

Abstract
The structural features that determine the state-dependent interaction of local anaesthetics with voltage-operated sodium channels are still a matter of debate. We have studied the blockade of sodium channels by 2,6-dimethylphenol, a phenol derivative which resembles the aromatic tail of lidocaine, etidocaine, and bupivacaine.
The effects of 2,6-dimethylphenol were studied on heterologously (HEK 293) expressed rat neuronal (rat brain IIA) and human skeletal muscle (hSkM1) sodium channels using whole-cell voltage-clamp experiments.
2,6-Dimethylphenol was effective in blocking whole-cell sodium inward currents. Its potency was comparable to the potency of lidocaine previously obtained with similar protocols by others. The IC50 at −70 mV holding potential was 150 and 187 μM for the skeletal muscle and the neuronal isoform, respectively. In both isoforms, the blocking potency increased with the fraction of inactivated channels at depolarized holding potentials. However, the block achieved at −70 mV with respect to −150 mV holding potential was significantly higher only in the skeletal muscle isoform. The estimated dissociation constant Kd from the inactivated state was 25 μM and 28 μM in the skeletal muscle and the neuronal isoform, respectively. The kinetics of drug equilibration between resting and inactivated channel states were about 10 fold faster compared with lidocaine.
Our results show that the blockade induced by 2,6-dimethylphenol retains voltage-dependency, a typical feature of lidocaine-like local anaesthetics. This is consistent with the hypothesis that the ‘aromatic tail' determines the state-dependent interaction of local anaesthetics with the sodium channel.
Keywords: sodium channels, voltage-operated; binding site, local anaesthetic; local anaesthetic; lidocaine; structure
Introduction
Voltage-sensitive sodium channels are responsible for increased sodium permeability during the rapidly rising phase of the action potential in nerve, skeletal muscle, neuroendocrine, and heart cells. In response to membrane depolarization the channels open from the resting, closed state and then inactivate spontaneously. Mediating excitability in different tissues, they represent important target sites for local anaesthetic, anti-epileptic, and antiarrhythmic drugs. The structural units that are characteristic of class I antiarrhythmic drugs and local anaesthetics are a hydrophobic aromatic group connected via an intermediate chain to a hydrophilic amine group (Ehring et al., 1988). It has long been debated which part of the molecule determines its state-dependent interaction with the sodium channel. The approach of dissecting the lidocaine molecule into phenol and diethylamide has provided some evidence that the ‘aromatic tail' accounts for certain aspects of lidocaine block (Zamponi & French, 1993), but did not take into account the fact that the aromatic group of the parent compound actually resembles a methylated phenol derivative (see Figure 1).
Figure 1.
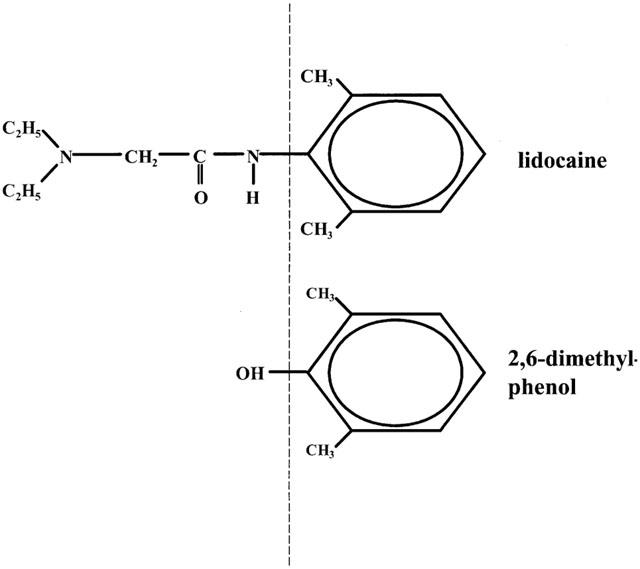
Structures of lidocaine and the phenol derivative that most resembles its aromatic tail, 2,6-dimethylphenol.
Although phenol block mimicked the slow block of cardiac sodium channels seen with lidocaine, the blocking potency was an order of magnitude lower and skeletal muscle sodium channels were only minimally affected (Zamponi & French, 1993).
The objective of our in vitro study was to determine the blocking potency and the kinetics of drug equilibration between inactivated and resting states for 2,6-dimethylphenol, which is a structural analogue of the aromatic tail of lidocaine, bupivacaine, and etidocaine.
We studied the effects of 2,6-dimethylphenol on heterologously expressed α-subunits of rat brain IIA and human skeletal muscle (hSkM1) sodium channels. The α-subunit is the primary pore-forming subunit of voltage-operated sodium channels and functions as an ion channel when expressed alone (Catterall, 1992). α-subunits of rat brain IIA and human skeletal muscle (hSkM1) sodium channels have normal gating characteristics (with respect to experiments in native tissue) when expressed in the background of a mammalian cell line (Chahine et al., 1994; Sarkar et al., 1995). The rat brain IIA form is most abundant in adult brain (Beckh et al., 1989) and hSkM1 is the principal voltage-gated sodium channel expressed in adult human skeletal muscle (George et al., 1992).
