

Abstract
In the present study we investigated the effect of glucocorticoids on the activation of renal tubular epithelial cells, which are thought to play an important role in inflammatory processes in the kidney.
Activation of renal epithelial cells by IL-1, TNF-α or CD40L resulted in increased production of cytokines and chemokines. Both in the renal epithelial cell line HK-2 and in primary cultures of human proximal tubular epithelial cells (PTEC) production of IL-6, IL-8 and monocyte chemotactic protein 1 (MCP-1) was not inhibited by glucocorticoids, independent of the stimulus.
In contrast, dexamethasone strongly inhibited cytokine production by immortalized renal fibroblasts and an airway epithelial cell line (A549).
Stimulation of renal epithelial cells resulted in activation of NF-κB, a pivotal transcription factor in the regulation of cytokine genes, as was shown by IκB-α degradation and increased DNA-binding activity. In contrast to dexamethasone, addition of the NF-κB inhibitors pyrrolidine dithiocarbamate (PDTC) and n-tosyl-l-phenylalanine chloromethyl ketone (TPCK) completely abolished cytokine and chemokine production.
Renal epithelial cells express abundant levels of the functional glucocorticoid receptor alpha (GRα) isoform and low levels of the inhibitory beta isoform (GRβ).
In conclusion, cytokine production by renal epithelial cells is insensitive to the inhibitory effects of glucocorticoids. The lack of dexamethasone-mediated inhibition was specific for renal epithelial cells and could not be explained by an increased expression of the glucocorticoid receptor beta isoform.
Keywords: Cytokines, chemokines, renal epithelial cells, dexamethasone, glucocorticoid receptor, NF-κB
Introduction
Influx of monocytes and T-cells into the tubulo-interstitial area is a hallmark of many renal inflammatory diseases. Rather than being a passive target, resident proximal tubular epithelial cells (PTEC) are thought to play an active role in the inflammatory process via production of inflammatory mediators (van kooten & Daha, 2001). It was shown before that the cytokines IL-1 and TNF-α and the cell surface molecule CD40L play an important role in T-cell-mediated activation of PTEC (Kuroiwa et al., 2000). Stimulation of PTEC in vitro with these cytokines or CD40L results in increased cytokine and chemokine production, including IL-6, IL-8, MCP-1 and RANTES (Deckers et al., 1998; Pattison et al., 1994; van kooten et al., 1997; 1999). Furthermore, expression of adhesion molecules and MHC class II is increased (Kirby et al., 1993).
Glucocorticoids are potent suppressors of the immune system and are therefore used as therapeutic treatment in a broad range of autoimmune and inflammatory diseases. One of the major anti-inflammatory effects of glucocorticoids is suppression of cytokine production (Almawi et al., 1998). Glucocorticoids can inhibit gene transcription via negative glucocorticoid responsive elements (Tsai & O'malley, 1994). However, several genes that are negatively regulated by glucocorticoids, including cytokine genes, do not contain a negative response element in their promoter. Expression of these genes is inhibited via glucocorticoid-induced repression of transcription factors, including AP-1 and NF-κB (Cato & Wade, 1996). Many anti-inflammatory effects of glucocorticoids have been linked to their ability to inhibit the activation of NF-κB (Lee & Burckart, 1998; Mckay & Cidlowski, 1999).
NF-κB is a pro-inflammatory transcription factor that regulates the expression of many proteins involved in inflammation, including cytokines (TNF-α, IL-6), chemokines (IL-8, MCP-1, RANTES) and adhesion molecules (ICAM-1, VCAM-1) (Barnes & Karin, 1997; Lee & Burckart, 1998). The classical form of NF-κB is a heterodimer of p50/p65 (Barnes & Karin, 1997; Ghosh et al., 1998). In the cytoplasm, NF-κB is kept inactive through binding to its inhibitor, IκB-α. Stimulation of cells with cytokines, like IL-1 and TNF-α results in phosphorylation and subsequent degradation of IκB-α. The released NF-κB translocates to the nucleus to activate transcription of target genes (Ghosh et al., 1998; Karin & Ben-Neriah, 2000).
In the present study we investigated the effect of dexamethasone on cytokine and chemokine production by activated human renal epithelial cells and found that these cells were insensitive to the inhibitory action of dexamethasone. We also investigated the role of NF-κB in cytokine production by PTEC and the expression of the glucocorticoid receptor to obtain more insight in the mechanism of the observed steroid-insensitivity.
Methods
Cell culture
PTEC
Primary human PTEC were isolated from kidneys not suitable for transplantation and from pre-transplant biopsies (van kooten et al., 1997). Cell monolayers were cultured on a matrix of heat-inactivated foetal calf serum (ΔFCS; Gibco BRL/Life Technologies Inc., Paisley, U.K.) in DMEM/HAMF12 (Bio-Whittaker, Walkersville, MD, U.S.A.) supplemented with insulin (5 μg ml−1), transferrin (5 μg ml−1), selenium (5 ng ml−1), hydrocortisone (36 ng ml−1), tri-iodothyronine (40 pg ml−1), epidermal growth factor (10 ng ml−1, all from Sigma Chemical Co., St. Louis, MO, U.S.A.) and 100 U ml−1 penicillin/100 μg ml−1 streptomycin (Gibco) (Detrisac et al., 1984). Specific outgrowth of PTEC was confirmed by morphological appearance and immunofluorescence staining as described (van kooten et al., 1997). Cells were used between passage 2–6 of culture.
