

Abstract
This review examines the biological significance, therapeutic potential and mechanism(s) of action of a range of nitric oxide-releasing non-steroidal anti-inflammatory drugs (NO-NSAID) and related nitric oxide-releasing donating drugs (NODD). The slow release of nitric oxide (NO) from these compounds leads to subtle changes in the profile of pharmacological activity of the parent, non-steroidal anti-inflammatory drugs (NSAID). For example, compared with NSAID, NO-NSAID cause markedly diminished gastrointestinal toxicity and improved anti-inflammatory and anti-nociceptive efficacy. In addition, nitroparacetamol exhibits hepatoprotection as opposed to the hepatotoxic activity of paracetamol. The possibility that NO-NSAID or NODD may be of therapeutic benefit in a wide variety of disease states including pain and inflammation, thrombosis and restenosis, neurodegenerative diseases of the central nervous system, colitis, cancer, urinary incontinence, liver disease, impotence, bronchial asthma and osteoporosis is discussed.
Keywords: Nitric oxide, non-steroidal anti-inflammatory drug, gastrointestinal, ulcers, pain, inflammation, colitis, cancer, asthma, liver
Introduction
Nitric oxide releasing non-steroidal anti-inflammatory drugs (NO-NSAID) are a novel group of drugs with potential therapeutic applications in a variety of clinical conditions. They are synthesized by the ester linkage of an NO-releasing moiety to conventional non-steroidal anti-inflammatory drugs (NSAID) such as aspirin (NO-aspirin), flurbiprofen (NO-flurbiprofen), naproxen (NO-naproxen), diclofenac (nitrofenac) and ibuprofen (NO-ibuprofen), amongst others. The structures of some of the more widely studied NO-NSAID are shown in Figure 1.
Figure 1.
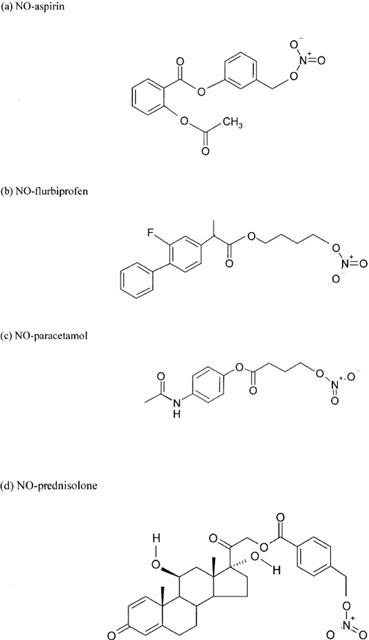
Chemical structures of various NO-donating drugs.
At present, conventional NSAID are the mainstay for the treatment of inflammatory disease. However, these drugs have serious side effects in the gastrointestinal tract which limit their therapeutic usefulness. The NO-NSAID may therefore be considered as the lastest addition to a long list (e.g. enteric-coating of tablets, pro-drugs and selective cyclo-oxygenase-2 (COX-2) inhibitors) of therapeutic attempts to overcome the gastric injury caused by NSAID. The capacity of NO-NSAID to release NO appears to reduce the gastrointestinal toxicity of the parent NSAID in animals (Wallace et al., 1994a, b). Several mechanisms are considered to underlie the protective effect of NO in the stomach including vasodilation of local mucosal blood vessels, inhibition of leukocyte adhesion and inhibition of caspase enzyme activity. Evidence for the relative lack of gastrotoxicity of NO-NSAID and for the mechanism(s) involved will be discussed.
Recently, a series of new compounds have been synthesized in which a NO-releasing group has been linked to parent molecules which are both chemically and pharmacologically unrelated to NSAID. The strategy has been to identify novel molecules with an improved profile of pharmacological activity either in terms of enhanced therapeutic efficacy or reduced side effects. Several of these compounds (hereinafter referred to as nitric oxide donating drugs or NODD) have been described. Examples include nitroparacetamol (NO-paracetamol), nitroprednisolone (NO-prednisolone) andnitromesalamine (NO-mesalamine).
The aim of this review is to outline the biological significance and mechanism(s) of action of NO-NSAID and NODD and to evaluate the potential therapeutic significance of these novel agents.
Catabolism and pharmacokinetics of NO-NSAID and NODD: the NO ‘release' mechanism
The breakdown of NO-NSAID and NODD to yield NO and the corresponding ‘parent' compound is a pivotally important first step in determining the time course and spectrum of pharmacological activity of this class of compounds. NO-NSAID and NODD are chemically stable agents and NO release is therefore achieved enzymatically following exposure to biological tissues. In this way, it may be suggested that NO-NSAID perhaps best resemble organic nitrates which also require metabolic activation prior to NO release. The process is summarized in Figure 2.
Figure 2.
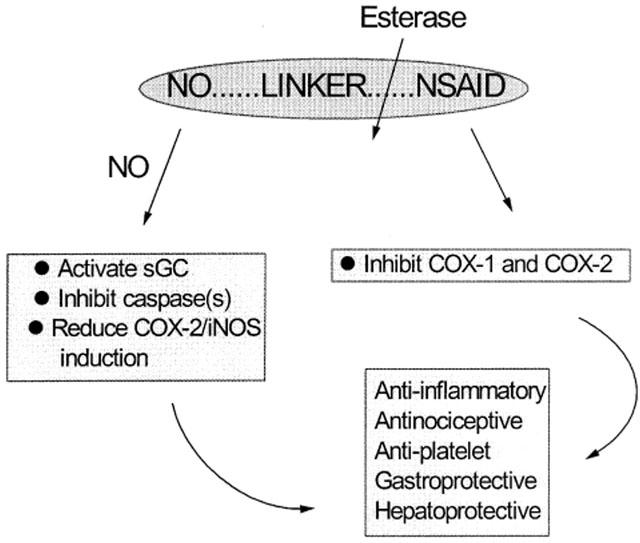
The enzymatic catabolism of NO-NSAID. Some of the potential molecular targets for NO and NSAID and their major pharmacological effects are indicated.
The precise identity of the enzymes involved is not yet clear, but the bulk of the evidence obtained to date suggests that release of the NO moiety from NO-NSAID and NODD occurs as a result of the activity of esterase enzymes (Burgaud et al., 2002; Cirino et al., 1995; Keeble et al., 2001a). Indirect evidence for the involvement of cytochrome P450 in NO-NSAID breakdown has also been presented (Grosser & Schroder, 2000).
Esterases are a large and diverse group of enzymes. Furthermore, individual esterases exhibit varied substrate specificity and are capable of hydrolyzing both endogenous and exogenous esters with a very wide range of structures. The involvement of esterases in NO-NSAID catabolism is suggested not only on theoretical grounds (the nitroxybutyl moiety is linked to the parent compound by an ester bond) but also by direct experimental evidence. For example, flurbiprofen is a more potent inhibitor of the aggregation of human washed platelets in vitro than is NO-flurbiprofen. However, this potency advantage was abolished when purified esterase enzyme was added to the platelet suspension thereby releasing anti-aggregatory NO from its linkage with NO-flurbiprofen (Cirino et al., 1995).
Estimates of the catabolism of NO-NSAID and NODD suggest that the rate of NO release from these compounds both in vitro and in vivo is slow in comparison with other categories of NO donors such as sodium nitroprusside (SNP) and S-nitroso-N-acetyl-D,L-penicillamine (SNAP).
In vitro, incubation of NO-mesalamine in buffer, in the presence of colonic tissue, led to a gradual increase in the concentration of NOX (i.e. nitrate plus nitrite) over a 30 min period reaching a plateau at 60 min. In stark contrast, incubation of an equimolar concentration of SNP under identical in vitro conditions released more NO (five times greater than that produced from NO-mesalamine) considerably more quickly (i.e. within 5 min) (Wallace et al., 1999a). The absolute percentage of NO-mesalamine/SNP catabolized in these experiments was not determined.
The slow rate of catabolism of NO-NSAID in vitro has been confirmed by incubation of NO-flurbiprofen in human whole blood followed by assay for flurbiprofen and NO-flurbiprofen by high performance liquid chromatography (HPLC). Prior to commencing incubation, only intact NO-flurbiprofen was detectable, after incubation for 1 h, flurbiprofen concentration increased to 35% that of NO-flurbiprofen and only by 24 h had almost complete conversion to flurbiprofen taken place (Santini et al., 1996).
In vivo, increases in NOX have been detected in rat plasma for up to 10 h after oral administration of NO-aspirin (Cuzzolin et al., 1996). In separate experiments, administration of equimolar doses of either aspirin or NO-aspirin to rats resulted in the appearance of salicylate in plasma within 1 and 2 h, respectively. The amount of salicylate generated by NO-aspirin was about 45% of that produced by aspirin over a 6 h period suggesting that catabolism of NO-aspirin was still incomplete even at this late time stage (Fiorucci et al., 1999b). Broadly similar data has been obtained by Carini et al. (2001) who noted detectable levels of nitrosylhaemoglobin in plasma 1 h after oral administration of NO-aspirin to rats with peak levels occurring at 4–6 h. Intact nitrofenac has also been identified in plasma for several hours following administration to rats (Benoni et al., 1995).
The relatively slow onset of some of the pharmacological effects of NO-NSAID also imply slow catabolism to NO and the corresponding parent NSAID. For example, pre-incubation of NO-flurbiprofen in human blood for 0, 1 or 3 h prior to stimulation with thrombin resulted in a time-dependent increase in inhibition of platelet aggregation (Cirino et al., 1995). Furthermore, NO-aspirin was a less effective inhibitor of platelet COX activity than aspirin after 1 h preincubation but showed similar inhibitory potency at longer time periods (3–24 h) which was probably due to lack of NO-aspirin catabolism at the 1 h time point (Cuzzolin et al., 1996)
In summary, it seems reasonable to suggest that the relatively tardy rate of NO cleavage from NO-NSAID and NODD distinguishes these compounds from ‘classical' NO donor drugs.
It is important to realise that a number of key questions about this crucial step in the biology of NO-NSAID have yet to be resolved. For example, do all esterases catabolize these compounds or is just a specific sub-set of these enzymes responsible? Precisely where are these enzymes located? Are they extracellular or intracellular? Do NO-NSAID pass across cell membranes – an obvious requirement if the esterases are intracellular? Do tissues/cells/species differ in their esterase activity and what effect does this have on their biological properties?
Blood plasma contains high esterase activity and it is therefore reasonable to assume that this may be a major site for NO release. However, if this is the case then why do NO-NSAID not affect blood pressure? It might perhaps be argued that NO is released from these compounds as a ‘slow trickle' into the bloodstream and is consequently rapidly sequestered by binding to haemoglobin (or other macromolecules) or by reacting with other free radicals such as superoxide anion. However, if such binding (and subsequent biological inactivation) occurs, then how does the NO contribute to the biological effects (e.g. anti-inflammatory, hepatoprotective – see below) of these compounds?
If the NO released is converted to a more stable form (e.g. nitrosylhaemoglobin, Carini et al., 2001), as discussed above, then may this explain the longevity of some of the biological effects of NO-NSAID? Indeed, nitrosohaemoglobin has been proposed as one of the elements (along with the erythrocyte membrane transporter, anion-exchanger-1) which might enable NO to ‘circulate' (Pawloski et al., 2001). Is it possible that NO released from NO-NSAID is also carried around the bloodstream in this way? Interestingly, paracetamol (and some other NSAID) bind avidly to erythrocytes (Pang et al., 1995). If NO-paracetamol (and other NO-NSAID) retain this ability then the high concentrations achieved locally in the vicinity of erythrocytes may facilitate NO uptake and carriage by these cells.
At this stage, the answers to these various important questions remain elusive. It is to be hoped that clarification of these issues will lead to a better understanding of the pharmacology of these compounds.
Pharmacological effects of NO-NSAID
The majority of the early pharmacological studies with NO-NSAID focused on their effect on gastrointestinal function most particularly on stomach ulcer formation and haemorrhage. It was soon realized that NO-NSAID usage was associated with fewer gastrointestinal side effects than NSAID and attention was therefore switched to an evaluation of their effect in clinical conditions in which NSAID have proven efficacy. The clinically beneficial actions as well as the adverse side effects of NO-NSAID and NSAID are compared in the following sections.
NO-NSAID and the gastrointestinal tract
Gastrointestinal damage is an important side effect of NSAID which has been attributed to inhibition of gastric COX-1 activity leading to loss of cytoprotective prostaglandin (mainly PGI2 and PGE2) formation. NSAID are the most commonly self-administered class of drug and gastrointestinal side effects cause an estimated 16,000 deaths and 107,000 cases of hospitalization each year in the U.S.A. (Fries et al., 1998).
One of the earliest experimental findings with NO-NSAID was their ability to ‘spare' the gastrointestinal tract after either acute or chronic use in animals. For example, a single dose of naproxen (80 mg/kg) produced lesions in the rat stomach within 24 h of administration. In contrast, an equimolar dose of NO-naproxen was completely devoid of ulcerogenic activity (Davies et al., 1997). Similar results have now been reported following administration to rats of single doses of NO-aspirin (Takeuchi et al., 1998a, b; Fiorucci et al., 1999a), nitrofenac (Conforti et al., 1993; Wallace et al., 1994a), NO-indomethacin (Takeuchi et al., 2001) and NO-ketoprofen (Wallace et al., 1994a). Furthermore, the damage to the rat jejunum/ileum following a single injection of indomethacin was not apparent in animals given a molar equivalent dose of NO-indomethacin (Mizoguchi et al., 2001). Injection of NO-flurbiprofen also failed to damage the rat small intestine whilst extensive haemorrhagic lesions are observed in animals treated with similar doses of flurbiprofen (Bertrand et al., 1998; Somasundaram et al., 1997). From this, it would appear that acute administration of NO-NSAID is associated with markedly less toxicity than the parent NSAID, both in the small intestine and in the stomach.