Methods
Transfection and cell culture
Stably transfected HEK 293 (human embryonic kidney cell) lines expressing either the α-subunit of rat brain IIA or human skeletal muscle (hSkM1) sodium channels were a gift from Professor Lehmann-Horn, Ulm, Germany. The expression vector pRc/CMV (Invitrogen, San Diego, USA) was used for mammalian transfection (Mitrovic et al., 1994). Transfection was performed using calcium phosphate precipitation (Graham & Van der Eb, 1973). Permanent expression was achieved by selection for resistance to the aminoglycoside antibiotic geneticin G418 (Life Technology, Eggenstein, Germany) (Mitrovic et al., 1994). Successful channel expression was verified electrophysiologically. The clones have been used in several investigations (Haeseler et al., 2000; Mitrovic et al., 1994; Sarkar et al., 1995).
Solutions
2,6-Dimethylphenol (FLUKA, Deisenhofen, Germany) was prepared as a 1 M stock solution in ethanol, light-protected, and stored in glass vessels at −20°C. The stock solution was dissolved directly in bath solution immediately before experiments. Drug-containing vials were vigorously vortexed for 60 min. The solution was applied via a glass-polytetrafluoroethylene perfusion system and a stainless steel superfusion pipette. The bath solution contained (mM) NaCl 140, MgCl2 1.0, KCl 4.0, CaCl2 2.0, HEPES 5.0, dextrose 5.0. Patch electrodes contained (mM) CsCl2 130, MgCl2 2.0, EGTA 5.0, HEPES 10. All solutions were adjusted to 290 mosm l−1 by the addition of mannitol and to pH 7.4 by the addition of CsOH. The effects of the diluent ethanol corresponding to higher drug concentrations had been tested in a previous study up to a maximum ethanol concentration of 17.4 mM corresponding to a drug concentration of 1000 μM. At this concentration, the block caused by ethanol is less than 20% (Haeseler et al., 2001).
Electrophysiology
Standard whole-cell voltage-clamp experiments (Hamill et al., 1981) were performed at 20°C. Each experiment consisted of test recordings with the drug present at only one concentration, and of drug-free control recordings before and after the test. At least three experiments were performed at each concentration.
For data acquisition and further analysis, we used the EPC9 digitally-controlled amplifier in combination with Pulse and Pulse Fit software (HEKA Electronics, Lambrecht, Germany). The EPC9 provides automatic subtraction of capacitive and leakage currents by means of a prepulse protocol. The data were filtered at 10 kHz and digitized at 20 μs per point. The input resistance of the patch pipettes was 2.0–3.5 MΩ and the capacitances of the cells were 9–15 pF; the residual series resistance (after 50% compensation) was 1.2–2.5 MΩ. Experiments with a rise in series resistance were rejected. To minimize time-dependent shifts in the voltage-dependence of steady-state inactivation (Wang et al., 1996), all test experiments were performed within 5 min of patch rupture. Under these experimental conditions, time-dependent hyperpolarizing shifts in control conditions were less than −2 mV (Haeseler et al., 2000). Voltage-activated currents were studied by applying different voltage-clamp protocols described in the Results section or in the appropriate figure legends.
Drug effects on the peak current amplitude were assessed at a holding potential close to a normal resting membrane potential in physiological conditions (−70 mV) (Hodgkin & Horowicz, 1959), and at hyperpolarized membrane potentials (−150 mV). The residual sodium current (INa+) in the presence of drug (with respect to the current amplitude in control solution) was plotted against the applied concentration of the drug [C]. All data are presented as means±s.d. When the effects were concentration-dependent, the averaged data were fitted using the Hill equation (Equation 1), yielding the concentration for half-maximum channel blockade (IC50) and the Hill coefficient nH.
Statistical analysis
Each cell was exposed to one test concentration only. Thus, the results obtained in different test concentrations were independent from each other. A one-sample t-test was applied to analyse the statistical significance of differences in blocking effects at different holding potentials and of gating parameter changes in the presence of drug with respect to the starting value. To address problems related to multiple hypotheses testing, we applied the method of multiple comparisons with priori ordered hypotheses (Maurer, 1995). The method is based on the assumption that, if the null hypothesis can be rejected, there is a positive monotonic relation between concentration and effect. As a consequence, the hypotheses to be tested are ordered in advance, i.e. in our study, starting with the concentration where the largest difference in the effect was to be expected (1000 μM for differences in blocking potency at −70 vs −150 mV holding potential and 500 μM for differences in gating parameter changes with respect to control. In the 1000 μM concentration, the residual sodium current in the presence of drug was too small to allow analysis of gating parameters). If the null hypothesis could be rejected for the highest concentration, the effects of the next, lower concentration were evaluated. The statistical evaluation was stopped as soon as the first insignificant result was obtained. The advantage of this procedure, compared with other approaches to multiple testing, is, that the level of type I error is kept the same for each statistical test. In our study, the null hypothesis was rejected when P<0.05.
