

Abstract
Studies in isolated cells overexpressing ACE and bradykinin type 2 (B2) receptors suggest that ACE inhibitors potentiate bradykinin by inhibiting B2 receptor desensitization, via a mechanism involving protein kinase C (PKC) and phosphatases. Here we investigated, in intact porcine coronary arteries, endothelial ACE/B2 receptor ‘crosstalk' as well as bradykinin potentiation through neutral endopeptidase (NEP) inhibition.
NEP inhibition with phosphoramidon did not affect the bradykinin concentration-response curve (CRC), nor did combined NEP/ACE inhibition with omapatrilat exert a further leftward shift on top of the ≈10 fold leftward shift of the bradykinin CRC observed with ACE inhibition alone.
In arteries that, following repeated exposure to 0.1 μM bradykinin, no longer responded to bradykinin (‘desensitized' arteries), the ACE inhibitors quinaprilat and angiotensin-(1-7) both induced complete relaxation, without affecting the organ bath fluid levels of bradykinin. This phenomenon was unaffected by inhibition of PKC or phosphatases (with calphostin C and okadaic acid, respectively).
When using bradykinin analogues that were either completely or largely ACE-resistant ([Phe8Ψ(CH2-NH)Arg9]-bradykinin and [ΔPhe5]-bradykinin, respectively), the ACE inhibitor-induced shift of the bradykinin CRC was absent, and its ability to reverse desensitization was absent or significantly reduced, respectively. Caveolar disruption with filipin did not affect the quinaprilat-induced effects. Filipin did however reduce the bradykinin-induced relaxation by ≈25–30%, thereby confirming that B2 receptor-endothelial NO synthase (eNOS) interaction occurs in caveolae.
In conclusion, in porcine arteries, in contrast to transfected cells, bradykinin potentiation by ACE inhibitors is a metabolic process, that can only be explained on the basis of ACE-B2 receptor co-localization on the endothelial cell membrane. NEP does not appear to affect the bradykinin levels in close proximity to B2 receptors, and the ACE inhibitor-induced bradykinin potentiation precedes B2 receptor coupling to eNOS in caveolae.
Keywords: ACE, bradykinin, B2 receptor, caveolae, coronary artery, neutral endopeptidase, nitric oxide
Introduction
Bradykinin accumulation is believed to contribute to the beneficial effects of angiotensin-converting enzyme (ACE) inhibitors in hypertension and heart failure, although elevated bradykinin levels have not always been found during ACE inhibitor treatment (Campbell et al., 1999). Recent studies in isolated cells propose that ACE inhibitors potentiate bradykinin beyond blocking its hydrolysis, by inhibiting desensitization of its receptor (Minshall et al., 1997; Benzing et al., 1999). The mechanism behind this phenomenon is currently unclear, but may involve ‘crosstalk' between ACE and bradykinin type 2 (B2) receptors (Minshall et al., 1997; Benzing et al., 1999; Marcic et al., 1999; 2000; Marcic & Erdös, 2000). Although these findings were subsequently confirmed in intact coronary arteries (Danser et al., 2000; Tom et al., 2001; Mombouli et al., 2002), it has recently been proposed, on the basis of experiments performed in isolated perfused rat Langendorff hearts (Dendorfer et al., 2000) and rabbit jugular veins (Dendorfer et al., 2001) that inhibition of ACE in the immediate vicinity of B2 receptors (e.g., in caveolae) is a more likely explanation for the potentiation of bradykinin by ACE inhibitors than a direct interaction between ACE and B2 receptors. In support of this concept, aminopeptidase P inhibition resulted in a similar leftward shift of the bradykinin concentration-response curve as ACE inhibition (Dendorfer et al., 2000), and no ACE inhibitor-induced leftward shift was observed when studying B2 receptor-mediated vasoconstriction in response to ACE-resistant bradykinin analogues (Dendorfer et al., 2001; Gobeil et al., 2002). However, the latter data were obtained in endothelium-denuded vessels, i.e. a preparation that lacks the B2 receptor- and ACE-expressing endothelial cells that are responsible for the ACE inhibitor-induced potentiation of B2 receptor-mediated coronary vasorelaxation (Danser et al., 2000; Tom et al., 2001; Mombouli et al., 2002).
Neutral endopeptidase 24.11 (NEP) is a membrane-bound metalloprotease with a catalytic unit similar to that of ACE. It is widely distributed in endothelial and vascular smooth muscle cells (Llorens-Cortes et al., 1992; Soleilhac et al., 1992; Graf et al., 1993; Wang et al., 1994; Gonzalez et al., 1998). NEP catalyzes the degradation of a number of endogenous vasoactive peptides, including bradykinin and angiotensin (Graf et al., 1993; Kokkonen et al., 1999; Raut et al., 1999; Blais et al., 2000). Recent studies have shown that combined inhibition of ACE and NEP with so-called vasopeptidase inhibitors is more cardioprotective than ACE inhibition alone (Rouleau et al., 2000; D'uscio et al., 2001).
It was the aim of the present study to further investigate the possibility that the ACE inhibitor-induced potentiation of bradykinin in intact (i.e., endothelium-containing) porcine coronary arteries has a non-metabolic origin. First, we studied whether bradykinin potentiation also occurs with NEP inhibitors, and whether dual ACE/NEP inhibition results in even further potentiation of bradykinin. Second, we studied the effect of protein kinase C (PKC) and phosphatase inhibition (with calphostin C and okadaic acid, respectively) on the ACE inhibitor-induced bradykinin potentiation, since such inhibition was found to fully eliminate the ACE/B2 receptor crosstalk in Chinese hamster ovary (CHO) cells (Marcic & Erdös, 2000). Third, we studied whether potentiation occurs when using truly ACE-resistant bradykinin analogues. This is particularly important, since most studies that investigated ACE-B2 receptor crosstalk so far made use of a bradykinin analogue (Hyp3-Tyr(Me)8-bradykinin) that was recently shown not to be ACE-resistant at all (Dendorfer et al., 2001; Gobeil et al., 2002). Finally, in view of the possibility that ACE and B2 receptors co-localize in caveolae, we studied the effect of caveolar disruption (with filipin, cyclodextrin or nystatin) on the ACE inhibitor-induced bradykinin potentiation.
Methods
Chemicals
Angiotensin-(1-7) (Ang-(1-7)), bradykinin, calphostin C, captopril, cyclodextrin, 9,11-dideoxy-11α, 9α-epoxymethano-prostaglandin F2α (U46619), filipin, S-nitroso-N-acetylpenicillamine (SNAP), nystatin, okadaic acid, phosphoramidon, and substance P were purchased from Sigma (Zwijndrecht, The Netherlands). Quinaprilat was a kind gift of Dr H. van Ingen, Parke-Davis, Hoofddorp, The Netherlands. Omapatrilat was a kind gift of Dr N.C. Trippodo, Bristol-Myers Squibb, Princeton, NJ, U.S.A. [Phe8ψ(CH2-NH)Arg9]-bradykinin (PA-bradykinin) was bought from Calbiochem (Breda, The Netherlands). [ΔPhe5]-bradykinin (DP-bradykinin) was synthesized by a solid-phase method by means of the Boc-strategy (Reissmann et al., 1996). All chemicals were dissolved in water, with the exception of calphostin C and phosphoramidon, which were dissolved in dimethylsulphoxide, and of filipin, which was dissolved in ethanol.