Similar results have been obtained in animals exposed chronically to NO-NSAID. Thus, long-term administration of flurbiprofen (twice daily, 1 week) caused weight loss in rats associated with reduction in haematocrit and significant gastrointestinal bleeding. In contrast, NO-flurbiprofen at an equimolar dose had no such deleterious effect (Wallace et al., 1994a). Furthermore, chronic treatment of rats with naproxen caused gastric damage which was not evident in NO-naproxen injected animals (Cuzzolin et al., 1995). Very similar results have been obtained using nitrofenac and diclofenac (Cuzzolin et al., 1994). Recently, some interesting findings in animals with experimentally-induced cirrhosis (due to N-nitrosodiethylamine injection, Kato et al., 2001), Freund's adjuvant-induced arthritis (Kato et al., 2001) or diabetes (Tashima et al., 2000) have been described. In all of these pathological states, the stomach is exquisitely sensitive to the toxic activity of aspirin with even very low doses causing pronounced gastric haemorrhagic lesions. In contrast, molar equivalent dose of NO-aspirin in these diseased animals exhibited only very marginal or no gastrotoxic activity.
In addition to reduced gastrointestinal toxicity relative to NSAID, NO-NSAID also exert a positive effect on the gastric damage caused by other noxious stimuli as well as ameliorating pre-existing gastric lesions. For example, NO-flurbiprofen significantly restored gastric blood flow which was reduced by bacterial lipopolysaccharide (LPS) administration. This vascular event was associated with a concomitant reduction in the LPS-induced gastric damage in these animals (Wallace et al., 1995a). Similarly, in a rat model of haemorrhagic shock, NO-aspirin reduced the fall in gastric blood flow throughout the shock period (Wallace et al., 1997). Furthermore, NO-aspirin also dose-dependently decreased the severity of HCl/ethanol-induced stomach lesions in rats (Takeuchi et al., 1998a). In all of these cases, the parent NSAID were without significant beneficial effect. Finally, in a model of hypothermia-induced haemorrhagic gastric damage, pre-treatment with indomethacin or aspirin significantly worsened the severity of the lesions observed whilst NO-aspirin had no pro-ulcerogenic activity (Ukawa et al., 1998).
From the foregoing it seems very clear that NO-NSAID exhibit little gastric toxicity per se and indeed reduce ulceration produced by other causes in a variety of animal models. Perhaps of even greater interest is the finding that NO-NSAID may also be of value for the healing of pre-existing lesions. Thus, daily administration of nitrofenac (day 7 to 14) after stomach ulcer induction by HCl/ethanol treatment significantly accelerated ulcer healing. As expected, the non-NO releasing parent compound, diclofenac, did not alter ulcer area in the stomach in these experiments (Elliott et al., 1995). Furthermore, both indomethacin and aspirin pre-treatment delayed the healing of thermal cauterization-induced stomach ulcers whilst an equimolar dose of NO-aspirin actually accelerated the healing process (Ukawa et al., 1998).
It is therefore tempting to speculate that NO-NSAID may be of value in the treatment of existing ulcers or, at the very least, are likely to be of greater therapeutic benefit than classical NSAID for the treatment of inflammatory disease in patients with pre-existing gastric damage. To this end, NO-naproxen (AZD3582) has recently been reported to cause markedly less gastroduodenal injury (measured as erosions/ulcers by endoscopy) than did treatment with an equimolar dose of naproxen in human volunteers. Although further clinical studies are required it would appear therefore that the gastric sparing effect of NO-naproxen previously observed in animals also occurs in man (Hawkey et al., 2002).
Mechanisms underlying gastrointestinal tolerance to NO-NSAID
Whilst an abundance of experimental evidence has accumulated testifying to the reduced gastrointestinal toxicity of NO-NSAID (and indeed highlighting the possibility that these agents may even protect against ulcer formation) the precise mechanism(s) involved remain a mystery. Over the years, numerous explanations have been proposed. Some of these are described below.
Vasodilatation of the gastric vasculature
As noted above, administration of NSAID is associated with inhibition of prostaglandin (notably PGI2 and PGE2) formation in the stomach mucosa. These prostaglandins normally protect the mucosal lining against injurious stimuli by a number of mechanisms one of which is dilatation of mucosal blood vessels. In the presence of NSAID, vasodilator PGI2 and PGE2 production is diminished leading to constriction of mucosal blood vessels with the potential for ischaemia, leukocyte entrapment and subsequently for diminished mucosal functionality leading to stomach ulceration and haemorrhage.
With this in mind it is perhaps not surprising that the earliest proposed mechanism for the lack of gastric toxicity of NO-NSAID was NO-mediated mucosal vasodilatation to counteract the vasoconstrictor effect of the parent NSAID. Several lines of evidence support this proposal. For example, in anaesthetized rats, flurbiprofen constricted mesenteric post-capillary venues by 16.6% whilst NO-flurbiprofen dilated these vessels by 6.7% (Wallace et al., 1994a). Similarly, diclofenac administered i.p. in the rat produced a gradual reduction in gastric blood flow to about 50% of the basal value after 60 min whilst nitrofenac did not affect blood flow (Wallace et al., 1994b) suggesting that NO released from nitrofenac counteracted the vasoconstriction brought about by diclofenac.
A vasodilator effect of NO released following NO-NSAID administration most probably plays a significant part in minimizing the ulcerogenic potential of these compounds. Intriguingly, as discussed later, NO-NSAID have relatively weak vasorelaxant activity in vitro and do not affect blood pressure in vivo. It is therefore conceivable that gastric mucosal blood vessels dilate after oral ingestion of NO-NSAID because they are exposed to much higher concentrations of NO than other vascular beds. However, the possibility that the mucosal vasculature is more sensitive to released NO or cleaves NO-NSAID to NO more rapidly (perhaps due to higher endogenous esterase activity) warrants further study.
Inhibition of leukocyte adhesion
Since NSAID-induced gastric damage is a neutrophil-dependent process (Lee et al., 1992; Wallace et al., 1990; Elliot & Wallace, 1998), the effect of NO-NSAID on neutrophil margination in the gastric mucosal vasculature has also been investigated as a possible explanation for the lack of gastric toxicity of this class of compounds.
Administration of flurbiprofen, but not NO-flurbiprofen, significantly increased leukocyte adherence to mesenteric post-capillary venules in anaesthetized rats (Wallace et al., 1994a). Reduced leukocyte infiltration has also been shown by measurement of myeloperoxidase (MPO) activity, which is significantly reduced by NO-aspirin, NO-flurbiprofen and NO-naproxen, more than their respective parent NSAID (Fiorucci et al., 1999a).
These effects on leukocyte function most probably reflect NSAID-mediated inhibition of endothelial COX-1 enzyme activity to decrease the production of anti-adhesive PGI2 (Gimbrone & Buchanan, 1982). Since NO (released from NO-NSAID), like PGI2, also inhibits neutrophil adhesion to the blood vessel wall (Lefer & Lefer, 1996) it is conceivable that NO ‘replaces' the lost PGI2 not only as a mucosal vasodilator but also as an endogenous inhibitor of neutrophil activation.
Whether NO-NSAID also affect other features of the complex interactions between endothelial cells and circulating leukocytes perhaps by influencing the generation or function of leukocyte adhesion molecules is not known. However, NO-NSAID have been shown to reduce transduction via the NF-κB pathway which is an important route to expression of leukocyte adhesion molecules (Roebuck & Finnegan, 1999) and thus an effect on leukocyte sticking to the endothelium cannot be discounted. Further experiments are clearly required to examine the molecular targets underlying this effect of NO-NSAID.
Caspase inactivation
Caspases are a family of cysteine proteases that resemble interleukin-1β (IL-1β) converting enzyme (ICE). These enzymes fall into two broad groups, i.e. caspase-1-like (including caspase-1, -4 and -5) and caspase-3-like enzymes (for reviews, see Thornberry, 1997; Villa et al., 1997). Caspase-1 is primarily involved in cytokine release, cleaving pro-IL-1β to produce IL-1β (Thornberry et al., 1992) and, to a lesser extent, IL-8/interferon-γ (IFN-γ)-inducing factor to IFN-γ (Ghayur et al., 1997). Activation of caspase-3-like enzymes is the major pathway involved in cytokine-induced apoptosis (for review, see Budihardjo et al., 1999). Studies using conventional NO donors have revealed that low levels of NO inhibit cell apoptosis by post-translational inactivation of caspases 1 and 3 (Dimmeler et al., 1997). These results suggested that NO-NSAID might inhibit caspase activity in the gastrointestinal tract.
As predicted, pretreatment of rats with aspirin, but not NO-aspirin, increased gastric mucosal activity of both caspases 1 and 3 (Fiorucci et al., 1999a). The precise mechanism is not clear but NO-NSAID such as NO-aspirin may cause post-translational inactivation of caspases in a direct manner by S-nitrosylating cysteine residues in the enzyme core (Fiorucci et al., 1999a, b; Fiorucci, 2001). In separate experiments, exposure of gastric mucous cells to flurbiprofen resulted in concentration–dependent apoptosis whilst low concentrations of NO-flurbiprofen inhibited apoptosis and caspase-3 activity in these cells (Johal & Hanson, 2000). Moreover, NO-aspirin has been shown to protect gastric chief cells from TNF-α-induced toxicity by activating cGMP-dependent pathways that lead to inactivation of caspase-3 (Fiorucci et al., 1999b). The precise nature of the cGMP-dependent mechanisms are, as yet, unclear, but the soluble guanylyl cyclase inhibitor, 1H-[1,2,4] oxadiazolo (4,3-a] quinoxalin-1-one (ODQ), partially reversed the effect of NO-aspirin on caspase-3 activity (Fiorucci et al., 1999b). Clearly, the involvement of guanylyl cyclase/cGMP in the effect of NO-NSAID on caspase activity may be construed as evidence that the inhibition observed is NO-dependent.
Overall, the inactivation of caspase(s) appears to be an important factor in the gastrointestinal tolerability of NO-NSAID. The finding that NO-NSAID inactivate caspases raises the exciting possibility that these compounds may be of therapeutic use in other pathophysiological conditions in which apoptosis underlies the progression of the disease, e.g. neurodegenerative disease and liver disease. This possibility is discussed in more detail later.
Plasma NSAID concentration
As noted previously, the cleavage of NO-NSAID to NO and free NSAID is a relatively slow process and therefore the delivery of NSAID into plasma after NO-NSAID administration in vivo might be expected to be delayed. If this is indeed the case then it might be argued that the lack of significant gastric side effects associated with NO-NSAID use may be secondary to an altered pharmacokinetic profile of the parent NSAID.
Certainly, administration of single doses of NO-NSAID in the rat results in plasma levels of NSAID that are greatly reduced, i.e. 40–50%, compared with those observed after injecting equimolar doses of the parent compounds (Fiorucci et al., 1999b). This evidence supports the possibility that lower plasma NSAID concentrations following NO-NSAID administration have a bearing on their enhanced gastrointestinal tolerability. However, experiments using higher doses of NO-NSAID have shown that this is not the case. For example, NO-naproxen (116 mg/kg) caused no gastric injury in rats even though this dose actually delivers a higher plasma level of naproxen than that produced by naproxen (30 mg/kg) which is toxic to the stomach (Davies et al., 1997). Similarly, administration of NO-aspirin (450 mg/kg) resulted in significantly higher plasma salicylate levels than similar injection of aspirin (150 mg/kg) within 3 h of administration, but still did not caused mucosal injury when compared with aspirin alone (Fiorucci et al., 1999b).
Overall, a considerable body of evidence supports the view that NO-NSAID are less damaging to the gastrointestinal tract than are the parent NSAID. Indeed, NO-NSAID may prove to be gastroprotective in their own right. It is probably unhelpful even to try and ‘single out' one mechanism of action. It is more likely that this beneficial response to NO-NSAID results from a combination of biological effect of the released NO on blood vessels, circulating blood elements and mucosal cells in situ.
NO-NSAID and inflammation
Since NSAID are widely used anti-inflammatory agents it is perhaps not surprising that the anti-inflammatory activity of NO-NSAID has been a major focus for research with these compounds over recent years. Indeed, a number of different inflammatory disease states have been targeted in this period. Much of the basic work with NO-NSAID has involved the use of animal models of either acute (e.g. carrageenan-induced hindpaw oedema) or chronic (e.g. Freund's adjuvant-induced arthritis) inflammation with more recent studies on the potential application of these new drugs to specific diseases such as colitis and Alzheimer's disease. What is known of the effect of NO-NSAID in each of these conditions is described below. The possible mechanism(s) underlying the anti-inflammatory activity of NO-NSAID are also discussed.
Anti-oedema and anti-arthritis effects
NO-NSAID (like NSAID) exhibit anti-inflammatory activity. However, the relative potency of NO-NSAID (and the parent NSAID), varies with the drug chosen and with the model employed.
Thus, nitrofenac suppressed carrageenan-induced rat hindpaw oedema with a potency similar to that of diclofenac (Conforti et al., 1993; Wallace et al., 1994b). NO-naproxen and naproxen (Davies et al., 1997) and NO-indomethacin and indomethacin (Takeuchi et al., 2001) also caused similar anti-oedema effects in this inflammatory model. NO-aspirin and NO-flurbiprofen produced equal and, at some time points, greater inhibition of carrageenan-induced rat hindpaw oedema when compared with the molar equivalent doses of their parent compounds (Al-Swayeh et al., 1999; 2000a). Chronic treatment with NO-naproxen or naproxen (Cicala et al., 2000; Cuzzolin et al., 1995) and nitrofenac or diclofenac (Cuzzolin et al., 1994) elicited similar anti-inflammatory activity in a Freund's adjuvant model of arthritis in the rat. Finally, NO-paracetamol was significantly more potent (on a molar basis) than paracetamol as an inhibitor of carrageenan-induced rat hindpaw oedema (Al-Swayeh et al., 2000b).