Results
Resting state affinity
We studied a total of 65 cells. Average currents in the control experiments following depolarization from −150 mV to 0 mV were −3.4±0.7 nA for the skeletal muscle isoform and −2.6±1.5 nA for the neuronal isoform. Maximum inward currents elicited by 10 ms pulses from either −150 mV or −70 mV to 0 mV were reversibly suppressed by 2,6-dimethylphenol in a concentration-dependent manner. The 2,6-dimethylphenol-induced blockade was rapid in onset (less than 30 s of application) and readily reversible on wash-out. Normalized currents (with respect to control) derived from at least three different experiments for each drug concentration were averaged to establish concentration-response plots (see Figure 2). The amount of block achieved depended on the holding potential from which the depolarization was started. Hill fits to the averaged data yielded IC50 values of 185 and 150 μM at −70 mV, and 481 and 558 μM at −150 mV holding potential for the neuronal and skeletal muscle isoforms respectively. The calculated values for their Hill coefficients nH were 0.9 and 1.1 (−70 mV), and 1.0 and 1.2 (−150 mV), respectively. However, the difference in blocking potency at −70 mV with respect to −150 mV was significant only in the skeletal muscle isoform (see Figure 2).
Figure 2.
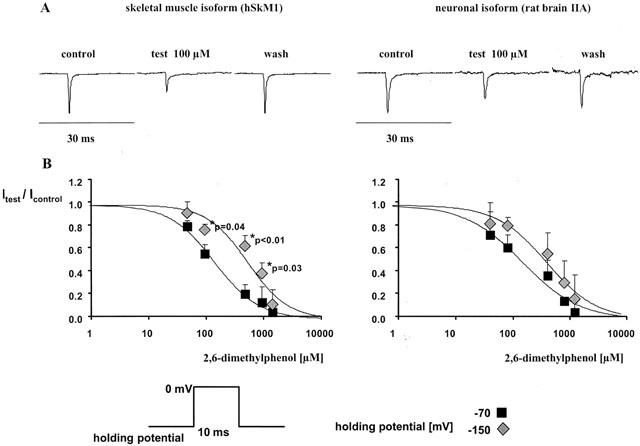
(A) Representative current traces after 10 ms depolarization from −70 mV to 0 mV in the absence (control and wash-out) and presence of 100 μM 2,6-dimethylphenol in the neuronal and skeletal muscle isoforms respectively. (B) Concentration-dependent reduction in test pulse current achieved by 2,6-dimethylphenol with respect to control (Itest/Icontrol, mean±s.d.) in both channel isoforms. Each cell was exposed to one test concentration only. The data were derived from at least three different experiments for each concentration tested. Depolarizing pulses to 0 mV (10 ms duration) were started from either −150 or −70 mV. *Indicates a significant increase in the peak current suppression at −70 mV holding potential (filled squares) compared to −150 mV (filled diamonds). Significant differences in the blocking potency at −70 with respect to −150 mV were detected in the skeletal muscle isoform only. Solid lines are Hill fits to the averaged data. The IC50 values derived from that fit were 150 and 558 μM at −70 mV and −150 mV holding potential in the skeletal muscle isoform and 185 μM and 481 μM at −70 and −150 mV in the neuronal isoform, respectively.
Inactivated state affinity
The increase in blocking potency at a holding potential of −70 mV, at which a fraction of the channels is inactivated, compared with −150 mV, at which all channels are expected to be in the resting state, suggests that the amount of block achieved by 2,6-dimethylphenol depends on the membrane potential and increases as the proportion of inactivated channels increases with respect to resting channels. The voltage-dependence of the block and the affinity for the inactivated state was further assessed using a double-pulse protocol. After brief depolarizations, sodium channels enter a fast-inactivated state, in which they cannot readily reopen. Currents elicited by test pulses (Itest) starting from different prepulse potentials (from −150 mV to −5 mV), normalized to the current elicited at the most hyperpolarized prepotential (−150 mV), represent the relative fraction of channels that have not been inactivated during the 50 ms inactivating prepulse. Boltzmann fits to the resulting current–voltage plots yield the membrane potential at half-maximum channel availability (V0.5) and the slope factor k (Equation 2).
In control conditions, the parameters of the Boltzmann fits reflect the voltage-dependence of the distribution between resting and fast-inactivated channels. Summarized control data showed that half of the channels were unavailable at −44.4±5 mV (rat brain IIA isoform) and at −61.5±5 mV (hSkM1) because of fast inactivation. The slope factors k were 7.9±1.1 and 7.4±1.0 respectively.
With exposure to 2,6-dimethylphenol, V0.5 was shifted considerably in the direction of more negative prepulse potentials; the degree of alteration showed concentration dependence. The slope factor k remained unchanged in the presence of drug. The drug-induced hyperpolarizing shifts reflect an additional reduction of channel availability in the voltage range of channel inactivation compared with −150 mV. Figure 3A illustrates the voltage shift in the availability curve as a result of the increase in blocking potency with membrane depolarization in the skeletal muscle isoform.
Figure 3.