HK-2
The immortalized PTEC-derived cell line HK-2 (Human Kidney-2) was kindly provided by M.P. Ryan, University College Dublin, Ireland (Ryan et al., 1994). Cells were cultured in RPMI 1640 (Gibco) supplemented with 10% ΔFCS and penicillin/streptomycin.
L-cells
Mouse fibroblast L-cells, non-transfected (L-orient) or stably transfected with CD40L (L-CD40L) (Garrone et al., 1995) were grown in IMDM (Bio-Whittaker), 10% ΔFCS and penicillin/streptomycin. For co-culture experiments, L-cells were irradiated (80 Gy) before addition to the target cells.
TK173
The human renal fibroblast cell line TK173 was established from normal human kidney and was kindly provided by F. Strutz, (Dept. of Nephrology and Rheumatology, Georg-August-University, Göttingen, Germany). Cells were cultured in IMDM/10% ΔFCS and penicillin/streptomycin (Muller et al., 1995).
Synovial fibroblasts
Primary synovial fibroblasts were cultured from the synovial tissue of patients undergoing total or partial knee or elbow surgery (Goossens et al., 1999). Synovial fibroblasts were cultured in IMDM/20% FCS and penicillin/streptomycin and used between passage 2–6 of culture.
A549
The human airway epithelial cell line A549 was cultured in RPMI 1640/10% ΔFCS and penicillin/streptomycin.
For passage of the cultures, cells were harvested by trypsinization with 0.02% (w v−1) EDTA/0.05% (w v−1) trypsin (Sigma).
Cytokine production
Production of IL-6, IL-8 and MCP-1 was measured in supernatants of stimulated cells after 48 h unless indicated otherwise. Prior to stimulation, cells were transferred to 48-well plates (Costar, Corning, NY, U.S.A.) at a density of 0.5×105 cells per well and serum-starved o/n. Cells were stimulated with IL-1α, TNF-α or CD40L-transfected L-cells in a 1 : 1 ratio. For inhibition experiments, cells were pre-incubated for 2 h with pyrrolidine dithiocarbamate (PDTC), dexamethasone and n-tosyl-1-phenylalanine chloromethyl ketone (TPCK). Cytokines or L-cells were added in the continuous presence of these inhibitors. Cell viability was checked with Trypan blue staining. The concentration of IL-6, IL-8 and MCP-1 in culture supernatants was measured by specific ELISA as described previously (van kooten et al., 1997).
Preparation of cell lysates
For preparation of whole cell-lysates, 1×106 cells were harvested and incubated in lysis buffer containing 20 mM Tris pH 7.4, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA and a protease inhibitor mix consisting of 5 u ml−1 Trasylol (Bayer, Leverkusen, Germany), 1 mM PMSF, 2 μg ml−1 antipain, 2 μg ml−1 chymostatin and 2 μg ml−1 leupeptin (all from Boehringer Mannheim, Germany) for 10 min on ice. After centrifugation for 15 min at 13,000 r.p.m., supernatants were collected and protein concentration was determined with a BCA protein assay reagent (Pierce, Rockford, IL, U.S.A.).
Western blot analysis
Whole cell extracts (20 μg) were separated on a 10% SDS–PAGE-gel and blotted to a PVDF-membrane (Millipore, Bedford, MA, U.S.A.). Membranes were blocked o/n in PBS/Tween/2% casein (PTC) at 4°C, incubated with antibodies to IκB-α or human glucocorticoid receptor for 1.5 h at room temperature and detected with horseradish peroxidase (HRP)-conjugated secondary antibodies. Antibodies were diluted in PTC. Blots were developed with a chemiluminescence substrate (Pierce) and detected with Hyperfilm ECL (Amersham Pharmacia Biotech, Buckinghamshire, U.K.). Densitometry was performed using a Stratagene Eagle-sight analysis programme. Efficiency of transfer was verified by staining with coomassie brilliant blue (Sigma).
Detection of NF-κB DNA-binding activity
Cells were incubated with 1 μM dexamethasone for 30 min and stimulated with 5 ng ml−1 IL-1 for 30 min. NF-κB DNA binding activity in whole cell lysates was measured with Trans-AM NF-κB p65 and p50 transcription factor assay kits (Active Motif Europe, Rixensart, Belgium) according to manufacturer's instructions. Briefly, 2 μg extract was added to 96-well plates coated with an oligonucleotide containing the NF-κB consensus site. Binding of NF-κB to the DNA was visualized by anti-p50 and anti-p65 antibodies that specifically recognize activated DNA-bound NF-κB. Antibody binding was detected with a secondary HRP-conjugated antibody and developed with TMB substrate. The reaction was stopped with 0.5 M H2SO4 and measured at 450 nm. Specificity of NF-κB activation was determined by competition experiments using NF-κB wild-type and mutant consensus oligonucleotides that were provided with the kit.