In addition to an anti-inflammatory effect in such models, NO-NSAID may influence other features of the inflammatory response. For example, chronic treatment of rats with NO-naproxen (but not naproxen) enhanced collagen deposition at the site of an inflammatory ‘wound' (caused by prior subcutaneous implantation of polyvinyl sponges) implying that at least this aspect of the healing process may be accelerated by the NO-NSAID (Muscara et al., 2000a, 2000b). In addition, NO-paracetamol and paracetamol exhibit similar anti-pyretic activity in rats challenged with LPS (Fiorucci et al., 2002).
The effect of NO-steroids on inflammatory disease has also been investigated. One of the major therapeutic targets for NSAID and/or steroids is rheumatoid arthritis. Pretreatment with NO-prednisolone has been shown to reduce all measured parameters of inflammation (e.g. leukocyte infiltration, chemokine generation) in a rat model of collagen-induced arthritis (Paul-Clark et al., 2002). NO-prednisolone also exhibited potent anti-inflammatory activity in a variety of other models of inflammation including zymosan-induced peritonitis and in a chronic model of granulomatous inflammation (Paul-Clark et al., 2000). In each of these various tests, NO-prednisolone proved to be significantly more potent than prednisolone on a molar basis.
Irritable bowel disease and colitis
Mesalamine (5-aminosalicylic acid) is one of the most commonly used drugs for the treatment of inflammatory bowel disorder (IBD) (Small & Schraa, 1994). It is believed to be the active moiety of sulphazalazine (Azad Khan et al., 1997). Although adverse effects are quite common with this drug they are mainly reversible and usually not serious (Watkinson, 1986). However, mesalamine is not effective in a large number of patients with IBD (Bresci et al., 1990; Watkinson, 1986) raising the possibility that a NO-releasing derivative of mesalamine may be of therapeutic benefit.
In a rat model of colitis, mesalamine caused a marked reduction in colonic damage score. Interestingly, NO-mesalamine was significantly more effective than mesalamine (Wallace et al., 1999a). The precise mechanism of the augmented effect of NO-mesalamine in this condition is not known. Both NO-mesalamine and mesalamine inhibited FMLP-induced leukocyte adherence to mesenteric vascular endothelium, yet only NO-mesalamine significantly reduced colonic tissue levels of MPO (a marker of neutrophil infiltration). Furthermore, the diameter of mesenteric venules was not affected by mesalamine but was significantly increased by NO-mesalamine, suggesting that vasodilatation by NO may have contributed to the reduction of colonic damage (Wallace et al., 1999a).
In separate experiments, neither NO-aspirin nor nitrofenac affected the severity of colonic damage in rats, but both drugs reduced colonic tissue MPO activity (Reuter et al., 1994; Wallace et al., 1999a). Unlike NO-mesalamine, NO-aspirin did not affect mesenteric vessel diameter in colitic rats, further indicating that the capacity of NO-mesalamine to cause vasodilatation may explain, at least in part, its protective effect.
More recently, it has also been demonstrated that NO-mesalamine, but not mesalamine, produces a concentration–dependent inhibition of caspase activity in colonic epithelial cells (Fiorucci et al., 2001a) and may therefore prevent apoptosis of these cells.
Inflammatory diseases of the CNS
Numerous reports in the literature suggest that chronic ingestion of NSAID is associated with a reduced risk of developing Alzheimer's disease (AD; Jenkinson et al., 1989; McGeer et al., 1990). NSAID can also slow the impairment of cognitive function in patients already diagnosed with AD (reviewed by Hull et al., 2000). It is now clear that chronic inflammatory processes take place in the brains of AD patients (reviewed by Rogers, 1995) leading to a decline in the number of cholinergic neurones within the basal forebrain. Significant inflammatory events that have been noted include the release of cytokines such as IL-1β and TNF-α (Griffin et al., 1998), and activation of caspase enzymes (Shimohama et al., 1999). Albeit protective in AD, the gastrointestinal side effects of NSAID limit their long-term usefulness in such patients.
Since NO-NSAID not only inhibit caspase activity thereby protecting cells against cytokine-induced apoptosis (Fiorucci et al., 1999a, b), but are also less toxic to the gastrointestinal tract than NSAID it is possible that these compounds may prove suitable for the treatment of AD. In a rat model of AD (intraventricular infusion of LPS) daily treatment with NO-flurbiprofen resulted in fewer reactive brain microglia than was apparent in animals treated with aspirin (Hauss-Wegrzyniak et al., 1999a). Interestingly, the same authors have also noted that the effect of NO-flurbiprofen on chronic neuroinflammation in this model is age-dependent and suggested that therapy with NO-NSAID should, preferably, begin from an early age in order to maximize the palliative effect of these compounds (Hauss-Wegrzyniak et al., 1999b). In a recent study, NO-flurbiprofen has been shown to reduce brain beta-amyloid in amyloid precursor protein plus presenilin-transgenic mice (a model of Alzheimer's disease). This was associated with activation of microglia which are presumed to be responsible for clearing beta-amyloid deposits. Interestingly, NO-aspirin proved more efficacious than either ibuprofen or the selective COX-2 inhibitor, celecoxib, in these experiments (Jantzen et al., 2002).
The neuroprotective effect of NO-NSAID has additionally been evaluated in a rat model of stroke/ischaemic brain damage. NO-aspirin (100 mg kg−1 i.p.) administered 10 min after permanent middle cerebral artery (MCA) occlusion afforded greater neuroprotection than either the powerful anti-excitatory drug, FK506 (1 mg kg−1), or aspirin (100 mg kg−1 i.p.) in rats with 24 h post-ischaemic survival (Fredduzzi et al., 2001). In these experiments, aspirin was not neuroprotective, strongly suggesting that NO release accounts for the protective action of NO-aspirin in this model. The mechanism(s) underlying this effect are not clear but it is possible that NO positively influenced the viability of cells at the margins of the ischaemic zone, i.e. the penumbra, by improving blood flow to this region and thereby reducing the size of the cerebral infarct, i.e. the area of dead tissue. Whether the ability of NO-NSAID to inhibit caspases and, thus, apoptosis (Fiorucci et al., 1999a, b) also contributes to the reduction in infarct size observed after treatment with NO-aspirin remains to be determined.
The mechanism of the anti-inflammatory activity of NO-NSAID
Whilst NO-NSAID exhibit anti-inflammatory activity in a variety of different tests the explanation for the enhanced activity of these compounds, observed with some compounds in some animal models, is unknown. A number of putative mechanisms have been put forward.
Firstly, there are a number of reports in the literature that NO-NSAID (but not the corresponding NSAID) inhibit cytokine formation. The ability of a range of NO-NSAID to inhibit caspase-1 (ICE) activity, thereby reducing the formation of pro-inflammatory IL-1β, has been discussed in a previous section of this review and provides a possible explanation for the reduced gastric damaging effect of these compounds (see Fiorucci et al., 1999a, b; Fiorucci, 2001). A similar action may contribute to the enhanced anti-inflammatory effect of NO-NSAID. Interestingly, the nitric oxide-releasing steroid, NO-prednisolone, also reduces IL-1β formation by LPS-treated human peripheral blood mononuclear cells. Prednisolone was considerably less potent in this assay suggesting that the causative agent was NO (Paul-Clark et al., 2000). IL-1β is not the only cytokine to be affected. NO-aspirin (but not aspirin) dose dependently inhibited interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α) formation by LPS-challenged human monocytes (Minuz et al., 2001b). It is therefore tempting to suggest that a NO-mediated inhibition of pro-inflammatory cytokine biosynthesis results from administration of both NO-NSAID and NO-releasing steroids. Whether this action is causally related to the anti-inflammatory effect of these drugs or is merely a secondary response to reduced inflammation brought about by other, unrelated mechanisms remains to be determined.
Bearing in mind the chemical nature of NO-NSAID, the possibility that the chemical modification of NSAID to yield the corresponding NO-NSAID interferes with the ability of the parent compound to inhibit COX activity should be considered. However, this is unlikely to be the case since pretreating animals with NO-NSAID (e.g. NO-flurbiprofen, nitrofenac, NO-aspirin) or equimolar doses of the corresponding parent NSAID caused similar inhibition of COX activity in tissues ex vivo (Bertrand et al., 1998; Wallace et al., 1994b; Reuter et al., 1994). In related experiments, nitrofenac and diclofenac were also equipotent as inhibitors of COX-1 and COX-2 (Reuter et al., 1994). It therefore seems unlikely that the incorporation of a NO-releasing ester moiety into NSAID creates a ‘super-NSAID' with enhanced COX enzyme inhibitory activity.
Although the enhanced anti-inflammatory activity of NO-NSAID is not explained by greater inhibition of COX enzyme activity an effect on the induction of COX-2 cannot be ruled out. For example, NO-aspirin and NO-paracetamol inhibit induction of COX-2 and inducible nitric oxide synthase (iNOS) in lipopolysaccharide (LPS)-pretreated cultured J774 macrophages (Churchman et al., 2001). NO-flurbiprofen has a similar effect to reduce iNOS expression both in J774 macrophages (Cirino et al., 1996) and in rat neutrophils (Mariotto et al., 1995). In all of these cases, the parent compounds (i.e. aspirin, paracetamol or flurbiprofen) are devoid of any such activity. At odds with the finding that NO-NSAID reduces iNOS expression in inflammatory cells is the observation that, in LPS-challenged brain microglial cultures, NO-flurbiprofen (but not flurbiprofen) actually increases expression of this enzyme (Ajmone-Cat et al., 2001). Whether this reflects differences in the response to NO of brain and peripheral macrophages remains to be determined.
Interestingly, there are several reports in the literature that NO may modulate NF-κB activity either by interfering with the phosphorylation of IκB (Peng et al., 1995) and/or by inhibition of NF-κB-DNA binding affinity by nitrosation of the p50 subunit (Matthews et al., 1996). Evidence for an effect of NO-NSAID on the NF-κB pathway has recently been presented (Cui et al., 2001).
Since NF-κB is also responsible in large measure for the cytokine-driven generation of leukocyte adhesion molecules (Roebuck & Finnegan, 1999) it is not inconceivable that NO release from NSAID may affect this process in such a way as to reduce neutrophil recruitment into the inflamed area. Such a possibility has already been raised in this review as one of the factors underlying the inhibition of neutrophil adhesion in gastric mucosal blood vessels as an explanation for the lack of gastric toxicity of these compounds. Clearly, further experiments to investigate this intriguing hypothesis are needed.
NO-NSAID: pain and hyperalgesia
One of the main therapeutic applications of NSAID is analgesia. PGE2 and PGI2, synthesized at the site of inflammation, have long been recognized to sensitize the firing of peripheral nociceptors (Ferreira, 1972). More recently, a permissive role for prostaglandins (synthesized by COX-2) in spinal pain perception has also been noted (e.g. Yamamoto & Nozaki-Taguchi, 1996; Yaksh et al., 2001). In either case, NSAID-induced inhibition of prostaglandin production accounts for the analgesic activity of these compounds in inflammatory pain and hyperalgesia.
Compared with the now established role for prostaglandins in pain perception, the part played by NO in this process is less clear (for review, see Luo & Cizkova, 2000). In the dorsal horn of the spinal cord, NO release promotes glutamate and neurokinin A release from afferent sensory nerve endings and in this way triggers central sensitization (‘wind up') of dorsal nociresponsive neurones. As such, NO donor drugs, acting in the spinal cord, might perhaps be expected to promote pain perception. In stark contrast, NO donors acting at peripheral sites have been demonstrated to reduce pain perception. Clearly, the precise role of NO in pain and hyperalgesia is likely to depend on a variety of factors.
With this in mind, it is of interest that acute NO-naproxen injection inhibited acetic acid-induced abdominal constrictions in the mouse at lower doses than naproxen. Similarly, chronic treatment with NO-naproxen resulted in greater anti-nociception in a Freund's adjuvant-induced rat model of arthritis than did identical treatment with naproxen (Cicala et al., 2000). Moreover, NO-aspirin (Al-Swayeh et al., 1999) and NO-paracetamol (Al-Swayeh et al., 2000, 2000) were markedly more effective in the mouse acetic acid-induced abdominal constriction test than the parent molecules and also produced greater anti-nociception in rats in which hyperalgesia was induced by intraplantar carrageenan injection (Al-Swayeh et al., 1999; 2000, 2000). In separate experiments, NO-paracetamol (but not paracetamol) pretreatment of rats reduced action potential generation and ‘wind-up' in dorsal horn neurones suggesting a spinal site of action of the released NO (Romero-Sandoval et al., 2002).
The rationale for the enhanced anti-nociceptive activity of NO-NSAID is not clear. The inability of naloxone to reduce the anti-nociceptive effect of NO-paracetamol (Romero-Sandoval et al., 2002) suggests that release of endogenous opioids are not involved. Alternative mechanisms must therefore be sought. NO, or one of its redox species, has been suggested to interact with, and reduce transmission via, NMDA receptors in the spinal cord (e.g. Lipton et al., 1994). Since glutamate promotes spinal pain perception by stimulating NMDA receptors, a reduction of transmission via these receptors would be expected to inhibit hyperalgesia. Clearly, the interaction between prostaglandins and NO in terms of pain perception both at peripheral sites of inflammation and in the spinal cord is complex and further studies will be required to determine how and under what experimental conditions these are affected by NO-NSAID.