Effects of 2,6-dimethylphenol on fast inactivated channels assessed by shifts in the steady-state availability curve. (A (upper panel)) Steady-state availability curves assessed by a two-pulse protocol in the absence (control, circles) and presence of 500 μM 2,6-dimethylphenol (triangles) in the skeletal muscle isoform. Each symbol represents the mean fractional current (n=3 for each concentration) elicited by a 4 ms test pulse to 0 mV, following a 50 ms inactivating prepulse from −150 mV to the indicated prepulse potential. Currents were normalized to maximum value (in each series at −150 mV prepotential). Solid lines represent the best Boltzmann fit (Eq. 2) to the data yielding the membrane potential at half-maximum channel availability (V0.5) and the slope factor k. Error bars are standard deviations. In the presence of 500 μM 2,6-dimethylphenol, currents were normalized either to the maximum in the presence of the drug (filled symbols) or to the maximum in the controls (empty symbols). Vertical arrows illustrate the increase in the peak current suppression induced by 500 μM 2,6-dimethylphenol at more depolarized holding potentials versus hyperpolarized holding potentials. This reduction in channel availability at depolarized prepotentials resulted in a voltage shift in the midpoints of the availability curve (ΔV0.5), indicated by the horizontal arrows. (B,C (lower panel)) Concentration-dependence of drug-induced negative shifts in the midpoints (ΔV0.5 [mV], mean±s.d.) of the steady-state availability plots relative to the starting values in both isoforms. As the diluent ethanol applied at a concentration corresponding to a drug concentration of 1000 μM induced a small shift of +5 mV, i.e. into the opposite direction in comparison with the 2,6-dimethylphenol-induced shifts, the data were not corrected for the ethanol shifts. The solid line is the least-squares fit to Equation 3. The IC50 at −150 mV holding potential entered as fit parameter was 481 μM for the neuronal and 558 μM for the skeletal muscle isoform, respectively, the Kd values estimated for the inactivated sodium channels were 28 μM for the neuronal and 25 μM for the skeletal muscle isoform, respectively.
In order to estimate the dissociation constant (Kd) of 2,6-dimethylphenol for the fast-inactivated state of both channel isoforms, we used a model developed by Bean et al. (1983) for the effects of lidocaine on Purkinje fibres (Equation 3).
ΔV0.5 is the shift in the midpoint in each drug concentration (mean, n>3), k is the mean value for the slope factor derived from Boltzmann fits to the current–voltage plots (see Equation 2), [C] is the applied concentration of drug, IC50−150 the concentration for half-maximum effect derived from the concentration-response plots at the −150 mV membrane potential described above, and Kd the dissociation constant for the drug from the inactivated state.
The model is based on the assumption that the higher degree of channel block achieved with consecutive membrane depolarization is determined by the apportionment of channels between resting and fast-inactivated states and by the different binding affinities of the blocking drug for the two channel states. Figures 3B and C show the concentration-dependence of the voltage shift in both isoforms and the fit of Equation 3 to the data. However, as already revealed by Bean's own data, the fit tends to overestimate the effects of lower drug concentrations, and to underestimate the effects of higher drug concentrations. Thus, the model should only be regarded as an approximation.
For 2,6-dimethylphenol, the estimated values of Kd derived from that fit were 28 and 25 μM for the neuronal and skeletal muscle isoforms respectively (see Figure 3B,C).
When the diluent ethanol was applied instead of the test solution at a concentration of 17.4 mM, corresponding to a concentration of 1000 μM of 2,6-dimethylphenol, ethanol did not shift the steady-state voltage dependence in a hyperpolarizing direction. Instead there was a small positive shift of 5.0±0.2 mV (n=3). The shifts observed in the presence of 2,6-dimethylphenol were not corrected for the ethanol shifts.
Time course of channel inactivation and recovery from inactivated channel block in the presence of 2,6-dimethylphenol
The time constant of channel inactivation τh was obtained by fitting a single exponential (Equation 4) to the decay of current during a 40 ms depolarization from −100 mV to 0 mV.
In control conditions, τh was 0.71±0.2 ms for rat brain IIA and 0.59±0.18 ms for hSkM1 sodium channels. 2,6-Dimethylphenol did not significantly accelerate the time course of channel inactivation in rat brain IIA channels; in hSkM1 channels the acceleration of the current decay was significant only in very high concentrations (500 μM). Values obtained for τh in the presence of the drug were: 0.74±0.1 and 0.57±0.03 ms in 100 μM 2,6-dimethylphenol and 0.51±0.3 and 0.48±0.07 ms (P=0.02) in 500 μM 2,6-dimethylphenol in the neuronal and skeletal muscle isoforms respectively. The acceleration of the current decay phase in the skeletal muscle isoform is illustrated in Figure 4A.
Figure 4.