Glucocorticoid receptor mRNA expression
Total RNA was isolated using RNAzolB (Campro, Veenendaal, The Netherlands) according to manufacturer's instructions. OD260/280 ratios were measured to determine the quantity and purity of RNA preparations. Fixed amounts of total cellular RNA (1 μg) were reverse transcribed into cDNA by oligo-dT priming using M-MLV reverse transcriptase (Gibco). Expression of GRα and GRβ was determined by RT–PCR with a common upstream primer (5′-CCT AAG GAC GGT CTG AAG AGC-3′) and two specific downstream primers: GRα, 5′-GCC AAG TCT TGG CCC TCT AT-3′ (450 bp) and GRβ: 5′-CCA CGT ATC CTA AAA GGG CAC-3′ (377 bp) (Honda et al., 2000). Expression of β-actin was used as a control. β-actin forward primer, 5′-CTA CAA TGA GCT GCG TGT GG-3′; β-actin reverse primer, 5′-AAG GAA GGC TGG AAG AGT GC-3′ (527 bp) (van kooten et al., 1999). PCR was performed under standard conditions (50 mM KCl, 10 mM Tris-HCl, pH 8.4, 0.06 mg ml−1 bovine serum albumin (BSA), 0.25 mM dNTPs, 25 pmol of each primer, 1 u of Taq polymerase; Perkin Elmer, Norwalk, CT, U.S.A.) with MgCl2 1.5 mM for GRα and β-actin and 1 mM for GRβ. The following scheme was used: 5 min 95°C, GRα/β: 40 cycli of 30 s 94°C, 30 s 60°C, 1 min 72°C; β-actin: 35 cycli of 1 min 95°C, 1 min 55°C and 1 min 72°C, 7 min 72°C. PCR-products were analysed on a 1% agarose gel containing etihidium bromide.
Materials
Recombinant human IL-1α and TNF-α were purchased from Preprotech, Rocky Hill, NJ, U.S.A. PDTC and TPCK were supplied by Sigma Chemical Co., St. Louis, MO, U.S.A. whereas dexamethasone was obtained from the Pharmacy of the Leiden University Medical Center, The Netherlands. Primary antibodies used for Western blot were rabbit anti-IκB-α antibody (sc-371; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and mouse anti-human glucocorticoid receptor (Transduction Laboratories, Lexington, KY, U.S.A.). The secondary antibodies were purchased from DAKO, Glostrup, Denmark.
Statistical analysis
Cytokine and chemokine production is presented as mean concentration±s.d. from representative experiments. Experiments were repeated at least three times. Cytokine production in primary cultures was analysed with a paired student's t-test. Data are shown as mean±s.e.means. Results were considered significant if P<0.05.
Results
IL-6 production by PTEC is not inhibited by dexamethasone
IL-6 production in primary cultures of PTEC from different isolations ranged from 2.8–14.1 ng ml−1 for baseline levels and was increased to 39.7–121.0 ng ml−1 upon stimulation with 5 ng ml−1 IL-1. Incubation of PTEC with increasing doses of dexamethasone (10−9–10−5 M) showed no significant effect on constitutive or IL-1-induced IL-6 production after 48 h (Figure 1A,B). Similar results were obtained after stimulation for 24 and 72 h (data not shown). Increasing doses of IL-1 induced a dose-dependent increase in IL-6 production. No effect of dexamethasone (1 μM) was observed on either low-dose or high-dose stimulation with IL-1 (Figure 1C), showing that the strength of the IL-1 signal is not masking the inhibitory capacity of dexamethasone. Routinely cells were incubated with dexamethasone for 2 h followed by 48 h stimulation with IL-1 in the continuous presence of dexamethasone. Even incubation with dexamethasone for up to 24 h prior to stimulation with IL-1 showed no effect on IL-1-induced IL-6 production (data not shown). In parallel experiments, the same dexamethasone showed a strong inhibitory action on monocytes as published before (Woltman et al., 2000a).
Figure 1.
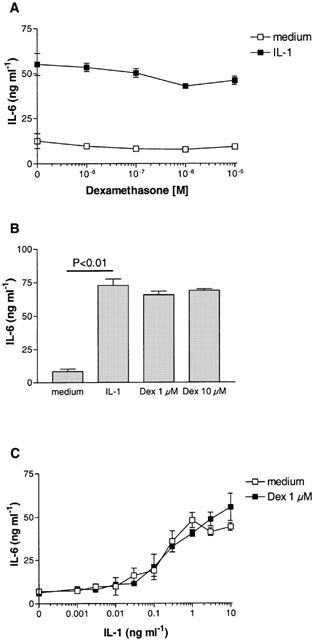
Effect of dexamethasone on IL-1-induced IL-6 production by PTEC. IL-6 production by primary human PTEC stimulated with 5 ng ml−1 IL-1 was not inhibited by increasing doses of dexamethasone. Data shown are mean±s.d. of one representative experiment (A) and mean±s.e.means of seven independent experiments (B). No effect of 1 μM dexamethasone was found on different doses of IL-1 (C). IL-6 production in culture supernatants was measured after 48 h by specific ELISA.
The absence of dexamethasone-mediated inhibition is specific for renal epithelial cells
As observed with primary cells, IL-1-induced IL-6 production by the PTEC-derived cell line HK-2 was not inhibited by dexamethasone (Figure 2A). In contrast, stimulation of the renal fibroblast cell line TK173 or primary synovial fibroblasts with IL-1 resulted in increased production of IL-6, which was completely inhibited by dexamethasone (Figure 2B,C). Similarly, IL-1-induced production of IL-8 by the airway epithelial cell line A549 was strongly inhibited by low doses of dexamethasone (Figure 2D).
Figure 2.