Platelets, thrombosis, atherosclerosis and restenosis
The use of aspirin for the prevention and treatment of cardiovascular disease, in particular thrombosis, is well established (Harter et al., 1979; Hirsh, 1979). Aspirin exerts its anti-thrombotic effects by irreversible inhibition of COX-1 thereby reducing formation of pro-aggregatory TXA2 by platelets and anti-aggregatory PGI2 by vascular endothelial cells (Schror, 1997). Whilst endothelial cells are able to synthesize new COX-1 enzyme, platelets cannot, and aspirin consequently ‘tips the balance' towards inhibition of platelet aggregation.
Aspirin is not universally effective in all animal models of thrombosis perhaps because it inhibits only one pathway of platelet aggregation thereby leaving alternative pathways (e.g. mediated by ADP and PAF) unaffected. However, NO is a potent inhibitor of platelet activation and adhesion to the vessel wall (see Ignarro, 1999). Thus, NO-aspirin may, at least in theory, be expected to exhibit a greater degree of inhibition of platelet function. In addition, the vasodilator activity of NO may assist in counteracting the vasoconstriction caused by platelet-derived mediators such as TXA2.
Increased anti-thrombotic potency of NO-aspirin, relative to aspirin, has been observed both in vitro and in vivo. Thus, NO-aspirin inhibited platelet aggregation in rat and human platelets in vitro with greater potency than aspirin (Lechi et al., 1996; Wallace et al., 1995b). Furthermore, in murine models of pulmonary thromboembolism, aspirin pretreatment proved to be effective only against collagen or adrenaline-induced thrombus formation and then only at the highest dose used (300 mg kg−1 i.p.). In contrast, a single (lower) dose of NO-aspirin (60–120 mg kg−1 i.p.) protected mice not only against death induced by i.v. injection of collagen or adrenaline but also following treatment with U46619 (a stable TXA2 analogue), thrombin and injection of rat swollen, hardened red blood cells (i.e. mechanically-induced thrombosis) (Momi et al., 2000). In the same study, NO-aspirin was also more effective than aspirin in reducing the collagen or adrenaline-induced fall in circulating platelets. That NO-aspirin was effective against mechanically-induced thrombus formation supports the possibility that vasodilatation by NO-aspirin may contribute to its protective effect in this model (Momi et al., 2000). Finally, it should be noted that anti-thrombotic activity in vivo is not restricted to NO-aspirin. Collagen-induced thromboembolism in the mouse can also be reduced by pre-treatment with NO-flurbiprofen. In these experiments, flurbiprofen was also effective but significantly less potent that NO-flurbiprofen (Cirino et al., 1995). Finally, NO-aspirin (but not aspirin) also reduces tissue factor expression and activity in LPS-challenged human monocytes (Minuz et al., 2001a, b). Whether this influences thrombosis in vivo remains to be determined.
As far as we are aware, the relative anti-platelet potency of NO-aspirin and aspirin in man has yet to be investigated. Such studies are of major importance since they are likely to provide valuable insights into the possibility of using NO-aspirin (or related drugs) for the treatment of thrombotic disease.
In the past, anti-thrombotic drugs such as aspirin have been used as standard treatment for patients undergoing percutaneous coronary angioplasty (PTCA) in an attempt to prevent restenosis. However, aspirin exhibits only modest therapeutic effects on restenosis (Terres et al., 1992) and gastrointestinal side effects limit its usefulness. Damage to coronary vessels during PTCA can result in concomitant impairment of endothelial function and with this in mind, it may be argued that evaluation of NO at the site of such an injury may prevent the consequences of impaired NO-dependent mechanisms. Thus, NO-aspirin might conceivably replace the ‘missing' NO and thereby provide a superior choice of therapy for this condition when compared to aspirin alone.
Indeed, in a model of restenosis in hypercholesterolaemic mice, NO-aspirin has been found to reduce restenosis to a greater degree than aspirin with this effect also occuring at lower doses (Napoli et al., 2001). In a follow-up study, NO-aspirin (but not aspirin) also reduced restenosis (due to carotid balloon injury) in aged rats (Napoli et al., 2002). In addition to NO-aspirin, a beneficial effect of NO-flurbiprofen in a rat model of vascular injury and restenosis has also been observed. In these experiments, NO-flurbiprofen (but not flurbiprofen) significantly reduced neointimal proliferation following PTCA. The biological effect of NO-flurbiprofen was correlated with an increase in plasma NOX concentration and it was therefore concluded that the benefit conferred was due to NO release from the compound (Maffia et al., 2002). In this context, it is of interest that effects of NO-NSAID on vascular smooth muscle proliferation have also been reported. Low concentrations of NO-aspirin (and other NO donors) inhibit growth of cultured rat aortic smooth muscle cells by a mechanism most probably related to S-nitrosylation of ornithine decarboxylase (Ignarro et al., 2001).
Interestingly, NO-aspirin reduced ischaemia-induced damage to the isolated perfused rabbit heart whilst aspirin and celecoxib exacerbated the resulting myocardial dysfunction (Rossoni et al., 2002). A similar protective effect on myocardial damage (determined as incidence of arrhythmias, infarct size and mortality) was apparent after pretreating anaesthetized rats (Rossoni et al., 2001) or rabbits (Rossoni et al., 2000) with NO-aspirin (and to a much lesser extent, aspirin). Furthermore, NO-aspirin also reduced myocardial injury following ischaemia and reperfusion in the pig (Wainwright et al., 2002). Whether this protective effect reflects coronary vasodilatation, neutrophil and/or platelet trapping in the coronary microcirculation, direct effects on cardiac cells or a combination of all three is not yet clear.
Overall, it would appear that NO-aspirin (and possibly other NO-NSAID) may merit consideration as alternatives to classical NSAID especially in situations when gastrointestinal intolerance to these compounds is a problem.
Cancer chemotherapy
The long-term use of NSAID significantly reduces the incidence of some forms of cancer notably colon cancer (for review, see Lupulescu, 1996; Bakhle, 2001). Various animal and human tumour tissues contain high concentrations of prostaglandins (Lavagna et al., 2001), which have well characterized tumour-promoting activities (see Lupulescu, 1996; Smythies, 1979). Such prostaglandins are derived from COX-2 enzyme activity (Lim et al., 2001; for review, see Watanabe et al., 2000) and act to inhibit apoptosis (Dubois et al., 1996). Inhibition of apoptosis improves the life span of cells, increasing the probability of mutations occuring and, thus, the likelihood of uncontrolled cell division. Inhibition of COX by NSAID therefore decreases tumour growth (for review, see Moore & Simmons, 2000). However, as has been noted for other clinical conditions, the gastrointestinal side effects of these drugs can limit their use in cancer chemoprevention and as such NO-NSAID might provide a safer alternative to the traditional NSAID.
Perhaps not surprisingly, NO-NSAID have been shown to affect cancer growth both in vivo and in vitro. For example, in an in vivo rat model of colonic adenocarcinoma, treatment with aspirin reduced the number of aberrant crypt foci induced by trinitrobenzene/sulphonic acid by 64%, whilst NO-aspirin was more effective (85% reduction) (Bak et al., 1998).
Similarly, in a cultured colonic adenocarcinoma cell line, nitrosulindac exerted a more potent inhibitory effect than sulindac on the growth of these cells (Lavagna et al., 2001). The superior effect of NO-sulindac again suggests that the NO moiety was, at least partly, responsible for the enhanced anti-neoplastic activity of this drug (Lavagna et al., 2001).
Overall, the NO moiety of NO-NSAID appears to exert an anti-proliferative activity which is independent of the effects of NSAID. Consequently, NO-NSAID appear to be more potent than the corresponding NSAID in the prevention of tumour growth. However, the exact mechanism(s) underlying this effect of NO has not, as yet, been determined.
Bone, calcium and osteoporosis
Prostaglandins have a regulatory effect on bone resorption. The principle effect of PGE2, is to stimulate both bone resorption and formation (for review, see Kawaguchi et al., 1995). Prostaglandins may play an important role in post-menopausal bone density reduction since oestrogen deficiency, which decreases bone turnover, has also been found to increase prostaglandin biosynthesis in bone (for review, see Kawaguchi et al., 1995). With this in mind, NSAID have been shown, in some models at least, to exert a positive effect on bone mass. For example, flurbiprofen depresses bone resorption in young rats without lowering bone formation (Jee et al., 1988) and decreases the resorption of bone in chronic destructive periodontal disease in beagles (Williams et al., 1985).
NO also has a positive effect on bone density (Van't Hof & Ralston, 2001) and the possibility has therefore been raised that NO-NSAID, by combining two potentially useful therapeutic entities, may be a good choice for the drug therapy of this condition. Ovariectomized B57B16 mice treated daily with flurbiprofen or vehicle over a 3 week period exhibited reduced bone mass density (BMD). In contrast, NO-flurbiprofen-treated mice showed little change in BMD (Armour et al., 2001). Furthermore, in vitro, NO-flurbiprofen strongly inhibited both basal and IL-1-stimulated osteoclast formation and resorption while flurbiprofen had little effect on osteoclast numbers and only partially reversed the effects of IL-1 (Armour et al., 2001).
Although still at a preliminary stage, the evidence available to date suggests that NO-NSAID may have a beneficial effect for the therapy of osteoporosis.
Vascular reactivity and blood pressure: NO-NSAID as atypical NO donors?
From the foregoing it will be clear that several of the pharmacological effects of NO-NSAID described to date may be accounted for by an effect on blood vessel tone or vascular perfusion. It is therefore especially important to evaluate here the interaction of these compounds with the cardiovascular system.
It is well established that NO is a potent vasodilator both in vitro and in vivo by activating soluble guanylyl cyclase (sGC) in vascular smooth muscle cells (Ignarro, 1999; Moncada, 1997) leading to accumulation of intracellular cGMP. It might therefore be expected that low doses of NO-NSAID would be able to relax blood vessels. However, this does not seem to be the case.
To date, there have been relatively few attempts to examine the vasorelaxant effect of NO-NSAID in vitro. Adami et al. (1996) reported that NO-flurbiprofen caused dose-dependent relaxation of precontracted rat aortic rings. In addition, we have recently shown that NO-aspirin, NO-prednisolone, NO-flurbiprofen and NO-paracetamol relax the noradrenaline-precontracted rat aorta and that NO-flurbiprofen is, additionally, vasodilator in the perfused rat renal vascular bed (Keeble et al., 2001b). Such a vasorelaxant effect is secondary to NO release since responses were potentiated by the cGMP phosphodiesterase inhibitor, zaprinast and reduced by ODQ. However, NO-NSAID are considerably less potent vasorelaxants than ‘classical' nitrovasodilators. In the noradrenaline-precontracted rat aorta, for example, SNP (EC50, 35.7 nM) was at least three orders of magnitude more effective than NO-NSAID (EC50s in the range, 71–688 μM) (Keeble et al., 2001b).
The relative lack of vasorelaxant effect of NO-NSAID in vitro is reflected in the inability of these compounds to reduce systemic blood pressure in normotensive animals in vivo. Thus, i.p. or p.o. administration of NO-NSAID to rats produced no significant change in blood pressure or heart rate over the following several hours whilst, in identical experiments, an equimolar dose of sodium nitroprusside resulted in severe hypotension causing death within 5 min (Wallace et al., 1994a). Furthermore, NO-aspirin did not affect blood pressure in the anaesthetized pig (Wainwright et al., 2002).
However, NO-NSAID are not totally devoid of effects on the vascular system. For example, as noted previously, single doses of several NO-NSAID (NO-flurbiprofen, NO-ketoprofen) dilate the gastric vasculature (Wallace et al., 1994a). Furthermore, a significant hypotensive response to NO-aspirin has been noted in hypertensive rats (Muscara et al., 2001). Chronic treatment with NO-NSAID has also uncovered a vasodilator effect of NO released from these compounds. Thus, long-term administration of either naproxen or NO-naproxen increased blood pressure in conscious rats (Muscara et al., 1998). However, the vasopressor effect of NO-naproxen was significantly less than that of naproxen implying that release of NO, at least partly, counteracted the hypertensive effect of naproxen. Similarly, the vasopressor effect of chronic L-NAME treatment in rats is potentiated by naproxen, but reduced by NO-naproxen (Muscara et al., 1998) and NO-aspirin (Muscara et al., 2001). Also, in a model of hypertension induced by occluding one renal artery, naproxen (administered daily for 2 weeks from 2 weeks after occlusion) significantly exacerbated the increase in blood pressure whilst, in contrast, NO-naproxen significantly reduced blood pressure (Muscara et al., 2000a, 2000b). Intriguingly, whilst NO-flurbiprofen has no effect on blood pressure of anaesthetized rats per se it has been reported to prevent the long-term (5 h) fall in blood pressure after injection of bacterial LPS (McLoughlin et al., 1999) and to reduce the hyporeactivity of aortic rings removed from LPS-challenged cirrhotic rats (Lefilliatre et al., 2001). The mechanism which underlies this protective effect of NO-flurbiprofen on the vasculature is not known but may involve inhibition of LPS-mediated vascular iNOS expression. Certainly, such observations are potentially of clinical significance for the treatment of conditions such as septic shock.
The mechanism(s) underlying the lack of vasodepressor activity of NO-NSAID in normotensive animals are not clear. However, the slow release of NO from these compounds may mean that the amount of ‘active' NO present at vascular smooth muscle sites at any one point in time is insufficient to cause widespread vasodilatation in the animal. Of course, this does not necessarily mean that changes in blood flow through individual vascular beds (e.g. the gastric mucosa) do not occur after NO-NSAID treatment. The ability of NO-NSAID to lower blood pressure in hypertensive animals cannot be explained at the present moment. It might be argued that an increase in blood pressure may, by some unknown mechanism, render vascular tissue more sensitive to NO released from these compounds. Further experiments to examine this possibility would be of value.