Tissue collection
Porcine coronary arteries were obtained from five 2–3 month-old pigs (Yorkshire x Landrace, weight 10–15 kg) that had been used in in vivo experiments studying the effects of α-adrenoceptor and calcitonin-gene related peptide receptor (ant)agonists or capsaicin under pentobarbital (600 mg, i.v.) anaesthesia (Willems et al., 2001), and from 38 pigs at the local slaughterhouse. The Ethics Committee of the Erasmus MC dealing with the use of animals for scientific experiments approved the protocol for this investigation. Arteries were either removed at the end of the experiment, or after the heart from the slaughterhouse had been brought to the laboratory in cold Krebs bicarbonate solution of the following composition (mM): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and glucose 8.3; pH 7.4. Vessels were stored overnight in cold, oxygenated Krebs bicarbonate solution. They were then cut into segments of approximately 4 mm length, suspended on stainless steel hooks in 15 ml-organ baths containing Krebs bicarbonate solution, aerated with 95% O2/5% CO2, and maintained at 37°C.
Organ bath studies
All vessel segments were allowed to equilibrate for at least 30 min and the organ bath fluid was refreshed every 15 min during this period. Changes in tissue contractile force were recorded with a Harvard isometric transducer (South Natick, MA, U.S.A.). The vessel segments, stretched to a stable force of about 15 mN, were exposed to 30 mM K+ twice. The functional integrity of the endothelium was verified by observing relaxation to 1 nM substance P after preconstriction with 1 μM U46619. Subsequently, the tissue was exposed to 100 mM K+ to determine the maximal contractile response to K+. The segments were then allowed to equilibrate in fresh organ bath fluid for 30 min. Thereafter, the following experiments were performed.
First, vessels were preincubated for 30 min in the presence or absence of 10 μM quinaprilat (ACE inhibitor) and/or 10 μM captopril (ACE inhibitor), 10 μM omapatrilat (vasopeptidase inhibitor), 1 μM phosphoramidon (NEP inhibitor), 10 μM Ang-(1–7) (inhibitor of ACE C-domain) (Tom et al., 2001), 4 μg ml−1 filipin, 2% cyclodextrin, or 20 μg ml−1 nystatin. Next, vessels were preconstricted with 1 μM U46619, and concentration-response curves were constructed to bradykinin, PA-bradykinin, or DP-bradykinin.
Second, vessels were pre-incubated for 30 min with or without 1 μM calphostin C, 0.5 μM okadaic acid, 4 μg ml−1 filipin, 2% cyclodextrin, or 20 μg ml−1 nystatin. Vessels were then preconstricted with 1 μM U46619 and exposed multiple times (to induce desensitization) to 0.1 μM bradykinin, 0.1 μM PA-bradykinin or 0.03 μM DP-bradykinin. Each next exposure was started as soon as the effect of the previous exposure had disappeared. When bradykinin no longer exerted a vasodilatory effect, 10 μM quinaprilat, 10 μM omapatrilat, or 1 μM phosphoramidon was added to the organ bath.
Finally, to study the metabolism of bradykinin under our experimental conditions, 1-ml fluid samples were taken from the organ bath at 0, 30, 60 and 120 min after the addition of the highest bradykinin concentration (1 μM) in the absence or presence of 10 μM quinaprilat. The samples were immediately supplemented with 1% trifluoroacetic acid, and stored until analysis at −70°C. Bradykinin was determined by high performance liquid chromatography and photometric detection at 210 nm (Dendorfer et al., 1997).
Statistical analysis
Data are given as mean±s.e.mean and expressed as a percentage of the contraction in response to U46619. Differences between arteries obtained from experimental pigs and from slaugterhouse pigs were not observed (data not shown), and data for the two groups were therefore combined. Concentration-response curves were analysed using the logistic function described by de lean et al. (1978) to obtain pEC50 (−10log EC50) values, EC50 representing the concentration at which 50% of the maximal relaxant effect has been reached. Statistical analysis was performed by Analysis of Variance (ANOVA), followed by post hoc evaluation according to Dunnett. P values <0.05 were considered significant.
Results
Potentiation of bradykinin by inhibitors of ACE and/or NEP
Bradykinin relaxed preconstricted porcine coronary arteries in a concentration-dependent manner (pEC50=7.95±0.03, n=5; Figure 1). Quinaprilat and omapatrilat, but not phosphoramidon, shifted the bradykinin concentration-response curve approximately 10 fold to the left (pEC50's respectively 9.22±0.05 (P<0.05 vs control), 9.28±0.08 (P<0.05 vs control) and 8.06±0.13, n=5 for each condition). These drugs were without effect in the absence of bradykinin, indicating that porcine coronary arteries do not contain endogenous bradykinin.
Figure 1.
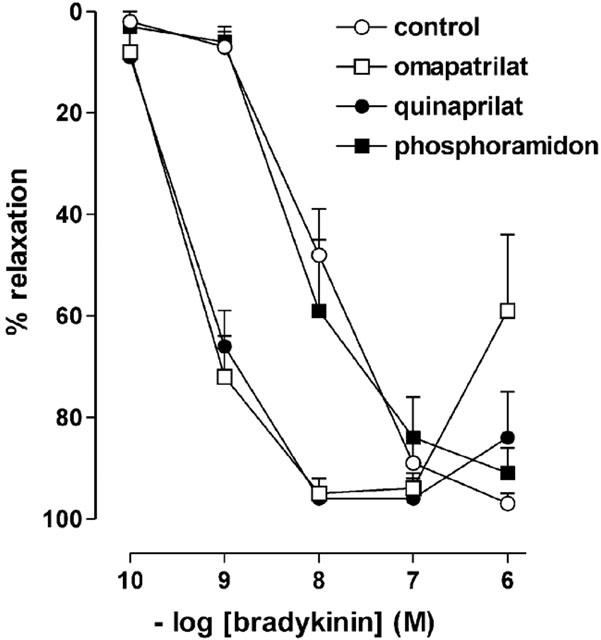
Relaxations of porcine coronary arteries, preconstricted with 1 μM U46619, to bradykinin in the absence (control) or presence of 10 μM quinaprilat, 10 μM omapatrilat or 1 μM phosphoramidon. Data (mean±s.e.mean of five experiments) are expressed as a percentage of the contraction induced by U46619.
Repeated exposure of preconstricted porcine coronary arteries to bradykinin produced progressively smaller responses (Figure 2, n=5). Its effects lasted less than 10 min, and contractility returned to preconstriction level within 15–20 min. The response to the third bradykinin dose was <50% of the response to the first bradykinin dose. Quinaprilat or omapatrilat, added to the organ bath after the effect of the third bradykinin dose had disappeared, completely restored the relaxant effect of bradykinin, whereas phosphoramidon was without effect (n=5 for each inhibitor). Bradykinin added to the organ bath after the effect of the enzyme inhibitors had disappeared exerted no effect (data not shown).
Figure 2.