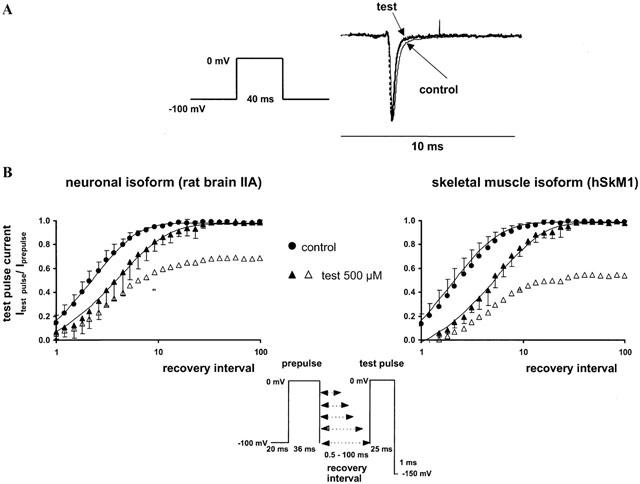
(A, upper panel) Acceleration of the sodium current decay phase by 500 μM 2,6-dimethylphenol in the skeletal muscle isoform. Representative current traces following a 40 ms depolarization to 0 mV in the control (solid line) and in the presence of 500 μM 2,6-dimethylphenol (test, dotted line). All currents were scaled to the same size for better comparison of the decay phase. (B, lower panel) Recovery from inactivation assessed by a two-pulse-protocol in the absence (control, circles) and presence of 500 μM 2,6-dimethylphenol (triangles). The abscissa represents the recovery time interval between prepulse and test pulse (up to 100 ms) on a logarithmic scale, and the ordinate represents the mean fractional current derived from at least three different experiments, having recovered from inactivation or inactivated channel block, respectively. Error bars are standard deviations. In the presence of 2,6-dimethylphenol, currents were normalized either to the prepulse in the presence of drug (filled symbols) or in the corresponding control experiment (empty symbols). Solid lines are bi-exponential fits (Equation 5) to the fractional current yielding the time constants of recovery, τrec. In the absence of the drug, recovery time constants τrec1 obtained for the neuronal and skeletal muscle isoform were 2.4±0.8 and 2.7±1.3 ms respectively. Additionally, we found a slow component τrec2 of 13.0±6.0 and 19.0±4.0 ms, which comprised 7 and 19% of the current amplitude. 2,6-dimethylphenol slightly increased the time constants of recovery from fast inactivation. τrec1 in the presence of 500 μM 2,6-dimethylphenol was 5.2±2.0 (P<0.01) and 6.0±1.6 ms (P=0.01); τrec2 was 17±8 and 20±10 ms. The amplitude of the slow component of recovery of about 20 ms increased up to 16±7 and 24±7% in the neuronal and skeletal muscle isoform in the presence of drug.
After inactivation, channel reopening is impossible until the channels recover from inactivation, a process that requires several ms after membrane repolarization. Further information about drug effects on the kinetics of drug dissociation from the fast-inactivated state can be derived from the rate at which the channels recover from inactivation in the presence of the drug. The time of membrane repolarization required to remove fast inactivation was assessed at −100 mV by a two-pulse protocol, with varying time intervals (up to 100 ms) between the inactivating prepulse and the test pulse. The time constants of recovery, τrec, were derived from biexponential fits to the fractional current after recovery from inactivation, plotted against the time interval between the inactivating prepulse and the test pulse (Equation 5).
In the absence of the drug, recovery time constants τrec1 obtained for the neuronal and skeletal muscle isoform were 2.4±0.8 and 2.7±1.3 ms respectively. Additionally, we found a slow component τrec2 of 13.0±6.0 and 19.0±4.0 ms, which comprised 7 and 19% of the current amplitude. 2,6-dimethylphenol slightly increased the time constants of recovery from fast inactivation. τrec1 in the presence of 500 μM 2,6-dimethylphenol was 5.2±2.0 (P<0.01) and 6.0±1.6 ms (P=0.01); τrec2 was 17±8 and 20±10 ms. The amplitude of the slow component of recovery of about 20 ms increased up to 16±7 and 24±7% (500 μM 2,6-dimethylphenol). This means that recovery from inactivated channel block at a hyperpolarized holding potential (−100 mV) should be too fast to accumulate relevant frequency-dependent block at stimulating frequencies lower than 50 Hz. Time course of recovery of sodium currents in the absence and presence of drug in both isoforms is illustrated in Figure 4B.
The accumulation of block during trains of depolarizing pulses suggests that the interval between pulses is too short to allow recovery of sodium channel availability. In order to derive an estimate of the kinetics of drug binding and unbinding during the interpulse interval, we applied series of 1–10 ms depolarizing pulses from −100 mV to 0 mV at high frequencies (10, 50 and 100 Hz). Frequency-dependent block was defined as the additional reduction in INa+ for the last pulse relative to the first pulse in a test train in the presence of drug. In control conditions, the amplitude of the last pulse relative to the first pulse in a test train was 99±2% at 10 and 50 Hz and 96±3% at 100 Hz, due to incomplete recovery from inactivation at very high stimulating frequencies. 2,6-dimethylphenol did not induce frequency-dependent block over 5% up to a stimulating frequency of 50 Hz. During a 100 Hz train, the additional fall relative to the first pulse was 6.0±0.2% (rat brain IIA) and 5.0±0.3% (hSkM1) in 500 μM 2,6-dimethylphenol.
Discussion
The main result of this study is that the potency of 2,6-dimethylphenol in blocking heterologously expressed voltage-operated sodium channels in skeletal muscle and brain is comparable to the potency of the parent compound lidocaine (Balser et al., 1996b; Fan et al., 1996; Pugsley & Goldin, 1998; Wright et al., 1997). In analogy to lidocaine-like local anaesthetics, the blocking potency of 2,6-dimethylphenol strongly depends on the kinetic state of the channel (Bean et al., 1983; Balser et al., 1996b; Fan et al., 1996; Wright et al., 1997; Scheuer, 1999). The estimated affinity of 2,6-dimethylphenol for the inactivated state, as revealed by the drug-induced voltage shifts in the availability curves (see Figure 3), was about 20 fold higher than the affinity for the resting state. Consequently, blocking effects of either lidocaine or 2,6-dimethylphenol are determined by the fraction of inactivated channels, which in turn depends on the respective membrane potential on the one hand and the isoform-specific voltage-dependence of inactivation on the other (Wright et al., 1997; 1999). At −70 mV holding potential, about 25% of the skeletal muscle channels undergo inactivation, yet only 5% of neuronal sodium channels do so. This explains why the difference in blocking potency at −70 mV with respect to −150 mV holding potential was significant in the skeletal muscle isoform, but not in the neuronal isoform.