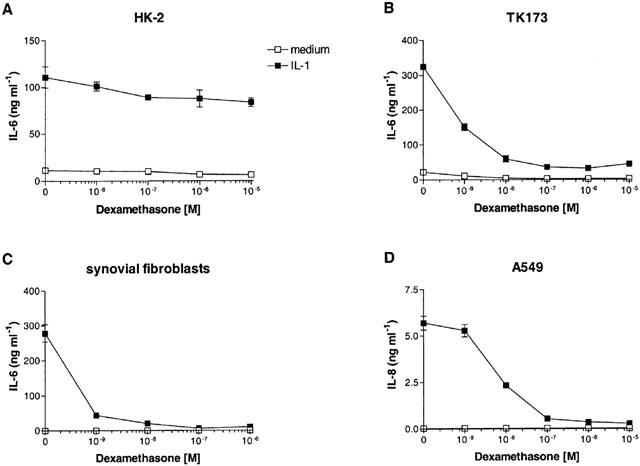
Effect of dexamethasone on different cell types. Dexamethasone was unable to inhibit cytokine production by the renal epithelial cell line HK-2, but strongly abolished cytokine production by a renal fibroblast cell line (TK173), primary human synovial fibroblasts and an airway epithelial cell line (A549). n=4.
Production of chemokines is insensitive to dexamethasone independent of the stimulus
As shown in Figure 3A, IL-1-induced production of the chemokines IL-8 and MCP-1 by primary PTEC was not inhibited by dexamethasone. Also, dexamethasone was unable to inhibit IL-6 production by PTEC that were activated with TNF-α (500 u ml−1) and CD40L (Figure 3B). Similar results were obtained for the production of IL-8 and MCP-1 in both primary cells and HK-2.
Figure 3.
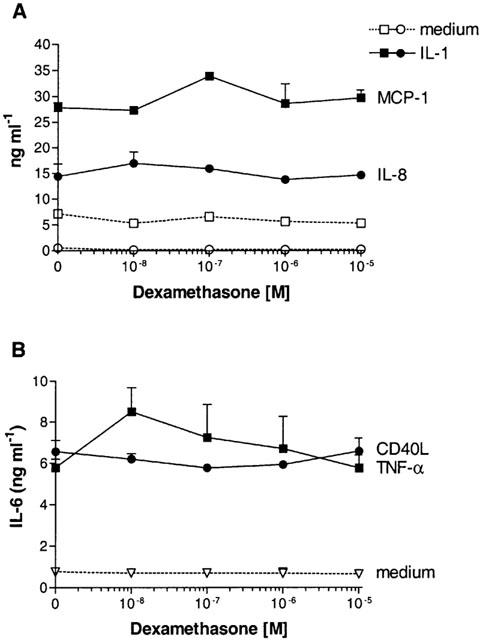
(A) In the same supernatants as described in Figure 1 the effect of dexamethasone on chemokine production by PTEC was determined. IL-1-induced production of IL-8 and MCP-1 was not inhibited by dexamethasone. (B) Representative experiment showing the effect of dexamethasone on PTEC that were activated by TNF-α (500 u ml−1) and CD40L. No inhibition of TNF-α- or CD40L-induced IL-6 production was found in five independent experiments.
Activation of PTEC induces NF-κB activation
Activation of primary PTEC with IL-1 induced degradation of IκB-α 15–60 min after stimulation (Figure 4A). Maximal degradation was observed after 30 min and IκB-α levels returned back to normal after 120 min. TNF-α and CD40L also induced degradation of IκB-α with similar kinetics, although less potent than IL-1 (respectively 48, 75 and 93.6% degradation as determined by densitometry). The lower band in the CD40L-stimulated cells is caused by the murine L-cells used for stimulation. Staining with the human and mouse reactive anti-IκB-α antibody showed that murine IκB-α migrated at a slightly smaller size than human IκB-α and remained stable over time.
Figure 4.
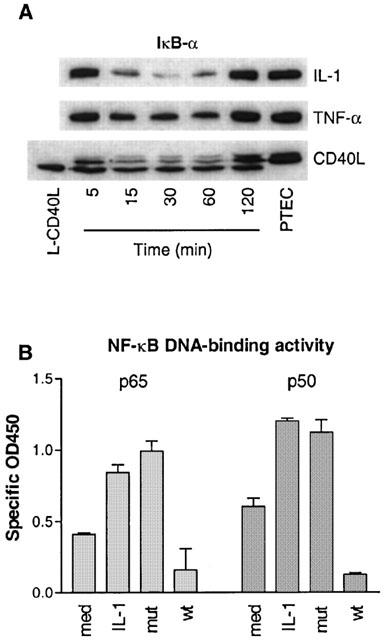
Assessment of NF-κB activation in activated PTEC. (A) Western blot analysis of IκB-α degradation in PTEC that were stimulated with IL-1 (5 ng ml−1), TNF-α (500 u ml−1) or CD40L for 5–120 min. Maximal IκB-α degradation occurred after 30 min. (B) Assessment of NF-κB DNA-binding activity using TransAm transcription factor assay kits as described in Methods. OD450 values were corrected for background levels. med, medium; mut, mutant and wt, wild-type consensus oligonucleotide. DNA-binding activity of p50 and p65 subunits was increased in HK-2 cells stimulated with 5 ng ml−1 IL-1 for 30 min. n=3.
We also analysed DNA-binding activity of NF-κB in the HK-2 cell line. In control cells, a basal level of NF-κB activity was observed, as shown by DNA-binding of the p65 and p50 subunits (Figure 4B). Stimulation of cells with IL-1 resulted in increased DNA-binding of p65 and p50. Pre-incubation of the plates with a wild-type NF-κB consensus oligonucleotide, but not a mutant oligonucleotide, completely prevented binding of p65 and p50 to the DNA.