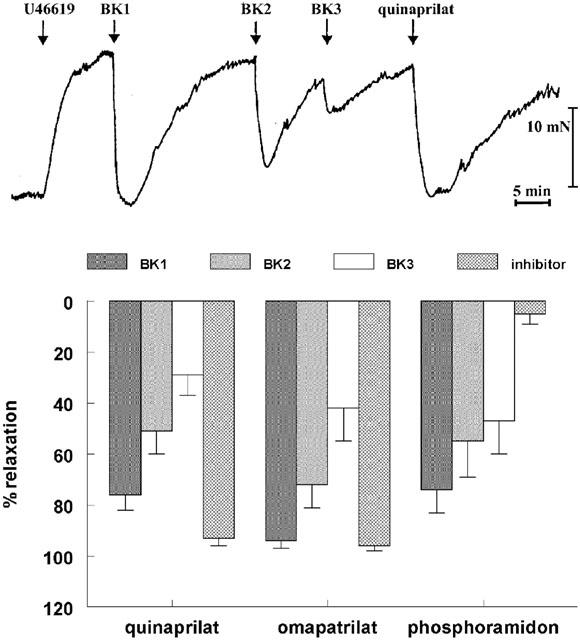
Relaxations of porcine coronary arteries, following preconstriction with 1 μM U46619, to three consecutive bradykinin doses (0.1 μM; BK1, BK2, BK3), followed by 10 μM quinaprilat, 10 μM omapatrilat, or 1 μM phosphoramidon. Top, original tracing; bottom, mean±s.e.mean of five experiments (data are expressed as a percentage of the contraction induced by U46619).
Does inhibition of phosphatases or PKC block the effect of quinaprilat in desensitized vessels?
The relaxant effect of quinaprilat in desensitized porcine coronary arteries was not altered in the presence of calphostin C or okadaic acid (Figure 3).
Figure 3.
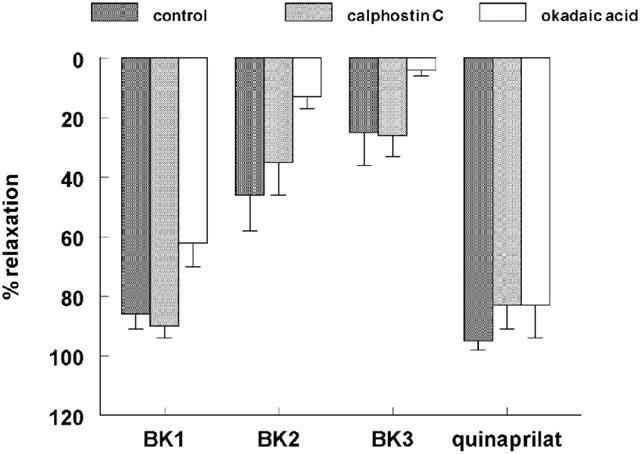
Relaxations of porcine coronary arteries, following preconstriction with 1 μM U46619, to three consecutive bradykinin doses (0.1 μM; BK1, BK2, BK3), followed by 10 μM quinaprilat, in the absence (control) or presence of 1 μM calphostin C or 0.5 μM okadaic acid. Data (mean±s.e.mean of five experiments) are expressed as a percentage of the contraction induced by U46619.
Do ACE inhibitors potentiate ACE-resistant bradykinin analogues?
In preconstricted porcine coronary arteries, Ang-(1–7), like quinaprilat, shifted the bradykinin concentration-response curve to the left (Figure 4), although its effect was smaller than that of quinaprilat (pEC50's 8.40±0.09 (n=7) and 8.85±0.06 (n=23) vs 7.94±0.05 for control (n=28); P<0.05). This relates to the fact that Ang- (1–7) inhibits the ACE C-domain only, whereas quinaprilat inhibits both the C- and N-domain of ACE (Tom et al., 2001). PA-bradykinin (pEC50 8.05±0.05; n=23) and DP-bradykinin (pEC50 8.64±0.19; n=6) relaxed preconstricted porcine coronary arteries to a similar degree as bradykinin (Figure 4). Their relaxant effects were not affected by either Ang-(1–7), quinaprilat, or captopril (Figure 4). Moreover, in the repeated exposure experiments, in contrast with bradykinin, PA-bradykinin induced relaxation only once, and the application of subsequent doses of PA-bradykinin (Figure 5), quinaprilat (Figure 5) or Ang-(1–7) (data not shown) exerted no effect. DP-bradykinin, like bradykinin, was capable of exerting multiple relaxations. However, the effect of quinaprilat in DP-bradykinin-desensitized vessels was significantly smaller than in bradykinin-desensitized preparations (Figure 5). Finally, adding 10 μM bradykinin to vessels that no longer responded to 0.1 μM bradykinin induced a vasorelaxation (74±10%, n=5) that was as large as that induced by quinaprilat.
Figure 4.
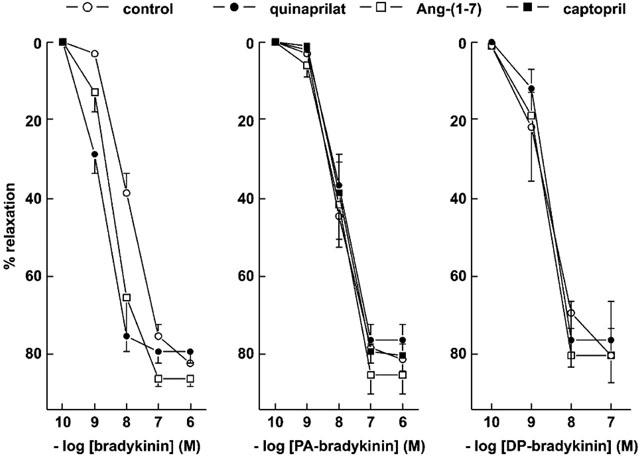
Relaxations of porcine coronary arteries, preconstricted with 1 μM U46619, to bradykinin, PA-bradykinin or DP-bradykinin in the absence (control) or presence of 10 μM quinaprilat, 10 μM captopril or 10 μM angiotensin-(1–7). Data (mean±s.e.mean of 5–28 experiments) are expressed as a percentage of the contraction induced by U46619.
Figure 5.
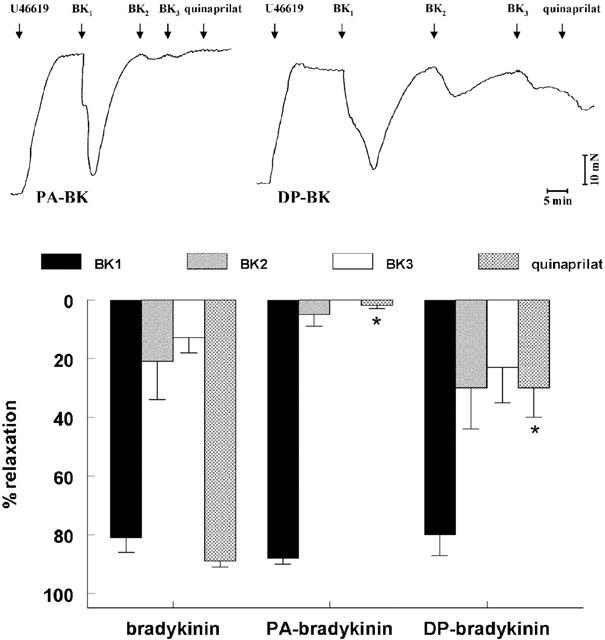
Relaxations of porcine coronary arteries, following preconstriction with 1 μM U46619, to three consecutive bradykinin, PA-bradykinin or DP-bradykinin doses (0.1, 0.1 and 0.03 μM, respectively; BK1, BK2, BK3), followed by 10 μM quinaprilat. Top, original tracings of experiments with PA-bradykinin and DP-bradykinin; bottom, mean±s.e.mean of 5–12 experiments (data are expressed as a percentage of the contraction induced by U46619). *P<0.01 vs bradykinin.