While 2,6-dimethylphenol parallels the effects of lidocaine with respect to its higher binding affinity to inactivated channels, the equilibration between resting and inactivated states occurs on a faster time scale. After membrane repolarization, recovery from inactivated channel block in the presence of 2,6-dimethylphenol is complete within 30–40 ms after membrane repolarization (see Figure 4), whereas lidocaine-bound channels require 300 ms for complete recovery (Fan et al., 1996). This rapid recovery from inactivated channel block explains the lack of use-dependent block seen with 2,6-dimethylphenol, in contrast to lidocaine, which elicits substantial use-dependent block during a 10 Hz train (Fan et al., 1996).
A similar lack of use-dependent block has been described for the local anaesthetic benzocaine. Studies on various benzocaine homologues support the hypothesis that the relatively fast dissociation of benzocaine from its receptor site, which accounts for this lack of use-dependent block, is related to its low molecular weight and neutral charge rather than to a hypothetical different binding site for benzocaine. Alkylation of functional groups at the phenyl ring, which increases the hydrophobicity and the molecular weight of the compound, enabled a benzocaine homologue to elicit use-dependent block (Quan et al., 1996). If the same holds for 2,6-dimethylphenol, we can assume that compounds with larger hydrophobic side chains will be more easily ‘trapped' within the hydrophobic binding domains of the local anaesthetic receptor site (Wang et al., 1993). Indeed, the anaesthetic propofol (2,6-diisopropylphenol), in which the methyl groups in the ortho position are replaced by bulkier isopropyl groups, elicited frequency-dependent block in neuronal sodium channels during a 5 Hz train (Rehberg & Duch, 1999). Thus, the presence or absence of use-dependent block by either 2,6-dimethylphenol and propofol or local anaesthetics reflects the physicochemical properties of the compounds and does not necessarily exclude the possibility that all compounds share a common local anaesthetic binding site on the sodium channel. Further studies should address the question of whether 2,6-dimethylphenol and lidocaine share the same binding site on the sodium channel. The identification of a common binding site might have important implications for the binding of other structurally closely related compounds that target voltage-operated sodium channels with high affinity, among them the anaesthetic propofol (Haeseler et al., 2001; Rehberg & Duch, 1999).
In a manner comparable to that of benzocaine (Quan et al., 1996), 2,6-dimethylphenol slightly accelerated the decaying phase of sodium currents during a depolarizing pulse in the skeletal muscle isoform. In the case of lidocaine, this effect was discernable only in mutated channels with severely impaired fast inactivation (Balser et al., 1996a). As the acceleration of the current decay was not observed in mutated channels where the inactivation mechanism was completely disrupted, the authors concluded that this effect was linked to the inactivation mechanism and could not be explained by open channel block. Thus, although we cannot rule out open channel block from our own data, the results obtained with 2,6-dimethylphenol are generally consistent with current models that describe the effects of local anaesthetics on the current decay phase as a result of drug-induced acceleration and stabilization of the inactivated state (Balser et al., 1996a). In the case of 2,6-dimethylphenol and benzocaine, this effect occurs on a time scale that is fast enough to interfere with the time course of normal channel inactivation.
Our results show that the block of sodium channels by 2,6-dimethylphenol mimics important features of sodium channel blockade by lidocaine and other local anaesthetics and antiarrhythmic drugs. We therefore suggest that the substituted benzene ring constitutes the smallest active compound of the lidocaine molecule that determines its state-dependent interaction with the sodium channel. As has been shown earlier using the example of phenylmethylamine and structurally related aromatic compounds (Zamponi & French, 1994), the interaction with voltage-operated sodium channels does not necessarily require the unsubstituted phenolic hydroxyl group.
In contrast, the substitution of this functionally active group at the benzene ring has important implications for additional effects of the compounds on different ion channels and receptors. While local anaesthetics and 2,6-dimethylphenol both block voltage-operated sodium channels, they differ profoundly in their effects on the major receptor for inhibitory neurotransmission in the mammalian brain, the γ- aminobutyric acid (GABAA-) receptor. Local anaesthetics like lidocaine, bupivacaine, procaine, benzocaine, and cocaine inhibit GABA-induced currents, a mechanism that might underlie their central nervous toxicity (Hara et al., 1995; Sugimoto et al., 2000; Ye et al., 1997). In contrast, 2,6-dimethylphenol and a series of other phenol derivatives potentiate currents elicited by low concentrations of GABA and activate Cl− currents through GABAA receptors in the absence of GABA (Krasowski et al., 2001; Mohammadi et al., 2001; Trapani et al., 1998). This effect, which is supposed to underlie the sedative properties of certain phenol derivatives in vivo (James & Glen, 1980), strongly depends on the hydrogen bond donor/acceptor properties of the phenolic hydroxyl group (Trapani et al., 1998). Consequently, structure–function analysis of the dual effects of phenol derivatives on voltage-operated sodium channels on the one hand and the GABAA receptor–ionophore complex on the other might yield compounds that combine local anaesthetic with sedative–anticonvulsant actions and make them an interesting alternative in the development of local anaesthetics and anticonvulsive drugs.