Role for NF-κB in IL-6 production by PTEC
Different NF-κB-inhibitors were used to study the role of NF-κB in cytokine production. Stimulation of PTEC in the presence of the anti-oxidant PDTC (3.75–30 μM) resulted in a strong dose-dependent inhibition of IL-6 production (Figure 5A). Similar results were found for production of IL-8 and MCP-1 (data not shown). This inhibition was not caused by toxicity, since PDTC showed no effect on viable cell counts as measured by Trypan blue staining. The proteosome inhibitor TPCK (2.5–10 μM) also inhibited IL-1-induced IL-6 production in a dose-dependent manner. Only incubation with the highest concentration of TPCK (10 μM) resulted in the induction of some cell death (Figure 5B).
Figure 5.
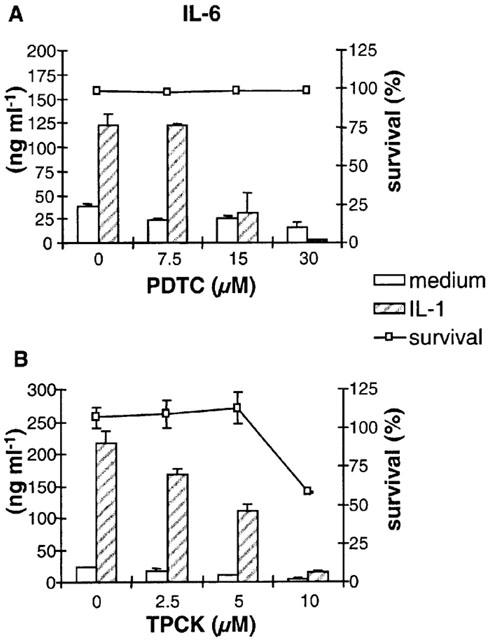
Role for NF-κB in cytokine production by PTEC. Stimulation of PTEC with 5 ng ml−1 IL-1 in the presence of increasing doses of PDTC (A) or TPCK (B) resulted in strong inhibition of IL-6 production. Data shown are mean±s.d. of one representative experiment out of four independent experiments. For TPCK, no inhibition was found when cells were treated with the dissolvent DMSO (data not shown). Cell viability was checked with Trypan blue staining.
Renal epithelial cells express the glucocorticoid receptor
Specific RT–PCR showed that mRNA for the functional glucocorticoid receptor (GRα) was strongly expressed by non-stimulated PTEC. Expression levels of GRα and the inhibitory counterpart GRβ were compared semiquantitatively, showing that although GRβ was expressed, mRNA for GRα was much more abundant (Figure 6A). Furthermore, Western blot analysis confirmed that GR protein was expressed in PTEC (Figure 6B). As a control for GR expression the steroid-sensitive cell line A549 was used.
Figure 6.
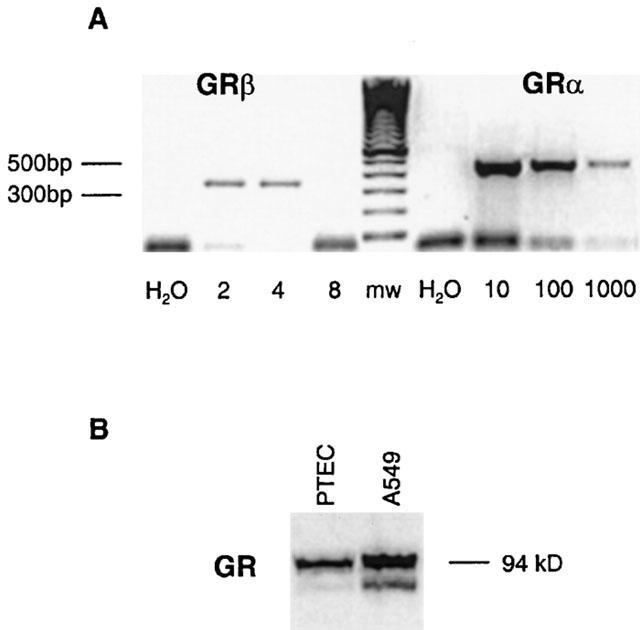
Expression of the glucocorticoid receptor by PTEC. (A) GRα and GRβ expression was determined by RT–PCR as described in Methods. For GRβ, cDNA was diluted 2–8 times; for GRα, cDNA was diluted 10–1000 times. mw, molecular weight marker. GRα mRNA was abundantly expressed in PTEC. (B) Representative Western blot analysis showing glucocorticoid receptor protein expression in PTEC and A549.
Discussion
In the present study we show that dexamethasone is unable to inhibit production of inflammatory mediators by renal tubular epithelial cells. Production of cytokines and chemokines by proximal tubular epithelial cells (PTEC) is thought to be critical for inflammatory processes in the kidney, including allograft rejection. Although immunosuppressive regimens in transplantation effectively inhibit acute allograft responses, the impact on chronic allograft rejection seems to be limited. Immunosuppression used to prevent allograft rejection usually includes glucocorticoids, which are known to have a profound effect on cytokine production by a variety of cell-types (Auwardt et al., 1998; John et al., 1997; Miyazawa et al., 1998). Unexpectedly, dexamethasone was unable to inhibit IL-6 production by activated primary human PTEC and the PTEC-derived cell line HK-2. Similarly, dexamethasone was unable to inhibit IL-1-induced production of the chemokines IL-8 and MCP-1. In contrast, dexamethasone strongly inhibited cytokine production in renal fibroblasts and lung epithelial cells, emphasising the cell-type specificity of glucocorticoid action.