In summary, whilst the underlying mechanisms remain unclear, it seems reasonable to suggest that the lack of cardiovascular activity of NO-NSAID in normotensive animals renders these compounds useful experimental tools with which to probe further the vascular pharmacology of NO. Furthermore, if the absence of significant vasodepressor effect of these compounds is confirmed in human subjects then NO-NSAID may provide a safer alternative than the parent NSAID for use in patients suffering from hypertension or other vascular states (e.g. angina pectoris, Raynaud's disease) in which localized vasoconstriction of vascular beds is to be avoided.
NO-donating drugs (NODD): a ‘second generation' of compounds with an ester-linked NO moiety and reduced side effects?
As mentioned previously, since the introduction of NO-NSAID a range of other compounds (unrelated to NSAID) have also been synthesized with an ester-linked NO moiety. Examples include nitric oxide releasing-paracetamol (NO-paracetamol), prednisolone (NO-prednisolone) and mesalamine (NO-mesalamine).
As with NO-NSAID, the NO moiety in NODD is released by esterase action and then acts to lessen the adverse side effects of such compounds. For example, it is well established that high concentrations of paracetamol cause severe and often fatal acute liver failure in man (Plevis et al., 1998). In studies in the rat, administration of paracetamol produced significant rises in plasma enzyme markers of liver injury (e.g. aspartate aminotransferase, alanine aminotransferase) after 6 h whilst administration of the molar equivalent dose of NO-paracetamol was devoid of hepatotoxic activity (Futter et al., 2001). Furthermore, recent work has revealed that NO-paracetamol is safe in mice with pre-existing liver disease and even protects against the hepatoxic effects of co-administered paracetamol (Fiorucci et al., 2002).
Similarly, the side effects of prednisolone (e.g. hypertension, osteoporosis) are very much less apparent with its NO-releasing derivative. For example, chronic treatment (3 weeks) of rats with NO-prednisolone resulted in a significantly lower blood pressure than in animals given the same molar dose of prednisolone (Di Filippo et al., 2000). Furthermore, incubation of NO-prednisolone with primary osteoclasts in vitro has been shown to cause a significant reduction in bone resorption when compared to identical treatment with prednisolone (di filippo et al., 2000) suggesting a bone-sparing effect of NO-prednisolone.
Incorporation of a NO-releasing moiety has therefore proved very successful in limiting the side effects of both paracetamol and prednisolone at least in laboratory animals. Further research is now necessary to investigate the effects of concomitant NO release on the side effects of other compounds and to determine whether similar reduction in side effects is seen in man.
Potential therapeutic applications of NODD
Treatment of impotence
Sildenafil is a potent and selective phosphodiesterase-5 (PDE5) inhibitor (see Corbin & Francis, 1999). The target organ for sildenafil is the penis where the drug elevates cGMP levels in the corpus cavernosum thereby enhancing the capacity of the penis to both erect and thereafter to remain erect (Goldstein et al., 1998). Although sildenafil is reasonably effective in diabetics with impotence it is less effective than in non-diabetic, impotent men (Palumbo et al., 2001; Rendell et al., 1999). Since impairment of NO release from nitrergic nerves can underlie impotence and endothelial dysfunction (with diminished NO release) is a common symptom of diabetes (Sullivan et al., 1998) it seems reasonable to propose that a NO releasing compound of sildenafil may present a better choice of anti-impotence drug for men with diabetes.
Although the direct testing of such a hypothesis in male volunteers has yet to be carried out some data to support this possibility has been obtained from experiments in animals. For example, sildenafil nitrate was some 5–10 times more potent than sildenafil in relaxing phenylephrine-precontracted strips of corpus cavernosum from hypercholesterolaemic rabbits (Riffaud et al., 2001). Moreover, in human corpus cavernosum tissue, sildenafil did not affect cGMP concentration in the absence of a NO donor whereas sildenafil nitrate increased cGMP concentrations in this preparation in a concentration-dependent manner in the absence of any other drug (Riffaud et al., 2001). It might be argued that the absence of ‘NO drive' (provided by the NO donor) in this animal model may be akin to the situation in diabetics.
Bronchial asthma
Salbutamol, a β2-adrenoceptor agonist, is widely used for the treatment of asthma (see Price & Clissold, 1989). It inhibits bronchoconstriction of airway smooth muscle in response to spasmogens such as histamine (Cockcroft et al., 1977). Since NO is also bronchodilator (Dupuy et al., 1992), a NO-releasing salbutamol compound may therefore provide a more powerful bronchodilator than salbutamol alone.
Recent work has revealed that salbutamol nitrate causes dose-related inhibition of histamine-induced bronchoconstriction in guinea-pigs (Toward et al., 2001. Indeed, at the highest dose of the compound used, histamine-induced responses were almost completely abolished. Salbutamol nitrate was more potent than salbutamol in this study.
In addition to β-adrenoceptor agonists, steroids are also a first line of therapy for the treatment of asthma (Suissa & Ernst, 2001. Accordingly, NO-steroids have also been studied for their effect in this disease. The anti-inflammatory activity of NO-prednisolone has been described earlier in this review. A NO-releasing moiety has now been attached to budesonide, dexamethasone and hydrocortisone to produce their respective NODD. All of these NO-releasing steroid derivatives possess more potent anti-inflammatory activity than their parent compounds in vitro which effect was coupled with a concentration-dependent bronchodilator activity in vivo (Tallet et al., 2002). Although the experiments carried out to date have concentrated largely on the response of airways smooth muscle, the data obtained is sufficiently encouraging to propose that NO-steroids may be able to alleviate the symptoms of asthma in vivo. However, even if this is not the case, the capacity of the NO moiety to limit the side effects of NO-steroids (di filippo et al., 2000; Paul-Clark et al., 2002) could render these compounds useful treatments for asthma.
Liver disease
The ability of NO-paracetamol to prevent the liver toxicity caused by paracetamol has been noted previously in this review (Futter et al., 2001; Fiorucci et al., 2002).
Other NODD have also been found to reduce hepatic injury. For example, concanavalin A-induced hepatitis is an immune disease caused by caspase activation and the subsequent release of cytokines, in particular IFN-γ (Küsters et al., 1996). NO-aspirin, but not aspirin, has been shown to produce S-nitrosylation/inhibition of caspases involved in cytokine production in concanavalin A-induced hepatitis (Fiorucci et al., 2000) and to protect against the resulting liver damage.
Ursodeoxycholic acid (UDCA) is a bile salt which is used in the treatment of liver disease (see Cirello & Zwas, 1994). However, long-term treatment of patients with this drug in the clinic does not prevent continuing bile duct destruction or the histological, virological and biochemical impairment of liver function in patients with primary biliary cirrhosis or virus-induced hepatitis C (Goulis et al., 1999). In murine models of concanavalin A-induced hepatitis, NO-UDCA, but not UDCA, protected against liver damage by inhibiting caspase enzyme activity and subsequent cytokine production (Fiorucci et al., 2001b). Thus, caspase inactivation appears to be an important factor in the prevention of this type of liver disease.
The release of NO from both paracetamol and ursodeoxycholic acid appears to reduce liver damage at least in these animal models. Whether a similar strategy might be employed to treat liver disease in main remains an open question.
Conclusions
Based on the evidence available in the literature and summarized in this review it may be concluded that NO-NSAID and NODD are novel compounds with a profile of pharmacological activity which is different from that of their respective parent molecules. Clearly, much has still to be learnt about these compounds. Further work to evaluate their effect in man is required as are additional studies to unravel the complexities of their mechanism of action. Despite the uncertainties it is certainly the case that NO-NSAID and NODD exhibit distinct advantages over their respective parent molecules. It is quite conceivable that these advantages may, ultimately, be exploited clinically. Perhaps even more importantly, these compounds are likely to prove useful ‘tools' with which to prove further the biological roles of NO within the body.
Acknowledgments
We would like to thank the British Pharmacological Society for financial support.
Abbreviations
- ADP
adenosine diphosphate
- AD
Alzheimer's disease
- BMD
bone mass density
- COX
cyclo-oxygenase
- FMLP
N-formyl-methionyl-leucyl-phenylalanine
- IBD
inflammatory bowel disorder
- IFN-γ
interferon-γ
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- ICE
interleukin-1β converting enzyme
- MCA
middle cerebral artery
- MPO
myeloperoxidase
- NO
nitric oxide
- NODD
nitric oxide donating drugs
- NO-NSAID
nitric oxide releasing non-steroidal anti-inflammatory drugs
- L-NAME
L-NG nitro arginine methyl ester
- NO-aspirin
nitroaspirin
- NO-flurbiprofen
nitroflurbiprofen
- NO-naproxen
nitronaproxen
- NO-prednisolone
nitroprednisolone
- nitrofenac
nitrodiclofenac
- NO-ibuprofen
nitroibuprofen
- NO-indomethacin
nitroindomethacin
- NO-mesalamine
nitromesalamine
- NO-UDCA
nitroursodeoxycholic acid
- NANC
non-adrenergic, non-cholinergic
- NSAID
non-steroidal anti-inflammatory drugs
- NOX
nitrate plus nitrite
- OC
osteoclast
- PTCA
percutaneous coronary angioplasty
- PAF
platelet activating factor
- ODQ
1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one
- SNP
sodium nitroprusside
- sGC
soluble guanylyl cyclase
- SNAP
S-N-acetylpenicillamine
- TNF-α
tissue necrosis factor-α
- UDCA
ursodeoxycholic acid
References
- ADAMI A., CUZZOLIN L., MINUZ P., CRIVELLENTE F., LECHI A., BENONI G. Vasodilating properties of a new non-steroidal anti-inflammatory drug, nitroflurbiprofen, on rat aortic rings. Pharmacol. Res. 1996;33:239–244. doi: 10.1006/phrs.1996.0033. [DOI] [PubMed] [Google Scholar]
- AJMONE-CAT M.A., NICOLINI A., MINGHETTI L. Differential effects of the nonsteroidal anti-inflammatory drug flurbiprofen and its nitric oxide-releasing derivative, nitroflurbiprofen, on prostaglandin E2, interleukin-1β, and nitric oxide synthesis by activated microglia. J. Neurosci. Res. 2001;66:715–722. doi: 10.1002/jnr.10038. [DOI] [PubMed] [Google Scholar]
- AL-SWAYEH O.A., CLIFFORD R.H., DEL SOLDATO P., MOORE P.K. A comparison of the anti-inflammatory and anti-nociceptive activity of nitroaspirin and aspirin. Br. J. Pharmacol. 2000a;129:343–350. doi: 10.1038/sj.bjp.0703064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AL-SWAYEH O.A., CLIFFORD R.H., MOORE P.K. Anti-oedema and anti-nociceptive effect of nitroprednisolone and nitroflurbiprofen. Br. J. Pharmacol. 1999;127:84. [Google Scholar]