Acknowledgments
We are indebted to Prof. Lehmann-Horn, Ulm, Germany, for providing us with transfected cells, to Birgitt Nentwig, Department of Anaesthesiology, Hannover, Germany, for taking care of the cell culture, to Dr Hans-Peter Reiffen, Department of Anaesthesiology, Hannover, Germany, for help with software problems, to Jobst Kilian and Andreas Niesel, Department of Neurology, Hannover, Germany, for technical support, to Dr Julian Grosskreutz, Department of Neurology, Hannover, Germany, for helpful discussions on the manuscript, and to Wolfgang Heyde, Clinical Pharmacy, for providing the stock solution of 2,6-dimethylphenol.
Abbreviations
- HEK 293
human embryonic kidney cell
- Kd
dissociation constant from the inactivated state
- rat brain IIA
neuronal sodium channel from adult rat brain
- hSkM1
human skeletal muscle sodium channel
- ΔV0.5
voltage shift in the midpoint of the steady-state availability curve
References
- BALSER J.R., BRADLEY NUSS H., ORIAS D.W., JOHNS D.C., MARBAN E., TOMASELLI G.F., LAWRENCE J.H. Local anesthetics as effectors of allosteric gating. J. Clin. Invest. 1996a;98:2874–2886. doi: 10.1172/JCI119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALSER J.R., BRADLEY NUSS H., ROMASHKO D.N., MARBAN E., TOMASELLI G.F. Functional consequences of lidocaine binding to slow-inactivated sodium channels. J. Gen. Physiol. 1996b;107:643–658. doi: 10.1085/jgp.107.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAN B.P., COHEN C.J., TSIEN R.W. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BECKH S., NODA M., LÜBBERT H., NUMA S. Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J. 1989;8:3611–3616. doi: 10.1002/j.1460-2075.1989.tb08534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CATTERALL W.A. Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 1992;72 Suppl:15–48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- CHAHINE M., BENNETT P., GEORGE A., HORN R. Functional expression and properties of the human skeletal muscle sodium channel. Pflügers Arch. 1994;427:136–142. doi: 10.1007/BF00585952. [DOI] [PubMed] [Google Scholar]
- EHRING G.R., MOYER J.W., HONDEGHEM L.M. Quantitative structure activity studies of antiarrhythmic properties in a series of lidocaine and procainamide derivatives. J. Pharmacol. Exp. Therapeut. 1988;244:479–492. [PubMed] [Google Scholar]
- FAN Z., GEORGE A.L., KYLE J.W., MAKIELSKI J.C. Two human paramyotonia congenita mutations have opposite effects on lidocaine block of Na+ channels expressed in a mammalian cell line. J. Physiol. 1996;496:275–286. doi: 10.1113/jphysiol.1996.sp021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEORGE A., KOMISAROF J., KALLEN R., BARCHI R. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Ann. Neurol. 1992;31:131–137. doi: 10.1002/ana.410310203. [DOI] [PubMed] [Google Scholar]
- GRAHAM F.L., VAN DER EB A.J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- HAESELER G., PETZOLD J., HECKER H., WÜRZ A., DENGLER R., PIEPENBROCK S., LEUWER M. Succinylcholine metabolite succinic acid alters steady-state activation in muscle sodium channels. Anesthesiology. 2000;92:1385–1392. doi: 10.1097/00000542-200005000-00029. [DOI] [PubMed] [Google Scholar]
- HAESELER G., STÖRMER M., BUFLER J., DENGLER R., HECKER H., PIEPENBROCK S., LEUWER M. Propofol blocks skeletal muscle sodium channels in a voltage-dependent manner. Anesth. Analg. 2001;92:1192–1198. doi: 10.1097/00000539-200105000-00021. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HARA M., KAI Y., IKEMOTO Y. Local anesthetics reduce the inhibitory neurotransmitter-induced current in dissociated hippocampal neurons of the rat. Eur. J. Pharmacol. 1995;283:83–89. doi: 10.1016/0014-2999(95)00293-t. [DOI] [PubMed] [Google Scholar]
- HODGKIN A.L., HOROWICZ P. The influence of potassium and chloride ions on the membrane potential of single muscle fibres. J. Physiol. 1959;148:127–160. doi: 10.1113/jphysiol.1959.sp006278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAMES R., GLEN J.B. Synthesis, biological evaluation, and preliminary structure-activity considerations of a series of alkylphenols as intravenous anesthetic agents. J. Med. Chem. 1980;23:1350–1357. doi: 10.1021/jm00186a013. [DOI] [PubMed] [Google Scholar]
- KRASOWSKI M.D., JENKINS A., FLOOD P., KUNG A.Y., HOPFINGER A.J., HARRISON N.L. General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of γ-aminobutyric acid (GABA) current at the GABAA receptor but not with lipid solubility. J. Pharmacol. Exp. Therapeut. 2001;297:338–351. [PubMed] [Google Scholar]