To investigate the effect of dexamethasone on other stimuli than IL-1, PTEC were stimulated with TNF-α and CD40L, which are known to induce cytokine and chemokine production. As was observed for IL-1, TNF-α- and CD40L-stimulated PTEC were insensitive to the inhibitory action of glucocorticoids. In contrast to our findings, dexamethasone has been shown to partially inhibit MCP-1 production in rat PTEC (Wang et al., 1999) or RANTES production by distal and proximal human tubular epithelial cells stimulated with a combination of IL-1β, TNF-α and IFN-γ (Baer et al., 2000). Both the origin of the renal epithelial cell or the nature of the stimulus (protein overload or IFN-γ) might contribute to this discrepancy. Using CD40L as a stimulus (van kooten et al., 1999), we found that RANTES production was not inhibited by dexamethasone (data not shown).
The transcription factor NF-κB is a key regulator of the expression of cytokine genes (Barnes & Karin, 1997; Lee & Burckart, 1998). In the present study we used degradation of IκB-α as a marker to show that CD40L, as well as IL-1 and TNF-α induced activation of the NF-κB signalling pathway. Furthermore, IL-1 induced specific binding of p65 and p50 NF-κB subunits to DNA, as was previously shown for CD40L (Woltman et al., 2000b). In line with these results, the NF-κB inhibitors PDTC and TPCK markedly reduced IL-6, IL-8 and MCP-1 production by PTEC, confirming a role for NF-κB. Glucocorticoid-mediated suppression of cytokine production is mainly attributed to inhibition of NF-κB (Lee & Burckart, 1998; Mckay & Cidlowski, 1999). Initially it was postulated that corticosteroids inhibit NF-κB activation by increasing the transcription of IκB-α (Auphan et al., 1995; Scheinman et al., 1995). Subsequent studies have suggested that glucocorticoids can prevent binding of NF-κB to DNA or interfere with the transactivating potential of NF-κB via protein–protein interaction with the p65 subunit (de bosscher et al., 2000; Ray & Prefontaine, 1994; Reichardt et al., 2001).
The lack of inhibition by dexamethasone suggests that in PTEC NF-κB signalling is insensitive to glucocorticoids, as has been shown in other cell systems (Amrani et al., 1999; Auwardt et al., 1998; Bourke & Moynagh, 1999). However, in these studies expression of adhesion molecules and cytokines is reduced by dexamethasone, while in activated human PTEC, all inflammatory mediators studied so far, including IL-6, RANTES, MCP-1, IL-8 and ICAM-1 seem to be insensitive to dexamethasone. In addition to the p50/p65 pathway of NF-κB activation, alternative pathways have been described (Bourke et al., 2000; Bours et al., 1993; Dechend et al., 1999; Thompson et al., 1995), which are regulated via different IκB-family members (IκB-β, IκB-ε, IκB-γ, p105, p100, Bcl-3) and lead to the activation of different NF-κB complexes (p50/p50 homodimers) or different subunits (c-rel, relB, p52). Although the functional role of these alternative pathways of NF-κB activation is not fully understood, they might play a role in dexamethasone-insensitive activation of NF-κB.
Alternatively, the lack of inhibition by dexamethasone could be explained by aberrant glucocorticoid receptor signalling. We have shown that glucocorticoid receptor mRNA and protein is expressed in renal epithelial cells. Furthermore we have excluded that lack of inhibition by dexamethasone was caused by over-expression of the glucocorticoid receptor beta isoform (GRβ) that could competitively inhibit the functional glucocorticoid receptor (GRα) as was suggested in other studies (Bamberger et al., 1995; Oakley et al., 1999). Unravelling the mechanism of the steroid-insensitivity of PTEC will be the subject of further studies.
In conclusion, we show that production of cytokines and chemokines by activated renal epithelial cells is insensitive to treatment with dexamethasone. These results emphasise the cell-type specific characteristics of glucocorticoid action. Although the mechanism of steroid-insensitivity of PTEC is not yet known, this finding might have important implications for use of glucocorticoids in treatment of acute inflammatory responses in the kidney. While glucocorticoid treatment might efficiently inhibit T-cell-mediated immune responses, the effect on PTEC activation might be limited. Considering the important role of PTEC in the amplification of the local immune response, this might allow for an ongoing low-grade immune response ultimately leading to chronic inflammation and fibrosis. The inability of dexamethasone to down-modulate the active state of PTEC therefore necessitates the development of novel immunosuppressive agents that are able to inhibit the pro-inflammatory role of PTEC during renal inflammation.
Acknowledgments
The authors would like to thank Dr S.J.P. Gobin (Dept. of Immunohaematology and Blood Transfusion, Leiden University Medical Center, The Netherlands) for helpful discussions. This work was financially supported by a grant from the Dutch Kidney Foundation (#C98.1749). C van Kooten is a fellow of the Royal Netherlands Academy of Arts and Sciences.