Bradykinin metabolism during incubation with porcine coronary arteries
Bradykinin disappeared mono-exponentially from the organ bath fluid (Figure 6), with a half life of 96±8 min (n=5). Quinaprilat marginally (P=NS) increased the half life to 126±12 min.
Figure 6.
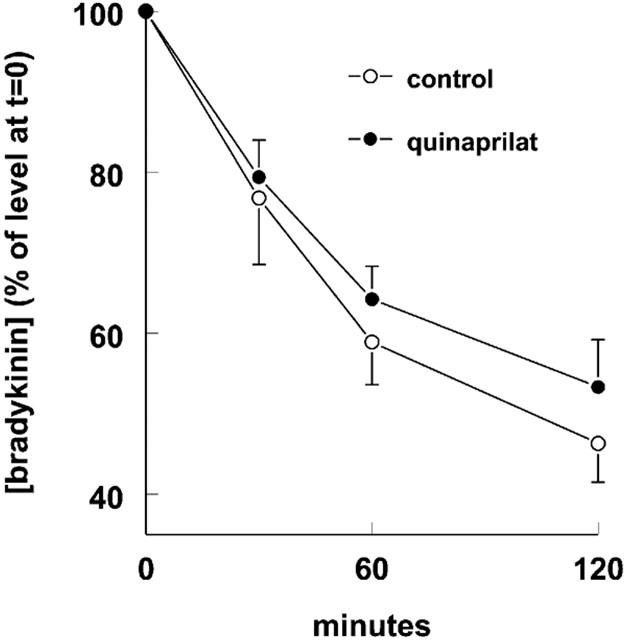
Metabolism of bradykinin by porcine coronary artery rings during incubation of 15 ml organ bath fluid at 37°C with 1 μM bradykinin in the absence (control) or presence of 10 μM quinaprilat. Data are mean±s.e.mean of five experiments and have been expressed as a percentage of the level at t=0 (0.70±0.04 and 0.82±0.04 μM without and with quinaprilat).
Effect of caveolar disruption on bradykinin potentiation by quinaprilat
Pretreatment with filipin reduced the maximal relaxant effect of bradykinin and PA-bradykinin by approximately 25–30% (Figure 7; P<0.05), but did not alter their potencies (7.63±0.19 (n=6) and 7.91±0.20 (n=7); P>0.05 vs without filipin). Filipin did not affect the response to the endothelium-independent relaxant SNAP (Figure 8). Neither cyclodextrin, nor nystatin (20 μg ml−1), affected the bradykinin, PA-bradykinin or SNAP concentration-response curves, although there was a tendency for nystatin at this concentration to reduce (P=NS) the maximal relaxant effect of SNAP. At a concentration of 50 μg ml−1, nystatin did significantly reduce the SNAP-induced relaxation (data not shown).
Figure 7.
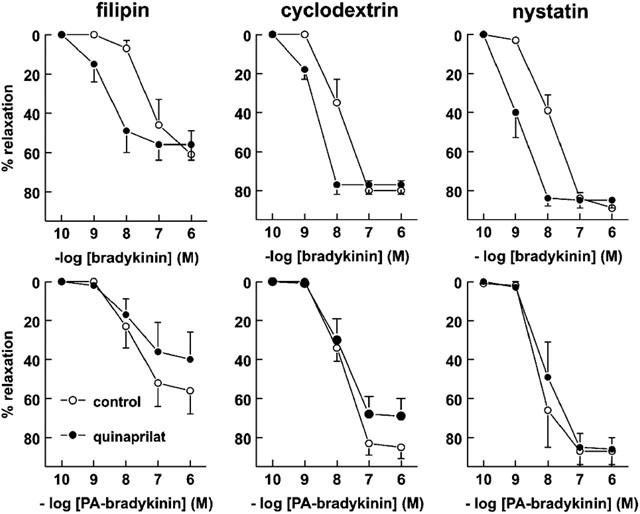
Relaxations of porcine coronary arteries, preconstricted with 1 μM U46619, to bradykinin (top panels) or PA-bradykinin (bottom panels) in the absence (control) or presence of 10 μM quinaprilat, following pretreatment with 4 μg ml−1 filipin, 2% cyclodextrin or 20 μg ml−1 nystatin. Data (mean±s.e.mean of 4–10 experiments) are expressed as a percentage of the contraction induced by U46619.
Figure 8.
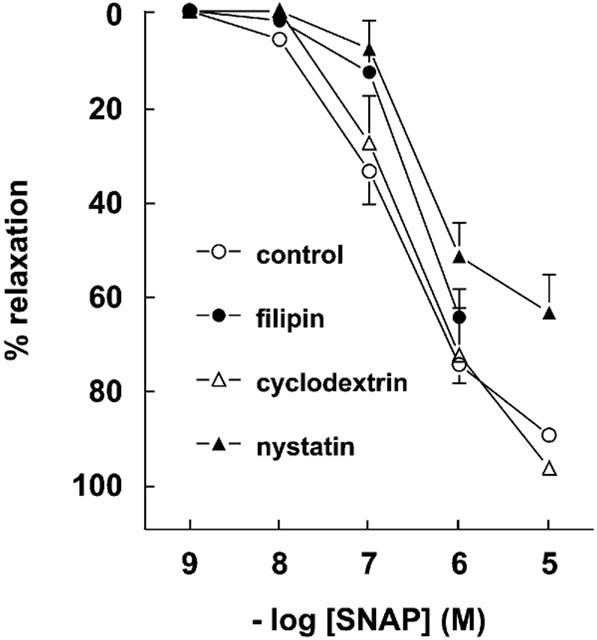
Relaxations of porcine coronary arteries, preconstricted with 1 μM U46619, to SNAP in the absence (control) or presence of 4 μg ml−1 filipin, 2% cyclodextrin or 20 μg ml−1 nystatin. Data (mean±s.e.mean of 5–21 experiments) are expressed as a percentage of the contraction induced by U46619.
Quinaprilat caused a leftward shift of the bradykinin concentration-response curve in the presence of all caveolae-disrupting agents, and the shift was fully comparable to that observed in the absence of these agents (Figures 4 and 7). Quinaprilat did not affect the PA-bradykinin concentration-response curve in the presence of filipin, cyclodextrin and nystatin.
Finally, none of the caveolar disrupting agents prevented the relaxant effect of quinaprilat in desensitized preparations (Figure 9).
Figure 9.
