed] [Google Scholar]
- SAKURA H., ÄMMÄLÄ C., SMITH P.A., GRIBBLE F.M., ASHCROFT F.M. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- SAKURA H., TRAPP S., LISS B., ASHCROFT F.M. Altered functional properties of KATP channel conferred by a novel splice variant of SUR1. J. Physiol. 1999;52:337–350. doi: 10.1111/j.1469-7793.1999.00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWANSTECHER M., LÖSER S., CHUDZIAK F., BACHMANN C., PANTEN U. Photoaffinity labelling of the cerebral sulfonylurea receptor using a novel radioiodinated azidoglibenclamide analogue. J. Neurochem. 1994;63:698–708. doi: 10.1046/j.1471-4159.1994.63020698.x. [DOI] [PubMed] [Google Scholar]
- SCHWANSTECHER M., SIEVERDING C., DORSCHNER H., GROSS I., AGUILAR-BRYAN L., SCHWANSTECHER C., BRYAN J. Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J. 1998;17:5529–5535. doi: 10.1093/emboj/17.19.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEGHERS V., NAKAZAKI M., DEMAYO F., AGUILAR-BRYAN L., BRYAN J. SUR1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J. Biol. Chem. 2000;275:9270–9277. doi: 10.1074/jbc.275.13.9270. [DOI] [PubMed] [Google Scholar]
- STURGESS N.C., KOZLOWSKI R.Z., CARRINGTON C.A., HALES C.N., ASHFORD M.L. Effects of sulphonylureas and diazoxide on insulin secretion and nucleotide-sensitive channels in an insulin-secreting cell line. Br. J. Pharmacol. 1988;95:83–94. doi: 10.1111/j.1476-5381.1988.tb16551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THÉVENOD F., CHATHADI K.V., JIANG B., HOPFER U. ATP-sensitive K+ conductance in pancreatic zymogen granules: block by glyburide and activation by diazoxide. J. Memb. Biol. 1992;129:253–282. doi: 10.1007/BF00232907. [DOI] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., PROKS P., TRAPP S., RYDER T.J., HAUG T., REIMANN F., ASHCROFT F.M. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., ZHAO C., TRAPP S., ASHCROFT F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- TUSNADY G.E., BAKOS E., VARADI A., SARKADI B. Membrane topology distinguishes a subfamily of the ATP-binding cassette (ABC) transporters. FEBS Lett. 1997;402:1–3. doi: 10.1016/s0014-5793(96)01478-0. [DOI] [PubMed] [Google Scholar]
- ZERANGUE N., SCHWAPPACH B., JAN Y.N., JAN L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]