Abbreviations
- GRα,β
glucocorticoid receptor alpha, beta
- MCP-1
monocyte chemotactic protein 1
- PDTC
pyrrolidine dithiocarbamate
- PTC
PBS/Tween/2% casein
- PTEC
proximal tubular epithelial cells
- RANTES
regulated upon activation, normal T-cell expressed and secreted
- TPCK
n-tosyl-l-phenylalanine chloromethyl ketone
References
- ALMAWI W.Y., HESS D.A., RIEDER M.J. Multiplicity of glucocorticoid action in inhibiting allograft rejection. Cell Transplant. 1998;7:511–523. doi: 10.1177/096368979800700602. [DOI] [PubMed] [Google Scholar]
- AMRANI Y., LAZAAR A.L., PANETTIERI R.A. Up-regulation of ICAM-1 by cytokines in human tracheal smooth muscle cells involves an NF-kappa B-dependent signaling pathway that is only partially sensitive to dexamethasone. J. Immunol. 1999;163:2128–2134. [PubMed] [Google Scholar]
- AUPHAN N., DIDONATO J.A., ROSETTE C., HELMBERG A., KARIN M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- AUWARDT R.B., MUDGE S.J., CHEN C.G., POWER D.A. Regulation of nuclear factor kappaB by corticosteroids in rat mesangial cells. J. Am. Soc. Nephrol. 1998;9:1620–1628. doi: 10.1681/ASN.V991620. [DOI] [PubMed] [Google Scholar]
- BAER P.C., SCHERBERICH J.E., BEREITER-HAHN J., GEIGER H. Induction of RANTES, HLA-DR, and intercellular adhesion molecule-1 on highly purified distal tubular cells from human kidney. Transplantation. 2000;69:2456–2459. doi: 10.1097/00007890-200006150-00045. [DOI] [PubMed] [Google Scholar]
- BAMBERGER C.M., BAMBERGER A.M., DE CASTRO M., CHROUSOS G.P. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J. Clin. Invest. 1995;95:2435–2441. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNES P.J., KARIN M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- BOURKE E., KENNEDY E.J., MOYNAGH P.N. Loss of Ikappa B-beta is associated with prolonged NF-kappa B activity in human glial cells. J. Biol. Chem. 2000;275:39996–40002. doi: 10.1074/jbc.M007693200. [DOI] [PubMed] [Google Scholar]
- BOURKE E., MOYNAGH P.N. Antiinflammatory effects of glucocorticoids in brain cells, independent of NF-kappa B. J. Immunol. 1999;163:2113–2119. [PubMed] [Google Scholar]
- BOURS V., FRANZOSO G., AZARENKO V., PARK S., KANNO T., BROWN K., SIEBENLIST U. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- CATO A.C., WADE E. Molecular mechanisms of anti-inflammatory action of glucocorticoids. Bioessays. 1996;18:371–378. doi: 10.1002/bies.950180507. [DOI] [PubMed] [Google Scholar]
- DE BOSSCHER K., VANDEN BERGHE W., VERMEULEN L., PLAISANCE S., BOONE E., HAEGEMAN G. Glucocorticoids repress NF-kappaB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3919–3924. doi: 10.1073/pnas.97.8.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DECHEND R., HIRANO F., LEHMANN K., HEISSMEYER V., ANSIEAU S., WULCZYN F.G., SCHEIDEREIT C., LEUTZ A. The Bcl-3 oncoprotein acts as a bridging factor between NF-kappaB/Rel and nuclear coregulators. Oncogene. 1999;18:3316–3323. doi: 10.1038/sj.onc.1202717. [DOI] [PubMed] [Google Scholar]
- DECKERS J.G., DE HAIJ S., VAN DER WOUDE F.J., VAN DER KOOIJ S.W., DAHA M.R., VANKOOTEN C. IL-4 and IL-13 augment cytokine- and CD40-induced RANTES production by human renal tubular epithelial cells in vitro. J. Am. Soc. Nephrol. 1998;9:1187–1193. doi: 10.1681/ASN.V971187. [DOI] [PubMed] [Google Scholar]
- DETRISAC C.J., SENS M.A., GARVIN A.J., SPICER S.S., SENS D.A. Tissue culture of human kidney epithelial cells of proximal tubule origin. Kidney Int. 1984;25:383–390. doi: 10.1038/ki.1984.28. [DOI] [PubMed] [Google Scholar]
- GARRONE P., NEIDHARDT E.M., GARCIA E., GALIBERT L., VAN KOOTEN C., BANCHEREAU J. Fas ligation induces apoptosis of CD40-activated human B lymphocytes. J. Exp. Med. 1995;182:1265–1273. doi: 10.1084/jem.182.5.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHOSH S., MAY M.J., KOPP E.B. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- GOOSSENS P.H., SCHOUTEN G.J., 'T HART B.A., BOUT A., BROK H.P., KLUIN P.M., BREEDVELD F.C., VALERIO D., HUIZINGA T.W. Feasibility of adenovirus-mediated nonsurgical synovectomy in collagen-induced arthritis-affected rhesus monkeys. Hum. Gene. Ther. 1999;10:1139–1149. doi: 10.1089/10430349950018139. [DOI] [PubMed] [Google Scholar]
- HONDA M., ORII F., AYABE T., IMAI S., ASHIDA T., OBARA T., KOHGO Y. Expression of glucocorticoid receptor beta in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology. 2000;118:859–866. doi: 10.1016/s0016-5085(00)70172-7. [DOI] [PubMed] [Google Scholar]
- JOHN M., HIRST S.J., JOSE P.J., ROBICHAUD A., BERKMAN N., WITT C., TWORT C.H., BARNES P.J., CHUNG K.F. Human airway smooth muscle cells express and release RANTES in response to T helper 1 cytokines: regulation by T helper 2 cytokines and corticosteroids. J. Immunol. 1997;158:1841–1847. [PubMed] [Google Scholar]