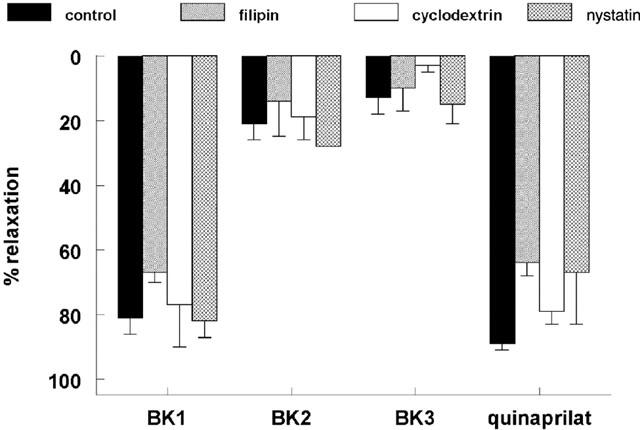
Relaxations of porcine coronary arteries, following preconstriction with 1 μM U46619, to three consecutive bradykinin doses (0.1 μM; BK1, BK2, BK3), followed by 10 μM quinaprilat, in the absence (control) or presence of 4 μg ml−1 filipin, 2% cyclodextrin or 20 μg ml−1 nystatin. Data are mean±s.e.mean of 5–12 experiments and have been expressed as a percentage of the contraction induced by U46619.
Discussion
The present study shows that the ACE inhibitor-induced potentiation of bradykinin in intact porcine coronary arteries has a metabolic origin. The explanation for this potentiation is therefore not that ACE and B2 receptors physically interact, as proposed on the basis of studies in isolated cells overexpressing ACE and B2 receptors (Minshall et al., 1997; Benzing et al., 1999; Marcic et al., 2000; Marcic & Erdös, 2000), but rather that ACE is located in close proximity of B2 receptors, thereby directly determining the bradykinin concentration in the micro-environment of the B2 receptor. As a consequence, the bradykinin concentrations seen by the receptor will only approach those in the organ bath when ACE is inhibited, a phenomenon that can be achieved instantaneously by adding an ACE inhibitor to the organ bath.
An extensive series of experiments, both in transfected cells (Minshall et al., 1997; Benzing et al., 1999; Marcic et al., 2000; Marcic & Erdös, 2000) and in intact vessel preparations (Danser et al., 2000; Tom et al., 2001; Mombouli et al., 2002), supports the idea that bradykinin potentiation by ACE inhibitors is an effect beyond hydrolysis. The data suggest that ACE inhibitors, in an PKC- and phosphatase-dependent manner (Marcic & Erdös, 2000), increase the number of cell surface B2 receptors, thereby preventing/reversing the rapid B2 receptor desensitization that normally occurs upon exposure to bradykinin (Minshall et al., 1997; Marcic et al., 1999, 2000; Marcic & Erdös, 2000; Bachvarov et al., 2001). In order to draw this conclusion, the studies depended on the use of ‘ACE-resistant' bradykinin analogues, in particular Hyp3-Tyr(Me)8-bradykinin. However, in a recent investigation Dendorfer et al. (2001) showed that rabbit ACE cleaves Hyp3-Tyr(Me)8-bradykinin at 71% of bradykinin degradation activity, and this finding was confirmed by Gobeil et al. (2002).
Potentiation of ACE-resistant bradykinin analogues by ACE inhibitors?
In the present study we therefore re-investigated the ACE inhibitor-induced bradykinin potentiation in porcine coronary arteries, using two bradykinin analogues, one (PA-bradykinin) of which was truly ACE-resistant, whereas the other (DP-bradykinin) was cleaved at 9% of bradykinin degradation activity (Dendorfer et al., 2001; Gobeil et al., 2002). We followed two approaches: (1) comparison of the bradykinin concentration-response curves in the absence and presence of an ACE inhibitor, and (2) the application of an ACE inhibitor to desensitized coronary arteries (i.e., arteries that no longer relaxed in response to 0.1 μM bradykinin, a concentration that normally causes complete relaxation). In the latter experimental setup, the addition of the ACE inhibitor immediately results in full relaxation, an effect that depends on the prior addition of bradykinin, since it does not occur in the absence of bradykinin (Danser et al., 2000). Apparently, at the time the ACE inhibitor is added (i.e., approximately 10 min after the last bradykinin dose), the organ bath still contains sufficient bradykinin to allow full relaxation. Our measurement of the bradykinin half life (>90 min) in the current study confirms this assumption. The quinaprilat-induced relaxation of desensitized preparations is not due to unspecific properties of this drug, since similar data have been obtained with a whole range of ACE inhibitors (Danser et al., 2000; Marcic et al., 2000; Gobeil et al., 2002).
The results of both experimental approaches indicate that the effect of ACE inhibition is absent or greatly reduced when using ACE-resistant bradykinin analogues. First, the 10 fold leftward shift of the bradykinin concentration-response curve that normally occurs in the presence of the ACE inhibitors quinaprilat (Danser et al., 2000), captopril (Tom et al., 2001) or perindoprilat (Mombouli et al., 2002) was absent when using PA-bradykinin or DP-bradykinin. Moreover, the leftward shift that we (Tom et al., 2001) and others (Gobeil et al., 2002) observed earlier in the presence of Ang-(1–7), also did not occur in combination with these agonists, indicating that this shift, too, has a metabolic origin. The Ang-(1–7)-induced shift is smaller than the shift induced by quinaprilat and captopril, because Ang-(1–7), at the high concentration that was applied in this study (10 μM), blocks the active centre of only one (the C-terminal domain) of the two homologous domains of the ACE molecule, whereas quinaprilat and captopril at this same concentration block both domains (Tom et al., 2001). These data therefore strongly argue against the idea that a non-ACE related effect of Ang-(1–7) (e.g., an effect mediated via the putative Ang-(1–7) receptor) underlies its bradykinin-potentiating capabilities (Fernandes et al., 2001).
Second, the above described quinaprilat-induced relaxation of desensitized coronary arteries did not occur following PA-bradykinin-induced desensitization, and was significantly reduced after DP-bradykinin-induced desensitization. Complete desensitization was already obtained after the application of 0.1 μM PA-BK, since the addition of subsequent PA-BK doses (increasing the PA-BK concentrations 2 and 3 fold) did not exert further effects. In contrast, desensitization induced by 0.1 μM bradykinin or 0.03 μM DP-bradykinin was less complete, as the application of a second and third dose of these agonists still induced modest relaxations. Desensitization is due to a reduction in the number of B2 receptors on the endothelial cell surface, and/or to exhaustion of post-receptor mechanisms (e.g., NO depletion). In support of the latter, desensitization occurred much more rapidly in the presence of an NO synthase inhibitor (Danser et al., 2000). The lack of effect of quinaprilat after PA-BK suggests that the B2 receptor resensitization that has been detected upon ACE inhibitor administration in isolated cells (Minshall et al., 1997; Marcic et al., 1999; 2000; Marcic & Erdös, 2000), either does not occur or is insufficient in intact coronary arteries.