- AL-SWAYEH O.A., FUTTER L.E., CLIFFORD R.H., MOORE P.K. Nitroparacetamol exhibits anti-inflammatory and anti-nociceptive activity. Br. J. Pharmacol. 2000b;130:1453–1456. doi: 10.1038/sj.bjp.0703509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARMOUR K.J., VAN'T HOF R.J., ARMOUR K.E., TORBERGSEN A.C., DEL SOLDATO P., RALSTON S.H. Inhibition of bone resorption in vitro and prevention of ovariectomy-induced bone loss in vivo by flurbiprofen nitroxybutylester (HCT1026) Arthritis. Rheum. 2001;44:2185–2192. doi: 10.1002/1529-0131(200109)44:9<2185::aid-art372>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- AZAD KHAN A.K., PIRIS J., TRUELOVE S.C. An experiment to determine the active moiety of sulphasalazine. Lancet. 1977;2:892–895. doi: 10.1016/s0140-6736(77)90831-5. [DOI] [PubMed] [Google Scholar]
- BAK A.W., MCKNIGHT W., LI P., CALIGNANO A., CIRINO G., WALLACE J.L. Cyclooxygenase-independent chemoprevention with an aspirin derivative in a rat model of colonic adenocarcinoma. Life Sci. 1998;62:PL367–PL373. doi: 10.1016/s0024-3205(98)00191-x. [DOI] [PubMed] [Google Scholar]
- BAKHLE Y.S. COX-2 and cancer: a new approach to an old problem. Br. J. Pharmacol. 2001;134:1137–1150. doi: 10.1038/sj.bjp.0704365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENONI G., TERZI M., ADAMI A., GRIGOLINI L., DEL SOLDATO P., CUZZOLIN L. Plasma concentration and pharmacokinetic parameters of nitrofenac using a simple and sensitive HPLC method. J. Pharm. Sci. 1995;84:93–95. doi: 10.1002/jps.2600840121. [DOI] [PubMed] [Google Scholar]
- BERTRAND V., GUIMBAUD R., SOGNI P., LAMRANI A., MAUPRIVEZ C., GIROUD J.P., COUTURIER D., CHAUVELOT-MOACHON L., CHAUSSADE S. Role of tumour necrosis factor-alpha and inducible nitric oxide synthase in the prevention of nitro-flurbiprofen small intestine toxicity. Eur. J. Pharmacol. 1998;356:245–253. doi: 10.1016/s0014-2999(98)00550-0. [DOI] [PubMed] [Google Scholar]
- BRESCI G., CARRAI M., VENTURINI G., GAMBARDELLA L. Therapeutic effectiveness and tolerance of 5-aminosalicylic acid in short term treatment of patients with ulcerative colitis at a low or medium phase of activity. Int. J. Tissue React. 1990;12:243–246. [PubMed] [Google Scholar]
- BUDIHARDJO I., OLIVER H., LUTTER M., LUO X., WANG X. Biochemical pathways of caspase activation during apoptosis. Ann. Rev. Cell Dev. Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- BURGAUD J.L., RIFFAUD J.P., DEL SOLDATO P. Nitric-oxide releasing molecules: a new class of drugs with several major indications. Curr. Pharm. Des. 2002;8:201–213. doi: 10.2174/1381612023396357. [DOI] [PubMed] [Google Scholar]
- CARINI M., ALDINI G., STEFANI R., ORIOLI M., FACINO R.M. Nitrosylhemoglobin, an unequivocal index of nitric oxide release from nitroaspirin: in vitro and in vivo studies in the rat by ESR spectroscopy. J. Pharm. Biomed. Anal. 2001;26:509–518. doi: 10.1016/s0731-7085(01)00478-2. [DOI] [PubMed] [Google Scholar]
- CHURCHMAN A., MALLINSON H., CLIFFORD R.H., FUTTER L.E., DEL SOLDATO P., MOORE P.K., BAYDOUN A.R. Regulation of COX-2 and iNOS expression by novel NO-NSAIDs. Inflammation Res. 2001;50:S211. [Google Scholar]
- CICALA C., IANARO A., FIORUCCI S., CALIGNANO A., BUCCI M., GERLI R., SANTUCCI L., WALLACE J.L., CIRINO G. NO-naproxen modulates inflammation, nociception and downregulates T cell responses in rat Freund's adjuvant arthritis. Br. J. Pharmacol. 2000;130:1399–1405. doi: 10.1038/sj.bjp.0703449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIRELLO N.W., ZWAS F.R. Ursodeoxycholic acid in the treatment of chronic liver disease. Am. J. Gastroenterol. 1994;89:1447–1452. [PubMed] [Google Scholar]
- CIRINO G., CICALA C., MANCUSO F., BAYDOUN A.R., WALLACE J.L. Flurbinitroxybutylester: A novel anti-inflammatory drug has enhanced anti-thrombotic activity. Thrombosis Res. 1995;79:73–81. doi: 10.1016/0049-3848(95)00092-6. [DOI] [PubMed] [Google Scholar]
- CIRINO G., WHEELER-JONES C.P., WALLACE J.L., DEL SOLDATO P., BAYDOUN A.R. Inhibition of inducible nitric oxide synthase expression by novel nonsteroidal anti-inflammatory derivatives with gastrointestinal-sparing properties. Br. J. Pharmacol. 1996;117:1421–1426. doi: 10.1111/j.1476-5381.1996.tb15301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COCKCROFT D.W., KILLIAN D.N., MELLON J.J., HARGREAVE F.E. Protective effect of drugs on histamine-induced asthma. Thorax. 1977;32:429–437. doi: 10.1136/thx.32.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONFORTI A., DONINI M., BROCCO G., DEL SOLDATO P., BENONI G., CUZZOLIN L. Acute anti-inflammatory activity and gastrointestinal tolerability of diclofenac and nitrofenac. Agents Actions. 1993;40:176–180. doi: 10.1007/BF01984058. [DOI] [PubMed] [Google Scholar]
- CORBIN J.D., FRANCIS S.H. Cyclic GMP phosphodiesterase-5: target of sildenafil. J. Biol. Chem. 1999;274:13729–13732. doi: 10.1074/jbc.274.20.13729. [DOI] [PubMed] [Google Scholar]
- CUI Z., DEL SOLDATO P., MOORE P.K., BAYDOUN A.R. Inhibition of NF-kB activation by nitro-paracetamol and nitro-aspirin in J774 macrophages. Inflammation Res. 2001;50:S152. [Google Scholar]
- CUZZOLIN L., ADAMI A., DEGAN M., CRIVELLENTE F., BONAPACE S., MINUZ P., BENONI G. Effect of single and repeated doses of a new nitroderivatve of acetylsalicylic acid on platelet TXA2 production in rats. Life Sci. 1996;58:PL207–PL210. doi: 10.1016/0024-3205(96)00038-0. [DOI] [PubMed] [Google Scholar]
- CUZZOLIN L., CONFORTI A., ADAMI A., LUSSIGNOLI S., MENESTRINA F., DEL SOLDATO P, BENONI G. Anti-inflammatory potency and gastrointestinal toxicity of a new compound, nitronaproxen. Pharmacol. Res. 1995;31:61–65. doi: 10.1016/1043-6618(95)80049-2. [DOI] [PubMed] [Google Scholar]
- CUZZOLIN L., CONFORTI A., DONINI M., ADAMI A., DEL SOLDATO P., BENONI G. Effects on intestinal microflora, gastrointestinal tolerability and anti-inflammatory efficacy of diclofenac and nitrofenac in adjuvant arthritis in rats. Pharmacol. Res. 1994;29:89–97. doi: 10.1016/1043-6618(94)80101-0. [DOI] [PubMed] [Google Scholar]
- DAVIES N.M., ROSETH A.G., APPLEYARD C.B., MCKNIGHT W., DEL SOLDATO P., CALIGNANO A., CIRONO G., WALLACE J.L. NO-naproxen vs. naproxen: ulcerogenic, analgesic and anti-inflammatory effects. Aliment. Pharmacol. Ther. 1997;11:69–79. doi: 10.1046/j.1365-2036.1997.115286000.x. [DOI] [PubMed] [Google Scholar]
- DI FILIPPO C., PERRETTI M., D'AMICO M., ROSSI M.Effects on blood pressure and heart rate of normotensive rats by chronic treatments with prednisolone 21-[(4′-nitrooxymethyl)benzoate] 20006811th International Conference on Advances in Prostaglandin and Leukotriene Research, Florence (Italy), June 3–4p
- DIMMELER S., HAENDELER J., NEHLS M., ZEIHER A.M. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1β-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J. Exp. Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUBOIS R.N., SHAO J., TSUJI M., SHENG H., BEAUCHAMP R.D. G1 delay in cells overexpressing prostaglandin endoperoxide synthase-2. Cancer Res. 1996;56:733–737. [PubMed] [Google Scholar]
- DUPUY P.M., SHORE S.A., DRAZEN J.M., FROSTELL C., HILL W.A., ZAPOL W.M. Bronchodilator action of inhaled nitric oxide in guinea-pigs. J. Clin. Invest. 1992;90:421–428. doi: 10.1172/JCI115877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELLIOTT S.N., MCKNIGHT W., CIRINO G., WALLACE J.L. A nitric oxide-releasing nonsteroidal anti-inflammatory drug accelerates gastric ulcer healing in rats. Gastroenterology. 1995;109:524–530. doi: 10.1016/0016-5085(95)90341-0. [DOI] [PubMed] [Google Scholar]
- ELLIOT S.N., WALLACE J.L. Neutrophil-mediated gastrointestinal injury. Can. J. Gastroenterol. 1998;12:559–568. doi: 10.1155/1998/398384. [DOI] [PubMed] [Google Scholar]
- FIORUCCI S. NO-releasing NSAIDs are caspase inhibitors. Trends Immunol. 2001;22:232–235. doi: 10.1016/s1471-4906(01)01904-4. [DOI] [PubMed] [Google Scholar]
- FIORUCCI S., ANTONELLI E., MENCARELLI A., PALAZZETTI B., ALVAREZ-MILLER L., MUSCARA M., DEL SOLDATO P., SANPAOLO L., WALLACE J.L., MORELLI A. A NO-releasing derivative of acetaminophen spares the liver by acting at several checkpoints in the Fas pathway. Br. J. Pharmacol. 2002;135:589–599. doi: 10.1038/sj.bjp.0704500. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- FIORUCCI S., ANTONELLI E., SANTUCCI L., MORELLI O., MIGLETTI M., FEDERICI B., MANNUCCI R., DEL SOLDATO P., MORELLI A. Gastrointestinal safety of nitric oxide-derived aspirin is related to inhibition of ICE-like cysteine proteases in rats. Gastroenterology. 1999a;116:1089–1106. doi: 10.1016/s0016-5085(99)70012-0. [DOI] [PubMed] [Google Scholar]
- FIORUCCI S., DISTRUTTI E., AJUEBOR M.N., MENCARELLI A., MANNUCCI R., PALAZZETTI B., DEL SOLDATO P., MORELLI A., WALLACE J. NO-mesalamine protects colonic epithelial cells against apoptotic damage induced by proinflammatory cytokines. Am. J. Physiol. 2001a;281:G654–G665. doi: 10.1152/ajpgi.2001.281.3.G654. [DOI] [PubMed] [Google Scholar]
- FIORUCCI S., MENCARELLI A., PALAZETTI B., DEL SOLDATO P., MORELLI A., IGNARRO L. A NO derivative of ursodeoxycholic acid protects aginst liver injury by inhibiting caspase activity. Proc. Natl. Acad. Sci. U.S.A. 2001b;98:2652–2657. doi: 10.1073/pnas.041603898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIORUCCI S., SANTUCCI L., ANTONELLI E., DISTRUTTI E., DEL SERO G., MORELLI O., ROMANI L., FEDERICI B., DEL SOLDATO P., MORELLI A. NO-Aspirin protects from T cell-mediated liver injury by inhibiting caspase-dependent processing of Th1-like cytokines. Gastroenterology. 2000;118:404–421. doi: 10.1016/s0016-5085(00)70223-x. [DOI] [PubMed] [Google Scholar]
- FIORUCCI S., SANTUCCI L., FEDERICI B., ANTONELLI E., DISTRUTTI E., MORELLI O., RENZO G.D., COATA G., CIRINO G., SOLDATO P.D., MORELLI A. Nitric oxide-releasing NSAIDs inhibit interleukin-1β converting enzyme-like cysteine proteases and protect endothelial cells from apoptosis induced by TNF-α. Aliment. Pharmacol. Ther. 1999b;13:421–435. doi: 10.1046/j.1365-2036.1999.00442.x. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H. Prostaglandins, aspirin-like drugs and analgesia. Nature. 1972;240:200–203. doi: 10.1038/newbio240200a0. [DOI] [PubMed] [Google Scholar]
- FREDDUZZI S., MARIUCCI G., TANTUCCI M., DEL SOLDATO P., AMBROSINI M.V. Nitro-aspirin (NC4016) reduces brain damage induced by focal cerebral ischaemia in the rat. Neurosci. Lett. 2001;302:121–124. doi: 10.1016/s0304-3940(01)01672-x. [DOI] [PubMed] [Google Scholar]
- FRIES J., SINGH G., RAMEY D. Clinical Significance and potential of selective COX-2 inhibitors. London: The William Harvey Press; 1998. The clinical epidemiology of non-steroidal anti-inflammatory drug gastropathy; pp. 557–573. [Google Scholar]
- FUTTER L.E., AL-SWAYEH O.A., MOORE P.K. A comparison of the effect of nitroparacetamol and paracetamol on liver injury. Br. J. Pharmacol. 2001;132:10–12. doi: 10.1038/sj.bjp.0703837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHAYUR T., BANERJEE S., HUGUNIN M., BUTLER D., HERZOG L., CARTER A., QUINTAL L., SEKUT L., TALANIAN R., PASKIND M., WONG W., KAMEN R., TRACEY D., ALLEN H. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- GIMBRONE M.A., BUCHANAN M.R. Interactions of platelets and leukocytes with vascular endothelium: in vitro studies. Ann. N.Y. Acad. Sci. 1982;40:171–183. doi: 10.1111/j.1749-6632.1982.tb25716.x. [DOI] [PubMed] [Google Scholar]
- GOLDSTEIN I., LUE T.F., PADMA-NATHAN H., ROSEN R.C., STEERS W.D., WICKER P.A. Oral sildenafil in the treatment of erectile dysfunction. N. Engl. J. Med. 1998;338:1397–1404. doi: 10.1056/NEJM199805143382001. [DOI] [PubMed] [Google Scholar]
- GOULIS J., LEANDRO G., BORROUGHS A.K. Randomised controlled trials of ursodeoxycholic-acid therapy for primary biliary cirrhosis: a meta-analysis. Lancet. 1999;354:1053–1060. doi: 10.1016/S0140-6736(98)11293-X. [DOI] [PubMed] [Google Scholar]