- KARIN M., BEN-NERIAH Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- KIRBY J.A., RAJASEKAR M.R., LIN Y., PROUD G., TAYLOR R.M. Interaction between T lymphocytes and kidney epithelial cells during renal allograft rejection. Kidney Int. 1993;39 Suppl.:S124–S128. [PubMed] [Google Scholar]
- KUROIWA T., SCHLIMGEN R., ILLEI G.G., MCINNES I.B., BOUMPAS D.T. Distinct T cell/renal tubular epithelial cell interactions define differential chemokine production: implications for tubulointerstitial injury in chronic glomerulonephritides. J. Immunol. 2000;164:3323–3329. doi: 10.4049/jimmunol.164.6.3323. [DOI] [PubMed] [Google Scholar]
- LEE J.I., BURCKART G.J. Nuclear factor kappa B: important transcription factor and therapeutic target. J. Clin. Pharmacol. 1998;38:981–993. doi: 10.1177/009127009803801101. [DOI] [PubMed] [Google Scholar]
- MCKAY L.I., CIDLOWSKI J.A. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr. Rev. 1999;20:435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- MIYAZAWA K., MORI A., OKUDAIRA H. Regulation of interleukin-1 beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by glucocorticoids. J. Biochem. (Tokyo) 1998;124:1130–1137. doi: 10.1093/oxfordjournals.jbchem.a022231. [DOI] [PubMed] [Google Scholar]
- MULLER G.A., FRANK J., RODEMANN H.P., ENGLER-BLUM G. Human renal fibroblast cell lines (tFKIF and tNKF) are new tools to investigate pathophysiologic mechanisms of renal interstitial fibrosis. Exp. Nephrol. 1995;3:127–133. [PubMed] [Google Scholar]
- OAKLEY R.H., JEWELL C.M., YUDT M.R., BOFETIADO D.M., CIDLOWSKI J.A. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanisms of action. J. Biol. Chem. 1999;274:27857–27866. doi: 10.1074/jbc.274.39.27857. [DOI] [PubMed] [Google Scholar]
- PATTISON J., NELSON P.J., HUIE P., VON LEUTTICHAU I., FARSHID G., SIBLEY R.K., KRENSKY A.M. RANTES chemokine expression in cell-mediated transplant rejection of the kidney [see comments] Lancet. 1994;343:209–211. doi: 10.1016/s0140-6736(94)90992-x. [DOI] [PubMed] [Google Scholar]
- RAY A., PREFONTAINE K.E. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:752–756. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REICHARDT H.M., TUCKERMANN J.P., GOTTLICHER M., VUJIC M., WEIH F., ANGEL P., HERRLICH P., SCHUTZ G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO, J. 2001;20:7168–7173. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RYAN M.J., JOHNSON G., KIRK J., FUERSTENBERG S.M., ZAGER R.A., TOROK-STORB B. HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 1994;45:48–57. doi: 10.1038/ki.1994.6. [DOI] [PubMed] [Google Scholar]
- SCHEINMAN R.I., COGSWELL P.C., LOFQUIST A.K., BALDWIN A.S.J. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- THOMPSON J.E., PHILLIPS R.J., ERDJUMENT-BROMAGE H., TEMPST P., GHOSH S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- TSAI M.J., O'MALLEY B.W. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- VAN KOOTEN C., DAHA M.R. Cytokine cross-talk between tubular epithelial cells and interstitial immunocompetent cells. Curr. Opin. Nephrol. Hypertens. 2001;10:55–59. doi: 10.1097/00041552-200101000-00009. [DOI] [PubMed] [Google Scholar]
- VAN KOOTEN C., GERRITSMA J.S., PAAPE M.E., VAN ES L.A., BANCHEREAU J., DAHA M.R. Possible role for CD40-CD40L in the regulation of interstitial infiltration in the kidney. Kidney Int. 1997;51:711–721. doi: 10.1038/ki.1997.102. [DOI] [PubMed] [Google Scholar]
- VAN KOOTEN C., VAN DER LINDE X., WOLTMAN A.M., VAN ES L.A., DAHA M.R. Synergistic effect of interleukin-1 and CD40L on the activation of human renal tubular epithelial cells. Kidney Int. 1999;56:41–51. doi: 10.1046/j.1523-1755.1999.00514.x. [DOI] [PubMed] [Google Scholar]
- WANG Y., RANGAN G.K., TAY Y.C., HARRIS D.C. Induction of monocyte chemoattractant protein-1 by albumin is mediated by nuclear factor kappaB in proximal tubule cells. J. Am. Soc. Nephrol. 1999;10:1204–1213. doi: 10.1681/ASN.V1061204. [DOI] [PubMed] [Google Scholar]
- WOLTMAN A.M., DE FIJTER J.W., KAMERLING S.W., PAUL L.C., DAHA M.R., VAN KOOTEN C. The effect of calcineurin inhibitors and corticosteroids on the differentiation of human dendritic cells. Eur. J. Immunol. 2000a;30:1807–1812. doi: 10.1002/1521-4141(200007)30:7<1807::AID-IMMU1807>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- WOLTMAN A.M., DE HAIJ S., BOONSTRA J.G., GOBIN S.J., DAHA M.R., VAN KOOTEN C. Interleukin-17 and CD40-ligand synergistically enhance cytokine and chemokine production by renal epithelial cells. J. Am. Soc. Nephrol. 2000b;11:2044–2055. doi: 10.1681/ASN.V11112044. [DOI] [PubMed] [Google Scholar]