How then should the ACE inhibitor-induced relaxation following bradykinin desensitization be explained? Quinaprilat does not increase the organ bath fluid levels of bradykinin. In the absence of significant B2 receptor resensitization, this leaves the possibility that quinaprilat affects the bradykinin levels that are seen by the receptor. This explanation implies that ACE is located in close proximity of B2 receptors, and thus, that normally the bradykinin levels in the vicinity of the receptor are below those in the organ bath. In view of the ≈10 fold leftward shift of the bradykinin concentration-response curve in the presence of quinaprilat, it seems reasonable to assume that the bradykinin levels in the micro-environment of the B2 receptor are also ≈10 fold lower than the levels in the organ bath. For a fully ACE resistant bradykinin analogue (PA-bradykinin) such a difference will not exist, whereas for an analogue that is degraded by ACE at <10% of its bradykinin degrading activity (DP-bradykinin) the difference will be much smaller than 10 fold. This explains why we did not observe a significant leftward shift of the DP-bradykinin concentration-response curve, and only a modest relaxation following the addition of quinaprilat to DP-bradykinin desensitized vessels. It also explains why desensitization was already complete after one PA-BK dose (resulting in a concentration of 0.1 μM in the organ bath as well as in the micro-environment of B2 receptors), whereas desensitization remained incomplete after the addition of three subsequent bradykinin doses (resulting in a final concentration of 0.3 μM in the organ bath, and of ≈0.03 μM in the micro-environment of B2 receptors). In fact, incomplete desensitization is a prerequisite for the quinaprilat-induced relaxation, since in completely desensitized preparations a 10 fold (or more) rise of the bradykinin levels in the micro-environment of the receptor will of course have no effect. In agreement with this concept, exposure of bradykinin-desensitized arteries to a 100 fold higher bradykinin concentration (10 μM) resulted in a similar relaxation as the addition of quinaprilat.
Blockade of phosphatases and PKC did not interfere with the quinaprilat-induced relaxation, whereas these agents did prevent the ACE inhibitor-induced bradykinin potentiation in transfected CHO cells that overexpress ACE and B2 receptors (Marcic & Erdös, 2000). One reason for this discrepancy might be that overexpression itself leads to interactions which do not occur at low expression levels.
ACE inhibitor-induced potentiation of B2 receptor-mediated vasoconstriction
Our study is the first to demonstrate a metabolic background for the ACE inhibitor-induced potentiation of bradykinin-mediated vasodilation in intact arteries, i.e, for an interaction at the level of the endothelial cells, the same cells that were used to demonstrate ACE-B2 receptor crosstalk in cell culture studies (Marcic et al., 1999; Marcic & Erdös, 2000). The present data are in full agreement with two previous studies, using similar experimental protocols, on the lack of potentiation of PA-bradykinin-mediated vasoconstriction in endothelium-denuded rabbit jugular veins by either ramiprilat (Dendorfer et al., 2001) or captopril (Gobeil et al., 2002). Thus, the concept of B2 receptor-ACE co-localization may also apply to vascular smooth muscle cells.
Bradykinin potentiation by NEP inhibitors?
NEP inhibition, in contrast with ACE inhibition, did not potentiate bradykinin in isolated porcine arteries, neither alone, nor on top of ACE inhibition. These findings contrast with reports on the wide presence of NEP in the vascular wall (Llorens-Cortes et al., 1992; Soleilhac et al., 1992; Dussaule et al., 1993; Graf et al., 1993; Wang et al., 1994; Gonzalez et al., 1998) and its important contribution to bradykinin metabolism in vivo (Kentsch & Otter, 1999; McClean et al., 2000; Rouleau et al., 2000). Thus, either NEP is not present in porcine coronary arteries and/or its contribution to bradykinin metabolism in this in vitro model is of limited importance. Earlier studies in porcine vessels oppose the former explanation (Krassoi et al., 2000; Miyamoto et al., 2002). The most likely explanation is therefore that NEP in intact porcine coronary arteries, unlike ACE, does not co-localize with B2 receptors, and thus that NEP inhibition does not increase the bradykinin levels in the micro-environment of B2 receptors. In support of this concept, bradykinin potentiation did occur following NEP inhibition when co-localization had been artificially induced by transfecting CHO cells with both NEP and B2 receptors (Deddish et al., 2002).
Co-localization of ACE and B2 receptors in caveolae?
Both ACE and B2 receptors have been demonstrated in caveolae (Haasemann et al., 1998; Benzing et al., 1999). Caveolae are small micro-invaginations of the plasma membrane enriched with caveolin that are involved in the compartmentalization of signalling molecules. For instance, B2 receptors interact with endothelial NO synthase in this compartment (Ju et al., 1998). The structural integrity of caveolae depends on cholesterol, and sterol-binding agents such as filipin, cyclodextrin and nystatin are therefore capable of disrupting caveolae (Rothberg et al., 1992; Schnitzer et al., 1994; Neufeld et al., 1996). Interestingly, a recent study demonstrated that caveolar disruption mimics endothelial dysfunction in atheromatous vessels (Darblade et al., 2001).
To address the possibility of ACE-B2 receptor co-localization in caveolae, we studied the bradykinin-potentiating effects of quinaprilat in coronary arteries that had been exposed to the above sterol-binding agents. Our data confirm that caveolar disruption results in endothelial dysfunction, since filipin reduced the maximal relaxant effect of both bradykinin and PA-bradykinin by ≈25–30%, without affecting the relaxations induced by the endothelium-independent agent SNAP. Cyclodextrin and nystatin did not affect the concentration-response curves of bradykinin and PA-bradykinin. Possibly therefore, the 40–50% reduction in caveolar abundance that has been reported to occur in rabbit aortic rings following exposure to 2% cyclodextrin (the same concentration that was used in the present study, and that resulted in a reduction of the effect of acetylcholine in rabbit aorta rings) (Darblade et al., 2001) is insufficient to affect B2 receptor-mediated relaxations, or the reduction in porcine coronary arteries is less than 40%. Furthermore, nystatin at a concentration of 20 μg ml−1 tended to reduce the SNAP-induced relaxations (Figure 9), and a significant reduction occurred at a concentration of 50 μg ml−1, thus not allowing us to investigate the effect of higher nystatin concentrations on the bradykinin concentration-response curves.
Importantly however, although caveolar disruption appeared to reduce the relaxant effect mediated by B2 receptors (for instance because of interference with their interaction with endothelial NO synthase in this compartment), it did not affect the leftward shift induced by quinaprilat (or the absence thereof in the case of PA-bradykinin), nor did it prevent the quinaprilat-induced relaxation in desensitized preparations. Based on these data, it therefore seems unlikely that ACE inhibition within caveolae underlies its bradykinin-potentiating effects. Apparently therefore, the ACE-B2 receptor co-localization occurs elsewhere, for instance in coated pits or non-caveolar lipid rafts.
Conclusion and perspective
Bradykinin potentiation by ACE inhibitors in porcine coronary arteries is a metabolic process based on the co-localization of ACE and B2 receptors on the endothelial cell membrane. NEP does not appear to be present in the micro-environment of coronary B2 receptors, and the ACE inhibitor-induced effect on bradykinin metabolism most likely does not occur in caveolae, i.e. it precedes the coupling of B2 receptors to endothelial NO synthase in this compartment. The co-localization of ACE and B2 receptors mimics the co-localization of ACE and AT1 receptors (Saris et al., 2002; Schuijt et al., 2002). Thus, ACE is located in a strategic position to allow maximal efficiency of B2 receptor and AT1 receptor stimulation.