- MAURER W.Multiple comparisons in drug clinical trials and preclinical assays: a priori ordered hypotheses Testing principles in clinical and preclinical trials 1995Stuttgart: Fischer; 3–8.ed. Vollmar, J. pp [Google Scholar]
- MITROVIC N., GEORGE A.L., HEINE R., WAGNER S., PIKA U., HARTLAUB U., ZHOU M., LERCHE H., FAHLKE C., LEHMANN-HORN F. K+-aggravated myotonia: destabilization of the inactivated state of the human muscle sodium channel by the V1589M mutation. J. Physiol. 1994;478:395–402. doi: 10.1113/jphysiol.1994.sp020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOHAMMADI B., HAESELER G., LEUWER M., DENGLER R., KRAMPFL K., BUFLER J. Structural requirements of phenol derivatives for direct activation of chloride currents via GABAA-receptors. Eur. J. Pharmacol. 2001;421:85–91. doi: 10.1016/s0014-2999(01)01033-0. [DOI] [PubMed] [Google Scholar]
- PUGSLEY M.K., GOLDIN A.L. Effects of bisaramil, a novel class I antiarrhythmic agent, on heart, skeletal muscle and brain Na+ channels. Eur. J. Pharmacol. 1998;342:93–104. doi: 10.1016/s0014-2999(97)01420-9. [DOI] [PubMed] [Google Scholar]
- QUAN C., MOK W.M., WANG G.K. Use-dependent inhibition of Na+ currents by benzocaine homologs. Biophys. J. 1996;70:194–201. doi: 10.1016/S0006-3495(96)79563-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REHBERG B., DUCH D. Suppression of central nervous system sodium channels by propofol. Anesthesiology. 1999;91:512–520. doi: 10.1097/00000542-199908000-00026. [DOI] [PubMed] [Google Scholar]
- SARKAR S.N., ADHIKARI A., SIKDAR S.K. Kinetic characterization of rat brain type IIA sodium channel α-subunit stably expressed in a somatic cell line. J. Physiol. 1995;488:633–645. doi: 10.1113/jphysiol.1995.sp020996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHEUER T. A revised view of local anesthetic action: What channel state is really stabilized. J. Gen. Physiol. 1999;113:3–6. doi: 10.1085/jgp.113.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUGIMOTO M., UCHIDA I., FUKAMI S., TAKENOSHITA M., MASHIMO T., YOSHIYA I. The α and γ subunit-dependent effects of local anesthetics on recombinant GABAA receptors. Eur. J. Pharmacol. 2000;401:329–337. doi: 10.1016/s0014-2999(00)00463-5. [DOI] [PubMed] [Google Scholar]
- TRAPANI G., LATROFA A., FRANCO M., ALTOMARE C., SANNA E., USALA M., BIGGIO G., LISO G. Propofol analogues. Synthesis, relationships between structure and affinity at GABAA receptor in rat brain, and differential electrophysiological profile at recombinant human GABAA receptors. J. Med. Chem. 1998;41:1846–1854. doi: 10.1021/jm970681h. [DOI] [PubMed] [Google Scholar]
- WANG D.W., GEORGE A.L., BENNETT P.B. Comparison of heterologously expressed human cardiac and skeletal muscle sodium channels. Biophys. J. 1996;70:238–245. doi: 10.1016/S0006-3495(96)79566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG G.K., SIMON R., BELL D., MOK W.M., WANG S.-Y. Structural derminants of quaternary ammonium blockers for batrachotoxin-modified Na+ channels. Mol. Pharmacol. 1993;44:667–676. [PubMed] [Google Scholar]
- WRIGHT S.N., WANG S.Y., KALLEN R.G., WANG G.K. Differences in steady-state inactivation between Na channel isoforms affect local anaesthetic binding affinity. Biophys. J. 1997;73:779–788. doi: 10.1016/S0006-3495(97)78110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WRIGHT S.N., WANG S.Y., XIAO Y.F., WANG G.K. State-dependent cocaine block of sodium channel isoforms, chimeras, and channels coexpressed with the β1 subunit. Biophys. J. 1999;76:233–245. doi: 10.1016/S0006-3495(99)77192-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE J.H., LIU P.L., WU W.H., MCARDLE J.J. Cocaine depresses GABAA current of hippocampal neurons. Brain Res. 1997;770:169–175. doi: 10.1016/s0006-8993(97)00782-8. [DOI] [PubMed] [Google Scholar]
- ZAMPONI G.W., FRENCH R.J. Dissecting lidocaine action: Diethylamide and phenol mimic separate modes of lidocaine block of sodium channels from heart and rat skeletal muscle. Biophys. J. 1993;65:2335–2347. doi: 10.1016/S0006-3495(93)81292-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAMPONI G.W., FRENCH R.J. Amine blockers of the cytoplasmatic mouth of sodium channels: a small structural change can abolish voltage dependence. Biophys. J. 1994;67:1015–1027. doi: 10.1016/S0006-3495(94)80567-3. [DOI] [PMC free article] [PubMed] [Google Scholar]