- GRIFFIN W.S.T., SHENG J.G., ROYSTON M.C., GENTLEMAN S.M., MCKENZIE J.E., GRAHAM D.I., ROBERTS G.W., MRAK R.E. Glial-neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle' in disease progression. Brain Pathol. 1998;8:5–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROSSER N., SCHRODER H. A common pathway for nitric oxide release from NO-aspirin and glyceryl trinitrate. Biochem. Biophys. Res. Commun. 2000;274:1255–1258. doi: 10.1006/bbrc.2000.3121. [DOI] [PubMed] [Google Scholar]
- HARTER H.R., BURCH J.W., MARJERUS P.W., STANFORD N., DELMEZ J.A., ANDERSON C.B., WEERTS C.A. Prevention of thrombosis in patients on hemodialysis by low-dose aspirin. N. Engl. J. Med. 1979;301:577–579. doi: 10.1056/NEJM197909133011103. [DOI] [PubMed] [Google Scholar]
- HAUSS-WEGRZYNIAK B., VRANIAK P., WENK G.L. The effects of a novel NSAID on chronic neuroinflammation are age dependent. Neurobiol. Aging. 1999b;20:305–313. doi: 10.1016/s0197-4580(99)00028-7. [DOI] [PubMed] [Google Scholar]
- HAUSS-WEGRZYNIAK B., WILLARD L.B., DEL SOLDATO P., PEPEU G., WENK G.L. Peripheral administration of novel anti-inflammatories can attenuate the effects of chronic inflammation within the CNS. Brain Res. 1999a;815:36–43. doi: 10.1016/s0006-8993(98)01081-6. [DOI] [PubMed] [Google Scholar]
- HAWKEY C., JONES J.I.W., ATHERTON C.T., SKELLY M.M., BEBB J.R., FAGERHOLM B.J., KARLSSON P., BJARNESON I.T.Gastrointestinal safety of AZD3582: A new chemical entity with a novel multi-pathway mechanism of action American Gastroenterological Society 2002. Digestive Diseases Week, ID 446, San Francisco, U.S.A
- HIRSH J. Anti-platelet drugs in thromboembolism. Postgrad. Med. 1979;66:119–123. doi: 10.1080/00325481.1979.11715250. [DOI] [PubMed] [Google Scholar]
- HULL M., LIEB K., FIEBLICH B.L. Anti-inflammatory drugs: a hope for Alzheimer's disease. Expert Opin. Invest. Drugs. 2000;9:671–683. doi: 10.1517/13543784.9.4.671. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J. Nitric oxide: a unique endogenous signalling molecule in vascular biology. Biosci. Rep. 1999;19:51–61. doi: 10.1023/a:1020150124721. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J., BUGA G.M., WEI L.H., BAUER P.M., WU G., DEL SOLDATO P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4202–4208. doi: 10.1073/pnas.071054698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANTZEN P.T., CONNOR K.E., DICARLO G., WENK G.L., WALLACE J.L., ROJIANI A.M., COPPOLA D., MORGAN D., GORDON M.N. Microglial activation and beta-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J. Neurosci. 2002;15:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JEE W.S., LI X.J., LI Y.L. Flurbiprofen-induced stimulation of periosteal bone formation and inhibition of bone resorption in older rats. Bone. 1988;9:381–389. doi: 10.1016/8756-3282(88)90120-2. [DOI] [PubMed] [Google Scholar]
- JENKINSON M.L., BLISS M.R., BRAIN A.T., SCOTT D.L. Rheumatoid arthritis and senile dementia of the Alzheimer's type. Br. J. Pharmacol. 1989;28:86–88. doi: 10.1093/rheumatology/28.1.86-b. [DOI] [PubMed] [Google Scholar]
- JOHAL K., HANSON P.J. Opposite effects of flurbiprofen and the nitroxybutyl ester of flurbiprofen on apoptosis in cultured guinea pig gastric mucous cells. Br. J. Pharmacol. 2000;130:811–818. doi: 10.1038/sj.bjp.0703383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWAGUCHI H., PILBEAM C.C., HARRISON J.R., RAISZ L.G. The role of prostaglandins in the regulation of bone metabolism. Clin. Ortho. 1995;313:36–46. [PubMed] [Google Scholar]
- KATO S., SUZUKI K., IKAWA H., KAMOIKE Y., TAKEUCHI K. Low gastric toxicity of nitric oxide-releasing aspirin, NCX-4016, in rats with cirrhosis and arthritis. Dig. Dis. Sci. 2001;46:1690–1699. doi: 10.1023/a:1010601520497. [DOI] [PubMed] [Google Scholar]
- KEEBLE J., AL-SWAYEH O.A., MOORE P.K. Vasorelaxant effect of nitric oxide releasing steroidal and nonsteroidal anti-inflammatory drugs. Br. J. Pharmacol. 2001b;133:1023–1028. doi: 10.1038/sj.bjp.0704161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEEBLE J., APOSTOLOU K., CLIFFORD R.H., FUTTER L.E., MOORE P.K. The catabolism of NO-NSAIDs and other nitric oxide-releasing adduct drugs in vitro. Proc. Western Pharmacol. Soc. 2001a;44:240. [Google Scholar]
- KÜSTERS S., GARTNER F., KÜNSTLE G., TIEGS G. Interferon gamma plays a critical role in T cell-mediated hepatic injury in mice. Gastroenterology. 1996;113:1315–1322. doi: 10.1053/gast.1996.v111.pm8690213. [DOI] [PubMed] [Google Scholar]
- LAVAGNA C., BURGAUD J.L., DEL SOLDATO P., RAMPAL P. Antiproliferative effects of nitrosulindac on human colon adenocarcinoma cell lines. Biochem. Biophys. Res. Commun. 2001;284:808–816. doi: 10.1006/bbrc.2001.5057. [DOI] [PubMed] [Google Scholar]
- LECHI C., GAINO S., TOMMASOLI R., ZULIANI V., BONAPACE S., FONTANA L., DEGAN M., LECHI A., MINUZ P. In vitro study of the anti-aggregating activity of two nitroderivatives of acetylsalicylic acid. Blood Coag. Fibrinol. 1996;7:206–209. doi: 10.1097/00001721-199603000-00024. [DOI] [PubMed] [Google Scholar]
- LEE M., ALDRED K., LEE E., FELDMAN M. Aspirin-induced acute gastric mucosal injury is a neutrophil-dependent process in rats. Am. J. Physiol. 1992;263:G920–G926. doi: 10.1152/ajpgi.1992.263.6.G920. [DOI] [PubMed] [Google Scholar]
- LEFER A.M., LEFER D.J. The role of nitric oxide and cell adhesion molecules on the microcirculation in ischaemia-reperfusion. Cardiovasc. Res. 1996;32:743–751. [PubMed] [Google Scholar]
- LEFILLIATRE P., SOGNI P., BERTRAND V., DEL SOLDATO P., PATERON D., MOREAU P., LEBREC D. Aortic hyporeactivity to norepinephrine induced by lipopolysaccharide in cirrhotic rats: beneficial effects of a non-steroidal anti-inflammatory drug coupled with a nitric oxide donor. J. Gastroenterol. Hepatol. 2001;16:70–78. doi: 10.1046/j.1440-1746.2001.02386.x. [DOI] [PubMed] [Google Scholar]
- LIM J.W., KIM H., KIM K.H. Nuclear factor-kappaβ regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab. Invest. 2001;81:349–360. doi: 10.1038/labinvest.3780243. [DOI] [PubMed] [Google Scholar]
- LIPTON S.A., SINGEL D.J., STAMLER J.S. Nitric oxide in the central nervous system. Prog. Brain Res. 1994;103:359–364. doi: 10.1016/s0079-6123(08)61149-8. [DOI] [PubMed] [Google Scholar]
- LUO Z.D., CIZKOVA D. The role of nitric oxide in nociception. Curr. Rev. Pain. 2000;4:459–466. doi: 10.1007/s11916-000-0070-y. [DOI] [PubMed] [Google Scholar]
- LUPULESCU A. Prostaglandins, their inhibitors and cancer. Prostaglandins Leukot. Essent. Fatty Acids. 1996;54:83–94. doi: 10.1016/s0952-3278(96)90064-2. [DOI] [PubMed] [Google Scholar]
- MAFFIA P., IANARO A., SORRENTINO R., LIPPOLIS L., MAIELLO F.M., DEL SOLDATO P., IALENTO A., CIRINO G. Beneficial effects of NO-releasing derivative of flurbiprofen (HCT-1026) in rat model of vascular injury and restenosis. Arterioscler. Thromb. Vasc. Biol. 2002;22:263–267. doi: 10.1161/hq0202.104064. [DOI] [PubMed] [Google Scholar]
- MARIOTTO S., CUZZOLIN L., ADAMI A., DEL SOLDATO P., SUZUKI H., BENONI G. Effect of a new non-steroidal anti-inflammatory drug, nitroflurbiprofen, on the expression of inducible nitric oxide synthase in rat neutrophils. Br. J. Pharmacol. 1995;115:225–226. doi: 10.1111/j.1476-5381.1995.tb15867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATTHEWS J.R., BOTTING C.H., PANICO M., MORRIS H.R., HAY R.T. Inhibition of NF-kappa DNA binding by nitric oxide. Nucleic Acids Res. 1996;24:2236–2242. doi: 10.1093/nar/24.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGEER P.L., MCGEER E.G., ROGERS J., SIBLEY J. Anti-inflammatory drugs and Alzheimer's disease. Lancet. 1990;335:1037. doi: 10.1016/0140-6736(90)91101-f. [DOI] [PubMed] [Google Scholar]
- MCLOUGHLIN C., KEEBLE J., MOORE P.K. Effect of nitroflurbiprofen on lipopolysaccharide-induced sepsis. Med. Inflam. 1999;8 Suppl 1:P16-1. [Google Scholar]
- MINUZ P., DEGAN M., GAINO S., MENEGUZZI A., ZULIANI V., SANTONASTASO C.L., DEL SOLDATO P., LECHI A. NCX 4016 (NO-aspirin) inhibits thromboxane biosynthesis and tissue factor experssion and activity in human monocytes. Med. Sci. Monit. 2001a;7:1573–1577. [PubMed] [Google Scholar]
- MINUZ P., DEGAN M., GAINO S., MENEGUZZI A., ZULIANI V., SANTONASTASO C.L., DEL SOLDATO P., LECHI A. NCX4016 (NO-aspirin) has multiple inhibitory effects in LPS-stimulated human monocytes. Br. J. Pharmacol. 2001b;134:905–911. doi: 10.1038/sj.bjp.0704326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIZOGUCHI H., HASE S., TANAKA A., TAKEUCHI K. Lack of small intestinal ulcerogenecity of nitric oxide-releasing indomethacin, NCX-530, in rats. Aliment. Pharmacol. Ther. 2001;15:257–267. doi: 10.1046/j.1365-2036.2001.00916.x. [DOI] [PubMed] [Google Scholar]
- MOMI S., EMERSON M., PAUL W., LEONE M., MEZZASOMA A.M., DEL SOLDATO P., PAGE C.P., GRESELE P. Prevention of pulmonary thromboembolism by NCX-4016, a nitric oxide-releasing aspirin. Eur. J. Pharmacol. 2000;397:177–185. doi: 10.1016/s0014-2999(00)00223-5. [DOI] [PubMed] [Google Scholar]
- MONCADA S. Nitric oxide in the vasculature: physiology and pathophysiology. Ann. N.Y. Acad. Sci. 1997;811:60–67. doi: 10.1111/j.1749-6632.1997.tb51989.x. [DOI] [PubMed] [Google Scholar]
- MOORE B.C., SIMMONS D.L. COX-2 inhibition, apoptosis and chemoprevention by nonsteroidal anti-inflammatory drugs. Curr. Med. Chem. 2000;7:1131–1144. doi: 10.2174/0929867003374273. [DOI] [PubMed] [Google Scholar]
- MUSCARA M.N., LOVREN F., MCKNIGHT W., DICAY M., DEL SOLDATO P., TRIGGLE C.R., WALLACE J.L. Vasorelaxant effects of a nitric oxide-releasing aspirin derivative in normotensive and hypertensive rats. Br. J. Pharmacol. 2001;133:1314–1322. doi: 10.1038/sj.bjp.0704209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUSCARA M.N., MCKNIGHT W., ASFAHA S., WALLACE J.L. Wound collagen deposition in rats: effects of a NO-NSAID and a selective COX-2 inhibitor. Br. J. Pharmacol. 2000a;129:618–686. doi: 10.1038/sj.bjp.0703112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUSCARA M.N., MCKNIGHT W., DEL SOLDATO P., WALLACE J.L. Effect of a nitric oxide-releasing naproxen derivative on hypertension and gastric damage induced by chronic nitric oxide inhibition in the rat. Life Sci. 1998;62:235–240. doi: 10.1016/s0024-3205(98)00072-1. [DOI] [PubMed] [Google Scholar]
- MUSCARA M.N., MCKNIGHT W., LOVREN F., TRIGGLE C.R., CIRINO G., WALLACE J.L. Antihypertensive properties of a nitric oxide-releasing naproxen derivative in two-kidney, one-clip rats. Am. J. Physiol. 2000b;279:H528–H535. doi: 10.1152/ajpheart.2000.279.2.H528. [DOI] [PubMed] [Google Scholar]
- NAPOLI C., ALDINI G., WALLACE J.L., DE NIGRIS F., MAFFEI R., ABETE P., BONADUCE D., CONDORELLI G., RENGO F., SICA V., D'ARMINETO F.P., MIGNONGA C., DE ROSA G. Efficacy and age-related effects of nitric oxide-releasing aspirin on experimental restenosis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1689–1694. doi: 10.1073/pnas.022639399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAPOLI C., CIRINO G., DEL SOLDATO P., SORRENTINO R., SICA V., CONDORELLI M., PINTO A., IGNARRO L.J. Effects of nitric oxide-releasing aspirin versus aspirin on restenosis in hypercholesterolaemic mice. Proc. Natl. Acad. Sci. U.S.A. 2001;98:2860–2864. doi: 10.1073/pnas.041602898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALUMBO F., BETTOCCHI C., SELVAGGI F.P., PRYOR J.P., RALPH D.J. Sildenafil: efficacy and safety in daily clinical experience. Eur. Urol. 2001;40:176–180. doi: 10.1159/000049769. [DOI] [PubMed] [Google Scholar]
- PANG K.S., BARKER K.S., SIMARD A., SCHWAB A.J., GORESKY C.A. Sulfation of acetaminophen by the perfused rat liver: the effect of red blood cell carriage. Hepatology. 1995;22:267–282. [PubMed] [Google Scholar]