Abbreviations
- ACE
angiotensin-converting enzyme
- Ang
angiotensin
- AT1
angiotensin II type 1
- B2
bradykinin type 2
- CHO
Chinese hamster ovary
- DP-bradykinin
[ΔPhe5]-bradykinin; NO, nitric oxide
- PA-bradykinin
[Phe8ψ(CH2-NH)Arg9]-bradykinin; S-nitroso-N-acetylpenicillamine (SNAP)
- U46619
9,11-dideoxy-11α, 9α-epoxymethano-prostaglandin F2α
References
- BACHVAROV D.R., HOULE S., BACHVAROVA M., BOUTHILLIER J., ADAM A., MARCEAU F. Bradykinin B2 receptor endocytosis, recycling, and down-regulation assessed using green fluorescent protein conjugates. J. Pharmacol. Exp. Ther. 2001;297:19–26. [PubMed] [Google Scholar]
- BENZING T., FLEMING I., BLAUKAT A., MÜLLER-ESTERL W., BUSSE R. Angiotensin-converting enzyme inhibitor ramiprilat interferes with the sequestration of the B2 kinin receptor within the plasma membrane of native endothelial cells. Circulation. 1999;99:2034–2040. doi: 10.1161/01.cir.99.15.2034. [DOI] [PubMed] [Google Scholar]
- BLAIS C., JR, FORTIN D., ROULEAU J.L., MOLINARO G., ADAM A. Protective effect of omapatrilat, a vasopeptidase inhibitor, on the metabolism of bradykinin in normal and failing human hearts. J. Pharmacol. Exp. Ther. 2000;295:621–626. [PubMed] [Google Scholar]
- CAMPBELL D.J., DUNCAN A.M., KLADIS A. Angiotensin-converting enzyme inhibition modifies angiotensin but not kinin peptide levels in human atrial tissue. Hypertension. 1999;34:171–175. doi: 10.1161/01.hyp.34.2.171. [DOI] [PubMed] [Google Scholar]
- DANSER A.H.J., TOM B., DE VRIES R., SAXENA P.R. L-NAME resistant bradykinin-induced relaxation in porcine coronary arteries is NO-dependent: effect of ACE inhibition. Br. J. Pharmacol. 2000;131:195–202. doi: 10.1038/sj.bjp.0703555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DARBLADE B., CAILLAUD D., POIROT M., FOUQUE M., THIERS J.C., RAMI J., BAYARD F., ARNAL J.F. Alteration of plasmalemmal caveolae mimics endothelial dysfunction observed in atheromatous rabbit aorta. Cardiovasc. Res. 2001;50:566–576. doi: 10.1016/s0008-6363(01)00251-6. [DOI] [PubMed] [Google Scholar]
- DE LEAN A., MUNSON P.J., RODBARD D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- DEDDISH P.A., MARCIC B.M., TAN F., JACKMAN H.L., CHEN Z., ERDÖS E.G. Neprilysin inhibitors potentiate effects of bradykinin on B2 receptor. Hypertension. 2002;39:619–623. doi: 10.1161/hy0202.103298. [DOI] [PubMed] [Google Scholar]
- DENDORFER A., REISSMANN S., WOLFRUM S., RAASCH W., DOMINIAK P. Potentiation of kinin analogues by ramiprilat is exclusively related to their degradation. Hypertension. 2001;38:142–146. doi: 10.1161/01.hyp.38.1.142. [DOI] [PubMed] [Google Scholar]
- DENDORFER A., VORDERMARK D., DOMINIAK P. Degradation of bradykinin by bovine tracheal epithelium and isolated epithelial cells. Br. J. Pharmacol. 1997;120:121–129. doi: 10.1038/sj.bjp.0700874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENDORFER A., WOLFRUM S., SCHAFER U., STEWART J.M., INAMURA N., DOMINIAK P. Potentiation of the vascular response to kinins by inhibition of myocardial kininases. Hypertension. 2000;35:32–37. doi: 10.1161/01.hyp.35.1.32. [DOI] [PubMed] [Google Scholar]
- D'USCIO L.V., QUASCHNING T., BURNETT J.C., LÜSCHER T.F. Vasopeptidase inhibition prevents endothelial dysfunction of resistance arteries in salt-sensitive hypertension in comparison with single ACE inhibition. Hypertension. 2001;37:28–33. doi: 10.1161/01.hyp.37.1.28. [DOI] [PubMed] [Google Scholar]
- DUSSAULE J.C., STEFANSKI A., BEA M.L., RONCO P., ARDAILLOU R. Characterization of neutral endopeptidase in vascular smooth muscle cells of rabbit renal cortex. Am. J. Physiol. 1993;264:F45–F52. doi: 10.1152/ajprenal.1993.264.1.F45. [DOI] [PubMed] [Google Scholar]
- FERNANDES L., FORTES Z.B., NIGRO D., TOSTES R.C., SANTOS R.A.S., CATELLI DE CARVALHO M.H. Potentiation of bradykinin by angiotensin-(1-7) on arterioles of spontaneously hypertensive rats studied in vivo. Hypertension. 2001;37:703–709. doi: 10.1161/01.hyp.37.2.703. [DOI] [PubMed] [Google Scholar]
- GOBEIL F., JR, HALLE S., BLAIS P.A., REGOLI D. Studies on the angiotensin-converting enzyme and the kinin B2 receptor in the rabbit jugular vein: modulation of contractile response to bradykinin. Can. J. Physiol. Pharmacol. 2002;80:153–163. doi: 10.1139/y02-014. [DOI] [PubMed] [Google Scholar]
- GONZALEZ W., SOLEILHAC J.M., FOURNIE-ZALUSKI M.C., ROQUES B.P., MICHEL J.B. Characterization of neutral endopeptidase in vascular cells, modulation of vasoactive peptide levels. Eur. J. Pharmacol. 1998;345:323–331. doi: 10.1016/s0014-2999(98)00038-7. [DOI] [PubMed] [Google Scholar]
- GRAF K., GRAFE M., BOSSALLER C., NIEHUS J., SCHULZ K.D., AUCH-SCHWELK W., FLECK E. Degradation of bradykinin by neutral endopeptidase (EC 3.4.24.11) in cultured human endothelial cells. Eur. J. Clin. Chem. Clin. Biochem. 1993;31:267–272. doi: 10.1515/cclm.1993.31.5.267. [DOI] [PubMed] [Google Scholar]
- HAASEMANN M., CARTAUD J., MÜLLER-ESTERL W., DUNIA I. Agonist-induced redistribution of bradykinin B2 receptor in caveolae. J. Cell. Sci. 1998;111:917–928. doi: 10.1242/jcs.111.7.917. [DOI] [PubMed] [Google Scholar]
- JU H., VENEMA V.J., MARRERO M.B., VENEMA R.C. Inhibitory interactions of the bradykinin B2 receptor with endothelial nitric-oxide synthase. J. Biol. Chem. 1998;273:24025–24029. doi: 10.1074/jbc.273.37.24025. [DOI] [PubMed] [Google Scholar]
- KENTSCH M., OTTER W. Novel neurohormonal modulators in cardiovascular disorders. The therapeutic potential of endopeptidase inhibitors. Drugs R. D. 1999;1:331–338. doi: 10.2165/00126839-199901040-00011. [DOI] [PubMed] [Google Scholar]
- KOKKONEN J.O., KUOPPALA A., SAARINEN J., LINDSTEDT K.A., KOVANEN P.T. Kallidin- and bradykinin-degrading pathways in human heart: degradation of kallidin by aminopeptidase M-like activity and bradykinin by neutral endopeptidase. Circulation. 1999;99:1984–1990. doi: 10.1161/01.cir.99.15.1984. [DOI] [PubMed] [Google Scholar]
- KRASSOI I., PATARICZA J., TORDAY L.L., KUN A., PAPP J.G. Improvement by phosphoramidon of damaged endothelial function in porcine coronary artery. Ann. Thorac. Surg. 2000;70:878–882. doi: 10.1016/s0003-4975(00)01632-5. [DOI] [PubMed] [Google Scholar]
- LLORENS-CORTES C., HUANG H., VICART P., GASC J.M., PAULIN D., CORVOL P. Identification and characterization of neutral endopeptidase in endothelial cells from venous or arterial origins. J. Biol. Chem. 1992;267:14012–14018. [PubMed] [Google Scholar]
- MARCIC B.M., DEDDISH P.A., JACKMAN H.L., ERDÖS E.G. Enhancement of bradykinin and resensitization of its B2 receptor. Hypertension. 1999;33:835–843. doi: 10.1161/01.hyp.33.3.835. [DOI] [PubMed] [Google Scholar]
- MARCIC B.M., DEDDISH P.A., SKIDGEL R.A., ERDÖS E.G., MINSHALL R.D., TAN F. Replacement of the transmembrane anchor in angiotensin I-converting enzyme (ACE) with a glycosylphosphatidylinositol tail affects activation of the B2 bradykinin receptor by ACE inhibitors. J. Biol. Chem. 2000;275:16110–16118. doi: 10.1074/jbc.M909490199. [DOI] [PubMed] [Google Scholar]
- MARCIC B.M., ERDÖS E.G. Protein kinase C and phosphatase inhibitors block the ability of angiotensin I-converting enzyme inhibitors to resensitize the receptor to bradykinin without altering the primary effects of bradykinin. J. Pharmacol. Exp. Ther. 2000;294:605–612. [PubMed] [Google Scholar]
- MCCLEAN D.R., IKRAM H., GARLICK A.H., RICHARDS A.M., NICHOLLS M.G., CROZIER I.G. The clinical, cardiac, renal, arterial and neurohormonal effects of omapatrilat, a vasopeptidase inhibitor, in patients with chronic heart failure. J. Am. Coll. Cardiol. 2000;36:479–486. doi: 10.1016/s0735-1097(00)00741-5. [DOI] [PubMed] [Google Scholar]
- MINSHALL R.D., TAN F., NAKAMURA F., RABITO S.F., BECKER R.P., MARCIC B., ERDÖS E.G. Potentiation of the actions of bradykinin by angiotensin I-converting enzyme inhibitors. The role of expressed human bradykinin B2 receptors and angiotensin I-converting enzyme in CHO cells. Circ. Res. 1997;81:848–856. doi: 10.1161/01.res.81.5.848. [DOI] [PubMed] [Google Scholar]
- MIYAMOTO A., MURATA S., NISHIO A. Role of ACE and NEP in bradykinin-induced relaxation and contraction of isolated porcine basilar artery. Naunyn Schmiedebergs Arch. Pharmacol. 2002;365:365–370. doi: 10.1007/s00210-002-0543-0. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., BALLARD K.D., VANHOUTTE P.M. Kininase-independent potentiation of endothelium-dependent relaxations to kinins by converting enzyme inhibitor perindoprilat. Acta Pharmacol. Sin. 2002;23:203–207. [PubMed] [Google Scholar]
- NEUFELD E.B., COONEY A.M., PITHA J., DAWIDOWICZ E.A., DWYER N.K., PENTCHEV P.G., BLANCHETTE-MACKIE E.J. Intracellular trafficking of cholesterol monitored with a cyclodextrin. J. Biol. Chem. 1996;271:21604–21613. doi: 10.1074/jbc.271.35.21604. [DOI] [PubMed] [Google Scholar]
- RAUT R., ROULEAU J.L., BLAIS C., JR, GOSSELIN H., MOLINARO G., SIROIS M.G., LEPAGE Y., CRINE P., ADAM A. Bradykinin metabolism in the postinfarcted rat heart: role of ACE and neutral endopeptidase 24.11. Am. J. Physiol. 1999;276:H1769–H1779. doi: 10.1152/ajpheart.1999.276.5.H1769. [DOI] [PubMed] [Google Scholar]
- REISSMANN S., SCHWUCHOW C., SEYFARTH L., PINEDA DE CASTRO L.F., LIEBMANN C., PAEGELOW I., WERNER H., STEWART J.M. Highly selective bradykinin agonists and antagonists with replacement of proline residues by N-methyl-D- and L-phenylalanine. J. Med. Chem. 1996;39:929–936. doi: 10.1021/jm9301954. [DOI] [PubMed] [Google Scholar]
- ROTHBERG K.G., HEUSER J.E., DONZELL W.C., YING Y.S., GLENNEY J.R., ANDERSON R.G. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- ROULEAU J.L., PFEFFER M.A., STEWART D.J., ISAAC D., SESTIER F., KERUT E.K., PORTER C.B., PROULX G., QIAN C., BLOCK A.J. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet. 2000;356:615–620. doi: 10.1016/s0140-6736(00)02602-7. [DOI] [PubMed] [Google Scholar]
- SARIS J.J., VAN DEN EIJNDEN M.M.E.D., LAMERS J.M.J., SAXENA P.R., SCHALEKAMP M.A.D.H., DANSER A.H.J. Prorenin-induced myocyte proliferation: no role for intracellular angiotensin II. Hypertension. 2002;39:573–577. doi: 10.1161/hy0202.103002. [DOI] [PubMed] [Google Scholar]
- SCHNITZER J.E., OH P., PINNEY E., ALLARD J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J. Cell. Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHUIJT M.P., DEVRIES R., SAXENA P.R., SCHALEKAMP M.A.D.H., DANSER A.H.J. Vasoconstriction is determined by interstitial rather than circulating angiotensin II. Br. J. Pharmacol. 2002;135:275–283. doi: 10.1038/sj.bjp.0704452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOLEILHAC J.M., LUCAS E., BEAUMONT A., TURCAUD S., MICHEL J.B., FICHEUX D., FOURNIE-ZALUSKI M.C., ROQUES B.P. A 94-kDa protein, identified as neutral endopeptidase-24.11, can inactivate atrial natriuretic peptide in the vascular endothelium. Mol. Pharmacol. 1992;41:609–614. [PubMed] [Google Scholar]
- TOM B., DE VRIES R., SAXENA P.R., DANSER A.H.J. Bradykinin potentiation by angiotensin-(1-7) and angiotensin-converting enzyme (ACE) inhibitors correlates with ACE C- and N-domain blockade. Hypertension. 2001;38:95–99. doi: 10.1161/01.hyp.38.1.95. [DOI] [PubMed] [Google Scholar]
- WANG L., SADOUN E., STEPHENS R.E., WARD P.E. Metabolism of substance P and neurokinin A by human vascular endothelium and smooth muscle. Peptides. 1994;15:497–503. doi: 10.1016/0196-9781(94)90212-7. [DOI] [PubMed] [Google Scholar]
- WILLEMS E.W., HEILIGERS J.P.C., DE VRIES P., TOM B., KAPOOR K., VILLALON C.M., SAXENA P.R. A61603-induced vasoconstriction in porcine carotid vasculature: involvement of a non-adrenergic mechanism. Eur. J. Pharmacol. 2001;417:195–201. doi: 10.1016/s0014-2999(01)00898-6. [DOI] [PubMed] [Google Scholar]