- PAUL-CLARK M.J., DEL SOLDATO P., FIORUCCI S., FLOWER R.J., PERRETTI M. 21-NO-prednisolone is a novel nitric oxide releasing derivative of prednisolone with enhanced anti-inflammatory properties. Br. J. Pharmacol. 2000;131:1345–1354. doi: 10.1038/sj.bjp.0703704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAUL-CLARK M.J., MANCINI L., DEL SOLDATO P., FLOWER R.J., PERRETTI M. Potent antiarthritic properties of a glucocorticoid derivative, NCX-1015, in an experimental model of arthritis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1677–1682. doi: 10.1073/pnas.022641099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAWLOSKI J.R., HESS D.T., STAMLER J.S. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- PENG H.B., RAJAVASHISTH T.B., LIBBY P., LIAO J.K. Nitric oxide inhibits macrophage-colony stimulating factor gene transcription in vascular endothelial cells. J. Biol. Chem. 1995;270:17050–17055. doi: 10.1074/jbc.270.28.17050. [DOI] [PubMed] [Google Scholar]
- PLEVIS J.N., SCHINA M., HAYES P.C. The management of acute liver failure. Aliment. Pharmacol. Ther. 1998;12:405–418. doi: 10.1046/j.1365-2036.1998.00320.x. [DOI] [PubMed] [Google Scholar]
- PRICE A.H., CLISSOLD S.P. Salbutamol in the 1980s. A reappraisal of its clinical efficacy. Drugs. 1989;38:77–122. doi: 10.2165/00003495-198938010-00004. [DOI] [PubMed] [Google Scholar]
- RENDELL M.S., RAJFER J., WICKER P.A., SMITH M.D. Sildenafil for treatment of erectile dysfunction in men with diabetes: a randomized controlled trial. JAMA. 1999;281:421–426. doi: 10.1001/jama.281.5.421. [DOI] [PubMed] [Google Scholar]
- REUTER B.K., CIRINO G., WALLACE J.L. Markedly reduced intestinal toxicity of a diclofenac derivative. Life Sci. 1994;55:PL1–PL8. doi: 10.1016/0024-3205(94)90083-3. [DOI] [PubMed] [Google Scholar]
- RIFFAUD J.P., BERNABE J., GIULIANO F., JONES R., JEREMY J.Pharmacological profile of sildenafil nitrate (NCX-911) in various models of penile erection 2001. Satellite Symposium, Nitric Oxide and COX: New Therapeutic Approaches to Inflammatory Disease
- ROEBUCK K.A., FINNEGAN A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J. Leukoc. Biol. 1999;66:876–888. doi: 10.1002/jlb.66.6.876. [DOI] [PubMed] [Google Scholar]
- ROGERS J. Inflammation as a pathogenic mechanism in Alzheimer's disease. Drug Res. 1995;45:439–442. [PubMed] [Google Scholar]
- ROMERO-SANDOVAL E.A., MAZARIO J., HOWAT D., HERRERO J.F. NCX-701 (nitroparacetamol) is an effective antinociceptive agent in rat withdrawal reflexes and wind up. Br. J. Pharmacol. 2002;135:1556–1562. doi: 10.1038/sj.bjp.0704589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSSONI G., BERTI F., COLONNA V.D., BERNAREGGI M., DEL SOLDATO P., BERTI F. Myocardial protection by the nitroderivative aspirin, NCX-4016 in vitro and in vivo experiments in the rabbit. Ital. Heart J. 2000;1:146–155. [PubMed] [Google Scholar]
- ROSSONI G., MANFREDI B., COLONNA V.D., BERNAREGGI M., BERTI F. The nitroderivative of aspirin, NCX-4016, reduces infarct size caused by myocardial ischaemia-reperfusion in the anaesthetised rat. J. Pharmacol. Exp. Ther. 2001;297:380–387. [PubMed] [Google Scholar]
- ROSSONI G., MUSCARA M.N., CIRINO G., WALLACE J.L. Inhibition of cyclooxygenase-2 exacerbates ischaemia-induced myocardial dysfunction in the rabbit. Br. J. Pharmacol. 2002;135:1540–1546. doi: 10.1038/sj.bjp.0704585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANTINI G., SCIULLI M.G., PANARA M.R., PADOVANO R., GIAMBERARDINO M., ROTONDO M.T., DEL SOLDATO P., PATRIGNANI P. Effects of flurbiprofen and flurbinitroxybutylester on prostaglandin endoperoxidases. Eur. J. Pharmacol. 1996;316:65–72. doi: 10.1016/s0014-2999(96)00640-1. [DOI] [PubMed] [Google Scholar]
- SCHROR K. Aspirin and platelets: the antiplatelet action of aspirin and its role in thrombosis treatment and prophylaxis. Semin. Thromb. Hemost. 1997;23:349–356. doi: 10.1055/s-2007-996108. [DOI] [PubMed] [Google Scholar]
- SHIMOHAMA S., TANINO H., FUJIMOTO S. Changes in caspase expression in Alzheimer's disease: comparison with development and aging. Biochem. Biophys. Res. Commun. 1999;256:381–384. doi: 10.1006/bbrc.1999.0344. [DOI] [PubMed] [Google Scholar]
- SMALL R.E., SCHRAA C.C. Chemistry, pharmacology, pharmacokinetics, and clinical applications of mesalamine for the treatment of inflammatory bowel disease. Pharmacotherapy. 1994;14:385–398. [PubMed] [Google Scholar]
- SMYTHIES J.R. Prostaglandins and cancer. Psychoneuroendocrinology. 1979;4:177–189. doi: 10.1016/0306-4530(79)90001-5. [DOI] [PubMed] [Google Scholar]
- SOMASUNDARAM S., RAFI S., JACOB M., SIGTHORSSON G., MAHMUD T., SHERWOOD R., PRICE A.B., MACPHERSON A., SCOTT D., WRIGGLESWORTH J.M., BJARNASON I. Intestinal tolerability of nitroxybutyl-flurbiprofen in rats. Gut. 1997;40:608–613. doi: 10.1136/gut.40.5.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUISSA S., ERNST P. Inhaled corticosteroids: impact on asthma morbidity and mortality. J. Allergy Clin. Immunol. 2001;107:937–944. doi: 10.1067/mai.2001.115653. [DOI] [PubMed] [Google Scholar]
- SULLIVAN M., THOMPSON C.S., MIKHAILIDIS D.P., MORGAN R.J., ANGELINI G.D., JEREMY J.Y. Differential alterations of prostacyclin, cyclic AMP and cyclic GMP formation in the corpus cavernosum of the diabetic rabbit. Br. J. Urol. 1998;82:578–584. [PubMed] [Google Scholar]
- TAKEUCHI K., MIZOGUCHI H., ARAKI H., KOMOIKE Y., SUZUKI K. Lack of gastric toxicity of nitric oxide releasing indomethacin, NCX-530, in experimental animals. Dig. Dis. Sci. 2001;46:1805–1818. doi: 10.1023/a:1010638528675. [DOI] [PubMed] [Google Scholar]
- TAKEUCHI K., SUZUKI K., YAMAMOTO H., ARAKI H., MIZOGUCHI H., UKAWA H. Cyclooxygenase-2 selective and nitric oxide-releasing nonsteroidal anti-inflammatory drugs and gastric mucosal responses. J. Physiol. Pharmacol. 1998a;49:501–513. [PubMed] [Google Scholar]
- TAKEUCHI K., UKAWA H., KONAKA A., KITAMURA M., SUGAWA Y. Effect of nitric oxide-releasing aspirin on gastric functional and ulcerogenic responses in rats: comparison with plain aspirin. J. Pharmacol. Exp. Ther. 1998b;286:115–121. [PubMed] [Google Scholar]
- TALLET D., DEL SOLDATO P., OUDART N., BURGAUD J.L. NO-steroids: potent anti-inflammatory drugs with bronchodilating activity in vitro. Biochem. Biophys. Res. Commun. 2002;290:125–130. doi: 10.1006/bbrc.2001.6192. [DOI] [PubMed] [Google Scholar]
- TASHIMA K., FUJITA A., UMEDA M., TAKEUCHI K. Lack of gastric toxicity of nitric oxide-releasing aspirin, NCX 4016, in the stomach of diabetic rats. Life Sci. 2000;18:1639–1652. doi: 10.1016/s0024-3205(00)00746-3. [DOI] [PubMed] [Google Scholar]
- TERRES W., HAMM C.W., RUCHELKA A., WEILEPP A., KUPPER W. Residual platelet function under acetylsalicylic acid and the risk of restenosis after coronary angioplasty. J. Cardiovasc. Pharmacol. 1992;19:190–193. doi: 10.1097/00005344-199202000-00006. [DOI] [PubMed] [Google Scholar]
- THORNBERRY N.A. The caspase family of cysteine proteases. Br. Med. Bull. 1997;53:478–490. doi: 10.1093/oxfordjournals.bmb.a011625. [DOI] [PubMed] [Google Scholar]
- THORNBERRY N.A., BULL H.G., CALACAY J.R., CHAPMAN K.T., HOWARD A.D., KOSTURA M.J., MILLER D.K., MOLINEAUX S.M., WEIDNER J.R., ANUNINS J. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- TOWARD T.J., LEWIS-LAKELIN M.B., BURGAUD J.L., BROADLEY K.J.Comparison of the inhibition of NO-salbutamol and salbutamol of the bronchoconstriction by histamine in conscious guinea-pigs 2001. Satellite Symposium, Nitric Oxide and COX: New Therapeutic Approaches to Inflammatory Disease
- UKAWA H., YAMAKUNI H., SHINICHI K., TAKEUCHI K. Effects of cyclooxygenase-2 selective and nitric oxide-releasing nonsteroidal anti-inflammatory drugs on mucosal ulcerogenic and healing responses of the stomach. Dig. Dis. Sci. 1998;43:2003–2011. doi: 10.1023/a:1018846912032. [DOI] [PubMed] [Google Scholar]
- VAN'T HOF R.J., RALSTON S.H. Nitric oxide and bone. Immunology. 2001;103:255–261. doi: 10.1046/j.1365-2567.2001.01261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILLA P., KAUFMANN S.H., EARNSHAW W.C. Caspases and caspase inhibitors. Trends Biochem. Sci. 1997;22:388–393. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- WAINWRIGHT C.L., MILLER A.M., WORK L.M., DEL SOLDATO P. NCX4016 (NO-aspirin) reduces infarct size and suppresses arrythmias following myocardial ischaemia/reperfusion in pigs. Br. J. Pharmacol. 2002;135:1882–1888. doi: 10.1038/sj.bjp.0704646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLACE J.L., CIRINO G., MCKNIGHT G.W., ELLIOTT S.N. Reduction of gastrointestinal injury in acute endotoxic shock by flurbiprofen nitroxybutylester. Eur. J. Pharmacol. 1995a;280:63–68. doi: 10.1016/0014-2999(95)00184-m. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., KEENAN C.M., GRANGER N. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Am. J. Physiol. 1990;259:G462–G467. doi: 10.1152/ajpgi.1990.259.3.G462. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., MCKNIGHT W., DEL SOLDATO P., BAYDOUN A.R., CIRINO G. Anti-thrombotic effects of a nitric oxide-releasing, gastric-sparing aspirin derivative. J. Clin. Invest. 1995b;96:2711–2718. doi: 10.1172/JCI118338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLACE J.L., MCKNIGHT W., WILSON T.L., DEL SOLDATO, P., CIRINO G. Reduction of shock-induced gastric damage by a nitric oxide-releasing aspirin derivative: role of neutrophils. Am. J. Physiol. 1997;273:G1246–G1252. doi: 10.1152/ajpgi.1997.273.6.G1246. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., MUSCARA M.N., MCKNIGHT W., DICAY M., DEL SOLDATO P., CIRINO G. In vivo antithrombotic effects of a nitric oxide-releasing aspirin derivative, NCX-4016. Thromb. Res. 1999b;93:43–50. doi: 10.1016/s0049-3848(98)00134-0. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., REUTER B., CICALA C., MCKNIGHT W., GRISHAM M.B., CIRINO G. Novel nonsteroidal anti-inflammatory drug derivatives with markedly reduced ulcerogenic properties in the rat. Gastroenterology. 1994a;107:173–179. doi: 10.1016/0016-5085(94)90074-4. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., REUTER B., CICALA C., MCKNIGHT W., GRISHAM M.B., CIRINO G. A diclofenac derivative without ulcerogenic properties. Eur. J. Pharmacol. 1994b;257:249–255. doi: 10.1016/0014-2999(94)90136-8. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., VERGNOLLE N., MUSCARA M.N., ASFAHA S., CHAPMAN K., MCKNIGHT W., DEL SOLDATO P., MORELLI A., FIORUCCI S. Enhanced anti-inflammatory effects of a nitric oxide-releasing derivative of mesalamine in rats. Gastroenterology. 1999a;117:557–566. doi: 10.1016/s0016-5085(99)70448-8. [DOI] [PubMed] [Google Scholar]
- WATANABE K., KAWAMORI T., NAKATSUGI S., WAKABAYASHI K. COX-2 and iNOS, good targets for chemoprotection of colon cancer. Biofactors. 2000;12:129–133. doi: 10.1002/biof.5520120120. [DOI] [PubMed] [Google Scholar]
- WATKINSON G. Sulphasalazine: a review of 40 years' experience. Drugs. 1986;32 suppl 1:1–11. doi: 10.2165/00003495-198600321-00003. [DOI] [PubMed] [Google Scholar]
- WILLIAMS R.C., JEFFCOAT M.K., KAPLAN M.L., GOLDHABER P., JOHNSON H.G., WECHTER W.J. Flurbiprofen: a potent inhibitor of alveolar bone resorption in beagles. Science. 1985;227:640–642. doi: 10.1126/science.3969553. [DOI] [PubMed] [Google Scholar]
- YAKSH T.L., DIRIG D.M., CONWAY C.M., SVENSSON C., LUO Z.D., ISAKSON P.C. The acute antihyperalgesic action of nonsteroidal, anti-inflammatory drugs and release of spinal prostaglandin E2 is mediated by the inhibition of constitutive spinal cyclooxygenase-2 (COX-2) but not COX-1. J. Neurosci. 2001;15:5847–5853. doi: 10.1523/JNEUROSCI.21-16-05847.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO T., NOZAKI-TAGUCHI N. Analysis of the effects of cyclooxygenase (COX)-1 and COX-2 in spinal nociceptive transmission using indomethacin, a non-selective COX inhibition, and NS-398, a COX-2 selective inhibitor. Brain Res. 1996;11:104–110. doi: 10.1016/s0006-8993(96)00817-7. [DOI] [PubMed] [Google Scholar]